

Abstract
The speciation of framework-interacting CuII sites in Cu-chabazite zeolite catalysts active in the selective catalytic reduction of NOx with NH3 is studied, to investigate the influence of the Al content on the copper structure and their reactivity towards a NO/O2 mixture. To this aim, three samples with similar Cu densities and different Si/Al ratios (5, 15 and 29) were studied using in situ X-ray absorption spectroscopy (XAS), FTIR and diffuse reflectance UV-Vis during pretreatment in O2 followed by the reaction. XAS and UV-Vis data clearly show the main presence of Z2CuII sites (with Z representing a framework negative charge) at a low Si/Al ratio, as predicted. EXAFS wavelet transform analysis showed a non-negligible fraction of proximal Z2CuII monomers, possibly stabilized into two 6-membered rings within the same cage. These sites are not able to form Cu-nitrates by interaction with NO/O2. By contrast, framework-anchored Z[CuII(NO3)] complexes with a chelating bidentate structure are formed in samples with a higher Si/Al ratio, by reaction of NO/O2 with Z[CuII(OH)] sites or structurally similar mono- or multi-copper Zx[CuIIxOy] sites. Linear combination fit (LCF) analysis of the XAS data showed good agreement between the fraction of Z[CuII(OH)]/Zx[CuIIxOy] sites formed during activation in O2 and that of Z[CuII(NO3)] complexes formed by reaction with NO/O2, further confirming the chemical inertia of Z2CuII towards these reactants in the absence of solvating NH3 molecules.
The speciation of the CuII sites in Cu-CHA catalysts for NH3-SCR is studied, to investigate the influence of the Al content on the structure and reactivity of these sites in a NO/O2 mixture.
1. Introduction
Selective catalytic reduction mediated by ammonia (NH3-SCR) is one of the leading technologies for the abatement of NOx from diesel vehicles and power plants.1,2 In the NH3-SCR reaction NOx species are converted to N2 and H2O, via reduction with ammonia in the presence of O2, according to the equation 4NO + 4NH3 + O2 → 4N2 + 6H2O. Cu-Exchanged chabazite (Cu-CHA) is the catalyst of choice in the after-treatment of diesel vehicle exhaust gases, able to meet the Euro VI regulation for heavy-duty diesel vehicles thanks to its hydrothermal stability up to 700 °C and to its high NOx conversion and good selectivity from 200 °C.3–5
The catalytic activity of Cu-CHA in NH3-SCR has been related to the redox activity of the CuII counterions, which stabilize the negative charge on the framework induced by the presence of framework Al atoms (hereafter indicated as Z−).6,7 Depending on the chemical composition (Cu/Al and Si/Al ratios)8 and distribution of framework Al heteroatoms,9–11 after thermal treatment in O2 these Cu ions can be stabilized by two or one framework negative charges, resulting in Z2CuII or Z[CuII(OH)] structures, respectively (Scheme 1a and b, respectively).12 The relative amount of the two sites has been computed as a function of the Si/Al and Cu/Al ratios, assuming random Al distribution (subject to Löwenstein's rule) and the occupation of all 2Al sites as Z2CuII, before occupation of 1Al sites as Z[CuII(OH)].8
Scheme 1. Pictorial representation of some of the various coordination motifs of monomeric and dimeric CuII ions observed or proposed in the literature: (a) Z2CuII; (b) Z[CuII(OH)]; (c) mono(μ-oxo)dicopper(ii) Z2[CuII2O]; (d) superoxo end-on monocopper(ii) Z[CuII(O2)·]; (e) trans-μ-1,2-peroxo dicopper(ii) (end-on) Z2[CuII2O2]. Structures (c)–(e) are examples of the possible mono- and multi-meric Cu(ii)-oxo species characterized by a tridentate coordination motif of the CuII centers, collectively referred to as Zx[CuIIxOy] in this work. Atom colour code: Cu, green; H, white; O, red; Si, grey; Al, yellow.

Recently, it has been reported that CuII sites stabilized at 1Al sites can exist in a variety of mono- and multi-meric Cu(ii)-oxo moieties, such as Z[CuII(O2)] or other structures collectively referred to as Zx[CuIIxOy] in the following (Scheme 1c–e).13–16 Available characterization results suggest a common tridentate coordination motif of the CuII centers at the Z[CuII(OH)] and Zx[CuIIxOy] sites, making them hardly distinguishable with common short-range structural techniques such as X-ray absorption spectroscopy (XAS).17
The catalytic cycle, where NO, NH3 and O2 are converted into N2 and H2O, can be ideally divided into two half cycles.18 In the reduction half cycle CuII is reduced to CuI by NO/NH3 with the release of N2 and H2O;19–21 the catalytic cycle is closed by re-oxidation of CuI to CuII by NO and O2. The reoxidation step, requiring activation of O2 by CuI ions, is crucial in the NH3-SCR reaction cycle, and is vigorously debated in the literature.21–29 The current understanding is that Cu-pair formation, facilitated by mobile CuI(NH3)2 complexes, makes the O2 activation possible,25,26,30 with the formation of side-on μ-η2,η2-peroxo diamino dicopper(ii) complexes.16,31 This mechanism is proposed for the reaction at low temperatures (≤200 °C), where the NH3 molecule is able to solvate and mobilize CuII and CuI ions. At higher temperatures, NH3 ligands are lost and Cu ions bind back to the zeolite framework with the subsequent loss of their mobility.8,25,32–36 NH3-Solvated Cu pairs have also been proposed to be catalytically relevant in the reduction half-cycle.19,20
Some aspects are still under debate in the oxidation half cycle regarding the role of NO in mediating the activation of O2,22–24,29 and which is the structure of the formed intermediates relevant for the catalytic cycle.18,19,27,28,37–41 Cu–(N O)–NH2 (copper nitrosamine) species, postulated for both the reduction and oxidation half cycles,18,39 have been recently detected using transient modulation excitation spectroscopic experiments.40 NO oxidation on CuII ions has been proposed to form unstable gas-phase HONO in equilibrium with surface nitrites, and where HONO is supposed to react with NH3 to release N2.38,42 Due to their instability, these key intermediates in the so-called ‘nitrite route’43 have been mainly detected using chemical trapping techniques.37,42 The role of HONO in the NH3-SCR cycle has also been invoked in recent DFT calculations, though with a different hypothesis about the role of Brønsted acid sites.19,39
The role of Cu-nitrates, formed by reaction with NO/O2 or NO2 in the oxidation half cycle,18,44,45 as key intermediates in the NH3-SCR reaction is still debated.44 Unlike HONO and nitrites, nitrates are stable and can be spectroscopically detected using both infrared and XAS.40,46–48 Cu-nitrates have been proposed to act as off-cycle resting states on the basis of time-resolved operando XAS measurements and DFT calculations.22,41 On the other hand, a nitrate/nitrite route has recently been proposed to be relevant for NH3-SCR on Cu-CHA below 350 °C, on the basis of a combined EPR/FTIR spectroscopic and DFT study.27 Whereas Zhang et al. used modulated FTIR measurements to conclude that no significant amount of nitrate is formed during the NH3-SCR cycle,49 Greenaway et al. showed some nitrate formation using a similar approach.40
In a previous study, we followed, using infrared spectroscopy, the formation of Cu-nitrates by reaction of NO/O2 at low temperatures (50 and 200 °C) on a set of Cu-CHA samples with different composition.47 The results indicated that the formation of Z[CuIINO3] involves only the fraction of the copper present as Z[CuII(OH)] (or Zx[CuIIxOy]), while Z2CuII sites are not affected, probably in relation to their higher stability.47 Differences in the vibrational fingerprints of the Z[CuIINO3] complexes were also observed in samples with different Si/Al and Cu/Al ratios.
Based on these premises, we have used XAS spectroscopy to determine the amount and structure of Cu-nitrates formed by reaction with NO/O2 in the Cu-CHA sample with a different distribution of tetra- and tri-coordinated framework CuII ions (Scheme 1). The changes in the local environment of the Cu ions were also followed via diffuse reflectance (DR) UV-Vis spectroscopy, and were monitored by combining qualitative XAS analysis with extended X-ray absorption fine structure (EXAFS) fitting and advanced wavelet transform (WT)-based signal visualization techniques. Quantitative considerations based on LCF analysis of XAS spectra corroborate the FTIR evidence that Z2CuII sites are not involved in the formation of Cu-nitrates, and that only Z[CuII(OH)] and Zx[CuIIxOy] stabilized at 1Al sites are involved in the formation of framework-interacting Z[CuIINO3] complexes, at variance with recent reports.27 Moreover, EXAFS WT analysis allowed us to obtain unprecedented information on the formation of multimeric Zx[CuIIxOy] species in the O2 pretreated samples, as a function of the chemical composition.
2. Experimental section
2.1. Materials
The three Cu-CHA catalysts studied in this work were prepared as discussed elsewhere.47Table 1 summarizes the chemical composition of the samples, including the Cu volumetric density and the fraction of Z[CuII(OH)] and Z2CuII sites predicted on the basis of the compositional phase diagram proposed by Paolucci et al.8
Chemical composition of the studied samples and predicted fraction of Z[CuII(OH)] and Z2CuII sites.
| Sample | Si/Al | Cu/Al | Cu wt% | Cu/1000 Å3 | Z-[Cu(OH)]I | Z2-CuII |
|---|---|---|---|---|---|---|
| CHA 0.2_15 | 15.1 | 0.2 | 1.3 | 0.20 | ∼0.6 | ∼0.4 |
| CHA 0.1_5 | 5.0 | 0.1 | 1.5 | 0.28 | 0 | 1 |
| CHA 0.6_29 | 29.2 | 0.6 | 1.7 | 0.28 | 0.9–1 | 0–0.1 |
2.2. Data collection
The in situ FTIR spectra were recorded in transmission mode using a PerkinElmer System 2000 infrared spectrophotometer, equipped with an MCT detector averaging 128 interferograms (recorded at 2 cm−1 resolution) for each spectrum. The zeolite powders were pressed in the form of self-supporting pellets of ca. 15 mg and placed inside a commercial FTIR reactor cell (AABSPEC, no. 2000-A multimode) with a controlled gas atmosphere and temperature. The FTIR reactor cell inlet line was heated to 130 °C. The reported spectra are background subtracted, using as a reference the spectrum of the zeolite measured at 200 °C before NO/O2 dosage.
The in situ diffuse reflectance (DR) UV-Vis-NIR spectra were recorded in the 2500–200 nm range (50 000–4000 cm−1) at 1 nm resolution using a Varian Cary 5000 spectrophotometer, equipped with an R928 PMT UV-Vis detector and a cooled PbS photocell NIR detector. Spectra were collected using a Praying Mantis® element, coupled with a low temperature (LT) reaction chamber. The sample was pelletized using a hydraulic press, successively chopped and sieved, selecting for the measurement the fraction between 150 and 300 μm. The reference spectrum was measured at room temperature (RT) using Teflon powder inserted in the same LT cell. Spectra are reported as the relative reflectance (R%) defined as:R% = Rsample/RreferenceX-ray absorption spectroscopy (XAS) data were collected at the BM23 beamline50 of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) using the Microtomo reactor cell.51 The Cu-CHA catalysts were prepared in the form of self-supporting pellets with masses optimized for XAS data collection in transmission mode. Cu K-edge XAS measurements were performed in transmission mode, employing a double-crystal Si(111) monochromator for the incident energy scan, a pair of flat Si mirrors at a 2.5 mrad angle for harmonics rejection, and ionization chambers to detect the incident (I0) and transmitted (I1,2) photons. A Cu metal foil was measured simultaneously using a third ionization chamber I2, for energy calibration purposes.52 XAS spectra of ∼20 min each (energy range 8800–9965 eV; pre-edge region energy step = 5 eV, edge region energy step = 0.3 eV, and constant k-space sampling Δk = 0.035 Å−1 in the EXAFS region; acquisition time of 1 s per point over the whole energy range) were measured under stationary conditions. The three corresponding μ(E) curves were averaged after checking the reproducibility among the consecutive acquisitions. The gas composition of the outlet gas from the reactor was continuously monitored by means of a mass spectrometer (data not reported).
The gas flow rate was 100 ml min−1 for XAS and 50 ml min−1 for FTIR and DR UV-Vis. In all the experiments, the measured catalyst was first pretreated in O2 at 400 °C for 60 min (heating rate 5 °C min−1), then cooled to 200 °C (cooling rate 3 °C min−1) in O2 and subsequently exposed to 1000 ppm NO and 10% O2 in a He diluent. The reaction was monitored over time, until steady-state conditions were reached.
Athena software (Demeter package)53 was used to align the XAS data using the corresponding Cu metal foil spectra and for normalization to unity of the edge jump. The same program was used for extraction of the χ(k) function. R-Space FT-EXAFS spectra were obtained by calculating the Fourier transform of the k2χ(k) functions in the (2.4–12.0) Å−1k-range.
2.3. EXAFS fitting
The EXAFS fits were carried out in R-space, in the 1.0–4.0 Å range, employing Artemis software from the Demeter package53 on the k2-weighted EXAFS spectra, Fourier-transformed in the 2.4–12.0 Å−1 range. For each scattering path, amplitudes and phases were calculated based on DFT-optimized models of the relevant Cu sites, depending on the experimental conditions and catalyst composition. Scattering paths of each model were parametrized using the same amplitude reduction factor (S02) fixed to 0.95, and the same energy shift (ΔE). The latter was refined during the fitting.
The EXAFS fitting procedure of the catalyst spectra after pretreatment in O2 was performed starting from the structural models of Z[CuIIOH] in 8r and Z2CuII in 6r, for Cu-CHA 0.6_29 and 0.1_5, respectively, based on the expected Cu speciation summarized in Table 1.8,54 The single-scattering (SS) paths involving first-shell O atoms from the zeolite framework (Ofw) and, when present, extra-framework (Oef) species, as well as second-shell framework Si/Al atoms (Tfw) were included in the fit. These SS paths have been parametrized with independent interatomic distances and Debye–Waller (DW) factors for each coordination shell. SS paths stemming from more distant Si and O atoms of the framework (fw) of up to 4 Å are included and parametrized as the second shell, by means of a global contraction/expansion factor αfw and a DW factor σfw2 increasing as the square root of the distance Reff,i of the ith scattering atom from the absorber (ΔRfw,i = αfwReff,i, σfw,i2 = ssfw(Reff,i/R0)1/2, where R0 indicates the shortest Reff for the group of paths).12 Finally, the possible contribution from multimeric copper species is accounted for in the fitting model by including a Cu–Cuef SS path with an initial Reff = 3.4 Å.13,16 For this SS contribution the coordination number NCu and interatomic distances RCu are refined, while the corresponding Debye–Waller factor σ2Cu is fixed to avoid excessively high correlations with the NCu parameter.
The EXAFS fit of the Cu-CHA 0.6_29 spectrum after exposure to the NO/O2 mixture is carried out starting from a structural model of Z[CuIINO3] in 8r, with the nitrate in a chelating bidentate configuration.18,46 The SS paths involving the atoms of the nitrate group were included in three subsequent coordination shells, namely O1NO3, NNO3, and O2NO3, with coordination numbers fixed based on the DFT model.46 A common DW factor was employed for both NNO3 and O2NO3 shells whereas the radial distances for NNO3 and O2NO3 were parametrized as a function of the first-shell O1NO3 distance based on the hypothesis of a fixed geometry for the [NO3]− unit. Importantly, intense collinear multiple-scattering paths involving the NNO3 and O2NO3 atoms of the [NO3]− unit and which are known to contribute in the 2.8–3.5 Å R-space range18,46 are included in the fitting model, and are parametrized with a dedicated DW factor σ2MS and radial shift parameters expressed as a function of those used for the relevant SS paths.
2.4. XANES linear combination fit (LCF)
Linear combination fit (LCF) analysis was applied to both normalized XANES spectra and k2χ(k) EXAFS functions to estimate the Cu-speciation in the Cu-CHA 0.2_15 catalyst (see Table 1). XANES and k-space EXAFS LCF analysis was performed in the 8970–9020 eV energy interval, and in the 2.4–12.0 Å−1k-range, respectively, using the Athena software.53 Each experimental spectrum μEXP(E) or k2χEXP(k) was fitted as a linear combination of two reference spectra, i.e., μLCF(E) = w1μREF1(E) + w2μREF2(E) and k2χLCF(k) = w1k2χREF1(k) + w2k2χREF2(k), optimizing the weights wi for each reference spectrum. The spectra of Cu-CHA 0.6_29 and Cu-CHA 0.1_5 measured under the same conditions (O2 pretreatment and NO/O2 interaction) were used as references. LCF analysis was performed by imposing the mass balance condition: 0 ≤ wi ≤ 1 and ∑iwi = 1. For each analyzed scan and working space, the corresponding LCF R-factor was computed to evaluate the LCF quality: an R-factor equal to 0 corresponds to the ideal reproduction of the experimental curve, i.e., μEXP(E) ≡ μLCF(E) or k2χEXP(k) ≡ k2χLCF(k).
EXAFS wavelet transform analysis
Wavelet transform (WT) is a signal processing methodology employed in the field of EXAFS analysis for the resolution of multiple overlapped scattering paths.55 When two or more atoms are localized at similar distances from the absorber, their signals in the direct space R overlap, hindering their discrimination through the classic Fourier analysis. However, their EXAFS contributions in k-space are a function of their backscattering amplitude factors F(k). This depends on the atomic number Z, so that heavy atoms give signals at higher wavenumbers than lighter ones. This can be exploited in the analysis of an experimental EXAFS spectrum through its WT representation, a two-dimensional correlation map of the signal, enabling the simultaneous localization of the EXAFS features in both k and R spaces.55
The continuous WT representation of a general kn-weighted EXAFS spectrum is defined as:
 |
1 |
This equation can be seen as the inner product between the kn-weighted EXAFS spectrum χ(k) and a mother wavelet ψ function (where the apex ψ* denotes the complex conjugate of ψ), decaying at zero for higher values of |k′|. The a variable is a scaling factor connected to the R-space through the relation: a = η/2R, while b involves translation of the wavelet function along the k axis: b = k.
In this work, due to the high similarity with an arbitrary EXAFS signal, the Morlet mother wavelet, expressed in the following form, was used:56
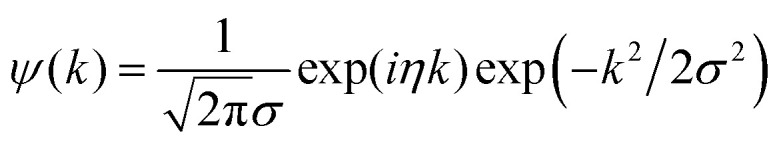 |
2 |
Here, i denotes the complex unit, while η and σ are the two parameters regulating the wavelet resolution in R and k spaces. Because the WT distributes the EXAFS information over k–R cells, usually named Heisenberg boxes, the selection of an appropriate set of these two parameters is critical and must be optimized in order to have the best visualization of the desired spectral features in both of the spaces. In particular, as described by Funke et al.,57 the condition for the optimum resolution at a given distance of interest Ri can be achieved requiring the conditions σ = 1 and η = 2Ri.
3. Results and discussion
3.1. Cu-nitrate formation followed by infrared spectroscopy
Fig. 1(a) reports the FTIR spectra measured on the series of Cu-CHA catalysts summarized in Table 1, after exposure to 1000 ppm of NO and 10% O2 at 200 °C. Based on the different chemical compositions (Cu/Al from 0.1 to 0.6 and Si/Al ratio from 5 to 29), these are expected to present different relative amounts of Z[CuII(OH)] (and/or other structurally similar CuII species at Z sites, such as Z[CuII(O2)]/Zx[CuIIxOy]) and Z2CuII sites, as reported in Table 1.58
Fig. 1. (a) In situ FTIR spectra of Cu-CHA 0.1_5 (yellow), Cu-CHA 0.2_15 (red) and Cu-CHA 0.6_29 (blue) exposed to 1000 ppm NO/10% O2 at 200 °C, after pretreatment in O2. Illustration of (a) chelating bidentate Z[CuII(NO3)], atom colour code: Cu, green; H, white; O, red; N, blue; Si, grey. (b) Schematic representation of a zeolite Brønsted site involved in a strong hydrogen bond with a H2O molecule. Spectra were subtracted from the zeolite spectrum before NO/O2 admission and normalized to the sample “optical thickness” and Cu content.

The spectra observed on samples Cu-CHA 0.6_29 and 0.2_15 show three main components at 1625, 1610 and 1575 cm−1, which have been assigned to the N O stretching vibration (νN O) of chelating bidentate Cu-nitrates Z[CuII(NO3)].47 The intensity ratio of the bands at 1625 and 1610 cm−1 is not constant in the two samples, indicating that they originate from different species. Indeed, a band around 1610 cm−1 in Cu-CHA catalysts was recently interpreted in terms of adsorbed NO2.27 In our previous work (focusing on a larger set of samples) we proposed that both bands are due to a similar chelating bidentate Z[CuII(NO3)] structure, and that the position of νN O is affected by the local environment, i.e. the presence of residual Brønsted sites or the electrostatic potential of the cage.47 The concomitant presence of other CuII-(N,O) species (e.g. bridging bidentate nitrates formed on Cu–Cu pairs and/or Z[CuII-(NO2)]), or physisorbed NOx (x = 1, 2) was excluded on the basis of the applied experimental conditions (i.e., a large excess of O2, reported to favour Z[CuII(NO3)] with respect to Z[CuII(NO2)]), on the study of the νNi O overtone mode47 and on the reactivity of the formed species towards other reagents (e.g., exposure to NO).46
Noticeably, almost no bands related to CuII–(N,O) moieties (very weak bands at 1575 and 1590 cm−1, shoulder) could be observed for the Cu-CHA 0.1_5 sample. Conversely, this is characterized by a broad feature centered at 1650 cm−1, which is due to highly perturbed OH groups involved in strong hydrogen bonds between Si(OH)Al Brønsted sites and water molecules (Fig. 1(b)), formed in the reaction 2NO + ½O2 + 2H+ → 2NO+ + H2O.59–62
The results summarized in this section point to the fact that Z[CuII(NO3)] moieties are only formed in samples that are expected to be characterized, after pretreatment in O2, by a significant fraction (∼60% in Cu-CHA 0.2_15, and almost 100% in Cu-CHA 0.6_29) of Z[CuII(OH)] sites (and/or other structurally similar CuII species at Z sites, generally referred to as Zx[CuIIxOy]). By contrast, a negligible amount of Z[CuII(NO3)] is observed in sample Cu-CHA 0.1_5, which is expected to have only Z2CuII sites (Table 1). This was also confirmed by experiments at a lower temperature (50 °C; see Fig. S1, ESI†). To check this hypothesis, which could be undermined by the uncertainty of the vibrational mode assignment,27 the experiments were repeated using XAS and UV-Vis techniques, which are more sensitive to the local coordination of the Cu ions.
3.2. Qualitative insight from XAS
3.2.1. Catalysts pretreated in O2
Fig. 2 shows the XAS spectra collected during the thermal treatment in 100% O2 for Cu-CHA 0.1_5 (left-hand panel), Cu-CHA 0.2_15 (middle panel) and Cu-CHA 0.6_29 (right-hand panel). The hydrated spectra are very similar, and are compatible with CuII ions in a pseudo-octahedron with five/six coordinated H2O molecules.36 During the thermal treatment in O2 (from light blue to grey/black curves) the water ligands are progressively removed, promoting the coordination of CuII ions to the CHA framework. The loss in coordination is clearly visible in the gradual decrease of the white-line intensity in the XANES spectra (Fig. 2 main panels). In parallel, the pre-edge 1s → 3d CuII transition at 8977 eV, and the CuII 1s → 4p rising-edge feature in the 8985–8990 eV range increase.
Fig. 2. Evolution of the in situ XAS spectra of Cu-CHA during pretreatment in 100% O2 from RT to 400 °C (from light blue to black curves, grey: intermediates). Main panels: Cu K-edge XANES spectra of Cu-CHA 0.1_5, Cu-CHA 0.2_15 and Cu-CHA 0.6_29; insets: moduli of the Fourier transform (FT) for the corresponding k2-weighted k2χ(k) EXAFS spectra.

In all the samples a very small fraction of CuI ions is detected by the corresponding 1s → 4p rising-edge feature at 8982 eV, at the end of the pretreatment in O2 (black curve, measured at 400 °C). In sample Cu-CHA 0.2_15, this feature first grows in parallel with a decrease in the intensity of the white line during heating, and then it decreases (grey to black lines). This phenomenon is less pronounced in the Cu-CHA 0.6_29 sample and it is not present in Cu-CHA 0.1_5. A similar transient formation of CuI has been observed in the Cu-CHA and Cu-mordenite samples during dehydration under oxidizing conditions.63 The higher tendency to reduction of the sample with an intermediate Si/Al ratio is coherent with the trend observed during self-reduction on a larger set of the Cu-CHA samples, which would imply a role of residual Brønsted sites in the CuII/CuI redox process.54
The final dehydrated CuII spectrum differs significantly among the studied catalysts. Cu-CHA 0.1_5 presents a sharper CuII 1s → 4p rising-edge peak, a narrower and more intense white-line region, with a well-defined peak at ∼8995 eV and peculiar broad post-edge features in the 9005–9015 eV range. These features can be assigned to Z2CuII moieties, as discussed elsewhere.8,54 Conversely, the spectrum of Cu-CHA 0.6_29 is characterized by broader and less intense white-line peaks, with the absence of defined features in the post-edge region. This agrees with the presence, in this catalyst, of the majority of CuII ions as Z[CuII(OH)] and/or Zx[CuIIxOy] moieties.63,64 The spectrum obtained upon dehydration of Cu-CHA 0.2_15 shows intermediate features between the other two: a lower white-line intensity with respect to Cu-CHA 0.1_5, but sharper and better-defined rising-edge peaks at ∼8985 and ∼8995 eV when compared with Cu-CHA 0.6_29, together with a broad feature in the 9000–9015 eV post-edge region, similar to Cu-CHA 0.1_5. This points to the presence of a mixture of sites, as expected on the basis of the theoretically predicted fraction of Z[CuII(OH)] and Z2CuII (∼0.6 and 0.4, respectively, see Table 1).
This different CuII speciation is further confirmed by the evolution of the EXAFS spectra (phase-uncorrected FT k2-weighted χ(k) spectra, insets in Fig. 2). In all Cu-CHA samples we observe a progressive decrease of the first-shell peak in the 1.0–1.8 Å range of the FT-EXAFS spectra, with the simultaneous growth of a second-shell peak in the 2.2–2.5 Å range. This agrees with the progressive loss in coordination when removing H2O ligands, and accounts for the coordination of CuII ions to the zeolite framework. In particular, the first-shell peak can be assigned to single scattering (SS) paths involving extra-framework (Oef) and framework (Ofw) O atoms, and the second-shell peak to the SS path of Al and Si atoms of the zeolite framework (Tfw) (Scheme 1). The intensity of both peaks is sensibly higher in Cu-CHA 0.1_5 (left hand inset in Fig. 2), and decreases in the order of 0.1_5 ≫ 0.2_15 > 0.6_29. This can be explained in terms of an ‘almost pure’ population of Z2CuII sites in Cu-CHA 0.1_5, since these ions are surrounded by four Ofw atoms in the first-shell and two Tfw atoms in the second-shell, at variance with Z[CuII(OH)]/Zx[CuIIxOy], which have only two first-shell Ofw and one Tfw (Al) atoms in the second-shell (see Scheme 1). While the population of Cu-CHA 0.6_29 can be described as ‘almost pure’ Z[CuII(OH)] sites (or structurally similar ions, such as Zx[CuIIxOy)]), Cu-CHA 0.2_15 again shows intermediate features in both the intensity and width of the EXAFS peaks, confirming a heterogeneous distribution of Z2CuII and Z[CuII(OH)]/Zx[CuIIxOy] sites.
3.2.2. EXAFS WT analysis for the catalysts pretreated in O2
Fig. 3 reports the moduli of the WTs associated with the EXAFS spectra for the O2 pretreated catalysts. Here the WTs are magnified in the 2–4 Å R-space range, corresponding to second-shell contributions from the framework and extra-framework atoms that cannot be discriminated using the conventional FT-EXAFS approach. The wavelet resolution parameters were fixed to σ = 1 and η = 7 in order to obtain the highest enhancement of the SS contributions involving the Cu–Cuef interaction, around 3.5 Å. For all the samples, the EXAFS WT representations are clearly split into two main lobes along the k-direction. The first one, localized in the k–R cell within 2–4 Å−1 and 2–2.6 Å is generally associated with the zeolite framework oxygens, characterized by a maximum in the F(k) curve at around 3 Å−1.16 The second WT lobe emerges at higher k values within 6–8 Å−1 and it extends in the R space up to 3.8 Å. Its morphology is complex, involving two main contributions. An intense peak located around k = 5.5–6.5 Å-1 and R = 2.5 Å is related to Si and Al framework atoms16 that cannot be discriminated through the WT analysis due to similar atomic numbers.65 At R values higher than 2.5 Å, the second lobe ridge extends within 7 and 8 Å−1, confirming the existence of a Cu–Cuef path as indicated by the FT-EXAFS fits reported in Section 3.2.1.
Fig. 3. WT representation of the EXAFS spectra associated with the Cu-CHA 0.1_5, Cu-CHA 0.2_15 and Cu-CHA 0.6_29 samples pretreated in O2. The WT resolution parameters σ and η are set to 1 and 7, respectively.

The intensity of each feature in the WTs follows the same trend of the second shell EXAFS FT magnitudes. The most intense WT features are observed for the Cu-CHA 0.1_5 sample dominated by Z2CuII sites with two Tfw atoms in the second shell. The sample Cu-CHA 0.6_29 shows less intense features, in accordance with the coordination motifs of both Z[CuII(OH)] and Zx[CuIIxOy] species (Scheme 1), with one Tfw (Al) in the second shell and a lower number of more distant O/Si framework atoms in the CHA 8r. Finally, the map of Cu-CHA 0.2_15 shows a morphology similar to that of Cu-CHA 0.6_29 with a slightly higher intensity of the WT second lobe, in agreement with the presence of a small fraction of Z2CuII sites, responsible for the enhancement of this WT feature.
3.2.3. Catalysts exposed to the NO/O2 mixture
Fig. 4 reports the spectra collected during exposure to 1000 ppm NO/10% O2 at 200 °C of Cu-CHA 0.1_5 (left-hand panel), Cu-CHA 0.2_15 (middle panel) and Cu-CHA 0.6_29 (right-hand panel) after the pretreatment in O2 described above. In agreement with the FTIR results described in Section 3.1, no changes in the XAS spectra of Cu-CHA 0.1_5 are observed while dosing NO/O2, except for a subtle intensity decrease in the weak CuI 1s → 4p rising-edge peak at 8982 eV. This confirms the oxidative ability of NO/O2 towards CuI ions,18,66 and the inability of the same mixture in forming nitrates with highly stable Z2CuII sites.47
Fig. 4. Evolution of the in situ XAS spectra of Cu-CHA during exposure to 1000 ppm NO/10% O2 at 200 °C (from grey to coloured curves). Main panels: Cu K-edge XANES spectra of Cu-CHA 0.1_5 (yellow), Cu-CHA 0.2_15 (red) and Cu-CHA 0.6_29 (blue); insets: k2-weighted k2χ(k) EXAFS curves.

Conversely, for both Cu-CHA 0.2_15 and Cu-CHA 0.6_29 catalysts (from grey to blue coloured, middle and right-hand panels of Fig. 4, respectively), the white-line intensity (XANES main panels) grows, pointing to an increase in the coordination of the Cu ions, which is confirmed by the increase of the first-shell peak in the EXAFS region (insets). The second-shell peak in the EXAFS region is largely unaffected for Cu-CHA 0.2_15 and it only slightly increases for Cu-CHA 0.6_29. This confirms that the formed species are still framework-anchored CuII moieties. As observed for Cu-CHA 0.1_5, the small fraction of CuI present at the end of the pretreatment in O2 is readily oxidized by the highly oxidative NO/O2 gas feed in both samples.
The type of nitrate species formed can be inferred from the growth of a third peak in the high-R EXAFS region, ascribable to chelating bidentate nitrate structures (Fig. 1(a)).18,46 This feature can indeed be related to an intense multiple scattering (MS) contribution, given by the peculiar linear arrangement of Cu–N–O atoms. A clear increase of a third-shell contribution during the reaction can be seen in the spectra of both Cu-CHA 0.2_15 and Cu-CHA 0.6_29, in the 2.7–3.5 Å range. These findings confirm our FTIR results, pointing to the formation of a sole type of Cu nitrate species on copper-exchanged chabazite by reaction of NO/O2 with the Z[CuII(OH)] and Zx[CuIIxOy] sites.
3.2.3.1. EXAFS WT analysis of the catalysts exposed to the NO/O2 mixture
The WT representations of the EXAFS spectra of the three samples after NO/O2 contact are reported in Fig. 5. In agreement with evidence from the FTIR (Section 3.1) and EXAFS spectra (Section 3.2.3), the EXAFS WT map of sample Cu-CHA 0.1_5 is very similar to that measured after treatment with O2 (reported in Fig. 3, see also the differential WT map in Fig. S4 (ESI†), showing negligible differences). This further confirms that the Z2CuII sites in this sample are not affected by the reaction with NO/O2.
Fig. 5. WT representation of the EXAFS spectra associated with the Cu-CHA 0.1_5, Cu-CHA 0.2_15 and Cu-CHA 0.6_29 samples exposed to the NO/O2 mixture. The WT resolution parameters σ and η are set to 1 and 7, respectively.

By contrast, the bilobed maps of Cu-CHA 0.6_29 and Cu-CHA 0.2_15 show differences with respect to the O2 pre-activated state reported in Fig. 3. The lobe ridge at low k-values is here mainly related to the contributions from the second shell O and N atoms, while the lobe peaking at k = 6.5 Å−1 and R = 2.5 Å is derived from Al/Si atoms. In Cu-CHA 0.6_29 a more intense ridge within 7 and 8 Å−1 in k and extending from 2.8 to 3.8 Å in R is present, which arises principally from the contributions of the MS paths involving the N and O atoms of the chelated NO3 group. This contribution overpowers the signal derived from a possible Cu–Cuef scattering contribution, which becomes slightly visible after 3 Å with a weak WT ridge at ca. 7.5–8 Å−1. A quantitative demonstration of this evidence is provided in Section S3.3 of the ESI.†
The WT representation for the Cu-CHA 0.2_15 sample maintains an intense second lobe feature although with a minor intensity if compared with the Cu-CHA 0.6_29 WT map, and a more intense first lobe, similar to Cu-CHA 0.1_5. These pieces of evidence suggest qualitatively that the Cu-CHA 0.2_15 sample is characterized by a mixture of two main structures, here represented by Z2CuII and Z[CuIINO3] dominating the 0.1_5 and 0.6_29 sample compositions, respectively. These findings are further corroborated by looking at the differential WT maps reported in Fig. S4 (ESI†). Herein, an intense contribution derived from the MS proper of the NO3 group, localized in the R-space around 3.1 Å (phase uncorrected), reaches its maximum intensity for the Cu-CHA 0.6_29 sample, while it is less pronounced for Cu-CHA 0.2_15. By contrast, the latter is characterized by a relatively more intense lobe at ca. 3.2 Å that is mainly related to the Cu–Al/Si/fw contributions. Further structural insights are obtained through the XAS quantitative analysis discussed in the next section.
3.3. Cu-speciation and structural insights from XAS quantitative analysis
3.3.1. Catalysts pretreated in O2
EXAFS fitting was carried out to corroborate the qualitative insights presented above. As discussed above, Cu-CHA 0.6_29 and Cu-CHA 0.1_5 pretreated in O2 should mainly contain Z[CuIIOH] sites (or similar three-fold coordinated mono- and/or multimeric CuII-oxo moieties that are hardly distinguishable by XAS, Scheme 1)17 and Z2CuII sites in 8r and 6r, respectively.
The EXAFS analysis results, summarized in Fig. 6 and Table 2, support this hypothesis, enabling a very good reproduction of the experimental spectra (R-factor < 1%) with physically reliable values of all the parameters optimized in the fits. In particular, as illustrated in Fig. 6(b and c), the EXAFS spectrum of pretreated Cu-CHA 0.6_29 is successfully reproduced by considering two sub-shells of O neighbors in the first-coordination sphere of the metal center, contributing in partial antiphase at 1.83 ± 0.02 Å and 1.95 ± 0.01 Å, for Oef and Ofw, respectively. The second maximum of the FT-EXAFS is properly modelled by the SS contribution of a single Tfw (Al) atom refined at 2.70 ± 0.02 Å from the Cu absorber. In the high-R range, we identify also a broad contribution from more distant framework O and Si atoms that is consistent with the preferential location of CuII species in the 8r of the CHA zeolite.
Fig. 6. (a) Structural model of Z[CuIIOH] sites in 8r, indicating the different scattering contributions employed in the fit for pretreated Cu-CHA 0.6_29. Atom colour code: red, oxygen; yellow, aluminium; grey, silicon; and green, copper. The atoms included in the fitting model are shown in ball-and-stick mode, while the others are visualized in stick mode. (b and c) Phase uncorrected (b) modulus and (c) imaginary part of the experimental and best fit FT EXAFS spectra for Cu-CHA 0.6_29 pretreated in O2. The experimental data are shown as black open circles and the best fits with red solid lines. The principal SS contributions included in the fitting model are reported as coloured solid lines vertically translated for the sake of clarity. (d) As part (a) but reporting the structural model of Z2CuII sites in 6r, employed in the fit for pretreated Cu-CHA 0.1_5. (e and f) As parts (b and c) but for Cu-CHA 0.1_5 pretreated in O2.

Results of the fits performed on the k2-weighted FT-EXAFS spectrum of Cu-CHA 0.6_29 and Cu-CHA 0.1_5 (transformed in the 2.4–12.0 Å−1k-range) pretreated in O2 at 400 °C and measured after cooling down to 200 °C in O2. Parameter values set in the fit are underlined. Coordination numbers for all coordination shells, except for the Cuef one, were fixed based on the structural models in Fig. 6(a) (Z[CuIIOH] 8r) and Fig. 6(d) (Z2CuII 6r).
| EXAFS parameters | Best-fit values Cu-CHA 0.6_29 Z[CuIIOH] 8r model | Best-fit values Cu-CHA 0.1_5 Z2CuII 6r model |
|---|---|---|
| N°par/N°ind | 10/18 | 9/18 |
| R factor | 0.0117 | 0.0065 |
| S 0 2 | 0.95 | 0.95 |
| ΔE (eV) | −2 ± 1 | −3.8 ± 0.8 |
| RO_ef (Å) | 1.83 ± 0.02 | — |
| 〈RO_fw〉 (Å) | 1.951 ± 0.01 | 1.959 ± 0.006 |
| 〈RT_fw〉 (Å) | 2.70 ± 0.02 | 2.799 ± 0.009 |
| α fw | −0.02 ± 0.01 | −0.003 ± 0.005 |
| RCu_ef (Å) | 3.36 ± 0.05 | 3.41 ± 0.02 |
| σO_fw2 (Å2) | 0.003 ± 0.002 | 0.004 ± 0.001 |
| σT_fw2 (Å2) | 0.009 ± 0.002 | 0.008 ± 0.001 |
| σfw2 (Å2) | 0.021 ± 0.008 | 0.012 ± 0.002 |
| σCu_ef2 (Å2) | 0.01 | 0.01 |
| N Cu_ef | 0.4 ± 0.3 | 1.2 ± 0.4 |
The differences noted in Sections 3.2.1 and 3.2.2 for the EXAFS of pretreated Cu-CHA 0.1_5 are rationalized through EXAFS fitting using the Z2CuII 6r model, proven to represent the dominant structural component stabilized in this catalyst upon pretreatment in O2. As seen in Fig. 6(e and f), the higher intensity of the first EXAFS peak with respect to Cu-CHA 0.6_29 is due to the in-phase SS contributions of four Ofw neighbors refined at an average distance of 1.959 ± 0.006 Å from the Cu center. Similarly, the enhanced intensity of the second-shell peak is linked to two equivalent Tfw atoms (1 Al and 1 Si), refined at a Cu–Tfw distance of 2.799 ± 0.009 Å. The Z2CuII 6r model also accounts for the contribution from more distant framework atoms, resulting in a broad but clearly detectable feature in the 2.5–3.0 Å range.
While minor (typically, <10% total Cu) contributions from different Cu-species cannot be excluded within the sensitivity of the technique, these fitting results clearly show how it is possible to safely connect two out of the three catalysts investigated in this work to the two main types of Cu-species predicted to form in Cu-CHA upon pretreatment under oxidizing conditions, namely Z[CuIIOH] species in 8r (or similarly three coordinated CuII ions) and Z2CuII species in 6r, for Cu-CHA 0.6_29 and 0.1_5, respectively.
Based on previous evidence16,65 and WT analysis results pointing to the presence of the framework coordinated multimeric Zx[CuIIxOy] species, in our EXAFS fitting models we also included a Cuef SS contribution with an estimated coordination number NCu_ef. Albeit with large fitting errors (as is expected for fractional contributions at high R values), the results in Table 2 support the presence of Cu–Cuef scattering for both Cu-CHA 0.6_19 and 0.1_15 after pretreatment in O2. For Cu-CHA 0.6_29, these are refined at an average distance of 3.36 ± 0.05 Å and could plausibly correspond to three-coordinated multicopper-oxo Zx[CuIIxOy] moieties (Scheme 1), involving less than half of the Cu centers in the catalyst. Interestingly, Cu-CHA 0.1_5 shows a higher NCu_ef value with respect to Cu-CHA 0.6_29, compatible with unity within the fitting errors, and a longer average Cu–Cuef distance of 3.41 ± 0.02 Å. In line with the WT analysis in Section 3.2.2 and the other fitting results, we tentatively assign these contributions to proximal Z2CuII ions, e.g., stabilized in two six-membered rings within the same d6r composite building unit of the CHA framework.
In order to reinforce the qualitative description of the experimental WT maps reported in Section 3.2.2, in Section S3.2 of the ESI† we show the WT plots of the single EXAFS contributions employed to fit the samples Cu-CHA 0.6_29 and Cu-CHA 0.1_5 after the pretreatment in O2. For both cases, it is possible to see that the WT first lobe is always reproduced by the contributions stemming from the first and second shells of framework and extra-framework oxygen atoms. By contrast, the second WT lobe is derived from the joined contributions of Al/Si (Tfw sites) and more distant framework atoms (fw), which constitute predominantly the first part of the lobe, while the one at higher k-values appears to be derived essentially from the Cu–Cuef interaction.
We will consider now in more detail the Cu-speciation in Cu-CHA 0.2_15. The qualitative observations presented above, together with the available literature on the topic, consistently point to the presence of a mixture of Z[CuIIOH] or Zx[CuIIxOy] and Z2CuII sites. In particular, the theoretical compositional phase diagram by Paolucci et al.8 predicts for this composition, ca. 60% Z[CuIIOH] and 40% Z2CuII (Table 1). To provide quantitative support for this hypothesis, we performed LCF analysis of the XAS spectra obtained for O2 pretreated Cu-CHA 0.2_15 using as references those of pretreated Cu-CHA 0.6_29 and 0.1_5 catalysts, which are representative of the two structural components. LCF was independently conducted on normalized XANES spectra in the energy space and on the k2χ(k) EXAFS function, as illustrated in parts (a) and (b) of Fig. 7, respectively. In both working spaces, LCF results were satisfactory and consistent within fitting errors. As detailed in Table 3, the analysis revealed the presence of ca. 65% Z[CuIIOH] and 35% Z2CuII.
Fig. 7. Comparison between experimental and best-fit curves for Cu-CHA 0.2_15 after pretreatment in O2, from LCF analysis of (a) the normalized XANES and (b) the k2χ(k) EXAFS spectra. The LCF residuals and the scaled components corresponding to Z[CuIIOH] and Z2CuII species are reported in both working spaces.

Quantitative results from LCF analysis for CHA 0.2_15 after pretreatment in O2 considering both the normalized XANES and the k2χ(k) EXAFS spectra.
| LCF parameters Cu-CHA 0.2_15 | XANES LCF | EXAFS LCF |
|---|---|---|
| Fraction Z[CuIIOH] 8r | 0.65 ± 0.02 | 0.66 ± 0.01 |
| Ref. Cu-CHA 0.6_29 | ||
| Fraction Z2CuII 6r | 0.35 ± 0.02 | 0.34 ± 0.01 |
| Ref. Cu-CHA 0.1_5 | ||
| LCF Rfactor | 0.00013 | 0.014 |
A two-component EXAFS fits for O2 pretreated Cu-CHA 0.2_15 were also attempted according to the methodology detailed in the Section S2.1 (ESI†). We performed an iterative three-step refinement procedure, using the LCF (k-space) results in Table 3 as the initial input and setting the Debye–Waller factors to the best-fit values independently refined for the relevant states in Cu-CHA 0.1_5 and Cu-CHA 0.6_29 (Table 2), while re-optimizing all the structural variables (see Table S1 and Fig. S2 (ESI†) for a detailed report). The analysis confirmed the LCF results, with negligible variations in the geometries of the Z[CuIIOH] and Z2CuII species hosted in 8r and 6r, respectively. In parallel, the optimized values for Cu–Cuef = 3.39 ± 0.02 Å and for NCu_ef = 0.8 ± 0.3 corresponded reasonably with the average of those retrieved from mono-component EXAFS fits for pretreated Cu-CHA 0.6_29 and Cu-CHA 0.1_5, consistent with the intermediate compositional characteristics of Cu-CHA 0.2_15.
3.3.2. Catalysts exposed to the NO/O2 mixture
XAS quantitative analysis was carried out on the spectra obtained upon exposure of pretreated samples to the NO/O2 mixture at 200 °C on samples of Cu-CHA 0.6_29 and Cu-CHA 0.2_15, where a significant formation of Cu-nitrates in the chelating bidentate configuration was observed. The initial input for the fit was built based on the Z[CuIINO3] 8r model illustrated in Fig. 8(a), for chelating bidentate Cu-nitrate species hosted at 1Al sites in the 8r of the CHA lattice (see Section 2.3 for details on the adopted fitting model).18,46
Fig. 8. (a) Structural model of Z[CuIINO3] sites in 8r, indicating the different scattering contributions employed in the fit for Cu-CHA 0.6_29 exposed to the NO/O2 mixture. Atom colour code: O, red; N, blue; Al, yellow; Si, grey; and Cu, green. The atoms included in the fitting model are shown in ball-and-stick mode, while the others are visualized in stick mode. (b, c) Phase uncorrected (b) modulus and (c) imaginary part of the experimental and best fit FT EXAFS spectra for Cu-CHA 0.6_29 exposed to the NO/O2 mixture. The experimental data are shown as black open circles and the best fits with red solid lines. The principal SS and MS contributions included in the fitting model are reported as coloured solid lines, vertically translated for the sake of clarity.

Fig. 8(b and c) compare the experimental and best-fit EXAFS spectra for Cu-CHA 0.6_29, highlighting the quality of the reproduction enabled by the employed structural model. The fit returned physically meaningful values for all the optimized parameters, underpinning how the chelating bidentate Cu-nitrate illustrated in Fig. 8(a) represents the main Cu-species formed in Cu-CHA 0.6_29 under the adopted experimental conditions. The corresponding fit results are detailed in Table 4.
Results of the fits performed on the k2-weighted FT-EXAFS spectrum of Cu-CHA 0.6_29 (transformed in the 2.4–12.0 Å−1k-range) after exposure to the NO/O2 mixture at 200 °C in O2. Parameter values set in the fit are underlined. Coordination numbers for all coordination shells, except for the Cuef one, were fixed based on the structural model in Fig. 6a (Z[CuIINO3] 8r).
| EXAFS parameters | Best-fit values Cu-CHA 0.6_29 Z[CuIINO3] 8r model |
|---|---|
| N°par/N°ind | 13/18 |
| R factor | 0.005 |
| S 0 2 | 0.95 |
| ΔE (eV) | −3 ± 1 |
| 〈RO_fw〉 (Å) | 1.87 ± 0.03 |
| 〈RO1_NO3〉 (Å) | 1.97 ± 0.02 |
| RT_fw (Å) | 2.72 ± 0.01 |
| α fw | −0.04 ± 0.01 |
| RCu_ef (Å) | 3.30 ± 0.08 |
| σO_fw2 (Å2) | 0.005 ± 0.003 |
| σO1_NO32 (Å2) | 0.004 ± 0.002 |
| σT_fw2 (Å2) | 0.006 ± 0.002 |
| σN/O2_NO32 (Å2) | 0.015 ± 0.002 |
| σfw2 (Å2) | 0.008 ± 0.002 |
| σCu_ef2 (Å2) | 0.01 |
| σ MS 2 | 0.009 ± 0.003 |
| N Cu_ef | 0.3 ± 0.3 |
The fit confirms the assignment of the third maximum visible in the 2.8–3.5 Å range of the (phase-uncorrected) FT-EXAFS spectra to a set of very intense (quasi-) collinear MS paths, labelled as MSNO3 in Fig. 8(a). These primarily involve the N and more distant O atoms of the nitrate unit, representing a diagnostic fingerprint for Cu-nitrates in the chelating bidentate binding mode. In addition, in the same R-space range, other very intense quasi-collinear MS contributions involve the first-shell O atoms belonging to the NO3− group and to the zeolite framework, in agreement with previous results on a Cu-CHA sample with a higher Al content (Si/Al = 15).46
However, differences slightly above the fitting errors are observed, especially in the refined interatomic distances between Cu and the framework atoms. EXAFS indicates small contractions of the Cu–Ofw and Cu–Tfw distances (of ca. 0.05 Å and 0.04 Å, respectively), for Cu-CHA 0.6_29 with respect to previously reported EXAFS values for Cu-CHA 0.5_15.46 Such subtle structural modifications, undoubtedly occurring within the same type of CuII-(N,O) species, could be connected to the different electrostatic environment and acidic Brønsted properties in the zeolite framework with different Si/Al ratios, which have a clear effect on the vibrational properties of the nitrate groups, as observed by FTIR.47
For consistency and based on the WT results in Fig. 5, also in this fit we included a Cu–Cuef contribution, using the same approaches discussed in Section 3.3.1. The fit returned an average Cu–Cuef interatomic distance of 3.30 ± 0.08 Å, with a NCu_ef value of 0.3 ± 0.3. While a minor contribution from Cu–Cuef scattering is found to be compatible with the experimental spectrum, the overlapping of such signals with the intense MSNO3 signals in the same R-space range hampers a conclusive assessment. Notably, if present at all, these paired Cu-ions are too far apart to support bridged Cu-nitrate species, requiring Cu–Cu distances of <3 Å.18
The final step in our analysis regarded the Cu-CHA 0.2_15 catalyst upon exposure to the NO/O2 mixture. As discussed in Sections 2.5 and 3.3.1, consistently with conventional and WT EXAFS qualitative analysis, this sample in the pretreated state contains a mixture of Z[CuIIOH] and Z2CuII sites, in a ca. 65% : 35% ratio. We thus attempted quantification of the Cu-speciation in this catalyst upon exposure to the NO/O2 mixture through LCF analysis using as references the spectra of pretreated Cu-CHA 0.1_5 (representative of Z2CuII unable to form Cu-nitrates) and of Cu-CHA 0.6_29 exposed to NO/O2 (representative of the Z[CuIINO3] species illustrated in Fig. 8(a)). As before, LCF analysis has been carried out on both normalized XANES and the k2χ(k) EXAFS curve, as shown in Fig. 9. While small discrepancies could be expected, also considering the slight differences in local coordination environment of the Z[CuIINO3] for different Si/Al ratios, the employed model guarantees a good reproduction of the experimental data, with R-factor values only slightly higher than those found for the pretreated sample (compare Tables 3 and 5). Importantly, the optimized percentages of Cu-species match perfectly, within LCF errors, those retrieved for the pretreated state: as detailed in Table 5, from both XANES and EXAFS LCF analysis, 66% of Cu-species are found as Z[CuIINO3] and the remaining 34% as (unreacted) Z2CuII.
Fig. 9. Comparison between experimental and best-fit curves for Cu-CHA 0.2_15 after exposure to the NO/O2 mixture at 200 °C from LCF analysis of (a) the normalized XANES and (b) the k2χ(k) EXAFS spectra. The LCF residuals and the scaled components corresponding to the Z[CuIINO3] and Z2CuII species are reported in both working spaces.

Quantitative results from LCF analysis for Cu-CHA 0.2_15 after exposure to the NO/O2 mixture at 200 °C considering both the normalized XANES and the k2χ(k) EXAFS spectra.
| LCF parameters Cu-CHA 0.2_15 | XANES LCF | EXAFS LCF |
|---|---|---|
| Fraction Z[CuIINO3] 8r | 0.66 ± 0.03 | 0.66 ± 0.02 |
| Fraction Z2CuII 6r | 0.34 ± 0.03 | 0.34 ± 0.02 |
| LCF Rfactor | 0.00018 | 0.016 |
The estimated relative abundance and the local structure/siting of Cu-species are further confirmed via a multi-component EXAFS fit (see Table S2 and Fig. S3, ESI†). The retrieved values of Cu–Cuef = 3.39 ± 0.02 Å and NCu_ef = 0.9 ± 0.3 are almost unchanged with respect to those found for Cu-CHA 0.2_15 after pretreatment. This further rules out the formation of ‘bridging’ Cu-nitrates, which would imply a significantly shorter Cu–Cu interatomic separation.
3.4. Cu-speciation and structural insight from XAS quantitative analysis
DR UV-Vis spectroscopy has been widely used in the study of Cu-CHA, to follow the changes in oxidation and coordination states of the Cu ions during thermal activation67 and in the presence of the NH3-SCR reactants.16,46,48 We have thus applied this technique to corroborate the results obtained by FTIR and XAS. Fig. 10 reports the spectra measured on the three samples after pretreatment in O2 (light curves) and subsequent interaction with NO/O2 (dark curves). The spectra can be ideally (and roughly) divided into two regions, with an intense absorption above ca. 25 000 cm−1 and a well-defined peak centered around 15 000 cm−1 (11 000 cm−1 for sample Cu-CHA 0.1_5), which are related to ligand-to-metal charge transfer (CT) and ligand field d–d transitions of CuII ions, respectively.67 The broad features at intermediate wavenumbers (centered around 20 500 and at 25 000 cm−1 in Cu-CHA 0.1_5 and Cu-CHA 0.6_29, respectively) are usually ascribed to a variety of Cu-oxo species in mono- and multimeric Zx[CuIIxOy] moieties.14,67–69
Fig. 10. DR UV-Vis spectra measured after pretreatment in O2 at 400 °C and subsequent cooling down to 200 °C (light curves) and after exposure to 1000 ppm NO/10% O2 at 200 °C (dark curves).

The spectra measured after pretreatment in O2 are in agreement with previous measurements under similar conditions.67 In more detail, the samples containing a significant fraction of Z[CuIIOH] and/or tri-coordinated Zx[CuIIxOy] moieties (Cu-CHA 0.6_29 and Cu-CHA 0.2_15, the middle and right-hand panels of Fig. 10) show the typical ‘quadruplet’ that is, an absorption with maxima around 19 700, 16 700, 13 700 and 11 600 cm−1. The corresponding d–d transition in the sample with a majority of Z2CuII (Cu-CHA 0.1_5, left panel) is centered around 11 000 cm−1 and does not show the typical quadruplet feature, in agreement with previous reports and theoretical calculations.67,68 This has been explained on the basis of the local geometry of the corresponding framework-anchored CuII sites.67 The intensity of these features cannot be used for semi-quantitative considerations. In fact, it has been proposed that the ‘quadruplet’ is the result of the contribution of a mixture of framework-coordinated mono and multimeric CuII-oxo species,68 and there is evidence that its extinction coefficient is extremely high, and likely sensitive to small variations in the local geometry.16
What is relevant for this work are the spectral changes observed after contacting the samples with the NO/O2 mixture (dark curves in Fig. 10). For both Cu-CHA 0.2_15 and 0.6_29 the reaction causes a significant modification in the d–d region, and a shift of the CT absorption, compatible with the formation of Cu-nitrates, as rationalized elsewhere.46,48 On the other hand, the only change observed in sample Cu-CHA 0.1_5 is the depletion of the broad absorption centered around 20 500 cm−1, while the d–d transition and CT absorption are hardly affected, in agreement with the high stability and inertia towards NO/O2 of the Z2CuII sites.
4. Conclusions
Qualitative and quantitative XAS analyses were carried out to quantify the relative amount of Z[CuIIOH] or other Zx[CuIIxOy] species hosted at 1Al sites and Z2CuII ions at 2Al sites in three samples with different chemical compositions (Si/Al and Cu/Al) and very similar Cu volumetric densities. The XAS data measured during thermal treatment in O2 up to 400 °C were found to be in very good agreement with the compositional diagram proposed by Paolucci et al.,8 based on the assumption that Al heteroatoms are statistically distributed in the framework and Z[CuIIOH] sites start to be filled after saturation of the most stable Z2CuII ones. The transient formation of a small fraction of CuI ions was also observed during the treatment in O2, in agreement with previous reports.63 Interestingly, this effect was more evident on the sample with an intermediate Si/Al ratio of 15, suggesting that the tendency to reduction of Z[CuIIOH] sites can be influenced by the presence of Brønsted sites, which are less abundant in sample Cu-CHA 0.6_29.
Aided by EXAFS WT analysis, we also investigated the presence of Cu–Cu contributions in the O2 pretreated samples, following recently acquired knowledge based on the application of Cu-CHA catalysts for the methane-to-methanol reaction, which are rarely considered in the field of the NH3-SCR reaction.16 Interestingly, Cu–Cu scattering contributions are detected in all the Cu-CHA samples upon pretreatment in O2, yet with composition-dependent structural differences. A combination of EXAFS WT analysis and EXAFS fitting based on available structural models led us to propose tridentate oxo-bridged CuII-dimers coexisting with Z[CuIIOH] sites in Cu-CHA 0.6_29 and 0.2_15, and proximal Z2CuII monomers, possibly stabilized into two adjacent 2Al-bearing 6r within the same cage, in Cu-CHA 0.1_5.
The XAS data acquired during exposure of the pretreated catalysts to the NO/O2 mixture confirm the formation of framework-interacting Z[CuIINO3] with a chelating bidentate structure only on samples characterized by a significant amount of Z[CuIIOH] sites (or analogues presented above). This confirms our FTIR and DR UV-Vis results, indicating that Z2CuII sites are unaffected by this reactivity, in relation to their high stability in 6r. Notwithstanding the different vibrational fingerprints observed using FTIR, the structure of the Z[CuIINO3] complexes formed in Cu-CHA 0.2_15 and 0.6_29 is conserved, as confirmed by EXAFS fitting analysis. A possible explanation of the different relative intensities of the infrared bands at 1625 and 1610/1575 cm−1 in the two samples could have been attempted by considering the recent assignment of the band at 1610 cm−1 to NO2 adsorbed on Cu ions.27 This interpretation can be excluded on the basis of our quantitative XAS data analysis. This in fact shows that all Cu ions (>90%, considering the sensitivity of the technique) are involved in Z[CuIINO3] formation in the sample characterized by ‘almost pure’ Z[CuIIOH] ions (Cu-CHA 0.6_29), while these complexes account for 66% of Cu ions in the sample (Cu-CHA 0.2_15) showing 65% of Cu ions as Z[CuIIOH] before NO/O2 dosage. These results point to the fact that all Z[CuIIOH] are able to form Cu-nitrates by reaction with NO/O2, at variance with what was recently proposed by Zhang et al.66
As a final comment, the only parameter influencing the formation of Cu-nitrates, under the explored experimental conditions, is the Si/Al and Cu/Al ratio, directing the distribution of Z[CuIIOH] and Z2CuII sites. The Cu volumetric density (similar for the three samples, and identical for Cu-CHA 0.1_5 and 0.6_29) does not influence this reactivity. This is not surprising, since the Cu density was shown to play a role when Cu ions were mobilized, thanks to the solvation effect of NH3. The NO/O2 mixture has a strong oxidizing activity, but it does not remove the Cu ions from their framework coordination unless NH3 is present when mixed-ligand complexes are formed.45
Author contributions
The manuscript was written through contributions of all the authors. All the authors have given approval to the final version of the manuscript.
Abbreviations
- CHA
Chabazite
- EXAFS
Extended X-ray absorption fine structure
- FTIR
Fourier transform infrared
- LCF
Linear combination fit
- MS
Multiple scattering
- SCR
selective catalytic reduction
- SS
Single scattering
- XANES
X-ray absorption near-edge spectroscopy
- WT
Wavelet transform
- XAS
X-Ray absorption spectroscopy
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
The ERC-SyG-2019 project CUBE: unravelling the secrets of Cu-based catalysts for C–H activation (ID 856446) is gratefully acknowledged. A. M. acknowledges project no. 2017KKP5Z PRIN-2017 MOSCATo (cutting-edge X-ray methods and models for the understanding of surface site reactivity in heterogeneous catalysts and sensors). Tommaso Selleri (European Commission Joint Research Centre) is gratefully acknowledged for his help during the XAS experiments. We are also grateful to Luca Bugarin for help in the XAS data analysis.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cp01754c
References
- Nova I. and Tronconi E., in Urea-SCR Technology for deNOx After Treatment of Diesel Exhausts, ed. H.-Y. Chen, Springer New York, 2014, pp. 123–147 10.1007/978-1-4899-8071-7_5 [DOI] [Google Scholar]
- Skalska K. Miller J. S. Ledakowicz S. Sci. Total Environ. 2010;408:3976–3989. doi: 10.1016/j.scitotenv.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Beale A. M. Gao F. Lezcano-Gonzalez I. Peden C. H. F. Szanyi J. Chem. Soc. Rev. 2015;44:7371–7405. doi: 10.1039/C5CS00108K. [DOI] [PubMed] [Google Scholar]
- Kwak J. H. Tonkyn R. G. Kim D. H. Szanyi J. Peden C. H. F. J. Catal. 2010;275:187–190. doi: 10.1016/j.jcat.2010.07.031. [DOI] [Google Scholar]
- Korhonen S. T. Fickel D. W. Lobo R. F. Weckhuysen B. M. Beale A. M. Chem. Commun. 2011;47:800–802. doi: 10.1039/C0CC04218H. [DOI] [PubMed] [Google Scholar]
- Deka U. Juhin A. Eilertsen E. A. Emerich H. Green M. A. Korhonen S. T. Weckhuysen B. M. Beale A. M. J. Phys. Chem. C. 2012;116:4809–4818. doi: 10.1021/jp212450d. [DOI] [Google Scholar]
- Gao F. Walter E. D. Kollar M. Wang Y. Szanyi J. Peden C. H. F. J. Catal. 2014;319:1–14. doi: 10.1016/j.jcat.2014.08.010. [DOI] [Google Scholar]
- Paolucci C. Parekh A. A. Khurana I. Di Iorio J. R. Li H. Albarracin Caballero J. D. Shih A. J. Anggara T. Delgass W. N. Miller J. T. Ribeiro F. H. Gounder R. Schneider W. F. J. Am. Chem. Soc. 2016;138:6028–6048. doi: 10.1021/jacs.6b02651. [DOI] [PubMed] [Google Scholar]
- Di Iorio J. R. Gounder R. Chem. Mater. 2016;28:2236–2247. doi: 10.1021/acs.chemmater.6b00181. [DOI] [Google Scholar]
- Di Iorio J. R. Li S. C. Jones C. B. Nimlos C. T. Wang Y. J. Kunkes E. Vattipalli V. Prasad S. Moini A. Schneider W. F. Gounder R. J. Am. Chem. Soc. 2020;142:4807–4819. doi: 10.1021/jacs.9b13817. [DOI] [PubMed] [Google Scholar]
- Di Iorio J. R. Nimlos C. T. Gounder R. ACS Catal. 2017;7:6663–6674. doi: 10.1021/acscatal.7b01273. [DOI] [Google Scholar]
- Borfecchia E. Lomachenko K. A. Giordanino F. Falsig H. Beato P. Soldatov A. V. Bordiga S. Lamberti C. Chem. Sci. 2015;6:548–563. doi: 10.1039/C4SC02907K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas D. K. Borfecchia E. Dyballa M. Pankin I. A. Lomachenko K. A. Martini A. Signorile M. Teketel S. Arstad B. Berlier G. Lamberti C. Bordiga S. Olsbye U. Lillerud K. P. Svelle S. Beato P. J. Am. Chem. Soc. 2017;139:14961–14975. doi: 10.1021/jacs.7b06472. [DOI] [PubMed] [Google Scholar]
- Ipek B. Wulfers M. J. Kim H. Goltl F. Hermans I. Smith J. P. Booksh K. S. Brown C. M. Lobo R. F. ACS Catal. 2017;7:4291–4303. doi: 10.1021/acscatal.6b03005. [DOI] [Google Scholar]
- Martini A. Alladio E. Borfecchia E. Top. Catal. 2018;61:1396–1407. doi: 10.1007/s11244-018-1036-9. [DOI] [Google Scholar]
- Negri C. Selleri T. Borfecchia E. Martini A. Lomachenko K. A. Janssens T. V. W. Cutini M. Bordiga S. Berlier G. J. Am. Chem. Soc. 2020;142:15884–15896. doi: 10.1021/jacs.0c06270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankin I. A. Martini A. Lomachenko K. A. Soldatov A. V. Bordiga S. Borfecchia E. Catal. Today. 2020;345:125–135. doi: 10.1016/j.cattod.2019.09.032. [DOI] [Google Scholar]
- Janssens T. V. W. Falsig H. Lundegaard L. F. Vennestrøm P. N. R. Rasmussen S. B. Moses P. G. Giordanino F. Borfecchia E. Lomachenko K. A. Lamberti C. Bordiga S. Godiksen A. Mossin S. Beato P. ACS Catal. 2015;5:2832–2845. doi: 10.1021/cs501673g. [DOI] [Google Scholar]
- Usberti N. Gramigni F. Nasello N. D. Iacobone U. Selleri T. Hu W. S. Liu S. J. Gao X. Nova I. Tronconi E. Appl. Catal., B. 2020;279:12. doi: 10.1016/j.apcatb.2020.119397. [DOI] [Google Scholar]
- Villamaina R. Iacobone U. Nova I. Ruggeri M. P. Collier J. Thompsett D. Tronconi E. ChemCatChem. 2020;12:3843–3848. doi: 10.1002/cctc.202000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. Kubota H. Amada T. Kon K. Toyao T. Maeno Z. Ueda K. Ohyama J. Satsuma A. Tanigawa T. Tsunoji N. Sano T. Shimizu K. ChemCatChem. 2020;12:3050–3059. doi: 10.1002/cctc.202000024. [DOI] [Google Scholar]
- Liu C. Kubota H. Toyao T. Maeno Z. Shimizu K. Catal. Sci. Technol. 2020;10:3586–3593. doi: 10.1039/D0CY00379D. [DOI] [Google Scholar]
- Falsig H. Vennestrøm P. N. R. Moses P. G. Janssens T. V. W. Top. Catal. 2016;59:861–865. doi: 10.1007/s11244-016-0560-8. [DOI] [Google Scholar]
- Chen L. Falsig H. Janssens T. V. W. Gronbeck H. J. Catal. 2018;358:179–186. doi: 10.1016/j.jcat.2017.12.009. [DOI] [Google Scholar]
- Gao F. Mei D. Wang Y. Szanyi J. Peden C. H. F. J. Am. Chem. Soc. 2017;139:4935–4942. doi: 10.1021/jacs.7b01128. [DOI] [PubMed] [Google Scholar]
- Paolucci C. Khurana I. Parekh A. A. Li S. C. Shih A. J. Li H. Di Iorio J. R. Albarracin-Caballero J. D. Yezerets A. Miller J. T. Delgass W. N. Ribeiro F. H. Schneider W. F. Gounder R. Science. 2017;357:898–903. doi: 10.1126/science.aan5630. [DOI] [PubMed] [Google Scholar]
- Moreno-Gonzalez M. Millan R. Concepcion P. Blasco T. Boronat M. ACS Catal. 2019;9:2725–2738. doi: 10.1021/acscatal.8b04717. [DOI] [Google Scholar]
- Ruggeri M. P. Selleri T. Nova I. Tronconi E. Pihl J. A. Toops T. J. Partridge W. P. Top. Catal. 2016;59:907–912. doi: 10.1007/s11244-016-0567-1. [DOI] [Google Scholar]
- Selleri T. Gramigni F. Nova I. Tronconi E. Appl. Catal., B. 2018;225:324–331. doi: 10.1016/j.apcatb.2017.11.068. [DOI] [Google Scholar]
- Paolucci C. Di Iorio J. R. Schneider W. F. Gounder R. Acc. Chem. Res. 2020;53:1881–1892. doi: 10.1021/acs.accounts.0c00328. [DOI] [PubMed] [Google Scholar]
- Oda A. Shionoya H. Hotta Y. Takewaki T. Sawabe K. Satsuma A. ACS Catal. 2020;10:12333–12339. doi: 10.1021/acscatal.0c03425. [DOI] [Google Scholar]
- Lomachenko K. A. Borfecchia E. Negri C. Berlier G. Lamberti C. Beato P. Falsig H. Bordiga S. J. Am. Chem. Soc. 2016;138:12025–12028. doi: 10.1021/jacs.6b06809. [DOI] [PubMed] [Google Scholar]
- Lezcano-Gonzalez I. Wragg D. S. Slawinski W. A. Hemelsoet K. Van Yperen-De Deyne A. Waroquier M. Van Speybroeck V. Beale A. M. J. Phys. Chem. C. 2015;119:24393–24403. doi: 10.1021/acs.jpcc.5b06875. [DOI] [Google Scholar]
- Giordanino F. Borfecchia E. Lomachenko K. A. Lazzarini A. Agostini G. Gallo E. Soldatov A. V. Beato P. Bordiga S. Lamberti C. J. Phys. Chem. Lett. 2014;5:1552–1559. doi: 10.1021/jz500241m. [DOI] [PubMed] [Google Scholar]
- Chen L. Janssens T. V. W. Skoglundh M. Grönbeck H. Top. Catal. 2019;62:93–99. doi: 10.1007/s11244-018-1095-y. [DOI] [Google Scholar]
- Borfecchia E. Negri C. Lomachenko K. A. Lamberti C. Janssens T. V. W. Berlier G. React. Chem. Eng. 2019;4:1067–1080. doi: 10.1039/C8RE00322J. [DOI] [Google Scholar]
- Selleri T. Ruggeri M. P. Nova I. Tronconi E. Top. Catal. 2016;59:678–685. doi: 10.1007/s11244-016-0543-9. [DOI] [Google Scholar]
- Ruggeri M. Nova I. Tronconi E. Top. Catal. 2013;56:109–113. doi: 10.1007/s11244-013-9937-0. [DOI] [Google Scholar]
- Chen L. Janssens T. V. W. Vennestrom P. N. R. Jansson J. Skoglundh M. Gronbeck H. ACS Catal. 2020;10:5646–5656. doi: 10.1021/acscatal.0c00440. [DOI] [Google Scholar]
- Greenaway A. G. Marberger A. Thetford A. Lezcano-González I. Agote-Arán M. Nachtegaal M. Ferri D. Kröcher O. Catlow C. R. A. Beale A. M. Chem. Sci. 2020;11:447–455. doi: 10.1039/C9SC04905C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marberger A. Petrov A. W. Steiger P. Elsener M. Krocher O. Nachtegaal M. Ferri D. Nat. Catal. 2018;1:221–227. doi: 10.1038/s41929-018-0032-6. [DOI] [Google Scholar]
- Ruggeri M. P. Selleri T. Colombo M. Nova I. Tronconi E. J. Catal. 2014;311:266–270. doi: 10.1016/j.jcat.2013.11.028. [DOI] [Google Scholar]
- Wang D. Zhang L. Kamasamudram K. Epling W. S. ACS Catal. 2013;3:871–881. doi: 10.1021/cs300843k. [DOI] [Google Scholar]
- Hammershøi P. S. Negri C. Berlier G. Bordiga S. Beato P. Janssens T. V. W. Catal. Sci. Technol. 2019;9:2608–2619. doi: 10.1039/C9CY00358D. [DOI] [Google Scholar]
- Negri C. Borfecchia E. Martini A. Deplano G. Lomachenko K. A. Janssens T. V. W. Berlier G. Bordiga S. Res. Chem. Intermed. 2021;47:357–375. doi: 10.1007/s11164-020-04350-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrsted C. Borfecchia E. Berlier G. Lomachenko K. A. Lamberti C. Bordiga S. Vennestrøm P. N. R. Janssens T. V. W. Falsig H. Beato P. Puig-Molina A. Catal. Sci. Technol. 2016;6:8314–8324. doi: 10.1039/C6CY01820C. [DOI] [Google Scholar]
- Negri C. Hammershøi P. S. Janssens T. V. W. Beato P. Berlier G. Bordiga S. Chem. – Eur. J. 2018;24:12044–12053. doi: 10.1002/chem.201802769. [DOI] [PubMed] [Google Scholar]
- Negri C. Borfecchia E. Cutini M. Lomachenko K. A. Janssens T. V. W. Berlier G. Bordiga S. ChemCatChem. 2019;11:3828–3838. doi: 10.1002/cctc.201900590. [DOI] [Google Scholar]
- Zhang Y. Peng Y. Li K. Liu S. Chen J. Li J. Gao F. Peden C. H. F. ACS Catal. 2019;9:6137–6145. doi: 10.1021/acscatal.9b00759. [DOI] [Google Scholar]
- Mathon O. Beteva A. Borrel J. Bugnazet D. Gatla S. Hino R. Kantor I. Mairs T. Munoz M. Pasternak S. Perrin F. Pascarelli S. J. Synchrotron Radiat. 2015;22:1548–1554. doi: 10.1107/S1600577515017786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellet D. Gorges B. Dallery A. Bernard P. Pereiro E. Baruchel J. J. Appl. Crystallogr. 2003;36:366–367. doi: 10.1107/S0021889803001158. [DOI] [Google Scholar]
- Bordiga S. Groppo E. Agostini G. van Bokhoven J. A. Lamberti C. Chem. Rev. 2013;113:1736–1850. doi: 10.1021/cr2000898. [DOI] [PubMed] [Google Scholar]
- Ravel B. Newville M. J. Synchrotron Radiat. 2005;12:537–541. doi: 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
- Martini A. Borfecchia E. Lomachenko K. A. Pankin I. A. Negri C. Berlier G. Beato P. Falsig H. Bordiga S. Lamberti C. Chem. Sci. 2017;8:6836–6851. doi: 10.1039/C7SC02266B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz M. Argoul P. Farges F. Am. Mineral. 2003;88:694–700. doi: 10.2138/am-2003-0423. [DOI] [Google Scholar]
- Timoshenko J. Kuzmin A. Comput. Phys. Commun. 2009;180:920–925. doi: 10.1016/j.cpc.2008.12.020. [DOI] [Google Scholar]
- Funke H. Scheinost A. C. Chukalina M. Phys. Rev. B: Condens. Matter Mater. Phys. 2005;71:094110. doi: 10.1103/PhysRevB.71.094110. [DOI] [Google Scholar]
- Paolucci C. Verma A. A. Bates S. A. Kispersky V. F. Miller J. T. Gounder R. Delgass W. N. Ribeiro F. H. Schneider W. F. Angew. Chem., Int. Ed. 2014;53:11828–11833. doi: 10.1002/anie.201407030. [DOI] [PubMed] [Google Scholar]
- Hadjiivanov K. Saussey J. Freysz J. L. Lavalley J. C. Catal. Lett. 1998;52:103–108. doi: 10.1023/A:1019059117488. [DOI] [Google Scholar]
- Rivallan M. l. Ricchiardi G. Bordiga S. Zecchina A. J. Catal. 2009;264:104–116. doi: 10.1016/j.jcat.2009.03.012. [DOI] [Google Scholar]
- Chen H. Y. Kollar M. Wei Z. H. Gao F. Wang Y. L. Szanyi J. Peden C. H. F. Catal. Today. 2019;320:61–71. doi: 10.1016/j.cattod.2017.12.022. [DOI] [Google Scholar]
- Henriques C. Marie O. Thibault-Starzyk F. Lavalley J. C. Microporous Mesoporous Mater. 2001;50:167–171. doi: 10.1016/S1387-1811(01)00444-9. [DOI] [Google Scholar]
- Borfecchia E. Pappas D. K. Dyballa M. Lomachenko K. A. Negri C. Signorile M. Berlier G. Catal. Today. 2019;333:17–27. doi: 10.1016/j.cattod.2018.07.028. [DOI] [Google Scholar]
- Pappas D. K. Borfecchia E. Dyballa M. Pankin I. A. Lomachenko K. A. Martini A. Signorile M. Teketel S. Arstad B. Berlier G. Lamberti C. Bordiga S. Olsbye U. Lillerud K. P. Svelle S. Beato P. J. Am. Chem. Soc. 2017;139:14961–14975. doi: 10.1021/jacs.7b06472. [DOI] [PubMed] [Google Scholar]
- Martini A. Signorile M. Negri C. Kvande K. Lomachenko K. A. Svelle S. Beato P. Berlier G. Borfecchia E. Bordiga S. Phys. Chem. Chem. Phys. 2020;22:18950–18963. doi: 10.1039/D0CP01257B. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Wu Y. Peng Y. Li J. Walter E. D. Chen Y. Washton N. M. Szanyi J. Wang Y. Gao F. J. Phys. Chem. C. 2020;124:28061–28073. doi: 10.1021/acs.jpcc.0c07971. [DOI] [Google Scholar]
- Negri C. Signorile M. Porcaro N. G. Borfecchia E. Berlier G. Janssens T. V. W. Bordiga S. Appl. Catal., A. 2019;578:1–9. doi: 10.1016/j.apcata.2019.03.018. [DOI] [Google Scholar]
- Li H. Paolucci C. Khurana I. Wilcox L. Goltl F. Albarracin-Caballero J. D. Shih A. J. Ribeiro F. H. Gounder R. Schneider W. F. Chem. Sci. 2019;10:2373–2384. doi: 10.1039/C8SC05056B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordanino F. Vennestrom P. N. R. Lundegaard L. F. Stappen F. N. Mossin S. L. Beato P. Bordiga S. Lamberti C. Dalton Trans. 2013;42:12741–12761. doi: 10.1039/C3DT50732G. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.