

Abstract
Nucleic acids are utilized in many therapeutic modalities, including gene therapy, but their ability to trigger host immune responses in vivo can lead to decreased safety and efficacy. In the case of adeno-associated viral (AAV) vectors, studies have shown that the genome of the vector activates Toll-like receptor 9 (TLR9), a pattern recognition receptor that senses foreign DNA. Here, we engineered AAV vectors to be intrinsically less immunogenic by incorporating short DNA oligonucleotides that antagonize TLR9 activation directly into the vector genome. The engineered vectors elicited markedly reduced innate immune and T cell responses and enhanced gene expression in clinically relevant mouse and pig models across different tissues, including liver, muscle and retina. Subretinal administration of higher-dose AAV in pigs resulted in photoreceptor pathology with microglia and T cell infiltration. These adverse findings were avoided in the contralateral eyes of the same animals that were injected with the engineered vectors. However, intravitreal injection of higher-dose AAV in macaques, a more immunogenic route of administration, showed that the engineered vector delayed but did not prevent clinical uveitis, suggesting that other immune factors in addition to TLR9 may contribute to intraocular inflammation in this model. Our results demonstrate that linking specific immunomodulatory non-coding sequences to much longer therapeutic nucleic acids can “cloak” the vector from inducing unwanted immune responses in multiple, but not all, models. This “coupled immunomodulation” strategy may widen the therapeutic window for AAV therapies as well as other DNA-based gene transfer methods.
Single Sentence Summary:
Incorporating TLR9-inhibitory sequences in the AAV vector genome inhibits immunogenicity and enhances transgene expression in multiple animal models.
INTRODUCTION
Gene therapy holds exciting promise for treating many genetic disorders, but host immune responses pose a major challenge for in vivo gene transfer (1). Whereas adeno-associated viral (AAV) vectors are known to be less immunogenic than other viral vectors such as adenoviruses, preclinical and clinical studies have demonstrated dose-dependent inflammation, which can reduce efficacy and lead to dose-limiting toxicity (2–8). Examples include ocular AAV therapies leading to a permanent reduction in visual acuity following intraocular inflammation (9–11) and liver-directed therapies resulting in liver transaminase elevation, which coincides with decreased factor IX (FIX) transgene expression, likely due to AAV capsid-specific cytotoxic T cells destroying transduced hepatocytes (12, 13). More recently, deaths have been reported in a high dose systemic AAV gene therapy trial for children with X-linked myotubular myopathy (14), suggesting that we are still learning about AAV-mediated toxicity and immune responses.
Several studies have demonstrated a central role for Toll-like receptor 9 (TLR9), an immune sensor of DNA, in detecting AAV genomes and triggering innate immune and CD8+ T cell responses (15–18). TLRs are a family of innate immune sensors conserved across mammalian species that are found on endosomal or plasma membranes of immune cells (19). TLR9 normally senses DNA from pathogenic viruses and bacteria that contains unmethylated cytosine-phosphate-guanine (CpG) motifs, which also are present in AAV vector genomes. CpG binding to TLR9 promotes its dimerization and activates TLR9 signaling via the signaling adaptor MyD88, leading to induction of type I interferons and pro-inflammatory cytokines. Innate immune responses, such as interferon induction, trigger an antiviral state among cells, while inflammation recruits other immune cells to the site of infection and primes adaptive immune responses (15, 16). In addition to TLR9, TLR2 is implicated in sensing the AAV capsid (20).
One strategy for blocking TLR9 activation is the administration of specific short single-stranded DNA oligonucleotides (typically 12 – 24 nt) that antagonize TLR9 activation (hereby termed “TLR9i” for TLR9-inhibitory). Structural studies recently described two different TLR9i sequences that formed stem-loop structures that fit snugly into the interior of the ring structure of TLR9, preventing dimerization and activation (21). Furthermore, the binding site of TLR9i oligonucleotides on TLR9 partially overlapped with that of CpG DNA, and the TLR9i oligonucleotide tested possessed higher binding affinity for TLR9 than CpG DNA. TLR9i sequences have been tested extensively in the literature as a standalone agent co-administered with inflammatory DNA ligands, but this strategy likely requires a large amount of oligonucleotide to inhibit all TLR9 molecules, which we speculate may be less feasible for clinical applications. Here, we hypothesized that the immune-inhibitory activity of short TLR9i sequences remained functional in a much longer strand of DNA and could be utilized to dampen immune responses to AAV vectors by directly incorporating them into the vector genome.
RESULTS
TLR9i sequences efficiently inhibit inflammation when applied in cis with CpG-containing oligonucleotides
To test this hypothesis, we first selected multiple TLR9i sequences described in the literature and individually linked each of them directly to a 24 nt-long highly inflammatory oligodeoxyribonucleotide (ODN), ODN 2006, which contained 4 CpG sites (22), resulting in fusion constructs hereafter termed “ODN 2006-TLR9i”. Using a TLR9 reporter cell assay, we observed that treatment of cells with ODN 2006-TLR9i oligonucleotides (with the ODN 2006 sequence at the 5’ end) resulted in markedly reduced NF-κB response compared to ODN 2006 linked to two separate control sequences not expected to stimulate or inhibit TLR9 (control1 or control2), demonstrating that TLR9i sequences are able to function in cis (fig. S1A and see table S1 for sequences). Insertion of a 5 nt intervening sequence (AAAAA) between ODN 2006 and selected TLR9i sequences (ODN 2088 or TTAGGG) yielded similar results, suggesting TLR9i sequences also can function when they are distal to CpG motifs (fig. S1B). When the order of ODN 2006 and the covalently linked TLR9i sequence was reversed so that the TLR9i sequence was at the 5’ end, a similar reduction in NF-κB response, as well as a reduction in the pro-inflammatory cytokine IL-8, were observed (fig. S1C). We identified a condition in which ODN 2006-control2 (in cis) gave comparable NF-κB response as ODN 2006 and control2 co-administered at the same concentration (in trans) and observed that ODN 2006-TTAGGG blocked ~80% of induced NF-κB response, while co-administration of ODN 2006 and TTAGGG only inhibited ~35% of induced NF-κB response (fig. S1D). Thus, incorporation of a TLR9i sequence within the same molecule as an inflammatory DNA sequence could be more effective at dampening inflammation than administering it as a separate molecule. ODN TTAGGG, a well-known TLR9i sequence derived from mammalian telomeres (23), did not inhibit the activation of TLR7 or TLR2/6 (sensors of microbial RNA and lipoproteins, respectively) even at high concentration, supporting its specificity for TLR9 (fig. S1E and S1F).
Incorporation of io1 into a self-complementary AAV8 vector reduces interferon responses and macrophage infiltration in mouse liver and boosts factor IX expression
Next, we evaluated the ability of TLR9-inhibitory sequences incorporated within AAV genomes to reduce innate and adaptive immune responses to gene therapy in vivo. We used multiple clinically relevant target tissues and routes of administration in both small and large animal models: (1) liver following an intravenous injection in mice, (2) skeletal muscle following an intramuscular injection in mice, (3) retina following an intravitreal injection in mice, (4) retina following a subretinal injection in outbred pigs, and (5) retina following an intravitreal injection in outbred macaques. In addition, we tested primary human immune cells for innate immune responses in vitro.
We first inserted an oligonucleotide consisting of three copies of ODN TTAGGG separated by short AAAAA linkers (see table S1 for sequence and Fig. 1A for vector organization, hereafter termed “io1” for “inflammation-inhibiting oligonucleotide 1”) into the 5’ untranslated region (UTR) of a self-complementary (sc) AAV vector plasmid encoding human factor IX (FIX) under the control of a liver-specific transthyretin promoter (16). We chose scAAV vectors for proof-of-concept as they have been shown to trigger TLR9-dependent innate immune responses in the liver of mice efficiently after intravenous (i.v.) administration (16).
Figure 1. Innate immune response and human factor IX expression in mice following intravenous AAV8 administration.
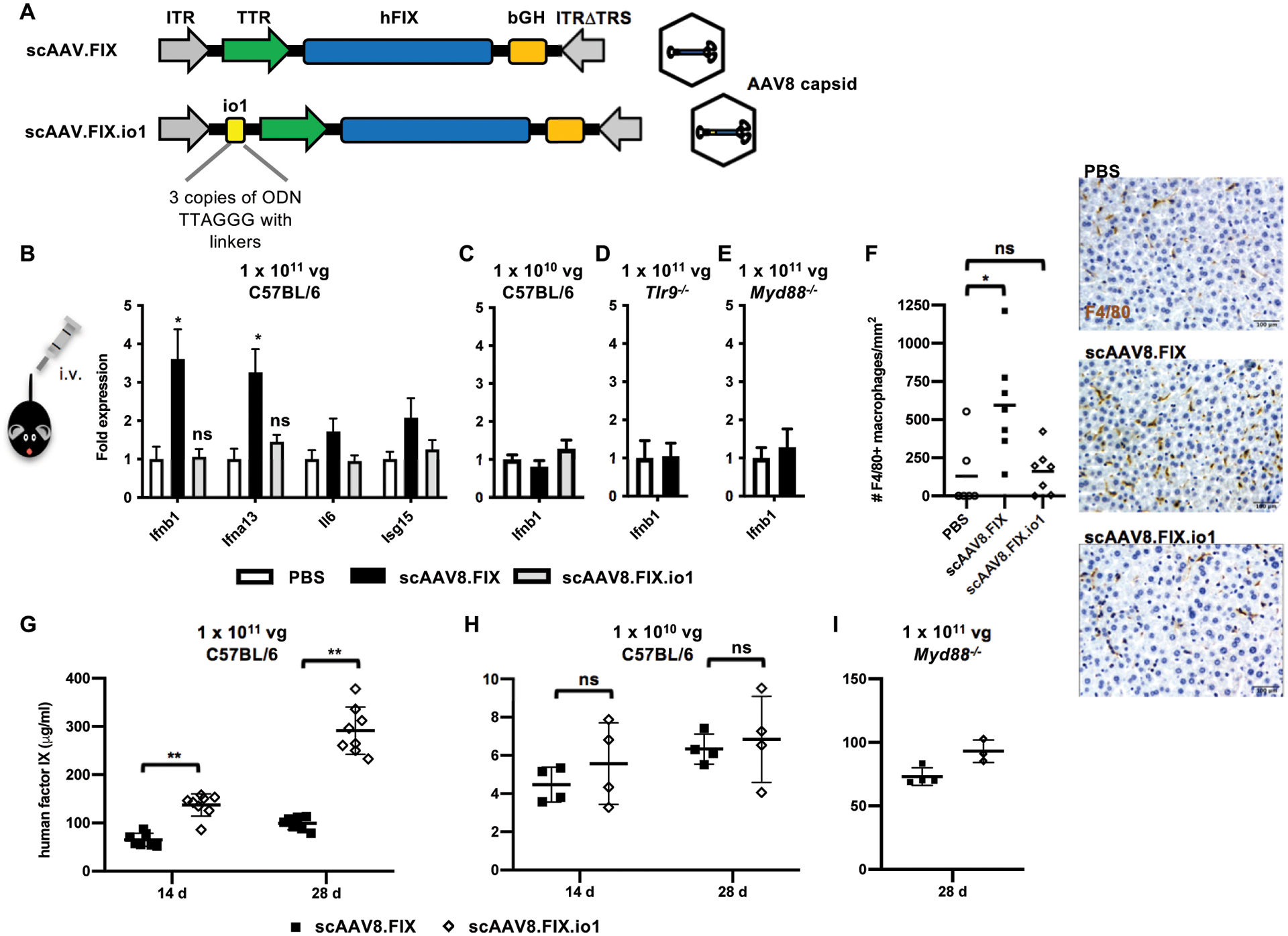
(A) Schematic diagram of vector organization of scAAV.FIX and scAAV.FIX.io1. The io1 sequence is not expected to be transcribed or translated due to its placement in an untranslated region upstream of the promoter. (B) Innate immune response in liver assayed by qPCR 2 h after 1 × 1011 vg AAV8 administration in C57BL/6 mice. n = 6 – 7 animals per condition. (C) Innate immune response in liver assayed by qPCR 2 h after 1 × 1010 vg AAV8 administration in C57BL/6 mice. n = 4 – 5 animals per condition. (D) Innate immune response in liver assayed by qPCR 2 h after 1 × 1011 vg AAV8 administration in Tlr9−/− mice. n = 4 animals per condition. (E) Innate immune response in liver assayed by qPCR 2 h after 1 × 1011 vg AAV8 administration in Myd88−/− mice. n = 4 animals per condition. PBS injection was set to 1-fold expression for each gene. Data shown are mean ± s.e.m. per condition. * p<0.05 by two-tailed Mann-Whitney test and compared against PBS condition. ns, not significant, p>0.05. (F) F4/80+ macrophage infiltration in C57BL/6 mouse liver 2 h after 1 × 1011 vg AAV8 administration. Data shown are mean of n = 6 – 7 animals per condition. * p<0.005 by two-tailed Mann-Whitney test. Scale bar, 100 μm. (G) Human factor IX in plasma of C57BL/6 mice at indicated time points after 1 × 1011 vg AAV8 administration. n = 7 – 8 animals per condition. (H) Human factor IX in plasma of C57BL/6 mice at indicated time points after 1 × 1010 vg AAV8 administration. n = 4 animals per condition. (I) Human factor IX in plasma of Myd88−/− mice at indicated time points after 1 × 1011 vg AAV8 administration. n = 3 – 4 animals per condition. Data shown are mean ± s.d. per condition. ** p<0.005 by two-tailed Mann-Whitney test. ITR, inverted terminal repeat; TTR, transthyretin promoter; hFIX, human factor IX; bGH, bovine growth hormone poly(A) signal; TRS, terminal resolution site.
As expected, systemic administration of 1 × 1011 vector genomes (vg) (equivalent to ~4 × 1012 vg/kg, a therapeutically relevant dose in the clinic) of scAAV8.FIX in mice stimulated moderate type I interferon (Ifnb1 and Ifna13) gene expression in the liver at 2 hours post-injection (hpi), the peak time point as previously described (16). In contrast, the same dose of scAAV8.FIX.io1 elicited no detectable innate immune response in the liver (Fig. 1B). scAAV8.FIX.io1 treatment also did not elevate interferon gene expression compared to PBS treatment 4 hpi (fig. S2). Administration of 10-fold lower doses (1 × 1010 vg) of either scAAV8.FIX or scAAV8.FIX.io1 did not trigger interferon responses in the liver, consistent with the immune response being AAV-dose dependent (Fig. 1C). We confirmed with Tlr9−/− and Myd88−/− mice that the innate immune response to self-complementary AAV was indeed dependent on the TLR9/MyD88 pathway (Fig. 1D and 1E), as previously shown in the literature (15–17). Whereas scAAV8.FIX stimulated macrophage infiltration of the liver, scAAV.FIX.io1 avoided immune cell infiltration (Fig. 1F).
When we studied transgene expression, we observed that scAAV8.FIX.io1 enhanced human FIX in plasma nearly 3-fold compared to scAAV8.FIX 28 days post-injection (dpi) (Fig. 1G), consistent with published findings that Tlr9−/− mice expressed more FIX than wild-type mice after AAV treatment and substantiating an important role for TLR9-mediated immune responses in AAV transgene expression (16). In experimental conditions where an innate immune response was not triggered (administration of 10-fold lower doses of AAV or administration to Myd88−/− mice), scAAV8.FIX.io1 did not lead to an enhancement in FIX expression (Fig. 1H and 1I), suggesting that scAAV8.FIX and scAAV8.FIX.io1 do not have inherently different potencies. We observed the development of comparable amounts of neutralizing antibody (NAb) titers at 28 dpi against AAV8 for both scAAV8.FIX and scAAV8.FIX.io1, in agreement with previous observations that Tlr9−/− and wild-type mice develop similar antibody titers by this time point (16) (fig. S3A–D). Together, these results indicated that incorporation of io1 into an scAAV vector evaded innate immune responses in the liver and enhanced transgene expression following intravenous administration.
Incorporation of io2 into an AAVrh32.33 vector reduces anti-capsid T cell responses and cytotoxic T cell infiltration in mouse muscle and enhances gene transfer
Whereas dose-dependent immune responses are well-appreciated in systemic AAV applications in the clinic, robust T cell responses are generally not observed in mice upon intravenous AAV administration. Similarly, in our intravenous scAAV8.FIX experiments, we did not detect appreciable CD8+ T cell responses against the AAV8 capsid in an IFN-γ enzyme-linked immune absorbent spot (ELISpot) assay (fig. S4). To test the effect of our strategy on anti-capsid T cell responses, we modeled a more immunogenic condition in mice by utilizing intramuscular delivery and the AAVrh32.33 capsid (hereafter referred to as rh32.33), a uniquely immunogenic capsid in mice. This combination has been shown to elicit robust CD8+ T cell responses against rh32.33 capsid and local infiltration of cytotoxic T cells into muscle in a TLR9-dependent manner (17, 24). We tested single-stranded AAV vectors, which are more widely used because of their larger coding capacity. We observed that at a dose of 1 × 1010 vg injected into the quadriceps muscle, rh32.33.GFP triggered a range of CD8+ T cell responses against an immunodominant capsid epitope at 21 dpi, with 7 of 10 animals showing positive T cell reactivity (Fig. 2A, left panel). In contrast, rh32.33.GFP.io2, which harbors inflammation-inhibiting oligonucleotide 2 (io2) designed for single-stranded vectors (see table S1 for sequence and fig. S5A for design considerations and vector organization), showed little CD8+ T cell response (1 of 10 animals positive) and was not statistically different from PBS treatment. We confirmed with Myd88−/− mice that development of CD8+ T cell responses against rh32.33 capsid depended on MyD88(15, 17) (Fig. 2A, middle panel). Cytotoxic T cell infiltrates have been observed in muscle biopsies of patients receiving intramuscular AAV gene therapy (25–27). Thus, we further characterized immune cell infiltration into the local tissue environment. We observed robust CD8+ T cell infiltration into muscle samples from rh32.33.GFP-injected animals, and approximately one-third were positive for granzyme B − a marker for activated cytotoxic T cells − while no CD8+ T cell infiltration was observed in any of the eight rh32.33.GFP.io2-injected muscle samples, suggesting that the presence of io2 prevented capsid-directed CD8+ T cell responses and infiltration (Fig. 2B–D).
Figure 2. Immune responses to single-stranded vectors in mouse skeletal muscle in vivo and human PBMCs in vitro.
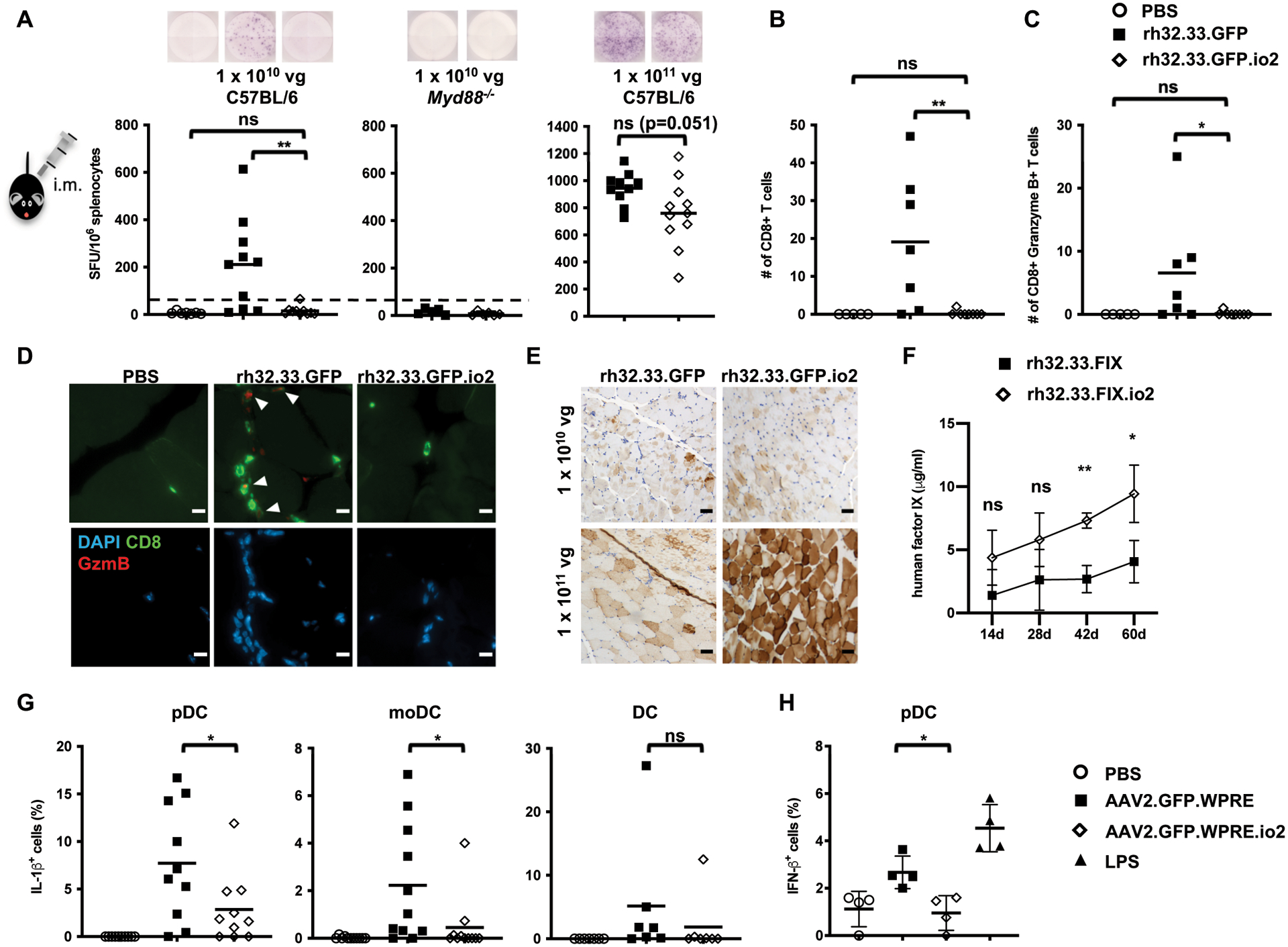
(A) CD8+ T cell responses to rh32.33 capsid 21 d after intramuscular injections. Representative images of the ELISpot well are shown. Dotted line (50 SFU/106 splenocytes) indicates cutoff for a positive T cell response. (B) Number of CD8+ T cells in the muscle sections (four fields per sample) of C57BL/6 mice for PBS and 1 × 1010 vg rh32.33 vectors. (C) Number of CD8+ Granzyme B+ T cells in the muscle sections (four fields per sample) of C57BL/6 mice for PBS and 1 × 1010 vg rh32.33 vectors. (D) Representative immunohistochemical images of CD8 and Granzyme B staining in the muscle sections of C57BL/6 mice for PBS and 1 × 1010 vg rh32.33 vectors. White arrows indicate double positive cells. Scale bar, 10 μm. (E) Representative images of GFP expression (brown) by immunohistochemistry staining in muscle sections of C57BL/6 mice. Scale bar, 50 μm. (F) Human FIX in plasma of C57BL/6 mice at indicated time points following 1 × 1011 vg rh32.33 vector administration. Data shown are mean ± s.d. of n = 4 animals per condition. (G) Intracellular cytokine staining of IL-1β in specific DC populations 24 h after infection of primary human PBMCs from different donors (n = 13). (H) Intracellular cytokine staining of IFN-β 24 h after infection of primary human PBMCs from different donors (n = 7). Some donor PBMCs did not respond to AAV or LPS stimulation (no innate immune response induced over PBS-treated samples) and are not shown. Data shown are mean, with each symbol representing an animal or a donor. n = 4 – 13 animals or donors per condition as indicated. *p<0.05 and **p<0.005 by two-tailed Mann-Whitney test for (A-D), two-way ANOVA with Sidak’s post-hoc test for (F), two-tailed Wilcoxon matched-pairs signed ranked test for (G), and two-tailed Student’s t-test for (H). ns, not significant, p>0.05. SFU, spot forming units.
At a higher dose of 1 × 1011 vg, both rh32.33.GFP and rh32.33.GFP.io2 elicited extremely high capsid-directed T cell responses (~800 – 1000 SFU/million splenocytes) (Fig. 2A, right panel). rh32.33.GFP.io2 muscle sections exhibited higher GFP expression than those with the unmodified vector by immunohistochemical analysis, suggesting that io2 might be partially protective against T cell responses at the higher dose (Fig. 2E). To facilitate quantification of transgene expression, we tested a similar dose of a single-stranded vector expressing human FIX under the control of a ubiquitous CAG promoter (rh32.33.FIX or rh32.33.FIX.io2) intramuscularly, and likewise observed that the engineered vector augmented human FIX in vivo (Fig. 2F). Together, these results indicated that incorporation of io2 into a single-stranded AAV vector reduced CD8+ T cell responses and infiltration and enhanced transgene expression following intramuscular AAV administration.
Engineered AAV2 vector diminishes inflammatory and innate immune responses in primary human plasmacytoid and monocyte-derived dendritic cells
Plasmacytoid dendritic cells (pDCs) are critical for mounting a TLR9/MyD88-dependent early innate immune response to AAV vectors (15) and they subsequently orchestrate anti-capsid CD8+ T cell responses in vivo by activating conventional DCs to cross-present capsid antigen (18). To further evaluate the effect of our TLR9-inhibitory approach on human innate immune responses, we tested primary human peripheral blood mononuclear cells from thirteen healthy donors with single-stranded AAV2 vectors (termed AAV.GFP.WPRE to distinguish it from the previous AAV.GFP vector) in vitro (28) (fig. S5B). AAV2.GFP.WPRE.io2 elicited lower IL-1β cytokine responses than the unmodified vector in pDCs from nine out of ten donors whose cells responded to AAV infection, and in monocyte-derived dendritic cells (moDCs) from nine out of eleven donors (Fig. 2G, fig. S6). Cells from only a few donors responded to AAV infection with IL-6 cytokine responses and we did not observe statistically significant differences between the two vectors (fig. S7). Furthermore, we found that AAV2.GFP.WPRE.io2 also elicited reduced IFN-β cytokine responses compared to AAV2.GFP.WPRE (Fig. 2H). These results underscored the value of our approach in evading innate immune responses in human pDCs.
Engineered vector reduces ocular T cell infiltration while augmenting number of transgene-expressing retinal cells and expression of transgene per cell in mice
The eye is often described as “immune-privileged” due in part to the presence of a blood-retina barrier that limits the entry of immune cells and a microenvironment rich in immunosuppressive factors. However, AAV dose-dependent intraocular inflammation and a few instances of persistent loss of visual acuity have been independently reported in clinical trials following subretinal AAV administration despite prophylactic immunosuppression (9–11). In addition to safety considerations, ocular inflammation also may reduce efficacy directly by inducing pathology in retinal cells, or indirectly by mandating the use of lower, sub-optimal doses.
To test if our TLR9-inhibitory strategy could be beneficial for ocular gene therapies, we started with intravitreal administration in mice, a more immunogenic route of administration in the eye compared to subretinal administration, but sparsely studied for immune responses beyond neutralizing antibody formation. Following intravitreal injection of 1 × 1010 vg of AAV2 vectors (fig. S5B), in vivo retinal imaging with non-invasive optical coherence tomography (OCT) clearly detected local inflammation ~10 dpi, featuring subtle optic disc swelling, retinal vascular changes and cellular infiltration of the vitreous cavity (29) (Fig. 3A and fig. S8A). This inflammatory response is consistent with the anatomical distribution and phenotype seen in ocular clinical trials but has not been reported before in mice (5). We dissociated eye tissues for flow cytometry analysis and surprisingly observed robust CD45+ leukocyte infiltrate, comprising CD3+ T cell and CD8+ T cell populations. These results are consistent with adaptive T cell responses in the eye. We took advantage of this model to test our strategy and found that AAV2.GFP.WPRE.io2 reduced ocular T cell numbers compared to AAV2.GFP.WPRE (Fig. 3B, fig. S8B and fig. S9).
Figure 3. Engineered vector reduces retinal infiltration and achieves greater GFP transgene expression following intravitreal injection in mice.
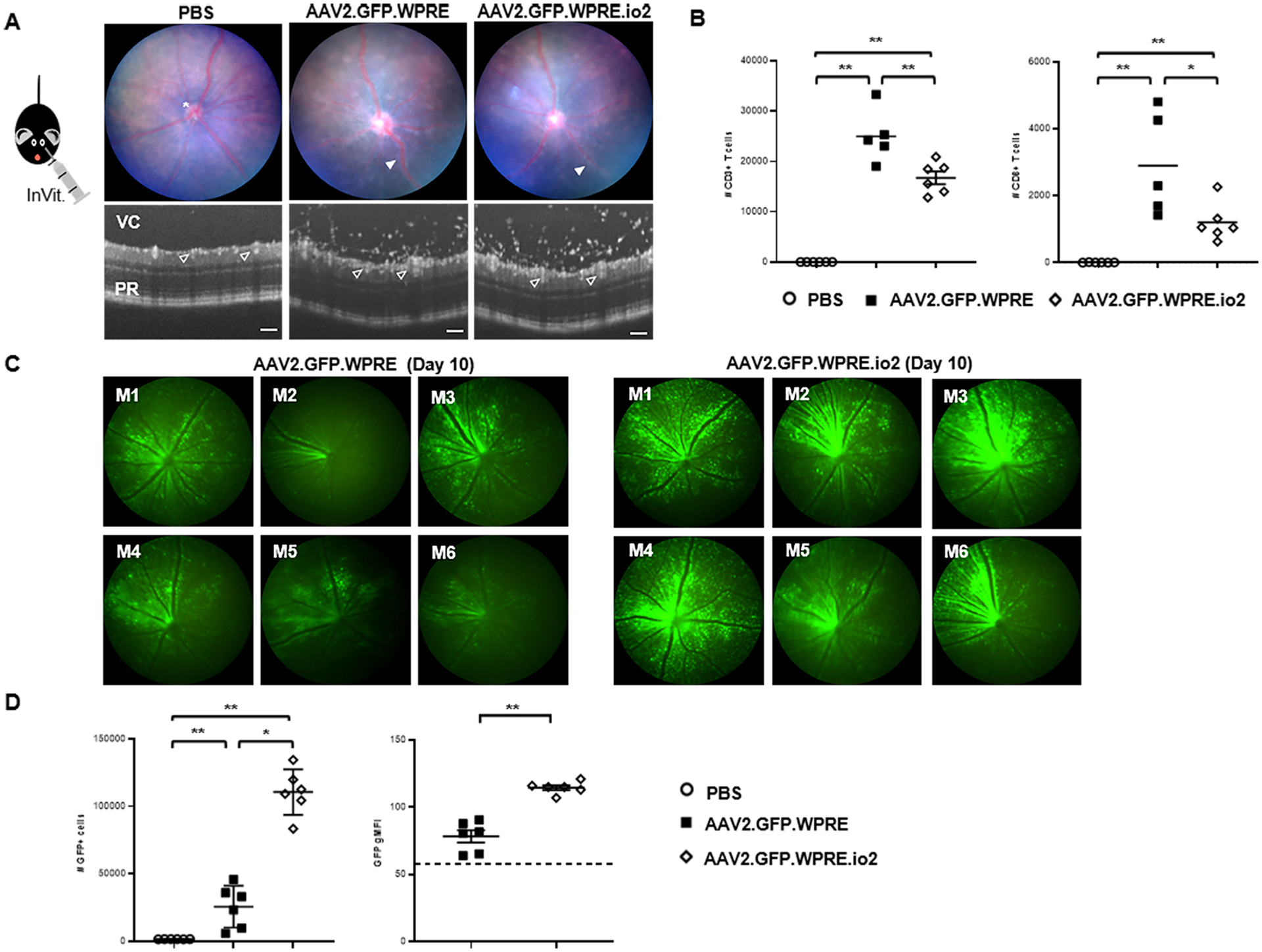
(A) Representative fundal and circular OCT scans of the retina 10 days after intravitreal injection with 2μl of titer-matched (5 × 1012 vg/mL) AAV2.GFP.WPRE, AAV2.GFP.WPRE.io2 or PBS control. Scale bars, 100 μm. Image annotations: optic disc (*), vasculitis (solid white arrow), vitreous cavity (VC), retinal vessels (open white arrow), photoreceptor layer (PR). By OCT, infiltrating cells entrapped within the optically empty vitreous gel above the retinal tissue are visualized as white dots. (B) At day 11 post-injection, eyes were dissected and processed by flow cytometry to identify and enumerate the absolute number of CD45+CD3+ and CD45+CD3+CD8+ T cell populations (n = 5–6 mice/group). Data shown are mean ± s.d., with each symbol representing a single eye. *p<0.05 and **p<0.005 by two-tailed Mann-Whitney test. (C) Standardized fluorescent in vivo retinal images captured on day 10 from AAV2.GFP.WPRE or AAV2.GFP.WPRE.io2 groups (n = 6 mice/group). (D) Flow cytometry of single retinas confirms higher numbers of GFP+ retinal cells and increased GFP geometric mean fluorescence intensity (gMFI) per individual cell. Dotted line indicates background (gMFI from normal retinal cell autofluorescence in PBS-injected eyes). Data shown are mean ± s.d., with each symbol representing a single eye. n = 6 eyes per group as indicated. *p<0.05 and **p<0.005 by two-tailed Mann-Whitney test.
Crucially, incorporation of io2 resulted in earlier, more extensive and greater sustained expression of GFP transgene in vivo observable by longitudinal intravital retinal imaging (Fig. 3C and fig. S10A). Flow cytometry analysis of dissociated retinas confirmed a multi-fold boost in the number of GFP+ retinal cells, and further revealed that GFP mean fluorescence intensity was higher on a per cell basis (Fig. 3D and fig. S10B). In addition, expression of the GFP transgene did not appear to impact the overall viability of retinal cells (fig. S10C). These observations on the magnitude and kinetics of transgene expression imply that evasion of TLR9-mediated immune responses enhanced viral transduction and/or survival of transduced retinal cells. Together, our results indicated that intravitreal AAV administration stimulated ocular T cell infiltration in mice, and that incorporation of io2 diminished local T cell numbers while allowing greater transgene expression by boosting both the number of GFP+ cells and the degree of GFP expression per transduced cell.
Subretinal injection of AAV8 in a pig model elicits cone photoreceptor pathology with microglia and T cell infiltration in the retina, which are evaded by engineered vector
Next, we studied subretinal AAV administration, which is a more commonly used route of administration in ocular gene therapy programs. In the mouse eye, subretinal AAV has been regarded as minimally immunogenic; hence we studied immune responses and pathology in the retina using a pig model, as large animal studies recapitulate intraocular inflammation observed in clinical trials (30, 31), and pig and human eyes share similarities in size and morphology (32). We injected a single-stranded AAV8 vector, AAV8.GFP or AAV8.GFP.io2 (fig. S5A, table S2 and fig. S11), and we selected an intermediate dose of 4 × 1011 vg per eye, based on published reports showing inflammation in patients at 1011 to 1012 vg per eye (9–11). To model a more immunogenic condition, systemic steroid treatment was not used.
Interestingly, we observed that at 6 weeks post-injection (wpi), in all five AAV-injected outbred pigs, AAV8.GFP led to a marked loss, shortening or altered morphology of cone outer segments as labeled by expression of opsins (Fig. 4A and fig. S12A), suggesting AAV-induced pathology in cone photoreceptors, the type of photoreceptors which are critical for high-acuity vision. In contrast, contralateral eyes injected with AAV8.GFP.io2 showed substantially better preservation of cone outer segments and appeared more morphologically similar to un-injected or vehicle-injected eyes, which was verified by quantification of the length of GFP+ cones (fig. S13A). We further confirmed these findings by performing cone arrestin staining which labels the entire cone photoreceptor (fig. S12B). In addition, we observed in two of five animals - animals 23585 and 23586 – apparent loss or retraction of cone pedicles (the synaptic terminals of cone photoreceptors important for transferring the light signal onto the dendrites of bipolar cells and horizontal cells) with AAV8.GFP; no such loss was observed in eyes injected with AAV8.GFP.io2 (see Fig. 4A and fig. S12B).
Figure 4. Engineered vector evades photoreceptor pathology and microglia and CD8+ T cell infiltration in subretinal-injected pig eyes.
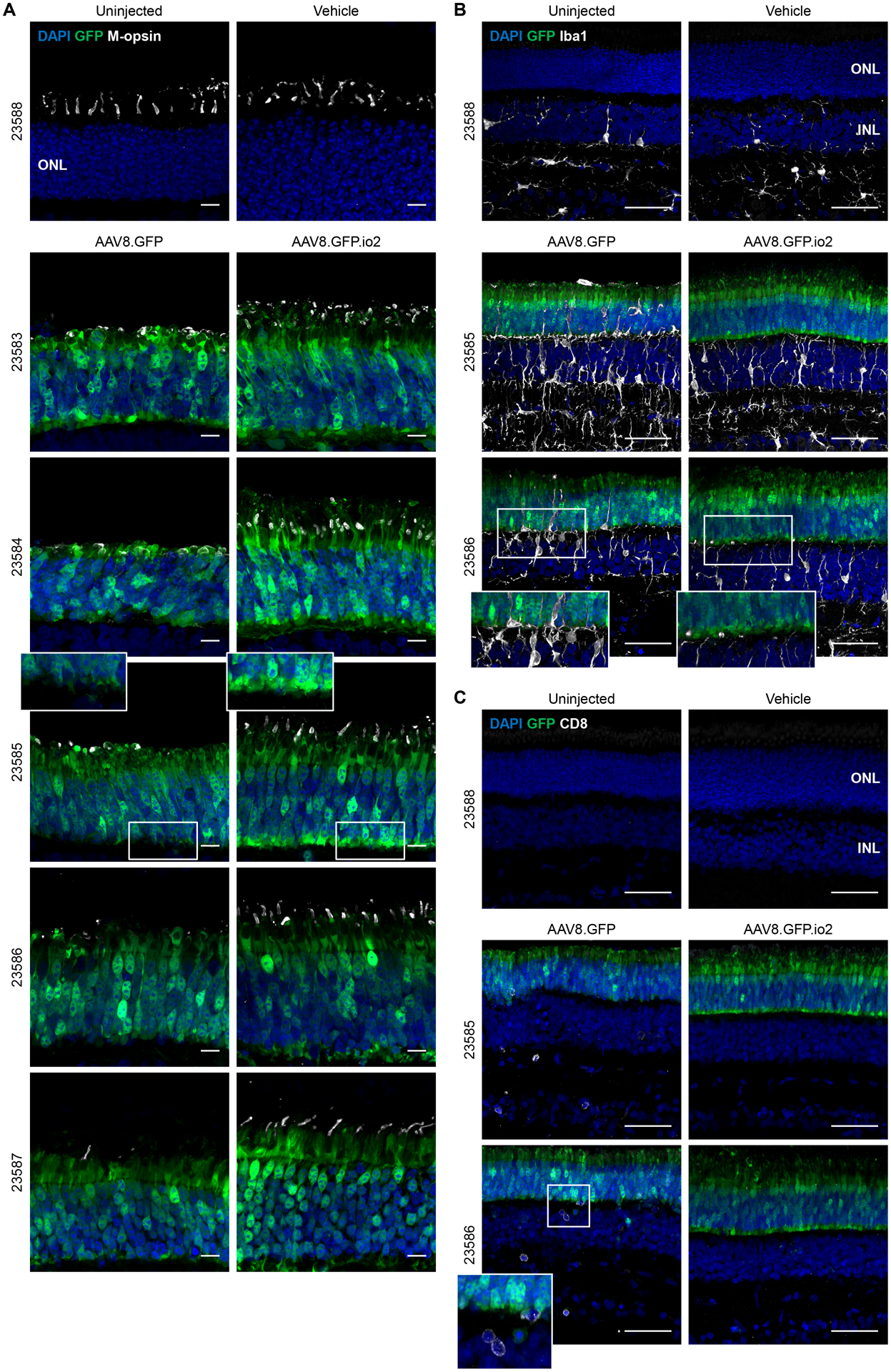
Immunohistochemical images of the ONL of pig retinas 6 weeks after subretinal injections. Each animal is indicated by an identification number and paired images are from the two treated eyes of each animal. (A) Outer segments of cone photoreceptors were visualized by anti-red-green (M) opsin staining. Scale bars, 10 μm. (B) Microglia proliferation and activation in the retina indicated by anti-Iba1 staining. Scale bars, 50 μm. (C) Cytotoxic T cell infiltration into the retina indicated by anti-CD8 staining. Scale bars, 50 μm. ONL, outer nuclear layer; Iba1, ionized calcium-binding adaptor protein 1.
In conjunction with the immunohistochemistry, we examined retinal images from in vivo OCT. The OCT imaging focused on areas of the fundus that began at the optic nerve head and ended ~ 13mm superior to the nerve head and centered between the two major ascending vascular processes. This strategy provided a broad assessment of the extent of damage to outer retinal lamination (see fig. S14 for definitions and examples). At euthanasia, the GFP+ area was located in the dissected eyecup and superimposed on the OCT fundus image (fig. S15B and S15C). In all injected eyes, there was an area of severe laminar damage surrounding the site of the subretinal injection, which was further surrounded by a region of non-severe laminar disruption (fig. S14). In the vehicle-injected retina, the laminar changes evident at 2 wpi were reduced at 6 wpi (fig. S14A and S15A). For two AAV-injected animals (23586 and 23587), GFP+ regions in both eyes were most completely visualized by OCT b-scan images (fig S15B and S15C). We analyzed these eyes as they provided the best estimates of areas of laminar changes over time. In both animals, laminar changes were similar between right (OD) and left (OS) eyes at 2 wpi (fig. S14B, S14C, S15B and S15C). At 6 wpi, both AAV8.GFP.io2-injected right eyes (OD) had reduced areas of severe laminar damage, similar to the vehicle treated eye. In contrast, the large areas of severe laminar damage were maintained in both AAV8.GFP-treated left eyes (OS). In addition, animal 23585 experienced severe vitritis at 2 wpi in its AAV8.GFP-treated eye, but not the contralateral eye treated with AAV8.GFP.io2 (fig. S16 and table S3). Together, these data demonstrated that subretinal administration of higher doses of AAV triggered photoreceptor pathology and the engineered vector reduced the induction of such pathology.
We next undertook a detailed analysis of immune responses at the cellular level in the pig retina at 6 wpi. Microglia are the resident innate immune cells of the retina and reports in the literature suggest that CNS microglia can respond to CpG ligands (33). Iba1 staining of un-injected and vehicle-injected eyes showed a ramified staining pattern outside the outer nuclear layer (ONL), consistent with resting microglia (Fig. 4B). Iba1 signal increased in all AAV-injected eyes (Fig. 4B), suggesting microglia activation and proliferation. Strikingly, the unmodified vector AAV8.GFP stimulated robust microglia infiltration into the ONL (extension of microglial processes into the photoreceptor layer, as well as infiltration of amoeboid-shaped cells, indicative of activated microglia) in animals 23585 and 23586, which was not observed for the engineered vector AAV8.GFP.io2 in the contralateral eyes (Fig. 4B and fig. S13B). In these same animals, we detected elevated numbers of CD8+ T cells infiltrating the outer and inner retina in the eye that received AAV8.GFP, but not in the contralateral eye receiving AAV8.GFP.io2 (Fig. 4C and fig. S13C). Interestingly, while both AAV8.GFP-treated eyes in animals 23585 and 23586 showed substantial microglia and T cell infiltration in the retina via histology, only the former presented with vitritis during clinical examinations, implying that clinically observable inflammation incompletely captures the extent of inflammation, particularly tissue-localized immune responses in the retina. Taken together (see summary in table S3), these data strongly suggest that, unlike the unmodified vector, the engineered vector carrying TLR9-inhibitory sequences could avoid eliciting undesirable innate and adaptive immune cell responses in the retina. We did not observe enhanced GFP expression in photoreceptors of AAV8.GFP.io2-treated eyes compared to AAV8.GFP (fig. S13C), suggesting that kinetics and impact of immune responses in the subretinal space are different than those in other tissues. It is possible that a longer in-life study may yield different results given the striking pathology and immune responses observed in AAV8.GFP eyes.
Engineered vector delays, but does not prevent, intraocular inflammation following intravitreal AAV2 administration in non-human primates
Encouraged by the benefits of io2 incorporation in the intravitreal AAV2 mouse and subretinal AAV8 pig studies, we next performed a non-human primate (NHP) study testing intravitreal administration of a therapeutically relevant vector encoding aflibercept. Aflibercept is composed of the binding domains of two human vascular endothelial growth factor (VEGF) receptors fused with the Fc region of human immunoglobulin gamma 1 (IgG1) and is approved for use to treat wet age-related macular degeneration. Five cynomolgus macaques per group were bilaterally injected with the single-stranded vector AAV2.aflibercept or AAV2.aflibercept.io2 at 1 × 1011 vg or 5 × 1011 vg, with or without prophylactic systemic steroid treatment (fig. S5C, fig. S17 and Fig. 5A). Three macaques received vehicle injections and served as negative controls. Clinical intraocular inflammation in the animals was tracked weekly via ophthalmic examinations and graded according to the SUN criteria system (score of 0 – 4) over the course of the 12-week study. Clinical uveitis was defined as an aqueous cell (AC) or vitreous cell (VC) score of 3 or higher that predicated need for treatment. All animals that experienced clinical uveitis were treated with systemic steroid administration until inflammation resolved.
Figure 5. Engineered vector may delay, but does not prevent, intraocular inflammation following intravitreal AAV2 administration in NHPs.
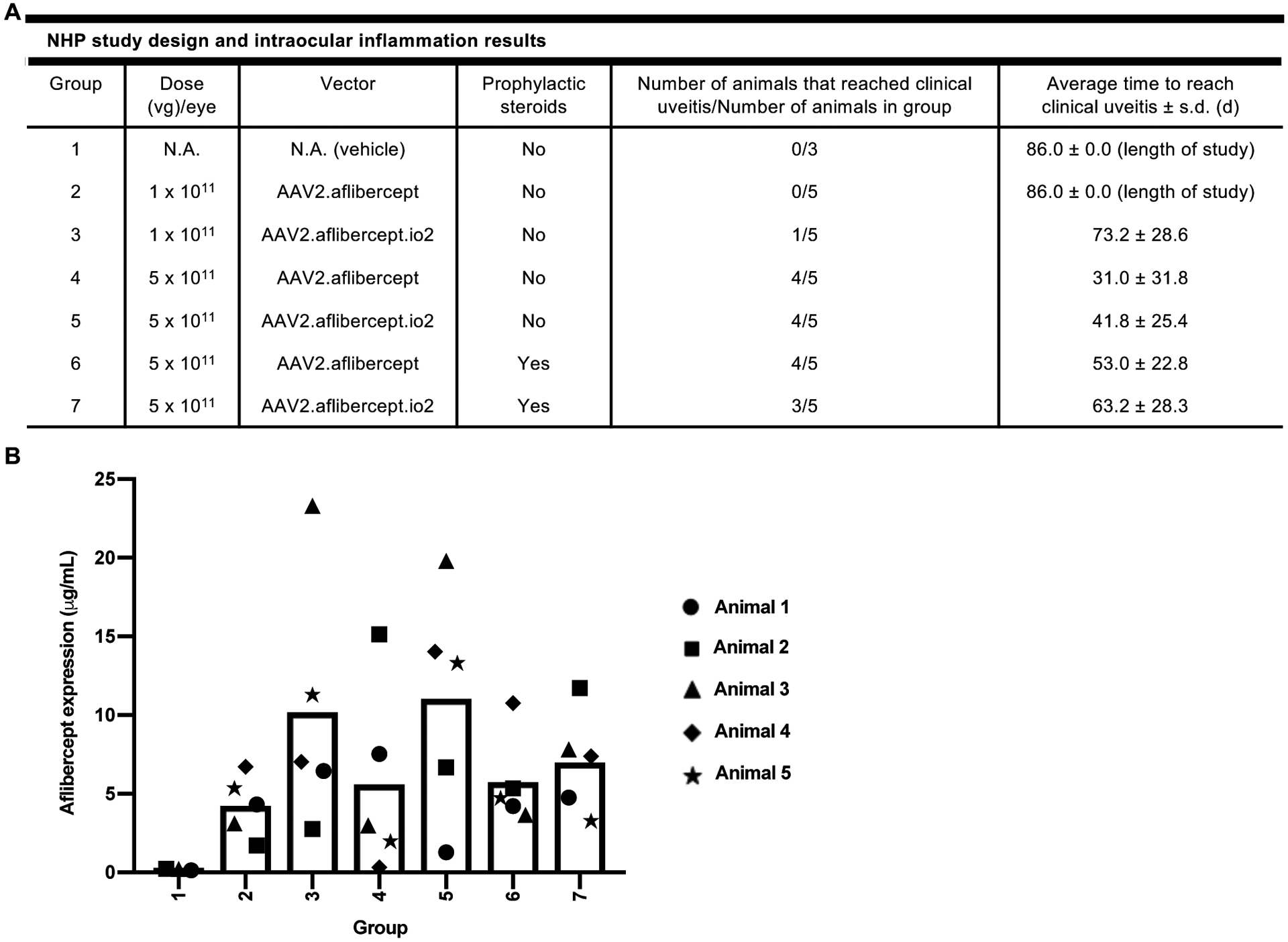
(A) Summary of NHP study design and key intraocular inflammation results. The indicated number of NHPs received bilateral intravitreal injections of AAV2.aflibercept or AAV2.aflibercept.io2 at 1 × 1011 vg or 5 × 1011 vg on Day 1, and intraocular inflammation was scored based on the SUN criteria system during the 12-week study. Group 6 and 7 animals received intramuscular injection of prophylactic systemic steroids on Days −1 and 6. Clinical uveitis was defined as AC or VC score of 3 or higher. A two-tailed Fisher’s Exact Test with stepdown Sidak adjustment was used to analyze number of animals that reached clinical uveitis while a two-tailed generalized linear model was used to analyze average time to reach clinical uveitis. No statistically significant differences were detected between groups. (B) Aflibercept concentrations (μg/mL) in NHP vitreous humors. Aflibercept concentrations were measured in NHP vitreous humor samples collected 12 weeks post-injection. For AAV-treated animals, animals 1 and 2 were male and animals 3 to 5 were female. For vehicle-treated animals, animal 1 was male and animals 2 and 3 were female. A univariate linear regression model with aflibercept expression as the outcome was used to compare vitreous aflibercept concentrations in vector-injected groups. No statistically significant differences were detected between groups.
As expected, uveitis occurred in a dose-dependent manner (Fig. 5A; compare Group 2 to 4 and Group 3 to 5), beginning 2 – 6 weeks after AAV dosing for the majority of animals that experienced inflammation. While AAV2.aflibercept.io2 did not reduce the incidence of clinical uveitis at 5 × 1011 vg (Fig. 5A; compare Group 4 to 5 and Group 6 to 7), incorporation of io2 at this dose delayed clinical uveitis by ~10 days both with and without prophylactic systemic steroids, although these differences did not reach statistical significance. Use of prophylactic systemic steroids similarly failed to reduce the incidence of clinical uveitis, but we did note a delay by ~22 days for both AAV2.aflibercept and AAV2.aflibercept.io2 (Fig. 5A, compare Group 4 to 6 and Group 5 to 7). The combination of io2 and prophylactic systemic steroids gave the latest onset of clinical uveitis at the 5 × 1011 vg dose (Fig. 5A, Group 7), suggesting that the effects of io2 and steroids were additive.
To measure transgene expression, we quantified aflibercept concentration in the vitreous humor of one eye of each animal upon euthanasia 12 weeks after AAV administration (Fig. 5B). We observed substantial variation in aflibercept concentrations among animals of each group and did not detect any statistically significant differences between groups. However, we did note that AAV2.aflibercept.io2 resulted in an approximately 2-fold enhancement in average aflibercept expression compared to AAV2.aflibercept at both the 1 × 1011 vg and 5 × 1011 vg doses (Fig. 5B, compare Group 2 to 3 and Group 4 to 5). The use of prophylactic systemic steroids did not boost aflibercept expression (Fig. 5B, compare Groups 6 and 7 to Groups 4 and 5). Comparing the 1 × 1011 vg and 5 × 1011 vg groups, there was no evidence of a dose-dependent increase in aflibercept concentrations (Fig. 5B; compare Groups 2 and 3 to Groups 4 and 5), in line with a previous intravitreal AAV study in NHPs showing that a 10-fold higher AAV dose only resulted in 2-fold more aflibercept expression (34). Together, these results suggest that the engineered vector might enhance transgene expression and delay clinical uveitis in NHP eyes. However, they also highlight that other immune factors in addition to TLR9 might contribute to intraocular inflammation in this highly immunogenic route of administration and model.
DISCUSSION
Our results demonstrated that the immune-inhibitory activity of short TLR9i sequences remained functional in a much longer strand of DNA, the AAV vector genome. During in vivo gene therapy, AAV can be taken up by TLR9+ immune cells such as plasmacytoid dendritic cells and the vector genome may become exposed during endosomal trafficking and detected by TLR9. We postulate that incorporation of TLR9i sequences reduces the immune response by out-competing CpG motifs in the same vector genome for TLR9 binding, and consequently prevents TLR9 dimerization and activation.
The TLR9-inhibitory approach described here can be easily and readily integrated into existing AAV vectors. To translate our results into the clinic, io1 or io2 could be incorporated into therapeutic vectors and characterized for safety and efficacy in preclinical studies. Furthermore, AAV vectors that have previously experienced robust immunogenicity during clinical testing may be good candidates for incorporating TLR9i sequences. Similarly, self-complementary AAV vectors have been shown to stimulate TLR9 more efficiently than single-stranded vectors in mice and may yield enhanced benefits with the TLR9-inhibitory approach. The described strategy could be further optimized for improved results by exploring the incorporation of more potent TLR9i sequences or more copies of TLR9i sequences, and testing different or multiple locations in the AAV vector genome. Finally, incorporation of TLR9i sequences may be broadly useful for reducing immunogenicity of both viral and non-viral DNA delivery, such as adenoviral vectors and lipid nanoparticles.
To date, many AAV clinical studies have relied on prophylactic or reactive corticosteroids to suppress immune responses such as intraocular inflammation and liver transaminase elevation. In addition, reducing or eliminating CpG content from parts of the AAV vector genome, such as the transgene coding region, to reduce TLR9 activation also has been explored. Future studies are needed to understand how our approach compares to corticosteroid treatment and CpG depletion for different routes of administration and vectors, and whether combining these approaches is beneficial.
There are some limitations to our study and the TLR9-inhibitory approach. Our experimental models tested the effects of io1 and io2 for 4 weeks to 3 months, a time period assumed to give stable or near-stable expression. Longer term studies would provide more information on durability of expression and the observed enhanced expressions. Moreover, while specific antagonism of TLR9 in AAV gene therapy has several benefits which we report in this study, our approach may not translate to complete suppression of all immune responses clinically, depending particularly on dose, route of administration and type of transgene. For example, we observed no impact on the formation of Nabs against AAV capsid and reduced effects on preventing anti-capsid T cell responses in highly immunogenic settings. Furthermore, incorporation of TLR9i sequences suppressed the extent of ocular T cell infiltrate induced by intravitreal AAV in mice, but the response was not eliminated. This may reflect incomplete TLR9 blockade with io2, or a potential influence of other non-TLR9 factors that are operative within and specific to the ocular environment. Future studies should seek to characterize intravitreal AAV-mediated inflammation in Tlr9 and Myd88 knock-out mice. Additionally, the approach may not block anti-transgene immune responses, as TLR9 is not thought to be essential for such responses. Finally, future studies should focus upon immunomodulatory strategies that synergize with the TLR9-inhibitory approach to further mitigate immune responses.
The immunogenicity challenges of intravitreal administration of AAV are well-recognized compared to subretinal administration (35). While the TLR9-inhibitory approach did not prevent clinical uveitis in NHPs at the higher dose of 5 × 1011 vg, our work marks a significant step in recognizing and addressing the inflammation. This may be surmounted in the future by identifying new factors mediating intraocular inflammation and combining immunomodulatory strategies like the TLR9-inhibitory approach. Limitations of the intravitreal NHP experiment include a single experiment of small and underpowered sample size per group to demonstrate a clinically significant effect, substantial intra-group variability in inflammation and expression, and a lack of understanding of inflammation at the tissue level via histopathology or cell immune phenotype characterization. Furthermore, the intravitreal NHP experiment utilized a secreted human transgene protein, and we did not attempt to measure if anti-drug antibodies (ADAs) formed in the NHPs. As there were less positive benefits of our approach with intravitreal delivery in NHPs compared to multiple mouse models and the pig model, it would be prudent to test our approach with other routes of administration in NHPs. Finally, the observation that prophylactic systemic steroids also delayed, but did not prevent, clinical uveitis may have important implications for clinical protocols for intravitreal AAV therapies.
Recent successes in AAV gene therapy in the clinic (4, 36) highlight the potential for in vivo gene therapy to treat multiple types of disorders. However, as the field moves beyond the most devastating diseases, there is a pressing need to address host immune responses (2–8). Furthermore, several ocular AAV gene therapy trials have reported intraocular inflammation at therapeutically relevant doses (see Figure S18 and S19 for analysis). In this study, we sought to develop AAV vectors that are intrinsically less immunogenic by exploiting the linkage of specific immunomodulatory non-coding sequences to much longer therapeutic nucleic acids to “cloak” the AAV genome from immune responses. We directly compared AAV vectors with or without TLR9i sequences in experiments encompassing different capsid serotypes, genome configurations, promoters and transgenes, target tissues, routes of administration and animal models, and showed marked reduction of immune responses without changing any other sequences in several models. This “coupled immunomodulation” strategy may offer a versatile, broadly applicable solution for different AAV therapies without impacting capsid or vector genome elements such as promoter choice, and may be a crucial part of the solution for highly immunogenic AAV therapies. Our approach may widen the therapeutic window of gene therapy, contribute to gene therapy applications, and guide the design of future nucleic acid-based therapeutics.
MATERIALS AND METHODS
Study design
The overall goal of this study was to determine if incorporating TLR9-inhibitory sequences in AAV vector genomes elicited reduced immune responses, and in certain models, increased transgene expression. The general approach was to select an experimental model with known immunogenicity in the literature, and directly compare an unmodified AAV vector to an engineered AAV vector under similar conditions in our laboratories. In most cases, vehicle injections served as a negative control to determine baseline immune responses.
For the intravitreal mouse study, laterality of injected eyes was randomized, and investigators were masked to the vector type throughout intervention and analysis.
For the subretinal pig study, the retinal surgeon performing the injections was blinded to the test article, and similarly, clinical examinations and scoring of inflammation were performed blinded. For the intravitreal NHP study, the veterinary ophthalmologist was masked to the test article when performing injections and scoring of inflammation. All other studies and experiments were not performed blinded.
All animal protocols were approved by respective institutions and committees. Cryopreserved peripheral blood mononuclear cells (PBMCs) from de-identified healthy donors were purchased from commercial providers with written informed consent from the donor and were used in accordance with Declaration of Helsinki principles. Sample size was chosen empirically based on results of previous studies. In general, three to six replicates for each condition was used per experiment, with precise numbers provided in the figure legends. All in vitro experiments were performed at least twice independently. Some data points were excluded from analyses due to: a sample with abnormal amplification curve during qPCR, an IFN-γ ELISpot well with too many spots to count for the plate reader, and lack of immune responses for some PBMC donors. Data file S1 contains all raw data including excluded data points.
Mice
7–9-week-old male C57BL/6J mice were purchased from the Jackson Laboratory. Tlr9−/− mice were a gift of D. Golenbock (UMass), who had originally received the mice from S. Akira (Osaka U.), and were bred at Harvard Medical School. Myd88−/− mice [B6.129P2(SJL)-Myd88tm1.1Defr/J] also were purchased from the Jackson Laboratory, then bred at Harvard Medical School. These mice were housed at Harvard Medical School under specific pathogen free conditions with food and water ad libitum. All mouse experimental protocols performed at Harvard (intravenous and intramuscular AAV experiments in this study) were approved by Harvard Medical School Institutional Animal Care and Use Committee.
6–8-week-old C57BL/6J male mice were purchased from Charles River Laboratories, Oxford, UK and housed at the University of Bristol Animal Services Unit under specific pathogen free conditions with food and water ad libitum. All procedures (intravitreal AAV experiments in this study) were conducted in concordance with the United Kingdom Home Office licence (PPL 30/3045) and were approved by the University of Bristol Animal Welfare and Ethical Review Group. The study also complied with the Association for Research in Vision and Ophthalmology (ARVO) Statement for Use of Animals in Ophthalmic and Vision Research.
AAV vectors
Self-complementary (sc) or single-stranded (ss) AAV vectors were used in this study and all vector genomes were flanked by AAV2 ITRs. Self-complementary AAV vectors lack the terminal resolution sequence in one ITR and allow earlier and increased transgene expression compared to single-stranded vectors. Unless indicated as self-complementary, all vectors were single-stranded. scAAV.FIX was a kind gift from R. Herzog (Indiana University School of Medicine, formerly U. of Florida) and has been described previously (16). scAAV.FIX expressed human factor IX (FIX) under the control of a liver-specific transthyretin mouse promoter and included a bovine growth hormone polyA sequence. AAV.GFP (single-stranded) was originally obtained from the Harvard DF/HCC DNA Resource Core (clone ID: EvNO00061595) and contained a CMV enhancer/promoter, human β-globin intron, eGFP, and β-globin polyA sequence (37). AAV.GFP.WPRE (single-stranded) was a kind gift from L. Vandenberghe (Massachusetts Eye and Ear Infirmary, MEEI) and contains a CMV promoter and woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) for eGFP expression. To distinguish the two single-stranded GFP vectors, we have denoted the former as AAV.GFP and the latter as AAV.GFP.WPRE. The single-stranded AAV.FIX vector was constructed by swapping out the CMV promoter and GFP transgene in AAV.GFP.WPRE for CAG promoter and human FIX respectively. Finally, the single-stranded AAV2.aflibercept vector was synthesized by Genscript and contains the CAG promoter, codon-optimized aflibercept with the human growth hormone signal peptide, and the rabbit β-globin polyA sequence.
To engineer scAAV.FIX, sequences were inserted into the unique KpnI site found immediately 5’ of the transthyretin promoter. 3 copies of ODN TTAGGG (TTTAGGGTTAGGGTTAGGGTTAGGG) were inserted, separated by AAAAA linkers (termed “io1” for inflammation-inhibiting oligonucleotide 1). A widely used ODN TTAGGG (manufactured by Invivogen, catalog code “tlrl-ttag”) harbored an additional T (in bold) compared to published studies and thus was included in the sequence. During the course of this study, Invivogen removed the additional T in their manufactured oligonucleotide (catalog code “tlrl-ttag151”). No differences in TLR9-inhibitory activity were observed with or without the inclusion of the additional T.
To engineer AAV.GFP, KpnI – [3 copies of ODN TTAGGG with linkers (positive sense)] – [3 copies of INH18 with linkers (negative sense)] – NheI (termed “io2” for inflammation-inhibiting oligonucleotide 2) was inserted immediately 5’ of the XbaI site just upstream of the right ITR. Again, AAAAA was used as a linker between copies of ODN TTAGGG and INH18. Similarly, to engineer AAV.GFP.WPRE and AAV.FIX, KpnI-io2 was inserted immediately 5’ of the XhoI site just upstream of the right ITR. AAV2.aflibercept and AAV2.aflibercept.io2 were synthesized in parallel with the io2 sequence positioned immediately 3’ of the KpnI site just upstream of the right ITR. As single-stranded AAV vectors have an equal chance of packaging positive or negative strands of the viral genome, we added both sense and anti-sense TLR9-inhibitory sequences. This strategy ensured that all packaged AAV genomes will carry copies of TLR9-inhibitory sequences in the right orientation.
AAV vectors were packaged into AAV2, AAV8 and AAVrh32.33 capsids and were purified by the viral vector core facility Gene Transfer Vector Core at MEEI/Harvard except for those noted otherwise below. Briefly, adenoviral helper plasmid, rep2-cap packaging plasmid, and transgene plasmid were transfected at a ratio of 2:1:1 (260 μg of adenoviral helper plasmid, 130 μg of rep2-cap packaging plasmid, 130 μg of transgene plasmid) into approximately 80% confluent HEK293 cells with polyethylenimine in ten-layer HYPERFlasks (Corning, CLS10030). PEI Max (Polysciences)/DNA ratio was maintained at 1.375:1 (w/w). The supernatant and cells were collected 72 h after transfection and cell lysates were formed by three sequential freeze-thaw cycles. The viral solution was incubated with 25 U/ml Benzonase (EMD Millipore) at 37°C for 1 h, and NaCl was added to a final concentration of 650 mM, then the viral solution was kept at 4 °C for overnight. Cell debris was removed by high speed centrifugation, then the supernatant was collected and run through Tangential Flow Filtration to concentrate the volume to around 10 mL and then run on an iodixanol gradient. Recovered AAV vectors were washed 3 times with final formulation buffer (FFB: 1 × PBS + 35 mM NaCl + 0.001% PF68) using Amicon 100K columns (EMD Millipore) and concentrated to ~600 – 1000 μl of FFB.
The viral titers were determined by digital droplet PCR (ddPCR) using primers directed against the promoter or poly-adenylation signal regions of the transgene cassette. The purity of vector preps was evaluated by running 1 × 1010 vg (viral genome) on an SDS-PAGE gel. In addition, vector preps had < 1EU/ml of endotoxin using a limulus amebocyte lysate assay (ToxinSensor Chromogenic LAL Endotoxin Assay Kit, Genscript). No substantial differences in viral yield (viral titer × volume) were observed between unmodified vectors and corresponding engineered vectors for >20 purifications, suggesting that insertion of the described sequences did not hamper viral packaging.
AAV2.GFP.WPRE and AAV2.GFP.WPRE.io2 were run on an iodixanol gradient as above and recovered vectors underwent an additional purification step using a heparin column and two washes of 25 ml of PBS plus 0.1 M NaCl as previously described (38). The eluate was then washed with FFB and concentrated as above. These vectors were originally produced for a different large animal study and were used on human PBMCs in vitro as they are highly purified. In addition, during the course of revising the manuscript, more vectors were made at Genethon for human PBMC testing. They were produced by triple transfection method as previously described (39). Briefly, HEK293 cells were transfected and harvested after 72h, followed by sonication and benzonase treatment. Vectors were purified by two successive ultracentrifugation rounds in cesium chloride density gradients. Vector titer was determined by quantitative real-time PCR (qPCR) and confirmed by SDS-PAGE, followed by SYPRO Ruby protein gel stain (Lonza, Rockland, ME). IL-1β/IL-6 staining experiments were performed with Harvard-produced vectors (iodixanol + column purification) and IFN-β staining experiments were performed with Genethon-produced vectors (cesium chloride purification).
AAVrh32.33.FIX and AAVrh32.33.FIX.io2 vectors (for intramuscular AAV mouse experiments), AAV2.GFP.WPRE and AAV2.GFP.WPRE.io2 vectors (for intravitreal AAV mouse experiments) and AAV2.aflibercept and AAV2.alifbercept.io2 vectors (for intravitreal NHP experiment) were produced by transient HEK293 cell transfection and cesium chloride sedimentation by the University of Massachusetts Medical School Viral Vector Core, as previously described (40). Vector preparations were titered by ddPCR, and purity was assessed by 4%–12% SDS-acrylamide gel electrophoresis and silver staining (Invitrogen) or Coomassie staining (Rockland Inc Cat# MB-023-1000). Aflibercept vectors and vehicle, as well as positive and negative control samples, were inoculated in growth media and incubated for 48 h (luria broth agar) or 2 weeks (soybean casein digest and fluid thiolglycollate medium liquid media), and sterility was assessed by observing microbial growth. Endotoxin testing showed < 1 EU/ml of endotoxin.
The percentage of full and empty capsids for preparations of AAV8.GFP and AAV8.GFP.io2 was determined by negative stain electron microscopy. Formvar/carbon-coated copper mesh grids (Electron Microscopy Sciences, Hatfield, PA) were glow discharged and loaded with 5 × 1010 vg of AAV8.GFP or AAV8.GFP.oligo in PBS for 5 minutes. Grids were washed twice in dH2O and stained with 0.5% uranyl acetate. After drying, grids were imaged on an FEI Tecnai T12 transmission electron microscope (FEI, Hillsboro, OR) equipped with a Gatan UltraScan 895 4k CCD (Gatan, Pleasanton, CA). 10 images for each sample were processed in ImageJ and particles counted based on the intensity of uranyl acetate staining. Empty capsids had a distinct dark staining in the center of the capsid. The percentage of full capsids was determined for each image and was reported as the average percentage and standard deviation across 10 images. Negative stain electron microscopy of AAV2.aflibercept and AAV2.aflibercept.io2 was performed at the University of Massachusetts Medical School Viral Vector Core using a 1% uranyl acetate stain. After removing excess stain and drying the grids, 6 images of each vector were processed to determine the percentage of full capsids and was reported as the average percentage with standard deviation across the 6 images.
Mouse liver studies in vivo
Adult C57BL/6J or Tlr9−/− or Myd88−/− mice were injected intravenously with 100 μl PBS or AAV8 self-complementary viruses (1 × 1010 vg or 1 × 1011 vg per animal) by tail vein injection as previously described (16). For innate immune responses, the animals were sacrificed 2 h later and a portion of the right median lobe of the liver was saved in RNAlater solution (Thermo Scientific) and a portion of the left median lobe was fixed in 10% formalin overnight and transferred to 70% ethanol.
For quantitative PCR (qPCR), total RNA was extracted from 10 – 30 mg of mechanically disrupted liver sample by using an RNA extraction kit (OMEGA Bio-Tek). Similar amounts of RNA were reverse transcribed into cDNA with a high-capacity RNA-to-cDNA kit (Thermo Scientific) and cDNA was assayed with quantitative PCR (qPCR) using TaqMan Fast Advanced Master Mix (Thermo Scientific) and commercially available pre-designed primers/probes with FAM reporter dye for the indicated target genes (IDT). Expression for each gene were calculated by normalizing against the housekeeping genes Actb or Gapdh using the ΔΔCT method and expressed as fold expression compared to saline-injected mice. The p values are calculated based on a two-tailed Mann-Whitney test comparing fold expression. All qPCR reactions were run on a realplex4 MasterCycler (Eppendorf).
For histology, liver samples were processed at Beth Israel Deaconess Medical Center histology core facility. The immunohistochemistry staining was performed on paraffin embedded mouse liver tissue sections that were cut, deparaffinized and hydrated before use. Antigen retrieval was performed by boiling the slides for 10 minutes in 10mM sodium citrate in a pressure cooker. The samples were stained for F4/80+ macrophages (1:50, ab16911, Abcam) overnight at 4°C and incubated with goat anti-rabbit IgG conjugated with HRP polymer (ab214880, Abcam) for an hour at room temperature. The slides were developed using DAB (Diaminobenzidine) metal enhanced kit (Vector lab, Cat#: SK4105), counter stained with hematoxylin (Thermo Fisher) and processed through dehydration steps before mounted in Permount mounting media. The slides were observed under the Zeiss Axio Zoom V16 microscope under a 1x/0.25 objective. Five random 25x brightfield images per sample slide were captured by AxioCam 506 digital camera using Zeiss Zen Pro imaging software. The macrophages were counted using the count tool feature in the Adobe Photoshop software and the data was calculated per millimeter square.
For hFIX expression, plasma (EDTA) samples were obtained 14 d and 28 d after AAV administration and hFIX expression was quantified by analyzing diluted plasma samples using an ELISA kit specific for human factor IX (ab188393, Abcam). Plasma from PBS-injected mice gave signals similar to that of a blank control, demonstrating specificity of the kit for human factor IX. The kit had a sensitivity of at least 0.78 ng/ml.
In vitro AAV neutralizing antibody (NAb) assays were performed on plasma samples 28 d after PBS or 1 × 1011 vg AAV8 administration as previously described (41) with the following modifications: plasma samples were heat-inactivated and diluted 1:40 in DMEM followed by five 3-fold serial dilutions. β-galactosidase activities in cell lysates were measured using the Galacto-StarTM One-Step β-galactosidase Reporter Gene Assay System (Thermo Fisher Scientific) on a Synergy HT microplate luminometer (BioTek). The transduction inhibition test samples at each dilution was normalized by comparing to naïve mouse serum (S-3509, Sigma-Aldrich) at the same dilutions.
Mouse muscle studies in vivo
Adult C57BL/6J or Myd88−/− mice were injected intramuscularly (i.m.) with 50 μl PBS or AAVrh32.33 single-stranded viruses (1 × 1010 or 1 × 1011 vg per animal) encoding GFP or hFIX in the left quadriceps muscle. 21 d later, the animals were sacrificed and a portion of the quadriceps was fixed in 10% formalin overnight and transferred to 70% ethanol. For histology, muscle samples were processed at Dana-Farber/Harvard Cancer Center Specialized Histopathology Services and Beth Israel Deaconess Medical Center histology core facilities, embedded in paraffin, and stained for CD8 (1:500, D4W2Z, CST) and granzyme B (1:500, polyclonal, catalog # AF1865, R&D). Antibodies were tagged with AlexaFluor 488 Tyramide or AlexaFluor 647 Tyramide (B40957 and B40958, Thermo Fisher). All immunohistochemistry was performed on the Leica Bond automated staining platform using the Leica Biosystems Refine Detection Kit with citrate antigen retrieval. Muscle sections also were stained for GFP (1:800, polyclonal, catalog # ab6556, Abcam) using the Leica Biosystems Refine Detection Kit (DAB chromogen) with EDTA antigen retrieval. For rh32.33.FIX injected mice, plasma samples were obtained 14 d, 28 d, 42 d and 60 d after 1 × 1011 vg AAV administration and hFIX expression was measured by ELISA as previously described.
IFN-γ T cell ELISpot assays
Spleens were harvested from C57BL/6J or Myd88−/− mice injected i.m. with scAAV8.FIX or AAVrh32.33 single-stranded vectors 21 d post-injection. Spleens were passed through a 70 μm cell strainer (Fisher Scientific), and dissociated cells were spun down. The cell pellet was treated with 1 ml of ACK lysing buffer (Life Technologies) to lyse red blood cells. To determine the number of cells secreting IFN-γ in response to antigenic stimulation, we used an IFN-γ ELISpot assay based on manufacturer’s instructions (R&D Systems). Briefly, 96-well plates were pre-blocked with RPMI growth media for 2 h at room temperature and rinsed twice with PBS. 5 × 105 splenocytes were seeded per well in T cell medium (DMEM supplemented with 10% heat-inactivated FBS, 1% penicillin/streptomycin, 10 mM HEPES buffer, 0.1 mM non-essential amino acids, 2mM sodium pyruvate and 10−6 M beta-mercaptoethanol, with 2 μg/ml of a CD8+ h2-kb restricted dominant epitope of AAVrh32.33 capsid (SSYELPYWM, purchased from Genemed Synthesis) or incubated with PMA/ionomycin as a non-specific positive control for 18 h, followed by staining. For scAAV8.FIX-treated animals, 2 μg/ml of an AAV8-derived immunodominant epitope NSLANPGIA (Genemed Synthesis) was used instead. ELISpot plates were evaluated in blinded fashion (ZellNet Consulting, Inc.) using an automated ELISpot reader system (KS ELISpot reader, Zeiss) with KS ELISpot software version 4.9.16. The plate evaluation process including the setup of optimal reading parameters followed the International guidelines on ELISpot plate evaluation (42).
Mouse intravitreal AAV injection and clinical assessment
Mice were anaesthetised using intraperitoneal injection of 90 μL/10 g body weight of a solution of Ketavet (Ketamine hydrochloride 100 mg/mL; Zoetis Ireland Ltd.) and Rompun (Xylazine hydrochloride 20 mg/mL; Bayer PLC) mixed with sterile water in the ratio of 0.6:1:8.4 respectively. Pupils were dilated with a single drop of 1% w/v Tropicamide (Chauvin Pharmaceuticals) prior to injection. 2μL volume of titer-matched AAV at 5 × 1012 vg/mL was delivered into the intravitreal space via the pars plana, using an operating microscope and a 33-gauge needle on a microsyringe under direct visualisation (Hamilton Company). The contralateral eye of each animal received a control injection of PBS. Immediately following injection, 1% Chloramphenicol ointment (Martindale Pharma, Wooburn Green, UK) was applied topically, and the animals were warmed on a heat-pad for recovery. Laterality of injected eyes was randomized, and investigators were masked to the vector type throughout intervention and analysis.
On selected days post-injection (dpi), pupils were dilated, and mice anaesthetised using isofluorane 2% (v/v) by inhalation for clinical assessment. The Micron IV retinal imaging microscope (Phoenix Research Laboratories) was used to capture OCT scans and color and fluorescence fundal images.
Isolation and flow cytometric assessment of mouse retinal immune cell infiltrate
Each eye was dissected in 100 μl ice-cold PBS with aqueous, vitreous and retina extracted using a limbal incision, lens removal and transfer into a 1.5 mL microcentrifuge tube. The tissue was mechanically dissociated by rapping the tube across a 80-well standard rack ten times before transfer into a 96-well 60 μm cell strainer plate (Merck Millipore). This was centrifuged at 1200 rpm for 5 minutes and the supernatant aspirated, and the remaining cell pellet resuspended in 0.1% BSA FACS buffer and transferred into a 96-well V-bottom plate for immuno-staining.
Cells were incubated with purified rat anti-mouse CD16/32 Fc block (1:50, 553142, [2.4G2], BD Biosciences (BD)) for 10 minutes at 4°C before incubation with fluorochrome-conjugated monoclonal antibodies against mouse cell surface markers including, CD45 [1:1500, 552848, [30-F11], BD), CD3 (1:40, 100355, [145–2C11], BioLegend), CD4 (1:100, 553051, [RM4–5], BD), CD8 (1:200, 126622, [YTS156.7.7], BioLegend), at 4°C for 20 minutes. Cells were washed and resuspended in 7-aminoactinomycin D (7AAD; Thermo Fisher) for dead cell exclusion. Cell suspensions were acquired using a fixed and stable flow rate for 2.5 minutes on a 4-laser Fortessa X20 flow cytometer (BD Cytometry Systems). Compensation was performed using OneComp eBeads (01-1111-41, ThermoFisher). Eight two-fold serial dilutions of a known concentration of AccuCheck Counting Beads (PCB100, ThermoFisher) were similarly acquired to construct a standard curve and calculate absolute cell numbers (43). Analysis was performed using FlowJo software (Treestar) (gating strategy as shown in fig. S9).
Subretinal injection of AAV in domestic pigs
All experimental protocols using pigs were performed at the University of Louisville, were approved by the University of Louisville Institutional Animal Care and Use Committee and adhered to the ARVO Statement for Use of Animals in Ophthalmic and Vision Research. Six 50-day-old wild-type domestic female pigs were purchased (Oak Hill Genetics). Surgery to inject AAV into the subretinal space was performed after a one-week acclimatization period at the University of Louisville AALAC-approved facility. Details have been published previously (44–46). Animals were sedated via intravenous administration of: Ketamine (10 mg/kg) and Dexmedetomidine (0.04 mg/kg) and treated with Atropine (0.25 mg/kg). An endotracheal tube was inserted and through it isoflurane was administered to achieve a surgical plane of anesthesia (1 – 3%). An IV line inserted in the ear vein delivered IV fluids (Lactated Ringers Solution with or without 5% dextrose; 10 – 15 mL/kg/h) to maintain blood pressure and normal glycemia (60 – 140 mg/dL). Body temperature was monitored every 30 min with a rectal thermometer and maintained via a heated procedure table. Heart and respiration rates and oxygen saturation were recorded every 10 min throughout the procedures and anesthesia adjusted to maintain a normal range for these physiological parameters.
After anesthesia and aseptic surgical preparation, we used a vitreoretinal surgical approach to gain access to the subretinal space (between the retina and the pigment epithelium at the back of the eye). A lateral canthotomy was performed to increase exposure in the surgical field. After insertion of an eyelid speculum, two 25 g trocars were placed at 1.5 mm posterior to the limbus; one in superior-nasal and the other in inferior-nasal quadrant. An anterior chamber fluid paracentesis was performed to make space for the injected volume. A light pipe was inserted into one trochar to help visualize the retina. A 41-gauge subretinal cannula needle was placed through the other trochar and used to make a local retinal detachment (bleb) in the dorsal retina, which included injection of inoculum (~75 μl). The inoculum contained either AAV8.GFP.io2 or AAV8.GFP in FFB, or FFB alone (vehicle). We injected either AAV8.GFP.io2 or AAV8.GFP into OD or OS of two pigs in each of three surgery sessions (total of 10 AAV-treated eyes, 1 FFB-injected eye and 1 uninjected control eye). After the injection, the light pipe, needle and the trochars were removed and the entry sites left to self-seal. At the end of surgery, the lateral canthotomy was sutured closed with 4–0 Nylon and antibiotic and steroid ointment was topically administered.
From 2 weeks post-injection (wpi) to 6 wpi, a complete clinical examination assessed the health of the retina at weekly intervals in anesthetized pigs. This included a slit lamp examination to inspect the anterior segment of the eye and to characterize damage to the cornea/lens, indirect ophthalmoscopy to inspect the health of the fundus and fundus photography to document the state of the retina, its optic nerve, the blood vessel pattern, and any damage that resulted from surgical procedures or viral expression. In addition, each eye was scored for inflammation of the eye/retina using the Standardization of Uveitis Nomenclature (SUN) classification (47). The retinal surgeon performing the injections was blinded to the test article, and similarly, clinical examinations and scoring of inflammation were performed blinded.
Prior to surgery and at 2 and 6 wpi, OCT (Bioptigen/Leica Biosciences) was performed to image the retinal layers of anesthesized pigs in vivo. Pupils were dilated and accommodation relaxed with topical applications of 2.5% phenylephrine hydrochloride and 1% Tropicamide. Lid specula held the eyelids open and corneas were wet throughout the imaging with artificial tears (Tears Again, OcuSoft, Inc). Using OCT b-scans we identified the retinotomy site and characterized the lamination pattern of the hypo- and hyper-reflective bands as a function of distance from the retinotomy site in both the axial and lateral dimensions. We defined two types of damage: severe and non-severe. Severe damage were areas where the hyper-reflective bands representing the RPE and photoreceptor inner/outer segments were disrupted. Non-severe disruption were areas where at least one of these hyper-reflective bands was present, but one or both were thinner and thus less defined than in areas without damage. The shape, location and size of these two types of damage were measured using software provided with the OCT system. Specifically, calipers were placed over severe and flanking non-severe damage in b-scans across the fundus and their areas were computed and summed over the entire extent of damage. These areas were then superimposed on the fundus image and the areas of damage compared to the areas of GFP expression (see below).
At 6 wpi (terminus), pigs were anesthetized and euthanized with a solution of Beuthanasia (390 mg pentobarbital sodium, 50 mg phenytoin sodium/ml; 1 mL/5 kg), and their eyes were enucleated. The cornea and lens were removed and the eyecup was dissected and fixed in 4% paraformaldehyde in PBS for 1 h at room temperature and then washed in PBS. Wholemount retinas were examined using a low power fluorescent microscope (Olympus MVX10) and the region of GFP+ expression was located and images acquired and plotted on the fundus images relative to the blood vessels and optic nerve head. The retina was dissected so that the piece used for histology included all of the GFP+ region, as well as GFP- flanking regions.
The pig retinal tissue was cryoprotected in graded sucrose solutions up to 30% sucrose in PBS, then embedded in a 1:1 mixture of 30% sucrose and optimal cutting temperature (OCT) compound (Tissue-Tek) followed by cryosectioning on a Leica CM3050S (Leica Microsystems). Transverse sections of retinal tissue were cut at 20 μm. For immunohistochemistry, tissue sections were first blocked with 5% donkey serum (if the secondary antibody was donkey-origin) or 5% goat serum (if the secondary antibody was goat-origin) in PBS with 0.1% Triton X-100 for 1 h at room temperature. Sections were then stained overnight at 4 °C in blocking solution with primary antibodies against red-green opsin (1:600, AB5405, EMD Millipore), blue opsin (1:200, AB5407, EMD Millipore), human cone arrestin (48) (1:10000), Iba1 (1:200, ab5076, Abcam) or CD8 (1:200, MCA1223GA, Bio-Rad), followed by staining for 2 h at room temperature with goat anti-rabbit, donkey anti-mouse, or donkey anti-goat AlexaFluor 594-labeled secondary antibodies (111-585-144, 715-585-150 and 705-586-147, all from Jackson ImmunoResearch) used at 1:1000 in PBS. Sections were then stained with 4’,6-diamidino-2-phenylindole (DAPI) for 5 min and mounted using Fluoromount-G (Southern Biotech). The slides were examined using a LSM710 laser scanning confocal microscope (Zeiss) with a 40x oil-immersion objective, and image processing was performed using ZEN software and ImageJ. For sections from AAV-injected eyes, care was taken to acquire representative images of GFP+ regions from the dorsal retina near, but not directly at, the retinotomy scar (where there is damage to photoreceptors from the injection). Images from the vehicle control eye were likewise acquired from the dorsal retina near the area of bleb formation. Similar laser settings were used when acquiring images of the two eyes of each animal.
To quantify the morphology of infected photoreceptors, we measured the inner segment plus outer segment (IS+OS) length of GFP+ cones, defined as the distance from the apex of the cone outer segment to the proximal edge of the nucleus. For each eye, the mean IS+OS length was calculated from 30 GFP+ cones over at least three different 20 μm sections. The number of Iba1+ cell bodies in the retina, Iba1+ processes extending into the ONL, and CD8+ cells infiltrating the retina also were quantified as described in fig. S13.
Intravitreal injection of AAV in NHPs
Experimental protocols using cynomolgus macaques (Macaca fascicularis) were performed at Charles River Laboratories and were approved by the Charles River Laboratories Institutional Animal Care and Use Committee. Thirty-three NHPs (2.5–3.5 years of age) with Nab titers in serum below 1:5 were used in the study. For AAV groups (five animals per group), two males and three females were dosed. For vehicle group (three animals), one male and two females were dosed. Animals were assigned randomly by weight to each group to achieve similar group mean body weights.
The vehicle and test articles were administered by a board-certified veterinary ophthalmologist once on Day 1 via bilateral intravitreal injection (1 × 1011 vg or 5 × 1011 vg at a dose volume of 80 μl/eye). The animal was sedated/anesthetized to effect and placed in dorsal recumbency. Topical proparacaine was applied to the eyes, the conjunctival fornices were flushed with a 1:50 dilution of betadine solution/saline, and the eyelid margins were swabbed with undiluted 5% betadine solution. The eyes were draped, wire eyelid speculums were placed, and a caliper was used to mark a spot 3.0 mm posterior to the limbus on the inferotemporal bulbar conjunctiva. The conjunctiva, at the marked spot, was swabbed with undiluted 5% betadine solution. A 31-gauge needle was gently inserted tangentially through the limbus and into the anterior chamber to collect 40 μl of aqueous humor. Conjunctival forceps were used to fixate the globe position while a 31-gauge needle, affixed to the injection syringe, was inserted at the marked spot, through the sclera, and advanced into the vitreous humor. The injection needle was positioned to face the posterior axis of the globe and 80 μl of vehicle or test article was delivered into the vitreous by slowly depressing the plunger. The needle was held in place for 30 seconds to lessen reflux of the injected material. Subsequently, the needle was removed, and the episcleral tissues approximated to the site of insertion were grasped with the conjunctival forceps to further lessen reflux of the injected material. The contralateral eye was prepared, and an identical injection procedure was performed. All animals received bilateral injections of the same test article, except for Animal 5 of Group 7, which only received 5 × 1011 vg AAV2.aflibercept.io2 injection in the right eye due to insufficient dosing solution (left eye was left uninjected). Some animals received prophylactic systemic immunosuppression in the form of an intramuscular injection of methylprednisolone (DepoMedrol 40 mg/animal) on Days -1 and 6.
Ophthalmic examinations were performed on Day 3 and weekly from Week 1 to Week 12 (termination) via slit-lamp biomicroscopy and indirect ophthalmoscopy. Uveitis was scored based on the SUN criteria system, assessing aqueous cells, aqueous flare, vitreous cells and vitreous haze. When AC or VC score was 3 or higher in an animal, the animal was treated with an intramuscular injection of methylprednisolone (40 mg/animal; up to 80 mg/animal if inflammation is severe). The injection was given weekly until AC = 0 and VC ≤ 1.
At termination, ocular tissues, including vitreous humor, from one eye of each animal were collected and frozen, while the other eye was fixed for potential future histological analysis.
ELISA for aflibercept
in vitro expression in HeLa cells
HeLa cells (3 × 104 cells/well in a 96-well plate) were transduced with AAV2.aflibercept or AAV2.aflibercept.io2 at 1 × 103 vg/cell, 1 × 104 vg/cell or 1 × 105 vg/cell in triplicate. Supernatants were harvested 48h post-transduction and analyzed for aflibercept expression (each sample run in duplicate). Briefly, high-binding 96-well ELISA plates (Corning 96 well EIA/RIA plates, Cat# 3590) were coated overnight at 4°C with purified recombinant human VEGF165 (Shenandoah Biotechnology, Cat# 100–44) diluted in DPBS. The next day, excess VEGF165 was removed and wells were washed 4 times with 1x PBS followed by blocking for 1 h at 37°C with 5% BSA in PBS. Blocking solution was removed and HeLa cell supernatants and standards containing aflibercept were added to the wells and incubated for 1 h at 37°C. Following incubation, wells were washed 4 times with 1x PBS. Bound aflibercept was detected using a HRP-conjugated goat anti-human IgG, Fc-specific antibody (Thermo Fisher, Cat# A18829) and ultra TMB substrate (Thermo Fisher, Cat# 34028). A standard curve was prepared for each run using purified aflibercept (BOC Sciences, CAS 862111-32-8) of known concentration diluted in DMEM + 10% FBS.
in vivo expression in NHP vitreous humor
NHP vitreous humor was analyzed as above with the following changes. After blocking solution was removed, NHP samples and standards containing aflibercept were added to the wells and incubated for 1 hour at 37°C. Following incubation, wells were washed 4 times with 1x PBS. Bound aflibercept was detected using a HRP-conjugated goat anti-human IgG, Fcγ fragment-specific antibody (Jackson ImmunoResearch, Cat# 109-035-170) diluted in 3% BSA in PBS and ultra TMB substrate. A standard curve was prepared for each run on each plate using purified aflibercept (BOC Sciences, CAS 862111-32-8) of known concentration diluted in cynomolgus macaque vitreous humor (BioIVT). NHP study samples were tested in 4 separate runs containing 3 dilutions/sample/run in technical duplicates.
Statistical analysis
Indicated statistical testing was performed as appropriate. Unpaired two-tailed Student’s t-tests were used to compare differences between two unpaired experimental groups in oligonucleotide testing in HEK293-TLR9 reporter cells in vitro. A two-tailed Mann-Whitney test or two-way ANOVA with Sidak’s post-hoc test or two-tailed Student’s t-test was used for most in vivo studies, as indicated. A two-tailed Wilcoxon matched pairs signed ranked test was used for human PBMC IL-1β/IL-6 experiments and a two-tailed Student’s t-test for IFN-β experiments. A two-tailed Fisher’s Exact Test with stepdown Sidak adjustment for number of animals that reached clinical uveitis, a two-tailed generalized linear model for average time to reach clinical uveitis, and a univariate linear regression model with aflibercept expression as the outcome were used for analysis of the intravitreal NHP study. A P value of <0.05 was considered statistically significant. No pre-specified effect size was assumed and in general three to six replicates, animals, or donors for each condition was used per experiment.
Supplementary Material
Figure S1. NF-kB response to single-stranded DNA oligonucleotides in reporter cells in vitro.
Figure S2. Innate immune response in mice 4 hpi.
Figure S3. Development of AAV8 neutralizing antibodies (NAb) after vector administration in mice.
Figure S4. CD8+ T cell responses to AAV8 capsid 21 d after intravenous injections in mice.
Figure S5. Engineered single-stranded vectors.
Figure S6. Gating strategy for the different DC subsets.
Figure S7. IL-6 immune responses to AAV2 vectors in primary human PBMCs.
Figure S8. Clinical inflammation in mouse eyes after intravitreal AAV injection.
Figure S9. Flow cytometry gating strategy for ocular infiltrating immune cell populations and GFP+ retinal cells from mice.
Figure S10. Time course of GFP expression and viability after intravitreal AAV injection in mice.
Figure S11. Characterization of AAV8 vectors used in pig study.
Figure S12. Engineered vector evades photoreceptor pathology in subretinal-injected pig eyes.
Figure S13. Quantification of retinal pathology in subretinal-injected pig eyes.
Figure S14. Outer retinal laminar pathology in pigs measured by OCT is consistent with retinal histology.
Figure S15. Engineered vector ameliorates outer retina laminar pathology in subretinal-injected pig eyes.
Figure S16. Vitritis in pig 23585.
Figure S17. Characterization of AAV2 vectors used in NHP study.
Figure S18. Ocular inflammation observed in subretinal AAV clinical trials.
Figure S19. Ocular inflammation observed in intravitreal AAV clinical trials.
Table S1. Oligonucleotide sequences used in this study.
Table S2. Study design to evaluate subretinal delivery of AAV vectors in wild-type pigs.
Table S3. Summary of findings in pig study, including histology and clinical examinations (inflammation scores).
Raw data
ACKNOWLEDGEMENTS
We thank members of the gene therapy community for their helpful discussion and members of the Harvard community for their critical comments on the manuscript. We also thank Dr. Roland Herzog for the gift of scAAV.FIX, Dr. Luk Vandenberghe for the gift of AAV.GFP.WPRE, and Dr. Cheryl Craft for the gift of the antibody against cone arrestin. We would like to acknowledge Dr. Adam Johnson for help with electron microscopy image acquisition, Thomas Ferrante for help with fluorescence microscopy image acquisition, Dr. Lay-Hong Ang for help on mouse immunohistochemistry, the Flow Cytometry Facility at the University of Bristol for assistance with flow cytometry and Dr. L. Sherwood and Dr. K. Powell and the large animal veterinary staff at the University of Louisville for technical support related to the work with the pigs. We also thank Jihye Ko for performing the ddPCR assay of AAV2.aflibercept and AAV2.aflibercept.io2 vectors, Dr. Darryl Patrick, Dr. Maritza McIntyre, Dr. Jeni Ton and Dr. Michael Lochrie for help with the design and execution of the intravitreal NHP study.
Funding:
Support for this work comes from the Wyss Institute for Biologically Inspired Engineering of Harvard University (Y.K.C. and G.M.C.) and NIH (RM1 HG008525, G.M.C.; EY026158, M.A.M.). S.K.W. was supported by a Howard Hughes Medical Fellowship. C.L.C. is an investigator of Howard Hughes Medical Institute. This work also was supported by the European Research Council Consolidator Grant (Grant Agreement No. 617432, F.M.), Kentucky Lions Eye Research Endowed Chair (M.A.M.) and grants from the National Eye Research Centre, (BRI 021, A.D.D.), UK and The Underwood Trust (8064, A.D.D.). The intravitreal NHP study was funded and executed by Ally Therapeutics, a venture capital funded startup.
Footnotes
Competing interests: Y.K.C. and G.M.C. are co-inventors on patent applications filed by Harvard University related to the described work (WO2017214378A1 “Engineered viral vector reduces induction of inflammatory and immune responses” and WO2019094548A1 “Compositions and methods for inhibiting viral vector-induced inflammatory responses”). Y.K.C., J.J.C., T.L. and G.M.C. are co-founders of Ally Therapeutics. G.M.C. also is a founder of Editas Medicine and eGenesis Bio (for the full disclosure list, please see http://arep.med.harvard.edu/gmc/tech.html). Y.K.C., S.K.W., J.J.C., M.K.W. and H.J.K. have consulted for Ally Therapeutics. J.J.C., M.K.W., W.J.N. and T.L. are currently employees of Ally Therapeutics. Y.K.C., J.J.C., M.K.W., W.J.N., T.L., G.G. and G.M.C. have equity holdings or stock options in Ally Therapeutics. A.D.D. has consulted for Ally Therapeutics, Affibody, Novartis and Genentech. C.L.C. is a founder of GenSight Biologics and serves on the Scientific Advisory Boards of the Institute of Ophthalmology, Basel and Sigilon Therapeutics. F.M. is currently an employee of Spark Therapeutics. G.G. is a co-founder of Voyager Therapeutics, Adrenas Therapeutics and Aspa Therapeutics. G.G. serves as a scientific consultant for AAVAA Therapeutics, Aevitas Therapeutics, Ally Therapeutics, CanBridge Pharma., Catalent, Decibel Therapeutics, Iveric bio., Modalis Therapeutics and Saturn Therapeutics. P.W.T. serves as a scientific consultant for Biogen and Shape Therapeutics on work not related to the present study. All other authors declare no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or the supplementary materials. We received the plasmid encoding scAAV.FIX from University of Florida under MTA and the plasmid encoding AAV.GFP.WPRE from Grousbeck Gene Therapy Center at Massachusetts Eye and Ear Infirmary. The plasmid encoding AAV.GFP is available on Addgene via MTA (plasmid #67634). Sequences of io1 and io2 and their insertion sites in these vector plasmids are described in Table S1 and Materials and Methods.
REFERENCES
- 1.Verma IM, Somia N, Gene therapy -- promises, problems and prospects. Nature 389, 239–242 (1997); published online EpubSep 18 ( 10.1038/38410). [DOI] [PubMed] [Google Scholar]
- 2.Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, Richman LK, Wilson JM, Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum Gene Ther 29, 285–298 (2018); published online EpubMar ( 10.1089/hum.2018.015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hordeaux J, Wang Q, Katz N, Buza EL, Bell P, Wilson JM, The Neurotropic Properties of AAV-PHP.B Are Limited to C57BL/6J Mice. Mol Ther 26, 664–668 (2018); published online EpubMar 7 ( 10.1016/j.ymthe.2018.01.018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, Lowes L, Alfano L, Berry K, Church K, Kissel JT, Nagendran S, L’Italien J, Sproule DM, Wells C, Cardenas JA, Heitzer MD, Kaspar A, Corcoran S, Braun L, Likhite S, Miranda C, Meyer K, Foust KD, Burghes AHM, Kaspar BK, Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med 377, 1713–1722 (2017); published online EpubNov 2 ( 10.1056/NEJMoa1706198). [DOI] [PubMed] [Google Scholar]
- 5.Cukras C, Wiley HE, Jeffrey BG, Sen HN, Turriff A, Zeng Y, Vijayasarathy C, Marangoni D, Ziccardi L, Kjellstrom S, Park TK, Hiriyanna S, Wright JF, Colosi P, Wu Z, Bush RA, Wei LL, Sieving PA, Retinal AAV8-RS1 Gene Therapy for X-Linked Retinoschisis: Initial Findings from a Phase I/IIa Trial by Intravitreal Delivery. Mol Ther 26, 2282–2294 (2018); published online EpubSep 5 ( 10.1016/j.ymthe.2018.05.025). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steven Pipe KS, Rajasekhar Anita, Everington Tamara, Poma Allen, Crombez Eric and Hay Charles RM, 101HEMB01 Is a Phase 1/2 Open-Label, Single Ascending Dose-Finding Trial of DTX101 (AAVrh10FIX) in Patients with Moderate/Severe Hemophilia B That Demonstrated Meaningful but Transient Expression of Human Factor IX (hFIX). Blood 130, 604 (2017). [Google Scholar]
- 7.Xiong W, Wu DM, Xue Y, Wang SK, Chung MJ, Ji X, Rana P, Zhao SR, Mai S, Cepko CL, AAV cis-regulatory sequences are correlated with ocular toxicity. Proc Natl Acad Sci U S A 116, 5785–5794 (2019); published online EpubMar 19 ( 10.1073/pnas.1821000116). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dalkara D, Byrne LC, Klimczak RR, Visel M, Yin L, Merigan WH, Flannery JG, Schaffer DV, In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med 5, 189ra176 (2013); published online EpubJun 12 ( 10.1126/scitranslmed.3005708). [DOI] [PubMed] [Google Scholar]
- 9.Bainbridge JW, Mehat MS, Sundaram V, Robbie SJ, Barker SE, Ripamonti C, Georgiadis A, Mowat FM, Beattie SG, Gardner PJ, Feathers KL, Luong VA, Yzer S, Balaggan K, Viswanathan A, de Ravel TJ, Casteels I, Holder GE, Tyler N, Fitzke FW, Weleber RG, Nardini M, Moore AT, Thompson DA, Petersen-Jones SM, Michaelides M, van den Born LI, Stockman A, Smith AJ, Rubin G, Ali RR, Long-term effect of gene therapy on Leber’s congenital amaurosis. N Engl J Med 372, 1887–1897 (2015); published online EpubMay 14 ( 10.1056/NEJMoa1414221). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dimopoulos IS, Hoang SC, Radziwon A, Binczyk NM, Seabra MC, MacLaren RE, Somani R, Tennant MTS, MacDonald IM, Two-Year Results After AAV2-Mediated Gene Therapy for Choroideremia: The Alberta Experience. Am J Ophthalmol 193, 130–142 (2018); published online EpubSep ( 10.1016/j.ajo.2018.06.011). [DOI] [PubMed] [Google Scholar]
- 11.Xue K, Jolly JK, Barnard AR, Rudenko A, Salvetti AP, Patricio MI, Edwards TL, Groppe M, Orlans HO, Tolmachova T, Black GC, Webster AR, Lotery AJ, Holder GE, Downes SM, Seabra MC, MacLaren RE, Beneficial effects on vision in patients undergoing retinal gene therapy for choroideremia. Nat Med 24, 1507–1512 (2018); published online EpubOct ( 10.1038/s41591-018-0185-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, Ozelo MC, Hoots K, Blatt P, Konkle B, Dake M, Kaye R, Razavi M, Zajko A, Zehnder J, Rustagi PK, Nakai H, Chew A, Leonard D, Wright JF, Lessard RR, Sommer JM, Tigges M, Sabatino D, Luk A, Jiang H, Mingozzi F, Couto L, Ertl HC, High KA, Kay MA, Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med 12, 342–347 (2006); published online EpubMar ( 10.1038/nm1358). [DOI] [PubMed] [Google Scholar]
- 13.Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, Chowdary P, Riddell A, Pie AJ, Harrington C, O’Beirne J, Smith K, Pasi J, Glader B, Rustagi P, Ng CY, Kay MA, Zhou J, Spence Y, Morton CL, Allay J, Coleman J, Sleep S, Cunningham JM, Srivastava D, Basner-Tschakarjan E, Mingozzi F, High KA, Gray JT, Reiss UM, Nienhuis AW, Davidoff AM, Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 365, 2357–2365 (2011); published online EpubDec 22 ( 10.1056/NEJMoa1108046). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson JM, Flotte TR, Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy. Hum Gene Ther 31, 695–696 (2020); published online EpubJul ( 10.1089/hum.2020.182). [DOI] [PubMed] [Google Scholar]
- 15.Zhu J, Huang X, Yang Y, The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J Clin Invest 119, 2388–2398 (2009); published online EpubAug ( 10.1172/JCI37607). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martino AT, Suzuki M, Markusic DM, Zolotukhin I, Ryals RC, Moghimi B, Ertl HC, Muruve DA, Lee B, Herzog RW, The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9-dependent innate immune responses in the liver. Blood 117, 6459–6468 (2011); published online EpubJun 16 ( 10.1182/blood-2010-10-314518). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faust SM, Bell P, Cutler BJ, Ashley SN, Zhu Y, Rabinowitz JE, Wilson JM, CpG-depleted adeno-associated virus vectors evade immune detection. J Clin Invest 123, 2994–3001 (2013); published online EpubJul ( 10.1172/JCI68205). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rogers GL, Shirley JL, Zolotukhin I, Kumar SRP, Sherman A, Perrin GQ, Hoffman BE, Srivastava A, Basner-Tschakarjan E, Wallet MA, Terhorst C, Biswas M, Herzog RW, Plasmacytoid and conventional dendritic cells cooperate in crosspriming AAV capsid-specific CD8(+) T cells. Blood 129, 3184–3195 (2017); published online EpubJun 15 ( 10.1182/blood-2016-11-751040). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beutler BA, TLRs and innate immunity. Blood 113, 1399–1407 (2009); published online EpubFeb 12 ( 10.1182/blood-2008-07-019307). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hosel M, Broxtermann M, Janicki H, Esser K, Arzberger S, Hartmann P, Gillen S, Kleeff J, Stabenow D, Odenthal M, Knolle P, Hallek M, Protzer U, Buning H, Toll-like receptor 2-mediated innate immune response in human nonparenchymal liver cells toward adeno-associated viral vectors. Hepatology 55, 287–297 (2012); published online EpubJan ( 10.1002/hep.24625). [DOI] [PubMed] [Google Scholar]
- 21.Ohto U, Shibata T, Tanji H, Ishida H, Krayukhina E, Uchiyama S, Miyake K, Shimizu T, Structural basis of CpG and inhibitory DNA recognition by Toll-like receptor 9. Nature 520, 702–705 (2015); published online EpubApr 30 ( 10.1038/nature14138). [DOI] [PubMed] [Google Scholar]
- 22.Hartmann G, Krieg AM, Mechanism and function of a newly identified CpG DNA motif in human primary B cells. J Immunol 164, 944–953 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Gursel I, Gursel M, Yamada H, Ishii KJ, Takeshita F, Klinman DM, Repetitive elements in mammalian telomeres suppress bacterial DNA-induced immune activation. J Immunol 171, 1393–1400 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Mays LE, Vandenberghe LH, Xiao R, Bell P, Nam HJ, Agbandje-McKenna M, Wilson JM, Adeno-associated virus capsid structure drives CD4-dependent CD8+ T cell response to vector encoded proteins. J Immunol 182, 6051–6060 (2009); published online EpubMay 15 ( 10.4049/jimmunol.0803965). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flotte TR, Trapnell BC, Humphries M, Carey B, Calcedo R, Rouhani F, Campbell-Thompson M, Yachnis AT, Sandhaus RA, McElvaney NG, Mueller C, Messina LM, Wilson JM, Brantly M, Knop DR, Ye GJ, Chulay JD, Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing alpha1-antitrypsin: interim results. Hum Gene Ther 22, 1239–1247 (2011); published online EpubOct ( 10.1089/hum.2011.053). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mingozzi F, Meulenberg JJ, Hui DJ, Basner-Tschakarjan E, Hasbrouck NC, Edmonson SA, Hutnick NA, Betts MR, Kastelein JJ, Stroes ES, High KA, AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood 114, 2077–2086 (2009); published online EpubSep 3 ( 10.1182/blood-2008-07-167510). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferreira V, Petry H, Salmon F, Immune Responses to AAV-Vectors, the Glybera Example from Bench to Bedside. Front Immunol 5, 82 (2014) 10.3389/fimmu.2014.00082). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuranda K, Jean-Alphonse P, Leborgne C, Hardet R, Collaud F, Marmier S, Costa Verdera H, Ronzitti G, Veron P, Mingozzi F, Exposure to wild-type AAV drives distinct capsid immunity profiles in humans. J Clin Invest, (2018); published online EpubOct 22 ( 10.1172/JCI122372). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chu CJ, Herrmann P, Carvalho LS, Liyanage SE, Bainbridge JW, Ali RR, Dick AD, Luhmann UF, Assessment and in vivo scoring of murine experimental autoimmune uveoretinitis using optical coherence tomography. PLoS One 8, e63002 (2013) 10.1371/journal.pone.0063002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramachandran PS, Lee V, Wei Z, Song JY, Casal G, Cronin T, Willett K, Huckfeldt R, Morgan JI, Aleman TS, Maguire AM, Bennett J, Evaluation of Dose and Safety of AAV7m8 and AAV8BP2 in the Non-Human Primate Retina. Hum Gene Ther 28, 154–167 (2017); published online EpubFeb ( 10.1089/hum.2016.111). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reichel FF, Dauletbekov DL, Klein R, Peters T, Ochakovski GA, Seitz IP, Wilhelm B, Ueffing M, Biel M, Wissinger B, Michalakis S, Bartz-Schmidt KU, Fischer MD, Consortium R-C, AAV8 Can Induce Innate and Adaptive Immune Response in the Primate Eye. Mol Ther 25, 2648–2660 (2017); published online EpubDec 6 ( 10.1016/j.ymthe.2017.08.018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sanchez I, Martin R, Ussa F, Fernandez-Bueno I, The parameters of the porcine eyeball. Graefes Arch Clin Exp Ophthalmol 249, 475–482 (2011); published online EpubApr ( 10.1007/s00417-011-1617-9). [DOI] [PubMed] [Google Scholar]
- 33.Olson JK, Miller SD, Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol 173, 3916–3924 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Kiss S, Grishanin R, Nguyen A, Rosario R, Greengard JS, Nieves J, Gelfman CM, Gasmi M, Analysis of Aflibercept Expression in NHPs following Intravitreal Administration of ADVM-022, a Potential Gene Therapy for nAMD. Mol Ther Methods Clin Dev 18, 345–353 (2020); published online EpubSep 11 ( 10.1016/j.omtm.2020.06.007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Verdera HC, Kuranda K, Mingozzi F, AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol Ther 28, 723–746 (2020); published online EpubMar 4 ( 10.1016/j.ymthe.2019.12.010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Russell S, Bennett J, Wellman JA, Chung DC, Yu ZF, Tillman A, Wittes J, Pappas J, Elci O, McCague S, Cross D, Marshall KA, Walshire J, Kehoe TL, Reichert H, Davis M, Raffini L, George LA, Hudson FP, Dingfield L, Zhu X, Haller JA, Sohn EH, Mahajan VB, Pfeifer W, Weckmann M, Johnson C, Gewaily D, Drack A, Stone E, Wachtel K, Simonelli F, Leroy BP, Wright JF, High KA, Maguire AM, Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet 390, 849–860 (2017); published online EpubAug 26 ( 10.1016/S0140-6736(17)31868-8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xiong W, MacColl Garfinkel AE, Li Y, Benowitz LI, Cepko CL, NRF2 promotes neuronal survival in neurodegeneration and acute nerve damage. J Clin Invest 125, 1433–1445 (2015); published online EpubApr ( 10.1172/JCI79735). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Auricchio A, Hildinger M, O’Connor E, Gao GP, Wilson JM, Isolation of highly infectious and pure adeno-associated virus type 2 vectors with a single-step gravity-flow column. Hum Gene Ther 12, 71–76 (2001); published online EpubJan 1 ( 10.1089/104303401450988). [DOI] [PubMed] [Google Scholar]
- 39.Collaud F, Bortolussi G, Guianvarc’h L, Aronson SJ, Bordet T, Veron P, Charles S, Vidal P, Sola MS, Rundwasser S, Dufour DG, Lacoste F, Luc C, Wittenberghe LV, Martin S, Le Bec C, Bosma PJ, Muro AF, Ronzitti G, Hebben M, Mingozzi F, Preclinical Development of an AAV8-hUGT1A1 Vector for the Treatment of Crigler-Najjar Syndrome. Mol Ther Methods Clin Dev 12, 157–174 (2019); published online EpubMar 15 ( 10.1016/j.omtm.2018.12.011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mueller C, Ratner D, Zhong L, Esteves-Sena M, Gao G, Production and discovery of novel recombinant adeno-associated viral vectors. Curr Protoc Microbiol Chapter 14, Unit14D 11 (2012); published online EpubAug ( 10.1002/9780471729259.mc14d01s26). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM, Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis 199, 381–390 (2009); published online EpubFeb 01 ( 10.1086/595830). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janetzki S, Price L, Schroeder H, Britten CM, Welters MJ, Hoos A, Guidelines for the automated evaluation of Elispot assays. Nat Protoc 10, 1098–1115 (2015); published online EpubJul ( 10.1038/nprot.2015.068). [DOI] [PubMed] [Google Scholar]
- 43.Chu CJ, Gardner PJ, Copland DA, Liyanage SE, Gonzalez-Cordero A, Kleine Holthaus SM, Luhmann UF, Smith AJ, Ali RR, Dick AD, Multimodal analysis of ocular inflammation using the endotoxin-induced uveitis mouse model. Dis Model Mech 9, 473–481 (2016); published online EpubApr ( 10.1242/dmm.022475). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noel JM, Fernandez de Castro JP, Demarco PJ Jr., Franco LM, Wang W, Vukmanic EV, Peng X, Sandell JH, Scott PA, Kaplan HJ, McCall MA, Iodoacetic acid, but not sodium iodate, creates an inducible swine model of photoreceptor damage. Exp Eye Res 97, 137–147 (2012); published online EpubApr ( 10.1016/j.exer.2011.12.018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ross JW, Fernandez de Castro JP, Zhao J, Samuel M, Walters E, Rios C, Bray-Ward P, Jones BW, Marc RE, Wang W, Zhou L, Noel JM, McCall MA, DeMarco PJ, Prather RS, Kaplan HJ, Generation of an inbred miniature pig model of retinitis pigmentosa. Invest Ophthalmol Vis Sci 53, 501–507 (2012); published online EpubJan 31 ( 10.1167/iovs.11-8784). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fernandez de Castro JP, Scott PA, Fransen JW, Demas J, DeMarco PJ, Kaplan HJ, McCall MA, Cone photoreceptors develop normally in the absence of functional rod photoreceptors in a transgenic swine model of retinitis pigmentosa. Invest Ophthalmol Vis Sci 55, 2460–2468 (2014); published online EpubApr 17 ( 10.1167/iovs.13-13724). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jabs DA, Nussenblatt RB, Rosenbaum JT; Standardization of Uveitis Nomenclature (SUN) Working Group, Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol 140, 509–516 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li A, Zhu X, Brown B, Craft CM, Gene expression networks underlying retinoic acid-induced differentiation of human retinoblastoma cells. Invest Ophthalmol Vis Sci 44, 996–1007 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. NF-kB response to single-stranded DNA oligonucleotides in reporter cells in vitro.
Figure S2. Innate immune response in mice 4 hpi.
Figure S3. Development of AAV8 neutralizing antibodies (NAb) after vector administration in mice.
Figure S4. CD8+ T cell responses to AAV8 capsid 21 d after intravenous injections in mice.
Figure S5. Engineered single-stranded vectors.
Figure S6. Gating strategy for the different DC subsets.
Figure S7. IL-6 immune responses to AAV2 vectors in primary human PBMCs.
Figure S8. Clinical inflammation in mouse eyes after intravitreal AAV injection.
Figure S9. Flow cytometry gating strategy for ocular infiltrating immune cell populations and GFP+ retinal cells from mice.
Figure S10. Time course of GFP expression and viability after intravitreal AAV injection in mice.
Figure S11. Characterization of AAV8 vectors used in pig study.
Figure S12. Engineered vector evades photoreceptor pathology in subretinal-injected pig eyes.
Figure S13. Quantification of retinal pathology in subretinal-injected pig eyes.
Figure S14. Outer retinal laminar pathology in pigs measured by OCT is consistent with retinal histology.
Figure S15. Engineered vector ameliorates outer retina laminar pathology in subretinal-injected pig eyes.
Figure S16. Vitritis in pig 23585.
Figure S17. Characterization of AAV2 vectors used in NHP study.
Figure S18. Ocular inflammation observed in subretinal AAV clinical trials.
Figure S19. Ocular inflammation observed in intravitreal AAV clinical trials.
Table S1. Oligonucleotide sequences used in this study.
Table S2. Study design to evaluate subretinal delivery of AAV vectors in wild-type pigs.
Table S3. Summary of findings in pig study, including histology and clinical examinations (inflammation scores).
Raw data
Data Availability Statement
All data associated with this study are present in the paper or the supplementary materials. We received the plasmid encoding scAAV.FIX from University of Florida under MTA and the plasmid encoding AAV.GFP.WPRE from Grousbeck Gene Therapy Center at Massachusetts Eye and Ear Infirmary. The plasmid encoding AAV.GFP is available on Addgene via MTA (plasmid #67634). Sequences of io1 and io2 and their insertion sites in these vector plasmids are described in Table S1 and Materials and Methods.