

Abstract
The start codon c.1A>G mutation in KLHL24, encoding ubiquitin ligase KLHL24, results in the loss of 28 N-terminal amino acids (KLHL24-ΔN28) by skipping the initial start codon. In skin, KLHL24-ΔN28 leads to gain of function, excessively targeting intermediate filament keratin-14 for proteasomal degradation and ultimately causing epidermolysis bullosa simplex (EBS). The majority of patients with EBS are also diagnosed with dilated cardiomyopathy (DCM), but the pathological mechanism in the heart is unknown. As desmin is the cardiac homolog of keratin-14, we hypothesized that KLHL24-ΔN28 leads to excessive degradation of desmin, resulting in DCM. Dynamically loaded engineered heart tissues (dyn-EHTs) were generated from human-induced pluripotent stem cell–derived (hiPSC-derived) cardiomyocytes from 2 patients and 3 nonfamilial controls. Ten-fold lower desmin protein levels were observed in patient-derived dyn-EHTs, in line with diminished desmin levels detected in patients’ explanted heart. This was accompanied by tissue dilatation, impaired mitochondrial function, decreased force values, and increased cardiomyocyte stress. HEK293 transfection studies confirmed KLHL24-mediated desmin degradation. KLHL24 RNA interference or direct desmin overexpression recovered desmin protein levels, restoring morphology and function in patient-derived dyn-EHTs. To conclude, presence of KLHL24-ΔN28 in cardiomyocytes leads to excessive degradation of desmin, affecting tissue morphology and function, which can be prevented by restoring desmin protein levels.
Keywords: Cardiology
Keywords: Heart failure, Ubiquitin-proteosome system, iPS cells

Introduction
Inherited cardiomyopathies are prevalent diseases with many cases of unknown etiology. New causative genes are frequently discovered. We believe we were the first to report a link between the heterozygous gain-of-function KLHL24:c.1A>G mutation (Met1Val) causing epidermolysis bullosa simplex (EBS) in skin and dilated cardiomyopathy (DCM) of the heart (1). Following our initial observation, around 33 patients with similar gain-of-function mutations leading to DCM have been reported (2, 3). Despite these publications, the causative effects of gain-of-function mutations in KLHL24 leading to cardiomyopathy have not yet been established.
The KLHL24:c.1A>G mutation is 1 of the 5 described methionine start codon mutations (c.1A>G, c.1A>T, c.2T>C, c.3G>A, and c.3G>T) in ubiquitin ligase KLHL24, resulting in the loss of 28 N-terminal amino acids (KLHL24-ΔN28) by skipping the initial start codon. In skin, it has been shown that KLHL24 functions as an ubiquitin ligase, forming a protein complex with CUL3 and RBX1 that mediates keratin 14 degradation. This process is tightly balanced through KLHL24 auto-ubiquitination. In contrast, the mutated protein KLHL24-ΔN28 is unable to undergo auto-ubiquitination and is consequently more stable. This results in excessive ubiquitination and therefore degradation of keratin 14, ultimately leading to skin fragility (4–7).
Keratins (KRT) belong to the protein family of intermediate filaments (IFs) and are essential for the keratinocyte cytoskeletal network in the skin. IFs provide cells with compliance to small deformations and simultaneously strengthen cells when subjected to stretch (8). Mutations in genes encoding different IFs have been associated with EBS and inherited cardiomyopathies, as they serve major regulatory roles in stretch-bearing tissues, such as skin and heart (9, 10). Desmin (DES) is the essential IF in cardiomyocytes and skeletal muscle cells (11). Desmin filaments extend the z-discs from one myofibril to another, forming a supportive latticework that stretches all the way from the sarcolemma to the nuclear envelope. They position, stabilize, and facilitate sensing and transferring of signals between nucleus, cell membrane, mitochondria, and myofibrils (12). When the IF network in the heart is compromised, mitochondrial dysfunction becomes one of the earliest and most prominent defects (10). Additionally, collapse of the desmin network results in nuclear delamination and intercalated disc abnormalities, which ultimately leads to loss of cardiac function. Indeed, many DES mutations, and mutations in other genes affecting the desmin network, have been implicated in these so-called desminopathies (10, 12–14).
Because desmin belongs to the same highly conserved family of intermediate filament proteins as keratins (15), we hypothesized that KLHL24-ΔN28 leads to excessive degradation of desmin in the heart, causing DCM. To investigate this hypothesis, we used human-induced pluripotent stem cells (hiPSCs) for the differentiation of patient-specific KLHL24WT/c.1A>G cardiomyocytes. To study the function of KLHL24 on cardiac tissue compliance (i.e., challenging the desmin network by stretch), a dynamically loaded engineered heart tissue (dyn-EHT) model was employed (16). Here, a polydimethylsiloxane (PDMS) strip is attached to engineered heart tissues (EHT) to simulate the loading (preload and afterload) experienced by the heart. Bending of the strip can be used to determine tissue contractile force via finite element modeling (FEM). This system can also be dynamically cultured, allowing the observation of the morphological and functional changes that occur during ventricular dilatation, which has been essential in revealing a clinically relevant phenotype in desmoplakin-mutated hiPSC-CMs (16). In the present study, we show that a gain-of-function mutation in KLHL24, leading to KLHL24-ΔN28 and resulting in excessive degradation of desmin in cardiomyocytes, thereby negatively impacting the morphology and function of dyn-EHTs, can be prevented by KLHL24 RNA interference or direct desmin overexpression.
Results
Familial cases of EBS with dilated cardiomyopathy.
For this study, 2 patients of Dutch ancestry were included, both carriers of the KLHL24:c.1A>G mutation (Figure 1A, II:3 and III:2) (1). This mutation leads to a truncated KLHL24 protein, KLHL24-ΔN28 (Figure 1B). Both patients had typical aplasia cutis congenita and skin fragility, diagnosed as EBS, that improved over time. In addition, the index patient (II:3) was diagnosed with a fast-progressing form of DCM at the age of 17, for which he underwent a heart transplantation at the age of 18. His son (III:2), age 18 years at the time of the present study, is still asymptomatic but his echocardiogram showed definite signs of DCM, characterized by left ventricular (LV) dilatation, reduced LV ejection fraction (LVEF), and impaired global longitudinal systolic strain (Figure 1, C and D).
Figure 1. Family tree and clinical characteristics of DCM and EBS.

(A) Family tree showing KLHL24 genotype-phenotype correlations, where +/– indicates KLHL24WT/c.1A>G genotype (heterozygous carrier for mutation c.1A>G in KLHL24) and –/– indicates normal KLHL24WT/WT genotype. NA indicates unknown genotype. Black boxes are members who have an EBS and dilated cardiomyopathy phenotype. (B) Schematic figure depicting the protein domains of KLHL24 and the shorter KLHL24-ΔN28 mutant. BTB: BR-C, ttk, and bab; BACK: BTB and C-terminal Kelch; KR: kelch repeat. (C) Echocardiographic image (long axis parasternal view) of the left ventricle of patient III:2. End-diastolic volume was increased (91 mL/m2; normal value < 75) and ejection fraction was reduced (0.51; normal value > 0.55). (D) Bull’s eye representation of echocardiographic analysis of different regions of the left ventricle with longitudinal strain imaging (normal value –20%) of patient III:2. The apex (central part) is relatively spared; in particular, the basal regions of the left ventricle (outer ring) show decreased strain values, indicating loss of contractile function. The family tree in A was updated after our initial observations (1).
Reduced desmin expression in the explanted heart and the generation of 3D cardiac tissues with the use of hiPSC-derived cardiomyocytes.
In the explanted heart of patient II:3, immunofluorescence analysis (IFA) revealed a lower desmin intensity than was observed in other diseased heart explants, including idiopathic DCM, arrhythmogenic cardiomyopathy, myocarditis, and ischemic heart failure (Figure 2A and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI140615DS1). Furthermore, structure of the intracellular desmin network appeared intact and no desmin-positive aggregates were observed in the explanted heart of patient II:3. hiPSCs of the 2 patients in addition to 1 familial (I:1) and 2 nonfamilial healthy controls were generated and validated (Supplemental Figure 2, A and B). Localization of KLHL24 by IFA was performed in hiPSC-derived control cardiomyocytes (hiPSC-CMs) cultured in 2D, where KLHL24 abundantly localized at the intersection of desmin with desmosomes (Figure 2B and Supplemental Figure 2C).
Figure 2. Analysis of ex vivo heart and morphology of in vitro patient-derived dyn-EHTs.

(A) IFA labeling of desmin in paraffin sections of the explanted heart of patient II:3 with DCM (KLHL24-ΔN28) versus sections of the heart from a patient with idiopathic DCM (named as WT, KLHL24). Scale bars: 50 μm. Panel A is part of a larger panel depicted in Supplemental Figure 1. (B) Co-IFA of KLHL24 with desmin and desmoplakin in control hiPSC-CMs cultured in 2D. The arrows point to the colocalized areas. Scale bars: 10 μm. (C) Side view of contractile EHTs (day 14) right after pulling tissues out of the PDMS molds and after 14 days of dynamic loading dyn-EHTs (day 28). Pictures are representative of all control tissues (KLHL24), derived of 3 control individuals in the upper panels and both patients in the mid and lower panels (KLHL24-ΔN28). Strips are in white and the images are an overlay of shots taken at diastole (red) and systole (green). Scale bars: 1.5 mm. (D) Significant differences in morphological parameters between control- and patient-derived tissues imaged at day 14 (EHTs) and day 28 (dyn-EHTs). n = 11 (d14) and n = 20 (d28) for control-derived tissues; n = 13 (d28) and n = 23 (d14) patient-derived tissues; ****P < 0.0001 (tissue uniformity: unpaired t test, compared with control dyn-EHTs; diastolic length: 2-way ANOVA, post hoc Sidak’s multiple comparison test compared control dyn-EHTs at day 28). (E) IFA labeling of desmin (Y20) and cardiac Troponin T on paraffin sections of control- and patient-derived dyn-EHTs at day 28. Images are representative for n = 4 stainings of tissues/group. Scale bars: 200 um. (F) Western blots depicting affected proteins measured in the 3 control- and 2 patient-derived dyn-EHTs at day 28 (quantified graphs available in Supplemental Material). (G) Overview TEM pictures of control- and patient-derived dyn-EHTs at day 28, emphasizing the swollen-like mitochondria (M) and electron denser glycogen granules (G) observed throughout patient-derived dyn-EHTs. Scales bars: 75 nm.
To study the function of KLHL24 on cardiac tissue compliance, the dyn-EHT model was employed using PDMS strips of 2 different thicknesses (130 μm or 260 μm), resulting in an approximately 8-fold difference in loading (16). Strikingly, culture of control-derived EHTs together with high loading (dyn-EHTs, 260 μm thick strips) resulted in elevated mRNA expression of KLHL24 and DES, compared with dyn-EHTs with low loading using 130 μm thick strips (Supplemental Figure 2D). Given this elevated expression in dyn-EHTs exposed to high loading, all further studies using patient and control-derived hiPSC-CM tissues were performed with 260 μm PDMS strips. Briefly, all EHTs were kept isometrically constrained in the well until day 14 to allow tissue formation (Supplemental Video 1), and dynamically loaded (dyn-EHTs) from day 14 (Supplemental Video 2, control) until day 28 (Supplemental Video 3, control) by removing the strip from its physically constrained environment.
Desmin is 10-fold reduced and affects morphology and function in patient-derived dyn-EHTs.
Morphologically, (Figure 2C and Supplemental Videos 4–7) during dynamic loading, patient-derived dyn-EHTs increased an additional 20% in diastolic length (11.6 ± 0.3 versus 9.5 ± 0.3 mm; P < 0.0001) compared with control (Figure 2D). As a measure of tissue uniformity, we divided the smallest by the largest cross-sectional area of tissues at baseline day 14 and 28. Control-derived tissues had a tissue uniformity ratio of 0.81 ± 0.03, suggesting a relatively uniform tissue cross-sectional area along their length, whereas patient-derived tissues displayed a ratio of 0.49 ± 0.05 (P < 0.0001), indicating presence of very thin sections of tissue flanked by thicker sections (Figure 2D). IFA results in patient-derived dyn-EHTs showed that desmin was able to form normal IF networks (10), similar to control networks, but the intensity of the desmin signal was highly reduced (Figure 2E and Supplemental Figure 3). Quantification of immune-blotting revealed that desmin protein levels were 10-fold lower in patient-derived dyn-EHTs compared with control (P < 0.001), while mRNA levels remained unaffected (Figure 2F and Supplemental Figure 4, A–C). The protein levels of desmin’s binding partners αβ-crystallin, plectin, and desmoplakin were not altered in dyn-EHTs, whereas actin, lamin A, and lamin C were increased (P < 0.05), which could be indicative of an adaptation to prevent nuclear delamination. Furthermore, phosphorylated GSK3β, associated with cardiomyopathy and desmin disassembly (17), was significantly increased in patient-derived dyn-EHTs (Figure 2F and Supplemental Figure 4, B and C). On ultrastructure, transmission electron microscopy (TEM) results showed more swollen-like mitochondria and electron denser glycogen storage vesicles in patient-derived compared with control-derived dyn-EHTs (Figure 2G). TEM also indicated that intercalated discs and myofibrils of patient-derived tissues were similar to control at day 28 (Supplemental Figure 4D).
No differences in functional parameters were observed between patient- and control-derived EHTs when exposed to elevated loading upon initial onset of dynamic culture at day 14 (Figure 3; day 14). However, after 14 days of dynamic loading, striking differences were observed, suggesting that exposure to higher levels of loading and increased culture time are needed to observe the changes between patient- and control-derived tissues (Supplemental Videos 5 and 7). First, dramatic differences in diastolic tissue stress were observed along the length of patient-derived dyn-EHTs, with clear stress concentrations at the regions with decreased thickness. In contrast, a more uniform diastolic tissue stress was observed in control-derived dyn-EHTs (Figure 3A; day 28). The peak contractile stress was in fact 6-fold higher in patient-derived dyn-EHTs (P < 0.001), which occurred because of the reduced cross-sectional area, even though the contractile force was similar to control (Figure 3B). Patient-derived dyn-EHTs furthermore displayed significantly decreased systolic force (0.67 ± 0.01 mN versus 0.76 ± 0.02 mN; P < 0.001), but due to their reduced cross-sectional area, corresponding peak systolic stress levels were 3-fold higher compared with control (P < 0.0001). The same observation was seen in diastolic force (0.61 ± 0.02 mN versus 0.72 ± 0.02 mN; P < 0.0001) and corresponding peak diastolic stress (P < 0.0001) (Figure 3C). Furthermore, the average fractional shortening (FS) during dynamic loading increased up to considerable levels (20%) in control- as well as patient-derived dyn-EHTs (Figure 3D), in agreement with the overall intact myofibril ultrastructure. Similarly to the observed differences in tissue stress and uniformity, regional FS varied widely along the length of patient-derived dyn-EHTs, whereas control-derived dyn-EHTs were quite uniform (Figure 3, D and E). These results suggest that patient-derived tissues develop nonuniform contractile activity upon exposure to dynamic loading.
Figure 3. Functional analysis of patient-derived EHTs/dyn-EHTs.

(A) Representative maps of regional diastolic stress using FEM in patient- and control-derived tissues at the onset and after 14 days of dynamic loading. Arrows point toward the thinnest sections with the highest stress. White dots represent the tissue attachment point with the PDMS strip. Scale bar: 10 mm. (B) Contractile force and corresponding peak contractile stress of control- and patient-derived tissues imaged at day 14 (EHTs) and day 28 (dyn-EHTs). n = 11 (d14) and n = 20 (d28) for control-derived tissues, n = 13 (d28) and n = 23 (d14) for patient-derived tissues. ****P < 0.0001 (2-way ANOVA, post hoc Sidak’s multiple comparison test compared with control-derived dyn-EHTs at day 28). (C) Diastolic and systolic force and corresponding peak diastolic and systolic stress. ***P < 0.001 (compared with control-derived dyn-EHTs at day 28); ****P < 0.0001 (compared with control-derived dyn-EHTs at day 28). (D) The average and regional FS of dyn-EHTs measured as percentages, where speckle tracking analysis is used to determine the latter. *P < 0.05 (2-way ANOVA, post hoc Sidak’s multiple comparison test comparing smallest diameters of dyn-EHTs at day 28). (E) Correlation analysis shows a negative correlation between FS and tissue diameter in patient-derived dyn-EHTs only.
Gain-of-function ubiquitin ligase KLHL24-ΔN28 targets desmin for excessive proteasomal degradation.
Desmin is a substrate for many different forms of posttranslational modification. Most of these lead to disassembly of filamentous desmin, but only ubiquitination leads to desmin degradation (18). First, we investigated high molecular weight (HMW) desmin bands in control- and patient-derived dyn-EHTs, using an antibody raised against the c-terminal epitope of desmin (clone Y66) that is unlikely to be masked by ubiquitinated site chains. As expected, the 52 kDa desmin band was highly reduced in patient-derived dyn-EHTs, while an increase was observed in HMW desmin bands, indicative of desmin ubiquitination (Figure 4A). Quantification revealed a 2.3-fold elevation of HMW desmin bands in patient-derived compared with control-derived dyn-EHTs (P < 0.001) and the ratio of HMW desmin/desmin was 15-fold higher in patient-derived dyn-EHTs (P < 0.001) (Supplemental Figure 5A). Second, by immunoprecipitation of desmin, we investigated direct ubiquitinated levels in hiPSC-CMs, which demonstrated an increase of ubiquitinated desmin in patient-derived extracts (Figure 4B). Transfection studies were performed to substantiate specific targeting of desmin by the KLHL24/RBX1/CUL3(4)–ubiquitin ligase complex. In HEK293A cells, no endogenous desmin or KLHL24 could be observed (Figure 4C). Transfection of cells with either desmin or KLHL24 plasmids successfully resulted in detection of exogenous protein. To study the impact of WT and truncated (KLHL24-ΔN28) KLHL24 proteins on desmin levels, we performed equal ratio cotransfections using desmin and ubiquitin ligase factors RBX1 and CUL3 in either the presence or absence of KLHL24 or KLHL24-ΔN28 plasmids. Coexpression with KLHL24-ΔN28 resulted in a strong reduction in desmin protein levels. This effect was pronounced when WT KLHL24 was coexpressed, and absent when a catalytically inactivate form of KLHL24-ΔN28 was coexpressed (Figure 4, C and D, and Supplemental Figure 5, B and C). Proteasomal inhibition, via bortezomib treatment, prevented the subsequent ubiquitination-induced desmin degradation, as shown by immune blot (Figure 4E). As the expression of KLHL24 seemed too excessive, we adjusted the desmin/KLHL24 transfection ratio from 1:1 to 5:1, in order to observe the difference in desmin ubiquitination between WT and mutant KLHL24. After performing immunoprecipitation of flag-tagged desmin, we observed the strongest expression of ubiquitinated desmin in the KLHL24-ΔN28 group, like in hiPSC-CMs (Supplemental Figure 5D). Based on these results, we have shown that loss of the first 28 amino acids of the KLHL24 protein leads to a more stable and therefore more abundant KLHL24 protein that ubiquinates desmin excessively.
Figure 4. Desmin ubiquitination levels in cardiomyocytes and HEK293A transfection studies.

(A) Western blots depicting the c-terminal-tagged desmin (Y66), showing both the HMW desmin levels as well as the normal (52 kDa) desmin levels in control- and patient-derived dyn-EHTs. NC, negative control; extracts of skin fibroblasts. Quantified levels are in the supplement. (B) Separate Western blots of desmin (Y66; rabbit) and ubiquitin (FK2; mouse) on different desmin immunoprecipitation fractions (IP antibody RD301; mouse) of hiPSC-CMs. The input levels used for the IP are depicted below on a separate gel containing desmin and GAPDH. (C) IFA staining of desmin and KLHL24 under different combinations of plasmid cotransfections in HEK293A cells (desmin/KLHL24 > 1:1 transfection ratio). Scale bars: 25 μm. (D) Western blots representative of the same cotransfection set-up depicted in (C). As all KLHL24 containing plasmids (KLHL24, KLHL24-ΔN28, and KLHL24-ΔN28–inactive) are flag-tagged, blots depict antibodies directed against both flag-tag and KLHL24 protein. Wild type KLHL24 protein runs at 68 kDa, whereas, KLHL24-ΔN28 and inactivated KLHL24-ΔN28 run at 65 kDa. Vinculin poses as loading control. All data are representative of 3 individual transfection experiments. Quantifications are in the supplement. (E) Western blots depicting the addition of bortezomib (400 nM Bz) to the cotransfections of CUL3, RBX1, and desmin with either KLHL24 and KLHL24-ΔN28 (desmin/KLHL24 > 1:1 transfection ratio). Numbers depict the ratio of desmin/vinculin.
Restoration of desmin protein levels rescues DCM phenotype in patient-derived dyn-EHTs.
To investigate if KLHL24 RNAi would restore desmin protein levels in patient-derived hiPSC-CMs, we generated short hairpin-mediated nontargeted (shNT) and KLHL24 (shKLHL24) mRNA knockdown hiPSC lines, using lentiviral constructs (Supplemental Figure 6A). To prove that desmin is a major contributor to the phenotype, we additionally generated patient hiPSC lines with a desmin overexpression construct (oeDES) (Supplemental Figure 6A). These lines were subsequently used for differentiation and generation of dyn-EHTs (Figure 5A and Supplemental Videos 8–10). Both desmin overexpression and shKLHL24 resulted in a 3- to 7-fold increase in desmin protein levels, respectively (P < 0.01) (Figure 5B and Supplemental Figure 6, B and C). The excessive tissue thinning observed in patient-derived dyn-EHTs was absent in shKLHL24 and oeDES tissues (P < 0.0001). In addition, both rescue models prevented most of the dilatation (10.4 ± 0.3 mm in shKLHL24 and 9.0 ± 0.2 mm in oeDES versus 11.5 ± 0.2 mm in shNT [P < 0.05]; with respect to 9.5 ± 0.3 mm in control [P = NS]). As a result, both the diastolic force (0.71 ± 0.01 in shKLHL24 and 0.70 ± 0.01 in oeDES versus 0.61 ± 0.02 in shNT [P < 0.01]; with respect to 0.72 ± 0.02 mN in control [P = NS]) and systolic force were restored (0.76 ± 0.02 in shKLHL24 and 0.74 ± 0.01 in oeDES versus 0.66 ± 0.01 in shNT [P < 0.05]; with respect to 0.76 ± 0.02 mN in control [P = NS]), fully returning the peak diastolic (P < 0.01) and systolic stress (P < 0.001) to values observed in control-derived dyn-EHTs (P = NS) (Figure 5C). This also resulted in restored peak contractile stress (P < 0.05) and normal variation in regional tissue diastolic stress in patient-derived dyn-EHTs containing KLHL24 RNAi or desmin overexpression, similar to control-derived dyn-EHTs (Figure 5, D and E).
Figure 5. Analysis of patient-derived dyn-EHTs with RNAi of KLHL24 or desmin overexpression.

(A) Representative side view pictures of patient-derived shNT, shKLHL24, and oeDES dyn-EHTs at day 28. Strips are in white and the images are overlays of shots at tissue diastole (red) and systole (green). Scale bars: 1.5 mm. (B) Desmin protein levels in shKLHL24 versus shNT and patient versus oeDES-derived patient dyn-EHTs, measured on blot (quantified graphs in the supplement). (C) Morphological and functional parameters of patient, shNT, shKLHL24, and oeDES compared with control-derived dyn-EHTs (n = ±15), shKLHL24 (n = 15), and oeDES (n = 10) patient tissues at day 28 (dyn-EHTs), relative to control (n = 20). #P < 0.05 (1-way ANOVA, post hoc Sidak’s multiple comparison test of patient shKLHL24 versus shNT dyn-EHTs); ##P < 0.01 (patient shKLHL24 versus shNT); ###P < 0.001 (patient shKLHL24 versus shNT); ####P < 0.0001 (patient shKLHL24 versus shNT); ^P < 0.05 (patient oeDES versus shNT); ^^P < 0.01 (patient oeDES versus shNT); ^^^^P < 0.0001 (patient oeDES versus shNT); **P < 0.01 (patient shKLHL24 versus control); ****P < 0.0001 (patient shKLHL24 versus control). (D) Peak contractile stress values of above dyn-EHTs. *P < 0.05 (1-way ANOVA, post hoc Sidak’s multiple comparison test of patient shKLHL24 versus control); #P < 0.05 (patient shKLHL24 versus shNT); ^^P < 0.01(patient oeDES versus shNT). (E) Representative maps of regional diastolic stress values derived from FEM in patient, shNT, shKLHL24, and oeDES compared with control-derived dyn-EHTs. White dots represent the tissue attachment point with the PDMS strip. Scale bar: 10 mm. (F) Western blots containing OXPHOS complex proteins (I–V) from patient-derived shNT compared with shKLHL24 dyn-EHTs and patient-derived oeDES compared with control-derived dyn-EHTs. mtHSP70 poses here as a specific loading control. Quantifications are in the supplement.
As the mitochondrial morphology was impaired and electron density of glycogen granules was increased in patient-derived dyn-EHTs, we investigated metabolic markers. On blot, shKLHL24 dyn-EHTs showed higher protein levels of oxidative phosphorylation complex III and IV compared with shNT patient-derived dyn-EHTs (P < 0.01), whereas these levels normalized in oeDES dyn-EHTs (Figure 5F and Supplemental Figure 6D). Direct measurements of glycolytic acidification and oxygen consumption rates of hiPSC-CMs corroborated these findings, showing normal basal (P < 0.05) and maximal respiratory capacities (P < 0.01) after rescue, during mitochondrial stress-testing (Supplemental Figure 7). In tissues, PDK4 mRNA expression increased (P < 0.05) in both shKLHL24 and oeDES patient-derived dyn-EHTs, while HADHB expression also increased in oeDES dyn-EHTs (P < 0.001). On the other hand, GLUT4 expression decreased in both rescue groups (P < 0.05), compared with both control and nonrescued patient-derived dyn-EHTs (Supplemental Figure 8). These data suggest that restoration of desmin protein levels enhances beta-oxidation while preventing excessive glucose utilization, as is normal for cardiac muscle.
Discussion
A cardio-cutaneous syndrome is a devastating, rare inheritable disease that affects both the skin and heart (19). In the current study, we explored a novel gene causative of a cardio-cutaneous syndrome, called KLHL24 (1, 2). We demonstrated with the use of 3D cardiac tissue engineering that a gain-of-function mutation in KLHL24 leads to excessive proteasomal degradation of desmin. This results in tissue dilatation, reduced contractile force, increased stress, and impaired mitochondrial function. With the use of RNAi of KLHL24 or overexpression of desmin, the morphology and function could be rescued. These results make KLHL24 RNAi a well-suited potential therapeutic strategy applicable for patients suffering from this genetic disorder.
A cardio-cutaneous syndrome is caused by dominant or recessive mutations, mostly in genes encoding for 2 desmosomal proteins, namely desmoplakin and plakoglobin (20). Both proteins are incorporated into cell–cell connecting structures called desmosomes and both are commonly shared by cardiac and cutaneous tissues (21). Desmosomes are also known for their intracellular anchorage of the IF network. In that sense, KLHL24 appears to be the first protein that regulates the turnover of both IF types, namely keratins and desmin. In our dyn-EHTs, we observed a difference of 15-fold in the protein ratio of desmin versus HMW desmin. Using immunoprecipitation of desmin, we showed more ubiquitinated desmin in patient-derived hiPSC-CMs than in controls. In addition to the 10-fold reduced functional desmin protein levels found in patient-derived cardiac tissues, our cotransfection studies substantiated the ability of KLHL24 and KLHL24-ΔN28 to degrade desmin through ubiquitination and proteosomal degradation. Overexpression of KLHL24 and KLHL24-ΔN28 in HEK cells clearly resulted in detectable protein levels, running at 68 kDa and 65 kDa respectively. The KLHL24-ΔN28 was more abundantly expressed than its WT counterpart, consistent with previously reported results (6). Our data are also in line with recent publications showing that loss of function mutations in KLHL24 are associated with increased desmin expression in myocardial biopsies and a hypertrophic cardiomyopathy phenotype (22, 23). Although our data indicates that KLHL24 could be detected throughout the cell by IFA, the antibody was not sensitive enough to detect endogenous levels of KLHL24 in hiPSC-CM or dyn-EHTs by immunoblot. However, the antibody was sensitive enough to detect exogenous overexpressed KLHL24. Similar sensitivity issues were encountered in patient primary keratinocytes and skin biopsies in previous studies (4, 5).
Desmin expression in heart muscle is low during development and increases only when cardiomyocytes near terminal differentiation (11). Clear compensatory proteins (e.g., nestin, a marker of early cardiac development; ref. 24) are set in place in the early phase of development, which complicates desmin research using relatively immature hiPSC-CMs (25). Cytoskeletal IFs are believed to be dispensable in 2D monolayer cultures, as their specific purpose is to maintain the integrity of 3D tissue structures, which makes current use of 2D cultures rather limited (24). To some degree, our dyn-EHT model bypassed these limitations by using dynamically loaded, more-mature cardiomyocytes in a 3D tissue environment, making it a well-suited model for genetic diseases that affect the compliance of the desmin network (i.e., desminopathies) (16).
In desminopathies, mitochondrial dysfunction is commonly the first and most prominent defect (10). When complete collapse of the desmin network occurs, nuclear deformations and intercalated disc mislocalizations follow, triggering apoptosis and fibrosis. Notably, in patients and in transgenic DES animal models, all myopathic DES mutations eventually lead to an accumulation of desmin-positive aggregates that contribute to the disease pathology (10, 24). Desmin knockout mice, on the other hand, develop all the aforementioned characteristics, except for the aggregates. They are fertile and develop normally, but all mice eventually develop DCM (26), again primarily due to mitochondrial failure. Furthermore, since these mice lack desmin, they are unable to resist physical stress, leading to intracellular disorganization of organelles. These features are believed to play the most prominent role in the development of DCM (10, 26).
There are several overlapping aspects between KLHL24-ΔN28–mediated desmin deficiency and desminopathies. First, tissues showed dilatation upon loading, similar to many desminopathies. Second, TEM revealed swollen-like mitochondria and electron denser glycogen storage vesicles in patient-derived dyn-EHTs. These results were confirmed by Seahorse experiments, which showed that the mitochondrial respiratory function is already impaired in patient hiPSC-CMs in 2D cultures. Third, some potential compensatory mechanisms were observed, such as the elevated protein levels of actin and nuclear lamins, indicating that patient-derived dyn-EHTs might be compensating to maintain contractile function and prevent events like nuclear delamination. Several of the desminopathy hallmarks are present in our model and show high resemblance to pathogenic human DES mutations and the desmin knockout mouse model. However, the KLHL24-ΔN28–induced pathology is also different in 2 ways. First, the activity of the proteasomal degradation pathways is distinctive. Desmin knockout mice do not express desmin, hence no proteasomal degradation of desmin takes place. In human desminopathies, the accumulating of desmin aggregates severely disrupts cellular protein quality control systems and in turn leads to impaired proteasomal activity. The pathology of KLHL24-ΔN28 is different from both aforementioned pathologies, as it displays an overactive proteasomal degradation pathway due to excessive breakdown of desmin. Second, in knockout mice the desmin network is absent, while in human disease, desmin aggregates ultimately contribute to collapse of this network (12). In our 3D model, the desmin network remained intact, both in patient-derived dyn-EHTs as well as in the patient explanted heart, despite 10-fold lower levels of desmin. This likely explains why several common desminopathy hallmarks, such as nuclear deformations, myofibril abnormalities, and intercalated disc mislocalizations, were not observed.
Our dyn-EHT model shows that the KLHL24-ΔN28–induced tissue dilatation and reduced uniformity underlies the lower systolic and diastolic forces, higher systolic and diastolic peak stress, and nonuniform tissue diastolic stress. Using FEM, patient-derived dyn-EHTs were shown to have a 6-fold increase in peak diastolic cardiomyocyte stress levels compared with controls. On a regional scale, tissue stress and FS varied widely in patient-derived dyn-EHTs, and a negative correlation was observed between shortening and diameter. In DCM as well as numerous other heart failure etiologies, left ventricular wall stress is frequently increased. Our data indeed closely resemble FEM of human left ventricular myocardial wall stress, which indicated that heart failure patients with reduced ejection fraction had significantly higher average cardiomyocyte stress than controls (27). In our previous research, we also used the dyn-EHT model in order to provoke disease in desmoplakin-mutated (DSP-mutated) hiPSC-CMs (16). Comparing the 2 cardio-cutaneous disease models, the etiology is quite different, even though mutations in both genes affect the stretch-bearing structural network of tissues. In DSP-mutated dyn-EHTs, cell–cell contacts were diminished, causing dilatation and partial loss of electrical continuity, ultimately reducing the contractile force and stress. In KLHL24-mutated dyn-EHTs, the intracellular compliance network was impaired, which resulted in tissue thinning in addition to dilatation, but the cell–cell contacts were preserved.
Further proof of the KLHL24-ΔN28–induced pathology was provided using either KLHL24 RNAi or direct desmin overexpression. Both rescue models prevented excessive tissue dilatation and reduced diastolic and systolic force values, in addition to fully alleviating the elevated diastolic, systolic, and contractile stress observed after dynamic loading. These results indicate that by restoring desmin expression, tissue stability is regained. Whether methionine gain-of-function mutations are causative for DCM in a systemic model remains to be further investigated.
KLHL24-targeted antisense oligonucleotide therapy in vivo has potential to be an effective treatment that could significantly benefit patient quality of life, while remaining within limits so as not to create hypertrophic cardiomyopathy by losing KLHL24 expression altogether.
Methods
Cell culture.
From human skin biopsies, fibroblasts were passaged using Trypsin and cultured in regular DMEM containing 15% FCS, 1% sodium pyruvate, 1% glutaMAX, 1% penicillin/streptomycin, 1% nonessential amino acids. Fibroblasts were cultured and reprogrammed to hiPSCs by means of nucleofection with 3 plasmids (pCXLE-hSK, pCXLE-hMLN, and pCXLE-hOCT3/4; Addgene plasmids 27078, 27079, and 27076; gifts from Shinya Yamanaka (Center for iPS Cell Research and Application, Kyoto University, Kyoto, Japan), according to previously published methods (28, 29). Each clonal line was karyotyped and maintained by single-cell passaging, as previously mentioned (16). From each human source, 3 lines were maintained and subsequently checked for pluripotency markers SSEA-4 and OCT3/4. hiPSCs lines were differentiated to cardiomyocytes and purified using lactate. Cardiomyocytes were cultured in 2D monolayers or in 3D tissues. All cell cultures were regularly investigated for mycoplasma contaminations.
Cardiac tissue culture.
Cardiomyocytes were used to generate EHTs, subjected to low loading with 130 μm strips versus high loading with 260 μm strips (dyn-EHTs). On day 22 after differentiation initiation, cardiomyocytes were used to generate dyn-EHTs, as previously described (16). In short, 90% cardiomyocytes were mixed with 10% healthy cardiac fibroblasts obtained from Lonza in an extracellular matrix gel (Matrigel and Collagen type I; Corning) into a designated PDMS mold with strip. After gelation, medium was added to cover the tissue. Cardiac tissues started contracting after 1 day. Fabricated cardiac tissues were kept constrained in a PDMS mold for 14 days to fully remodel and compact (EHTs). Hereinafter, tissues were pulled out of their constrained mold (dyn-EHTs) and kept in dynamic culture for an additional 14 days. Cardiac tissues maintained spontaneous contractile properties and responded to electrical pacing. Videos for functional analysis were generated at day 7, day 14, right after confinement, and at day 28, using a Zeiss Axio Zoom V16 microscope connected to a HRm camera and a Nikon SMZ18 microscope connected to a Nikon DS-Qi2 camera. Tissues were terminated and processed at day 28.
Cardiac tissue analysis.
Videos were analyzed according to a custom made MATLAB code, based on FEM, according to our previously published methods (16). Additional analysis included tissue uniformity assessment, regional strain tracking, and regional stress mapping. To determine the degree of tissue uniformity induced upon loading, the tissues’ smallest cross section area was divided by the largest cross section area Ø at day 14 (reference) and day 28 (after 14 days of dynamic loading). These numbers were used to determine whether this number decreased with loading, which is noted as the ratio of tissue uniformity: (smallest / largest Ø at d28) / (smallest / largest Ø at d14).
Regional FS was quantified using a modification of methods previously described for speckle tracking in echocardiography (30). Briefly, video frames of peak diastole and systole were isolated. Tissues were divided into 3 regions, each with approximately the same diameter across the longitudinal axis. Six speckles, or noticeable differences in gray values that could be tracked from diastole to systole, were identified in each region. Speckles were tracked using Fiji’s Manual Tracking Plugin. Regional FS was then calculated using the equation below for each pair of speckles, by subtracting the initial distance between each speckle (L0) from the final distance between each speckle (Lt) and dividing that by the initial distance between each speckle (L0). This resulted in 15 total measurements per region, which were averaged to get the FS in that region.
(equation 1)
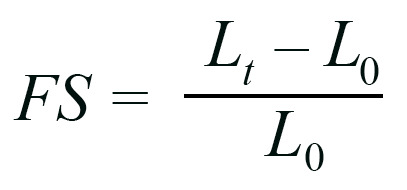
Peak contractile, diastolic, and systolic stress calculations were determined based on force values and cross-section area, assuming 100% cardiomyocyte content of whole-tissue cross-section, determined by the smallest measurable tissue diameter. In our previous method (16), we used 10% cardiomyocyte content for all tissues, based on histology results obtained from both control and DSP patient–derived dyn-EHTs, that were all uniform. However, KLHL24 patient–derived dyn-EHTs had varying degrees of cardiomyocyte content, therefore no assumptions of actual cardiomyocyte content could be made without doing histology on each individual tissue section.
Regional diastolic stress mapping was performed using finite element analysis with ANSYS Static Structural Software. First, representative tissue geometries were created in ANSYS Space Claim Software from binary images of tissues at diastole. This resulted in a model with accurate thickness displayed from the binary image. Since aerial images of the tissues were not taken, tissues from each group were assumed to have the same thickness in the z-dimension. Material properties were derived from diastolic stress-strain curves using a custom mounted optical force transducer (World Precision Instruments, SI-KG20). These values were fit to a first order Arruda-Boyce hyperelastic model, based on best fit to uniaxial test data. Tissues were uniaxially stretched in ANSYS software using the diastolic force obtained from the PDMS strip analysis. The optimal discretization of elements within the FE mesh was determined by slowly decreasing the mesh element size until little change in maximum equivalent (Von-Mises) stress was observed between subsequent modeling runs.
Lentiviral transduction and HEK transfection studies.
To generate 2 KLHL24 knockdown expression patient II:3 hiPSC lines, we combined the envelope (VSV-G) and packaging vector (pCMVΔ8.91) with a KLHL24-shRNA transfer pLKO1 vector (Sigma MISSION shRNA; TRCN0000323203). These lines were compared against 2 negative control II:3 hiPSC lines, for which we combined the envelope and packaging vectors with a nontargeted-shRNA transfer pLKO1 vector (Sigma MISSION NT-shRNA, catalog 4390843). To generate a DES overexpression (oeDES) patient II:3 hiPSC line, we combined the envelope and packaging vectors with a desmin (NM_001927.3) containing pReceiver-Lv156 vector (GeneCopoeia). First, HEK293T cells were transfected using Fugene HD to produce lentiviral particles. Viral particles were collected and hiPSC lines were transduced with addition of polybrene in 6 wp at 50% confluence. At 48 hours after transduction, successfully transduced hiPSCs were selected using puromycin.
For exogenous coexpression studies in HEK293A cells, desmin, CUL3, and RBX1 were cloned into the pcDNA3.1+ backbone. Another desmin, KLHL24, and KLHL24-ΔN28 and KLHL24-ΔN28/E355A/E535K/Y584C (KLHL24-ΔN28-inactivated) (4) were cloned into the pcDNA3.1+–N-DYK backbone. Plasmids were expanded by insertion into viable cell lines, LB-broth culture, and midi-prep isolation. HEK293A cells were grown in 12 wp until 70% confluency and cotransfected with 500 ng of each plasmid in different combinations using Fugene 6. Bortezomib was used to inhibit proteasomal degradation in a concentration of 400 nM for 24 hours. At 48 hours after transfection, cells were fixated with methanol/acetone (1:1) and immunolabeled, and proteins and RNA were harvested.
Immunofluorescence analysis.
Dyn-EHTs were fixated for 5 hours in 2% formalin, followed by incubation through a tissue transfer processor, and finalized by paraffin embedding. Paraffin-embedded explanted heart biopsies/dyn-EHTs were sectioned to histology slices of 5 μM using a microtome. Slices were deparaffinized and rehydrated through xylol/ethanol treatment. Histology slices were then blocked in 3% BSA/PBS for 1 hour at room temperature before incubation with primary antibody (Supplemental Table 1) in blocking buffer for 1 hour at room temperature or overnight at 4°C. Histology slices were then washed in PBS and incubated with secondary antibody in blocking buffer for 1 hour at room temperature. Histology slices were washed and mounted with DAPI before imaging.
Cultured cells were fixated and immunolabeled depending on the cell source and antibody (Supplemental Table 1). In short, hiPSCs were seeded on vitronectin-coated coverslips and fixated for 30 minutes with 4% paraformaldehyde at RT. Cardiomyocytes were seeded on Geltrex-coated coverslips and either fixated for 10 minutes with methanol/acetone (1:1 at –20°C) or 4% PFA (at 4°C). Formaldehyde-fixated cells were permeabilized with 0.3% Triton for 5 minutes. Cells were then blocked for 30 minutes with blocking buffer of 3% BSA/PBS containing 2% serum (host of secondary antibody). Cells were then incubated with primary (Supplemental Table 1) antibody in blocking buffer for 1 hour at room temperature or overnight at 4°C. Cells were washed and subsequently incubated with secondary antibody (Alexa 488 and/or 555) in blocking buffer for 1 hour at room temperature. Cells were washed and mounted with mounting media containing DAPI. All slides were imaged using a Leica DMI6000B fluorescence microscope.
Immunoprecipitation.
Cardiomyocytes were cultured as monolayers up to day 40 for desmin to become sufficiently expressed. Cells were treated with 10 μM Bortezomib (Sigma) for 6 hours before a 30-minute incubation in lysis buffer (20 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1% NP-40, 10 mM N-ethylmaleimide and protease inhibitors [Roche, 11873580001]) on ice. Lysates were centrifuged at 10,000g at 4°C for 10 minutes and the soluble fraction was used for immunoprecipitation (desmin clone RD301 [IgG2b] and a negative control [IgG]), using Magnetic Protein G SureBeads (Bio Rad). HEK293A cells were cultured in 60 mm dishes and transfected using different combinations of CUL3, RBX1, FLAG-tagged desmin, and KLHL24 plasmids (see above). Cells were treated with 400 nM Bortezomib (Sigma) for 24 hours before a 30-minute incubation in lysis buffer (20 mM Tris-HCl, pH 8.0, 1% NP-40, 10 mM N-ethylmaleimide and protease inhibitors [Roche 11873580001]) on ice. Lysates were centrifuged 10,000g at 4°C for 10 minutes and the soluble fraction was used for immunoprecipitation (anti-FLAG), using Magnetic Protein G SureBeads (Bio Rad). In short, antibodies (2–20 μg in lysis buffer) were bound to the magnetic beads for 1 hour at room temperature via rotation. Magnetic beads were magnetized and washed 3 times with lysis buffer. Protein concentrations in the soluble fraction were measured with the Pierce protein assay. Beads were incubated with soluble protein for 3 hours at 4°C via rotation. Magnetic beads were magnetized and washed 1 time with lysis buffer and 2 times with PBS-T. Finally, beads were boiled 1 time in sample buffer (62 mM Tris-HCl, 2,5% SDS, 1 mM EDTA, 10% glycerol, 5% β-mercaptoethanol and bromophenol blue) for 10 minutes at 90°C to elute the lysates. For cardiomyocytes only, 6 hours of 10 μM bortezomib treatment was not sufficient to restore desmin levels of patient cardiomyocytes to WT levels in control cardiomyocytes. Meanwhile, longer incubation periods eliminated their contractile properties and caused too much cell death. Therefore, while maintaining similar protein concentrations in the eluents during immunoprecipitation, we normalized eluents on gel electrophoresis for the amount of precipitated desmin afterwards.
Gel electrophoresis and Western blotting.
Total protein lysates were extracted from snap-frozen dyn-EHTs or cultured cells using a buffer containing 62 mM Tris-HCl, 2.5% SDS, and 1 mM EDTA protease inhibitor (Roche 11873580001), phosphatase inhibitor cocktail 3 (p2850; Sigma), and sodium orthovanadate. Protein concentrations were measured with the Pierce protein assay. All samples were diluted to 1.5 μg/μL with sample buffer (final solution contained 10% glycerol, 5% β-mercaptoethanol and bromophenol blue). Samples were heated for 5 minutes at 99°C, with the exception of OXPHOS proteins, to which no heating was applied. Proteins and immunoprecipitated fractions were separated in SDS-PAGE gels and transferred to PVDF or nitrocellulose membranes using tank or semidry blotting (depending on the protein size) for 2 hours. Membranes were washed and stained with Ponceau S. Membranes were then blocked with a polyvinylpyrrolidone-based (PVP-based) solution and incubated overnight at 4°C with primary (Supplemental Table 1) and secondary HRP-labeled (1 hour at RT) antibodies, before detecting with electrochemiluminescence. For each blot, either GAPDH or vinculin was used as loading control.
RT-PCR.
Dyn-EHTs were snap-frozen at day 28, prior to RNA isolation. Total RNA from hiPSCs, hiPSC-CMs, and dyn-EHTs was isolated by phenol-chloroform extraction (TRIzol, Sigma). cDNA was synthesized by reverse transcription, and real-time qPCRs were performed using SYBR Green (Sigma; see primer list in Supplemental Table 2). Relative expression levels were calculated using the 2-ΔΔCt method.
Transmission electron microscopy.
Dyn-EHTs were fixed with 2% glutaraldehyde in 0.1 M sodium cacodylate at 4°C. After post-fixation in 1% osmium tetroxide/1.5% potassium ferrocyanide (2 hours at 4°C), tissues were dehydrated using acetone and embedded in EPON epoxy resin. Seventy nanometer sections were cut longitudinal and transverse in cell direction and contrasted using 2% uranylacetate in water for 45 minutes followed by Reynolds lead citrate for 1 minute. Full nanotomy scans were generated as described by Sokol et al. (31). In short, the area of interest was scanned at 2.5 nm pixel resolution using the Zeiss supra 55 EM with Fibics (Canada) ATLAS software.
Seahorse assay.
Mitochondrial and glycolysis stress tests were performed on cardiomyocytes derived from II:3 shNT and shKLHL24 hiPSC lines. For each stress test, 7 individual experiments were performed (n = 7). Cardiomyocytes were randomly seeded (2D) on precoated Agilent XF24 Seahorse Cell Culture Microplates at a concentration of 100,000 cells/well. For each plate, 3 wells were kept without cells, as background. A stress test was performed 7 days after seeding. For the mitochondrial stress test, cells were washed and incubated according to Agilent’s instructions for 1 hour in a humid incubator without CO2 prior to testing. During the stress test, wells were injected using the Agilent Extracellular Flux Assay Kit with oligomycin (1 μM final concentration), followed by FCCP (carbonyl cyanide-p-trifluoromethoxyphenylhydrazone; 0.5 μM final concentration), and finally by antimycin A and rotenone (1 μM final concentrations). For the glycolysis stress test, cells were washed and incubated according to Agilent’s instructions in medium (pH 7.4), 2 hours in a humid incubator without CO2 prior to testing. During the stress test, wells were injected first with glucose (10 mM final concentration) using the Agilent Extracellular Flux Assay Kit, followed by oligomycin (1 μM final concentration), and finally by 2-DG (50 mM final concentration). After testing, microplates underwent a freeze-thaw cycle, and protein concentrations were measured using a Bio Rad protein quantification kit for normalization.
Statistics.
A Shapiro-Wilk normality test was performed on all data sets and sets were subsequently checked for outliers (robust regression and outlier removal Q = 1%), prior to analysis. For Figures 2 and 3 (and supporting supplemental figures), statistical analysis was based on n = 3 control-derived and n = 2 patient-derived individuals, supported by 3 hiPSC lines per patient per control and a minimum of n greater than 3 tissues per line. Statistically significant differences between functional tissue parameters were calculated using a 2-way ANOVA, with post hoc Sidak’s multiple comparisons test. Nonbinned Zeiss videos were used to assess regional FS. A Brown Forsythe test was used to assess the homogeneity of variances. To determine differences in regional FS based on tissue cross-section area, a 2-way ANOVA with post hoc Sidak’s multiple comparisons test was performed. For correlation analysis between regional FS and tissue diameter, a Spearman nonparametric correlation was performed. Statistically significant differences in small-set gene and protein expression levels were calculated using an unpaired t test or Mann-Whitney U test, depending on the outcome of the normality test performed. For Figure 4 (and supporting supplemental figures), statistical analysis of protein expression levels in tissues was based on an unpaired t test or Mann-Whitney U test, while a 1-way ANOVA was used for quantifying proteins in HEK-cell transfection studies. For Figure 5 (and supporting supplemental figures), statistical analyses were based on adding cardiac tissues derived from the 4 hiPSC lines treated either with shNT or shKLHL24 mRNA silencing constructs and 1 hiPSC line treated with a oeDES construct. Statistically significant differences between functional tissues parameters were calculated using a 1-way ANOVA, with post hoc Sidak’s multiple comparisons test. Statistically significant differences in protein expression levels were calculated using an unpaired t test or Mann-Whitney U test. Statistically significant differences in large-scale gene expression sets measured in all dyn-EHT groups were calculated using a 2-way ANOVA, corrected for multiple comparisons using FDR via the Benjamini and Hochberg method. For the Seahorse assay data, a 2-way ANOVA with post hoc uncorrected Fisher’s least significant difference test was performed. For all statistical tests, a P value less than 0.05 was considered significant. Specifics of each statistical test can be found in the figure legends. All statistics were generated using GraphPad Prism. All data are represented as mean ± SEM.
Study approval.
This study conforms to the Declaration of Helsinki. Approval of human participants was granted by the medical ethical committee of the University of Groningen (METc 2017/391) and written informed consent was received from all participants. Skin biopsies of family members and controls were taken at the dermatology department of the University Medical Center Groningen (UMCG). The residual fibroblast outgrowth cultures were used to generate hiPSCs in order to perform this study.
Author contributions
MCSCV, MCB, HHWS, and PVDM designed the research studies. MCSCV, JMB, AWF, PHRH, and KAG established the methods. MCSCV and JMB conducted the experiments. MCSCV, JMB, and DK acquired the data. MCSCV, JMB, and MPG analyzed the data. MCSCV wrote the manuscript. ALL performed critical data interpretation. MCB, MPVDB, AWF, GFHD, HHWS, and BDW provided reagents and expertise in the field. MCB, AWF, and PVDM collaborated on supported funding.
Supplementary Material
Acknowledgments
We thank Silke Oberdorf-Maass, Frouke Houtsma, and Christiane Baierl for their excellent technical contributions. In addition, we would like to acknowledge Annet Linders for contributing control-derived dyn-EHT data, Martijn F. Hoes for making control hiPSC lines available for this study, Rebecca M. Duffy for establishing part of the dyn-EHT groundwork, and Ivan Batalov for writing the MATLAB code, used for tissue tracking during contraction. Part of the work was performed at the UMCG Imaging and Microscopy Center (UMIC). Initial videos were shot at 23. European Research Institute of the Biology of Ageing, Laboratory of Stem Cell Regulation and Mechanisms of Regeneration, for which we express our gratitude. This work was supported by the Human Frontier Science Program (grant RGY 0071/2014 to AWF and PVDM), Vlinderkind (funding to MCB), and the European Research Counsel (STOP-HF [StG]; grant 715732, ERC-2016-STG to PVDM).
Version 1. 07/22/2021
In-Press Preview
Version 2. 09/01/2021
Electronic publication
Funding Statement
non, due to patient organization funding
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(17):e140615.https://doi.org/10.1172/JCI140615.
Contributor Information
Mathilde C.S.C. Vermeer, Email: m.vermeer@umcg.nl.
Maria C. Bolling, Email: m.c.bolling@derm.umcg.nl.
Jacqueline M. Bliley, Email: jbliley@andrew.cmu.edu.
Karla F. Arevalo Gomez, Email: k.f.arevalo.gomez@umcg.nl.
Mario G. Pavez-Giani, Email: mario.pavez@med.uni-goettingen.de.
Duco Kramer, Email: d.kramer@umcg.nl.
Pedro H. Romero-Herrera, Email: phromero@outlook.com.
B. Daan Westenbrink, Email: b.d.westenbrink@umcg.nl.
Gilles F.H. Diercks, Email: g.f.h.diercks@umcg.nl.
Maarten P. van den Berg, Email: m.p.van.den.berg@umcg.nl.
Adam W. Feinberg, Email: feinberg@andrew.cmu.edu.
Peter van der Meer, Email: p.van.der.meer@umcg.nl.
References
- 1.Yenamandra VK, et al. Cardiomyopathy in patients with epidermolysis bullosa simplex with mutations in KLHL24. Br J Dermatol. 2018;179(5):1181–1183. doi: 10.1111/bjd.16797. [DOI] [PubMed] [Google Scholar]
- 2.Schwieger-Briel A, et al. Epidermolysis bullosa simplex with KLHL24 mutations is associated with dilated cardiomyopathy. J Invest Dermatol. 2019;139(1):244–249. doi: 10.1016/j.jid.2018.07.022. [DOI] [PubMed] [Google Scholar]
- 3.Grilletta EA. Cardiac transplant for epidermolysis bullosa simplex with KLHL24 mutation-associated cardiomyopathy. JAAD Case Rep. 2019;5(10):912–914. doi: 10.1016/j.jdcr.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin Z, et al. Stabilizing mutations of KLHL24 ubiquitin ligase cause loss of keratin 14 and human skin fragility. Nat Genet. 2016;48(12):1504–1516. doi: 10.1038/ng.3701. [DOI] [PubMed] [Google Scholar]
- 5.He Y, et al. Monoallelic mutations in the translation initiation codon of KLHL24 cause skin fragility. Am J Hum Genet. 2016;99(6):1395–1404. doi: 10.1016/j.ajhg.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alkhalifah A, et al. Burnlike scars: a sign suggestive of KLHL24-related epidermolysis bullosa simplex. Pediatr Dermatol. 2018;35(3):e193–e195. doi: 10.1111/pde.13443. [DOI] [PubMed] [Google Scholar]
- 7.Lee JYW, et al. Mutations in KLHL24 add to the molecular heterogeneity of epidermolysis bullosa simplex. J Invest Dermatol. 2017;137(6):1378–1380. doi: 10.1016/j.jid.2017.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Herrmann H, et al. Intermediate filaments: primary determinants of cell architecture and plasticity. J Clin Invest. 2009;119(7):1772–1783. doi: 10.1172/JCI38214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coulombe PA, et al. Epidermolysis bullosa simplex: a paradigm for disorders of tissue fragility. J Clin Invest. 2009;119(7):1784–1793. doi: 10.1172/JCI38177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsikitis M, et al. Intermediate filaments in cardiomyopathy. Biophys Rev. 2018;10(4):1007–1031. doi: 10.1007/s12551-018-0443-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Capetanaki Y, et al. Desmin related disease: a matter of cell survival failure. Curr Opin Cell Biol. 2015;32:113–120. doi: 10.1016/j.ceb.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hnia K, et al. Desmin in muscle and associated diseases: beyond the structural function. Cell Tissue Res. 2015;360(3):591–608. doi: 10.1007/s00441-014-2016-4. [DOI] [PubMed] [Google Scholar]
- 13.Milner DJ, et al. The absence of desmin leads to cardiomyocyte hypertrophy and cardiac dilation with compromised systolic function. J Mol Cell Cardiol. 1999;31(11):2063–2076. doi: 10.1006/jmcc.1999.1037. [DOI] [PubMed] [Google Scholar]
- 14.Tse H-F, et al. Patient-specific induced-pluripotent stem cells-derived cardiomyocytes recapitulate the pathogenic phenotypes of dilated cardiomyopathy due to a novel DES mutation identified by whole exome sequencing. Hum Mol Genet. 2013;22(7):1395–1403. doi: 10.1093/hmg/dds556. [DOI] [PubMed] [Google Scholar]
- 15.Szeverenyi I, et al. The Human Intermediate Filament Database: comprehensive information on a gene family involved in many human diseases. Hum Mutat. 2008;29(3):351–360. doi: 10.1002/humu.20652. [DOI] [PubMed] [Google Scholar]
- 16.Bliley JM, et al. Dynamic loading of human engineered heart tissue enhances contractile function and drives desmosome-linked disease phenotype. Sci Transl Med. 2021;13(603):eabd1817. doi: 10.1126/scitranslmed.abd1817. [DOI] [PubMed] [Google Scholar]
- 17.Agnetti G, et al. Desmin modifications associate with amyloid-like oligomers deposition in heart failure. Cardiovasc Res. 2014;102(1):24–34. doi: 10.1093/cvr/cvu003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winter DL, et al. Posttranslational modifications of desmin and their implication in biological processes and pathologies. Histochem Cell Biol. 2014;141(1):1–16. doi: 10.1007/s00418-013-1148-z. [DOI] [PubMed] [Google Scholar]
- 19.Norgett EE. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9(18):2761–2766. doi: 10.1093/hmg/9.18.2761. [DOI] [PubMed] [Google Scholar]
- 20.Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol. 2004;13(4):185–194. doi: 10.1016/j.carpath.2004.03.609. [DOI] [PubMed] [Google Scholar]
- 21.Bolling MC, Jonkman MF. Skin and heart: une liaison dangereuse. Exp Dermatol. 2009;18(8):658–668. doi: 10.1111/j.1600-0625.2009.00908.x. [DOI] [PubMed] [Google Scholar]
- 22.Hedberg-Oldfors C, et al. Cardiomyopathy with lethal arrhythmias associated with inactivation of KLHL24. Hum Mol Genet. 2019;28(11):1919–1929. doi: 10.1093/hmg/ddz032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Danielsson O, et al. Hypertrophic cardiomyopathy and abnormal glycogen storage in heart and skeletal muscle associated with inactivation of KLHL24. Neuromuscul Disord. 2016;26(Supplement 2):S152 [Google Scholar]
- 24.Herrmann H, et al. Intermediate filaments: primary determinants of cell architecture and plasticity. J Clin Invest. 2009;119(7):1772–1783. doi: 10.1172/JCI38214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khudiakov A, et al. Generation of iPSC line from desmin-related cardiomyopathy patient carrying splice site mutation of DES gene. Stem Cell Res. 2017;24:77–80. doi: 10.1016/j.scr.2017.08.015. [DOI] [PubMed] [Google Scholar]
- 26.Milner DJ, et al. Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol. 1996;134(5):1255–1270. doi: 10.1083/jcb.134.5.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang ZJ, et al. Left ventricular diastolic myocardial stiffness and end-diastolic myofibre stress in human heart failure using personalised biomechanical analysis. J Cardiovasc Transl Res. 2018;11(4):346–356. doi: 10.1007/s12265-018-9816-y. [DOI] [PubMed] [Google Scholar]
- 28.Okita K, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8(5):409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 30.Lopez-Candales A, Hernandez-Suarez DF. Strain imaging echocardiography: what imaging cardiologists should know. Curr Cardiol Rev. 2017;13(2):118–129. doi: 10.2174/1573403X12666161028122649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sokol E, et al. Large-scale electron microscopy maps of patient skin and mucosa provide insight into pathogenesis of blistering diseases. J Invest Dermatol. 2015;135(7):1763–1770. doi: 10.1038/jid.2015.109. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.