

Abstract
Glioblastoma (GBM) remains among the deadliest of human malignancies, and the emergence of the cancer stem cell (CSC) phenotype represents a major challenge to durable treatment response. Because the environmental and lifestyle factors that impact CSC populations are not clear, we sought to understand the consequences of diet on CSC enrichment. We evaluated disease progression in mice fed an obesity-inducing high-fat diet (HFD) versus a low-fat, control diet. HFD resulted in hyperaggressive disease accompanied by CSC enrichment and shortened survival. HFD drove intracerebral accumulation of saturated fats, which inhibited the production of the cysteine metabolite and gasotransmitter, hydrogen sulfide (H2S). H2S functions principally through protein S-sulfhydration and regulates multiple programs, including bioenergetics and metabolism. Inhibition of H2S increased proliferation and chemotherapy resistance, whereas treatment with H2S donors led to death of cultured GBM cells and stasis of GBM tumors in vivo. Syngeneic GBM models and GBM patient specimens present an overall reduction in protein S-sulfhydration, primarily associated with proteins regulating cellular metabolism. These findings provide clear evidence that diet-modifiable H2S signaling serves to suppress GBM by restricting metabolic fitness, while its loss triggers CSC enrichment and disease acceleration. Interventions augmenting H2S bioavailability concurrent with GBM standard of care may improve outcomes for patients with GBM.
Keywords: Oncology, Stem cells
Keywords: Brain cancer

Introduction
Patients with glioblastoma (GBM) experience one of the most aggressive disease trajectories of all cancers (1, 2). Effective disease management is lacking due to the intrinsic heterogeneity and complexity of the disease. Heterogeneity at the cellular level includes multiple interacting populations of cancer stem cells (CSCs) (3–5), each capable of varying degrees of tissue invasion (6), proliferation (7, 8), and treatment resistance (9). Recent evidence indicates that metabolic heterogeneity confers an additional level of complexity and adaptability to evolving GBM tumors (10, 11). A growing number of enzymes and metabolic pathways have been highlighted that contribute to the maintenance and selection of CSC populations (12–14). Collectively, these studies suggest that metabolic adaptability enables CSCs to out-compete less-plastic tumor cells with clear metabolic dependencies. Specifically, while non–stem tumor cells may be limited to the glycolytic metabolism first described by Otto Warburg (15), the CSC phenotype enables cells to shift among various substrates, employing glycolysis, fatty acid oxidation, and amino and nucleic acid metabolism, depending on nutrient availability (16). As the nutrient landscape is profoundly altered through diet, we focused on understanding how an obesity-generating, high-fat diet (HFD) serves as a regulator of GBM progression and as a selective force for GBM CSC expansion.
HFD consumption alters numerous physiological systems, including the lipid composition within the brain (17) as well as the composition of the gut microbiome (18, 19) and its associated set of metabolites. HFD also modifies the cellular composition and function of the immune system (20). One system that is profoundly affected by HFD consumption and has been largely unexplored in the etiology of GBM is the synthesis of the gasotransmitter hydrogen sulfide (H2S), a byproduct of sulfur amino acid metabolism. H2S is an endogenously produced, bio-active metabolite (21). Three enzymes — cystathionine beta-synthase (CBS), cystathionine gamma-lyase (CGL), and mercaptopyruvate sulfurtransferase (MPST) — are responsible for enzymatic H2S production and are differentially expressed and active in a tissue-specific manner (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI138276DS1). Through a largely unknown mechanism, HFD consumption results in attenuation of these H2S-producing enzymes and therefore potently inhibits H2S production (22). Functionally, upon generation, H2S is quickly transferred to available cysteine residues in the form of a protein posttranslational modification referred to as S-sulfhydration or persulfidation (23). S-sulfhydration alters protein form and associated function; however, unlike nitrosylation or phosphorylation, the functional changes that result from S-sulfhydration are largely unknown (24). There are a limited number of studies on the association between H2S, S-sulfhydration and cancer (25–27). Evidence suggests that H2S serves as both a promoter and inhibitor of tumorigenesis in a tissue-specific manner (Supplemental Figure 2). For GBM, the limited information available suggests that H2S synthesis inhibits proliferation of cultured GBM tumor cells (28). Biochemical analysis indicates that the enzymatic activity of both CGL and MPST decreases with increasing astrocytoma grade and that these enzymes are essentially nonfunctioning in the context of GBM (29). To date, the loss of protein S-sulfhydration has gone entirely unexplored in the context of GBM.
HFD has been assessed through 2 types of research in the GBM field. First, metabolic dependency studies involve the use of ketogenic HFDs or metabolism pathway inhibitors to slow or stop disease progression by depriving tumors of critical energy sources (30–32). Second, epidemiological studies question whether obesity, brought about by the consumption of obesity-generating diets, serves as an initiator of GBM development (33, 34). The possibility that a long-term pattern of HFD consumption would exacerbate disease, changing the histological presentation and trajectory of GBM, has not yet been addressed. Thus, we compared GBM tumors developed in the brains of HFD-fed mice to those developed within the brains of animals fed a control diet. This led to a series of observations linking HFD consumption to alterations in the nutrient landscape of the brain, the CSC compartment of the tumor microenvironment, and the production and function of intracerebral H2S. We demonstrate a mechanistic connection between consumption of a HFD and the inhibition of H2S, which leads to enhanced metabolic fitness for GBM tumor cells. Importantly, we also demonstrate that H2S can be replaced to mitigate the progression of this disease.
Results
HFD consumption drives CSC enrichment and accelerates glioblastoma progression.
To test whether HFD modulates the growth and initiation of GBM, we employed both syngeneic mouse models (GL261, CT2A, KR158) and human patient–derived GBM models (hGBM 23, hGBM 124, hGBM 3691) in a series of in vivo experiments performed according to the schematic presented in Figure 1A. Experiments were initiated using animals of equivalent body mass and fat composition. Animals fed the HFD gained body mass (Supplemental Figure 3A) as a product of fat accumulation (Supplemental Figure 3B) over time throughout the duration of the experiment. Compared with control diet–fed mice, HFD-fed tumor-bearing animals experienced a significant reduction of overall survival (Figure 1, B–D, and Supplemental Figure 3C). Importantly, in the absence of GBM, HFD consumption does not limit survival. Under specific experimental conditions, a HFD has even been attributed to increased longevity (35) and protection against midlife mortality (36) in rodent models of aging. For each of the 3 syngeneic GBM models, these experiments were repeated across multiple cohorts in a limiting-dilution format using progressively fewer tumor cells at the time of intracerebral injection. Regardless of the initial cell dosage, a greater number of animals in the HFD group succumbed to disease during the course of the experiment, indicating that HFD induced a higher tumor initiation frequency compared with those that consumed the control diet. Specifically, 2- to 3-fold fewer tumor cells were required to initiate tumors that drove animals to the experimental endpoint under conditions of HFD consumption (Figure 1, E–G). Interrogation of the tumor microenvironment using standard immunofluorescence techniques revealed 3 contributing factors that helped to explain this enhanced tumor aggression. First, in accordance with the increased CSC frequency suggested by the in vivo limiting-dilution analysis, histological examination revealed a marked increase in the percentage of SOX2+ tumor cells within the brains of HFD-fed mice compared with mice fed the control diet (Figure 1, H–J). CSC enrichment was also verified in vitro using the patient-derived GBM models hGBM 23 and hGBM 3691, which were treated with escalating doses of the mono-unsaturated fatty acid oleic acid. Compared with vehicle-treated cells, lipid treatment resulted in dose-responsive expression increases in SOX2 and growth-associated protein 43 (GAP43), another CSC-associated protein (Supplemental Figure 3, D and E). Second, HFD consumption triggered a significant increase in tumor cell proliferation in vivo when compared with controls (Supplemental Figure 3, F–H). Third, necrosis was rarely present within the tumor microenvironment of HFD-fed mice. In contrast, sites of pseudopalisading necrosis were far more prevalent in tumor-bearing mice fed the control diet (Supplemental Figure 3, F and I–K). Thus, the combination of increased tumor cell proliferation, protection from cell death, and induction of the CSC phenotype helps to explain the truncated survival observed in tumor-bearing animals fed the HFD.
Figure 1. High-fat diet consumption drives CSC enrichment and accelerates glioblastoma progression.

(A) In vivo experimental design employed to test whether HFD consumption modifies GBM progression. (B–D) For the syngeneic GBM models CT2A and GL261, as well as the patient-derived GBM model hGBM 23, Kaplan-Meier survival analysis confirmed significant truncation of overall survival under conditions of HFD consumption compared with consumption of control diets. P values determined by log-rank survival analysis and experimental group (n) size noted above. (E–G) In vivo limiting dilution analysis was performed for the 3 syngeneic GBM models CT2A, GL261, and KR158. For each model, tumors were initiated using 50,000, 20,000, 15,000, 10,000, and 5000 cells per animal. P values were determined using the Walter and Eliza Hall ELDA portal (60) comparing the total number of endpoint animals in the HFD group versus the control diet group at the conclusion of each set of experiments. (H and I) Representative immunofluorescence micrographs of the CSC population observed in the GBM tumor microenvironment under HFD- versus chow-fed conditions. Scale bars: 75 μm. The CSC-associated transcription factor SOX2 was visualized in red; MCM2, visualized in green, identified the bulk tumor cell population; and nuclei were visualized in blue using DAPI. (J) SOX2 fluorescence intensity, normalized to the MCM2 fluorescence intensity, allowed us to measure CSC enrichment within the tumor microenvironment. Each dot represents the fluorescence intensity of the SOX2 signal divided by the fluorescence intensity of the MCM2 signal for each image. P value determined by unpaired t test.
HFD consumption drives stem cell phenotype enrichment.
We reasoned that HFD consumption may result in intracerebral lipid enrichment, which in turn may act directly (and/or indirectly) to increase proliferation and self-renewal within the tumor cell population. Therefore, matched tumor-bearing and non–tumor-bearing hemispheres were isolated from the brains of multiple HFD-fed and control diet–fed GBM-bearing animals at their experimental endpoints. To determine which lipid species were altered, we interrogated these specimens using mass spectrometry–based nontargeted lipidomic analysis (Figure 2A and Supplemental Table 1). When normalized to the set of chow-fed healthy specimens, we observed 2 modes of in vivo lipid alteration. First, we identified a variety of lipids enriched specifically within the tumors isolated from HFD-fed mice (Figure 2B). These lipid species were expressed at substantially greater levels within tumors of HFD-fed animals compared with the contralateral healthy tissues derived from the same mice as well as tissues isolated from control diet–fed mice. Second, we identified a separate set of lipids enriched generally within the brains of HFD-fed animals, regardless of the presence of tumor (Figure 2C). Expression of these species was significantly increased in the HFD-derived specimens compared with the control diet–fed specimens but was not different between the tumor-derived and contralateral specimens isolated from the HFD-fed mice. With the exception of 2 polyunsaturated ceramide species (HexCer 34:1;2 and HexCer 34:1;3), the lipids observed within the HFD, tumor-bearing brain were either saturated, mono-, or di-unsaturated lipid species, consistent with heavy consumption of a saturated fat–based diet. As ceramide lipids have a strong association with cellular stress (37, 38), which is commonly induced within the caustic growth zones (39, 40) of GBM, we omitted these species from our analysis. Based on this nontargeted lipid assessment, we concluded that HFD consumption induced accumulation of saturated fats within the brain and tumor microenvironment of HFD-fed animals. This nonstandard lipid accumulation, in turn, may have contributed to the hyperaggressive disease that presented in these animals. Rather than narrow our study to the function of an individual lipid, we hypothesized that increased proliferation, CSC phenotype induction, and/or CSC selection and expansion were driven by overall saturated lipid accumulation.
Figure 2. Consumption of HFD drives intracerebral lipid accumulation, promoting tumor cell viability and self-renewal.

(A) Heatmap representing the top 10 most abundant and the bottom 10 least abundant lipids of 216 total lipid species identified by untargeted lipidomic analysis. Four groups (HFD-fed, tumor-bearing hemisphere; HFD-fed, contralateral hemisphere; chow-fed, tumor-bearing hemisphere; and chow-fed, contralateral hemisphere) were compared; n = 5 specimens per group. Heatmap data were normalized to the chow-fed, contralateral hemisphere group. Two modes of lipid enrichment were noted. (B) Four saturated lipid species, including phosphatidylglycerol (PG) 36:0, were identified specifically within the tumors of the HFD-fed animals. P value determined by 1-way ANOVA. (C) Nine mono- or di-unsaturated lipid species, including PG 36:2, were identified within the HFD-fed brain and tumor. P value determined by 1-way ANOVA. In vitro treatment of either (D) KR158 or (G) CT2A with the mono-unsaturated fatty acid oleic acid 18:1 increased tumor cell viability in a dose-dependent manner. P value determined by 2-way ANOVA. (E and F) In vitro limiting dilution analysis conducted with KR158 indicated self-renewal enhancement resulting from exposure to excess oleic or linoleic acid. P values were determined using the Walter and Eliza Hall ELDA portal (60).
To test whether excess saturated lipid might increase tumor cell proliferation and self-renewal, we supplemented the standard growth media employed for the syngeneic GBM models with the mono- or di-unsaturated fatty acids oleic or linoleic acid. We then compared cellular growth and self-renewal in the presence of exogenous fatty acid with growth under control conditions. GBM cells grown in excess lipid were induced into a state of hyperproliferation (Figure 2, D and G) and exhibited increased self-renewal (Figure 2, E and F). Thus, in accordance with our hypothesis, we concluded that excess saturated lipid was acting directly on tumor cells and contributing to the enhanced GBM progression and increased CSC frequency observed in the context of HFD consumption.
While these findings indicated that saturated fats work directly on tumor cells, they did not rule out the possibility that lipid accumulation within the HFD-fed brain may have established an environment that selected for one or multiple stem-like populations. Our lipidomic profiling revealed a host of species that accumulated in the brains of HFD-fed animals regardless of the presence of a tumor. This finding reinforced the idea that long-term HFD consumption might shift the overall nutrient landscape of the brain, introducing a selective pressure for the enrichment of stem-like cells with an enhanced ability to forage and metabolize a diverse set of energy substrates, including lipids (41). To test whether HFD consumption established a stem cell–selective environment globally within the brain, we introduced a cohort of female, non–tumor-bearing C57BL/6J mice to ad libitum HFD, matched to a control cohort maintained on standard rodent chow. These differential diets were maintained for approximately 50 days, a time period roughly equal to the survival of HFD-fed GBM-transplanted mice. Using standard immunofluorescence techniques, we then carefully examined the subventricular zones (SVZs) of these differentially fed animals for the expansion of endogenous neural stem and progenitor cells (NSPCs). Staining for the stem cell–associated transcription factor SOX2 revealed a remarkable amplification of the NSPC fraction within the SVZs of the HFD-fed cohort compared with control animals (Supplemental Figure 4). Based on these data, we concluded that HFD consumption established an environment within the brain in which proliferation and self-renewal of stem-like tumor and NSPCs were selectively enhanced. Therefore, we continued to evaluate molecular mechanisms that are modulated by HFD feeding in general, rather than the actions of a specific lipid species.
The diet-modifiable metabolite hydrogen sulfide serves as a GBM tumor suppressor.
Long-term HFD consumption inhibits production of the gasotransmitter H2S, a byproduct of cysteine metabolism and a feature of the transsulfuration metabolic pathway (Figure 3A). While this diet-induced inhibition has been well documented in the livers of HFD-fed mice (22), we observed an approximate 50% reduction in H2S synthesis within the brains of HFD-fed tumor-bearing mice (Figure 3B). We began our investigation into H2S and glioma by analyzing data curated by The Cancer Genome Atlas (Figure 3, C–E) and the Chinese Glioma Genome Atlas (Supplemental Figure 5, A–C) to ascertain whether the expression of H2S synthesizing enzymes correlated with various types of glioma. Both data sets indicated that patients with GBM present with the lowest average expression of CBS and MPST, whereas the mRNA expression of CGL remains intact. Importantly, despite stable CGL transcript expression, recent work confirmed that the enzymatic function of both MPST and CGL was suppressed across multiple grades of astrocytoma and was entirely nonfunctional in the context of GBM (29). These findings suggest a tumor-suppressive role for H2S insofar as the shutdown of H2S synthesis confers a growth advantage to various pathologies within the glioma family (28). To test the hypothesis that H2S serves as a tumor suppressor specifically for GBM, we assessed the proliferation of cultured GBM cells treated with the potent and selective CGL inhibitor propargylglycine (PAG). Treatment with PAG inhibited H2S production in each of the syngeneic GBM models (Supplemental Figure 5D). Further, inhibition of H2S synthesis induced hyperproliferation (Figure 4, A–D) and protected against the cytotoxic effects of the standard-of-care chemotherapeutic temozolomide compared with vehicle controls (Supplemental Figure 5E). Because inhibition of H2S synthesis drove GBM cell proliferation, we reasoned that H2S replacement should suppress GBM tumor cell growth. We compared the in vitro IC50 value for sodium hydrosulfide (NaHS), a potent and fast-acting H2S donor, as well as for GYY 4137, a slow-releasing H2S donor, using multiple syngeneic and patient-derived GBM models and 2 liver cancer control cell lines (HepG2 and NCTC 1469) (Figure 4, A, E–G, and Supplemental Figure 6). We observed that patient-derived GBM cell viability was suppressed to a far greater degree than that of the mouse GBM or control liver cancer cell lines. These data support the conclusion that H2S is a diet-modifiable tumor suppressor of GBM. Additionally, these data suggest that HFD consumption sufficiently depleted this tumor suppressor, such that the HFD-fed mice experienced a hyperaggressive presentation of the disease.
Figure 3. HFD and gliomagenesis inhibit H2S production.

(A) Schematic detailing the generation of H2S as a byproduct of cysteine metabolism associated with MPST activity and the transsulfuration pathway. (B) H2S production analysis indicates that HFD consumption results in decreased H2S synthesis in the tumors of HFD-fed mice compared with tumors isolated from mice fed a control diet. Each well contains tumor tissue homogenate from separate and distinct experimental animals. P values determined by unpaired t test. (C and D) Human patient data curated by The Cancer Genome Atlas Low-Grade Glioma and GBM data set indicate that patients with GBM present with the lowest average expression of the H2S-generating enzymes CBS and MPST. (E) While the mRNA expression of CGL remained stable across glioma subtype, previously published biochemical analysis confirmed that this enzyme is nonfunctional in the context of GBM.
Figure 4. Inhibition of H2S production results in GBM tumor cell hyperproliferation.

(A) Schematic detailing how each chemical agent modifies production of H2S. (B–D) CellTiter Glo viability analysis confirmed that in vitro treatment with the CGL-selective inhibitor PAG increased GL261, KR158, and CT2A tumor cell viability compared with vehicle controls. P value determined by 2-way ANOVA. (E–G) H2S supplementation using the chemical donor sodium hydrosulfide (NaHS) or GYY 4137 resulted in selective viability reduction for human (hGBM 23) tumor cells compared with the non-GBM (NCTC 1469) liver cell line. IC50 values were determined based on nonlinear regression analysis. While representative IC50 curves for hGBM 23 and NCTC 1469 are depicted here, IC50 concentrations were determined for a total of 3 human GBM specimens, 2 syngeneic GBM specimens, and 2 non-GBM cell lines (Supplemental Figure 6).
HFD consumption but not obesity drives GBM acceleration.
There are a number of cancers, including hepatocellular carcinoma (HCC), that are accelerated by HFD consumption but also by the metabolic state that accompanies obesity regardless of diet (33, 34, 42). Our experimental animals were primed with HFD for 2 weeks prior to tumor introduction; however, their body fat percentage did not reflect obesity (≥25%) until much later (~3 weeks) in the experiment (Supplemental Figure 3B). Because obesity, and not diet, is the predominant clinical variable that is collected and used as an epidemiological benchmark, we wanted to understand the degree to which obesity contributed to GBM acceleration separate from HFD consumption. We therefore turned to the LepOB mutant mouse, which exhibits many of the hallmark physiological features of metabolic syndrome, including obesity (Supplemental Figure 7, A and B), as a result of hyperphagic consumption of standard rodent chow. We compared the overall survival of tumor-bearing LepOB mice and C57BL/6J mice, both fed a control, low-fat diet. Under conditions of obesity but in the absence of HFD, no GBM acceleration was observed. Overall survival was not significantly different between tumor-bearing LepOB and C57BL/6J mice (Supplemental Figure 7C). Further, analysis of the excised endpoint tumors revealed identical capacities for H2S production (Supplemental Figure 7D). Thus, inhibition of H2S synthesis, which accelerated GBM progression, required HFD consumption, and the metabolic profile associated with obesity was not sufficient to drive hyperaggression in GBM.
Enhanced cellular metabolism results from H2S inhibition.
To this point, we focused exclusively on experimental animals held under precisely controlled dietary conditions. Given this degree of control, we observed robust attenuation in the enzymatic production of H2S (Figure 3B). The degree to which H2S suppression translates to the human condition and the function that inhibition might serve remains a critical and unresolved question. To address this question, we collected and analyzed tissues from 5 patients with GBM and 5 noncancerous control brain specimens that had been flash frozen at the time of isolation. Initial examination for the ability to produce H2S revealed that GBM tissue produced approximately 50% of this critical tumor suppressor compared with the level observed in noncancerous control tissues (Figure 5A). Leveraging a modified biotin thiol assay to isolate S-sulfhydrated proteins coupled to protein mass spectrometry–based analysis, we further analyzed these specimens to generate differential cancer versus noncancer S-sulfhydrome profiles. Consistent with the reduced H2S production, we noted a dramatic decrease in the number of S-sulfhydrated proteins within the GBM specimens compared with controls (Figure 5B). S-sulfhydration loss affected more than 400 discrete proteins (Supplemental Table 2) mechanistically involved in multiple molecular pathways. We then stratified the S-sulfhydrated protein landscape into biochemical pathways using KEGG pathway analysis (Figure 5C). In the context of GBM, protein S-sulfhydration was dysregulated across multiple metabolic pathways. Carbon metabolism, pyruvate and amino acid metabolism, oxidative phosphorylation, and glycolysis were all significantly impacted through the loss of H2S signaling. Notably, we also observed depletion of S-sulfhydrated proteins (Supplemental Figure 5F and Supplemental Table 3) from syngeneic tumors isolated from the brains of HFD-fed mice compared with samples isolated from chow-fed control animals. In order to test whether H2S inhibition results in functional metabolic reprograming, we examined tumor cell energetics in the context of chemical inhibition of H2S production using the Agilent Seahorse Analyzer. We administered either the CGL-selective inhibitor PAG or vehicle to the syngeneic GBM models CT2A or KR158. After 3 consecutive passages in the presence of PAG or vehicle, we evaluated metabolic output before and after secondary administration of 5 μM oleic acid. For both GBM models, long-term inhibition of H2S synthesis increased bioenergetics measured both by oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) (Figure 5, D–G). These data support the conclusion that loss of H2S synthesis and signaling result in significant elevation of cellular metabolic function both at resting state and after exposure to the monounsaturated fat oleic acid. Taken together, these data indicate that loss of S-sulfhydration results in a broad-spectrum molecular reprogramming that enhances metabolism and bioenergetics, enabling the tumor to capitalize on the accumulating saturated fats that collect as a result of HFD consumption.
Figure 5. Gliomagenesis induces significant loss in H2S synthesis and signaling primarily associated with cellular metabolism.

(A) Analysis of H2S production confirms that human GBM tumors produce a lower amount of H2S than noncancerous control brain tissues. Each well contains brain or tumor tissue homogenate from separate biopsy specimens. P values determined by unpaired t test. (B) Volcano plot representing the LC-MS S-sulfhydration analysis reveals striking deficits in the posttranslational H2S signaling profile of human GBM as compared with noncancerous human brain tissue. (C) KEGG pathway analysis of the proteins that have undergone S-sulfhydration loss in the context of GBM identifies a broad-spectrum molecular reprogramming centered on GBM tumor cell metabolism. Inhibition of H2S synthesis drives cultured GBM cells into a state of enhanced cellular energetics. Long-term culture of the syngeneic GBM models KR158 and CT2A with PAG resulted in increased metabolic fitness and cellular energetics when compared with vehicle control conditions. Enhanced metabolism was evident at baseline and persisted after introduction of the fatty acid substrate oleic acid. Assessments of cellular metabolism and energetics were based on mitochondrial respiration (D and F), measured by the rate of oxygen consumption (OCR) as well as cellular glycolysis (E and G), measured by the extracellular acidification rate (ECAR). Seahorse Analyzer experiments were conducted in biological triplicate. P values determined by 2-way ANOVA.
H2S replacement arrests GBM progression in vitro and in vivo.
The connection between H2S production and diet has been well established in fields outside of cancer biology (43), and a partial mechanism by which HFD consumption inhibits H2S synthesis was recently described (21). In epithelial and endothelial tissues such as the liver, kidneys, and vasculature, HFD consumption resulted in decreased expression of critical H2S-synthesizing enzymes. Loss of enzyme expression and/or function resulted in decreased H2S synthesis and attenuation of protein S-sulfhydration. The precise mechanism by which saturated lipids directly regulate the expression of these key proteins remains unknown. To assess whether this potential mechanism was active in the brain in our in vivo experiments, we analyzed the protein expression of H2S-synthesizing enzymes and found that tumor-bearing mice fed a HFD presented a significant reduction in the protein expression of CBS compared with controls (Figure 6A). To more firmly establish a causal link between high-fat conditions and H2S, we performed in vitro rescue experiments in which we attempted to rescue the hyperproliferation induced by oleic acid by H2S replacement. Using 2 dosages, exogenous H2S was able to temper or completely abrogate lipid-induced hyperproliferation (Figure 6, B–E). Additionally, when we applied a similar experimental manipulation to tumors developing in vivo, we were encouraged to see that GBM tumor development was halted by daily administration of the chemical H2S donor NaHS. Specifically, in vivo H2S rescue resulted in stable tumor volume over time when compared with vehicle controls, which continually expanded throughout the experiment (Figure 6F). Further, H2S administration markedly reduced the protein expression of the CSC-associated factor SOX2 (Figure 6G). More broadly, these findings mechanistically link HFD consumption to the loss of GBM-suppressive H2S and also provide proof of concept for H2S replacement as a potential and unexplored treatment/management strategy against this incurable cancer. Additionally, these investigations clarify how H2S acts as a potent, nongenetic tumor suppressor, regulating cellular metabolism in order to resist GBM tumor cell proliferation and the emergence of the CSC phenotype.
Figure 6. In vitro and in vivo rescue experimentation links H2S to HFD and validates its tumor suppressive function.

To establish a clear connection between high-fat conditions and H2S, we investigated the protein expression of critical H2S-synthesizing enzymes. Protein analysis (A) confirmed a significant decrease in the protein expression of the H2S-synthesizing enzyme CBS in the context of high-fat feeding. In vitro rescue experiments using 2 concentrations of the H2S donor NaHS were performed while tracking cellular proliferation in real-time in the syngeneic GBM models KR158 (B and C) and CT2A (D and E). For these experiments, standard growth medium was supplemented with the following treatments: 5 μM oleic acid; vehicle; 200 μM or 500 μM NaHS; and 5 μM oleic acid + 200 μM or 500 μM NaHS. All in vitro experiments were conducted in biological triplicate. P values determined by 2-way ANOVA coupled to Dunnett’s multiple comparison test. Last, in vivo rescue (F) was demonstrated using flank hGBM 23 tumors that were established and treated daily with either the chemical H2S donor NaHS or vehicle. For these experiments, treatment was initiated once the flank tumors could be palpated and accurately measured for volume. Protein analysis (G) of endpoint flank tumors confirmed that H2S replacement resulted in marked reduction in the expression of SOX2.
Discussion
These findings confirm that HFD consumption accelerates and intensifies GBM in the preclinical experimental setting. The degree to which this finding extends to the human clinical condition remains an open question. As long-term dietary patterns are not accounted for as a variable in most clinical settings, epidemiological studies primarily question whether obesity, as measured by body mass index (BMI), represents a risk factor for GBM initiation. Such a link has been demonstrated for several cancers (33, 34); however, the connection between gliomagenesis and obesity has been inconsistent. For example, the recent completion of a massive meta-analysis leveraging clinical data from more than 10 million subjects led to the conclusion that overweight (BMI: 25–30 kg/m2) and obese (BMI > 30 kg/m2) statuses represent a risk factor for glioma development specifically for women (44). However, other reports fail to substantiate this finding (45, 46). Our own data indicate that HFD and not obesity functions as a GBM accelerant (Supplemental Figure 7). In combination with the H2S suppression reported here, it is worth noting that other diet-associated changes, such as hyperglycemia (47) and hyperinsulinemia (48), have been associated with GBM acceleration. Given the variety of compensatory mechanisms that maintain euglycemia, even long-term consumption of HFD rarely results in significant hyperglycemia (confirmed by the The Jackson Laboratory Mouse Phenome Database, Jaxpheno11). Severe hyperinsulinemia can present as early as 4 to 6 weeks on HFD as the pancreas overproduces insulin to overcome tissue insulin resistance. Based on these findings, it is conceivable that patients who consume a HFD long-term could experience a hyperaggressive disease trajectory and/or present disease that is more adaptable and therefore more challenging to treat. Recently, a diet-focused meta-analysis of 1.2 million subjects was presented involving self-reported dietary patterns. Once again, investigators were limited to interrogating this data set for risk factor identification, concluding that no specific diets served as risk factors for GBM initiation (46). Including factors such as progression-free survival and long-term dietary pattern will help clarify the degree to which obesogenic diets modulate the course of human GBM. Based on that additional level of understanding, we will be better able to evaluate diet as a prognostic indicator and manage our expectations for using diet as a tool to manage disease.
Our findings on HFD-induced adult cytogenesis (Supplemental Figure 4) suggest that HFD may facilitate GBM development through 2 non–mutually exclusive mechanisms. First, HFD consumption resulted in the expansion of the endogenous NSPC population, which has been identified as a potential cell of origin for GBM (49, 50). Combined with the right set of mutagenizing events, the presence of this more abundant and potentially vulnerable cell pool may lower the threshold required for malignant transformation and tumorigenesis. Second, it is conceivable that a subpopulation of NSPCs may be drawn into an evolving tumor, proximal to the SVZ to provide protection and support. This support may involve the production of oncogenic secreted factors and/or metabolites, which can then be used by the developing GBM. In support of this hypothetical NSPC-GBM interaction, multiple groups have demonstrated that exogenous NSPCs home to and infiltrate the GBM tumor microenvironment (43, 51). Additional investigations have substantiated that endogenous neural stem cells and their progeny migrate toward regions of CNS damage in response to ischemia (52, 53). While these efforts have primarily focused on this NSPC-to-GBM tropism as a therapeutic opportunity, it is equally possible that the tumor capitalizes on these interactions to drive tumorigenesis.
There are interesting overlaps between this study and work on the ketogenic diet as an anti-GBM intervention. Both lines of research embrace the idea that diet might have profound effects on the trajectory of disease. The ketogenic diet, which is commonly employed against epilepsy, is typically formulated as an extremely high-fat (90%), low-carbohydrate (5%) diet. Recently, it has been hypothesized that the glucose deprivation that results from strict adherence to the ketogenic diet might enable long-term GBM management by starving these tumors of their preferred metabolic substrate (31, 54). Unfortunately, a synthesis of recent work on GBM metabolism confirms that these tumors are not strictly dependent on glucose. Instead, subpopulations of tumor cells present a propensity for metabolic adaptability. A recent study of glucose starvation in GBM confirmed that while the bulk of the malignant population, made up of non–stem tumor cells, was clearly dependent on glucose, CSCs were metabolically plastic. They were uniquely capable of adapting to multiple metabolic substrates (12–14, 55), especially favoring fatty acid oxidation in the context of glucose starvation. While this strategy effectively eliminated the glucose-dependent non–stem tumor cell population, GBM CSCs bypassed this metabolic dependency and expanded into the treatment-induced vacancy (16). Consistently, we found that HFD consumption altered the nutrient landscape of the brain, specifically resulting in intracerebral accumulation of saturated lipids. As seen in the liver, this lipid excess inhibited H2S production and signaling, in turn driving CSC enrichment and disease acceleration. These findings caution against the use of the ketogenic diet as a long-term GBM management tool. The ketogenic diet represents a selective pressure, similar to a targeted chemo-, radio-, or immunotherapy. The inherent heterogeneity, complexity, and adaptability of GBM will ultimately drive the evolving tumor to sidestep any singular selective pressure.
Our findings also highlight a posttranslational modification that is profoundly altered in GBM and has received very little attention. H2S and S-sulfhydration have been a focus of research in the aging (56, 57), neurodegeneration (23), and metabolism fields; however, there is very little known about this metabolite in GBM tumors (28, 29). Our work reinforces the concept of H2S signaling as a tumor suppressor for GBM while simultaneously introducing numerous questions about the mechanisms by which this is achieved. One question, for example, is: What is the mechanism responsible for H2S signaling attenuation in GBM? Previous work established that attenuation of synthesis resulted from downregulation of CGL, CBS, and MPST. HFD consumption can decrease the expression of these enzymes at the protein level. Our work in the brain (Figure 6A) and previous work in the liver (22) identify H2S synthesizing enzyme downregulation; however, not all patients with GBM are long-term consumers of HFD. Synthesis attenuation can also result from enzyme loss of function. This has been confirmed in human GBM (29) but is not yet explained at the molecular mechanistic level. An additional question is: By what mechanism does loss of S-sulfhydration drive GBM progression? Here, it is worth noting that the functional consequences of S-sulfhydration have only been reported for a limited number of proteins. In a recent review, the functional changes that resulted from S-sulfhydration were highlighted for 43 cysteine residues present within only 25 total proteins (24). Thus, at this point, we can only speculate on the molecular changes that connect the loss of H2S signaling to increased tumor bioenergetics. A preliminary in silico examination revealed 3 metabolism-associated proteins: fatty acid–binding protein 3 (FABP3), enoyl-CoA hydratase 1 (ECH1), and ATP synthase peripheral stalk subunit OSCP (ATP5O), which established a presumptive signaling pathway that can be tested in the future for its capacity to increase metabolic fitness and fatty acid utilization based on the loss of S-sulfhydration (Supplemental Table 2). If loss of S-sulfhydration on these 3 proteins conferred enzymatic gain of function, then FABP3 would more effectively channel fatty acids into the cytosol of GBM tumor cells, ECH1 would enhance the lipid β-oxidation cycle, and ATP5O may increase a cell’s capacity for oxidative phosphorylation. Gain of function for these 3 proteins is consistent with functional changes that result from S-sulfhydration of factors that regulate metabolism and mitochondrial fitness (24). A final question is: Could this nontraditional, nongenetic tumor suppressor be replaced or supplemented in conjunction with standard of care to better manage GBM in human patients? There are a variety of chemical H2S donors, pharmaceutical H2S inducers, and diets that drive endogenous H2S production. A major challenge here will be one of bioavailability. For an H2S replacement strategy to work, the metabolite must be relatively stable and able to penetrate into the brain. While we are encouraged by our in vivo and in vitro H2S rescue data (Figure 6), currently available donors and inducers fall short of these standards. Nonetheless, we suggest that pursuing strategies to induce intracerebral H2S production and signaling may serve to limit GBM metabolic enhancement, making the disease a more receptive target for cytotoxic therapies.
In conclusion, we have demonstrated that consumption of an obesogenic HFD resulted in a shift in the nutrient profile of the brain. The resulting attenuation of tumor-suppressive and metabolism-suppressive H2S enabled adaptation to this lipid excess. These fundamental changes within the brain and tumor microenvironment induced CSC enrichment, heightened chemotherapy resistance, and accelerated GBM progression (Figure 7).
Figure 7. Consumption of HFD inhibits the tumor-suppressive activity of H2S, driving CSC enrichment and progression in GBM.

Our findings indicate that HFD consumption serves as an accelerant for GBM. HFD consumption drives accumulation of saturated, mono-and di-unsaturated fats within the brain. This lipid excess inhibits H2S production, which results in a wide-ranging attenuation of S-sulfhydration centered on tumor metabolism regulators. These linked intrinsic and extrinsic changes within the tumor result in the expansion of treatment-refractory CSCs, increased tumor cell proliferation, protection from necrotic cell death, and truncated overall survival.
Methods
For detailed reagent purchasing and use instructions, please refer to Table 1.
Table 1. Experimental reagents.

Cell culture.
The patient-derived GBM models hGBM 23, hGBM 124, hGBM 3832, and hGBM 3691, and the syngeneic mouse GBM models CT2A and KR158 were propagated under adherent culture conditions. Single-cell suspensions of 50,000 cells/mL were suspended in DMEM/F12 media enriched with N2 Supplement, recombinant human epidermal and fibroblast growth factors (rhEGF and rhFGF), and penicillin-streptomycin, and seeded into Geltrex-coated culture flasks.
The syngeneic mouse GBM model GL261 and the liver cancer control models HepG2 and NCTC 1469 were propagated under adherent culture conditions in DMEM/F12 supplemented with 10% FBS and penicillin-streptomycin.
For all cellular models, complete media including supplements were exchanged every other day until the cultures reached 85%–90% confluence. Cultures were passaged weekly using Accutase (StemCell Technologies, catalog 7920). PBS was used to wash, quench, and collect cells before replating. Cells were grown and maintained in humidified incubators held at 37°C and 5% CO2.
The patient-derived GBM models hGBM 23 and hGBM 124 were obtained from Erik Sulman (New York University, New York, New York, USA). The patient-derived GBM model hGBM 3691 was obtained from Jeremy Rich (University of Pittsburgh, Pittsburgh, Pennsylvania, USA). The syngeneic GBM models CT2A and KR158 were obtained from Loic Deleyrolle (University of Florida, Gainesville, Florida, USA).
Differential diet and intracerebral tumor cell implantation.
For comparative survival studies concerning high-fat versus control diets, 4-week-old female mice were purchased from Jackson Laboratories. Based on the patient epidemiological data, which indicated that overweight and obese statuses correlate with increased risk of gliomagenesis in women only, we elected to perform these in vivo studies exclusively in female mice. Mice were subdivided into 2 groups: a HFD-fed group and a control diet–fed group. For the syngeneic mouse models, C57BL/6J mice were used, and the control group was fed standard rodent chow. For the patient-derived GBM model, NOD-SCID mice were employed, and the control group was fed a low-fat, energy-balanced diet. Differential diets were introduced 2 weeks prior to tumor cell implantation and maintained throughout the postimplantation survival period. Diet was completely exchanged once per week to prevent spoilage. For tumor cell implantation, 6-week-old, diet-primed mice were anesthetized using inhaled isoflurane (2%–2.5%) and fit to a stereotaxic apparatus. Using an insulin syringe secured to a large probe holder, the 31-gauge needle was passed directly through the scalp and skull approximately 0.5 mm rostral and 1.8 mm lateral to the bregma. The needle was lowered 3.5 mm beneath the surface of the scalp, where a specific number of cells (50,000; 20,000; 15,000; 10,000; 5000) suspended in 5 μL DMEM/F12 was slowly injected. The needle was held in place an additional 60 seconds before a slow and measured removal. Based on initial power calculations, a sample size of 10 animals per diet (n = 10/group) was used for the syngeneic models CT2A and KR158 for each cell dose. For the syngeneic model GL261, a sample size of 16 animals per diet, per cell dose (n = 16/group) was used. Animals were monitored over time for changes in body mass, fat-to-lean mass composition using EchoMRI, and the presentation of the set of neurological and behavioral symptoms associated with end-stage brain cancer (Figure 1A).
In vivo H2S rescue.
These experiments were designed to compare a saline-treated (vehicle) control group with an NaHS-treated experimental group, both leveraging a sample size of 8 animals. hGBM 23 cells (1.0 × 106), suspended in 100 μL DMEM/F12 were injected into the subcutaneous space along the animal’s right rear flank. Tumors developed unimpeded over the course of the following 4 to 5 weeks to the point where a small, palpable, and visually clear mass developed. Digital caliper measurement of tumor length and width was collected every other day for a period of 11 days. Tumor volume was calculated according to the formula for an ellipsoid solid listed below. Animals received daily injections of either 5 mg/kg NaHS (solubilized in sterile saline) or vehicle directly into the developing tumor. Animals were anesthetized with 2% isoflurane in O2(gaseous, g) to ensure accurate measurement and precise control over the injection. The experiment was terminated, in accordance with Cleveland Clinic Lerner Research Institute IACUC policy, when the first set of control animals developed 2 cm3 tumors. At that point, tumors were harvested and flash frozen in N2(liquid, l) for downstream protein analysis.
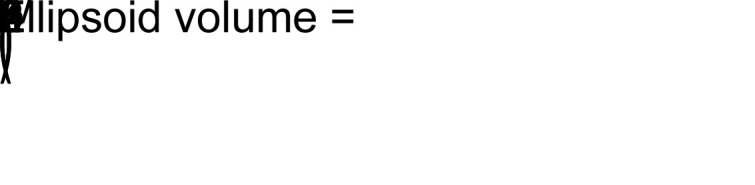
Equation 1
Tissue preparation and immunofluorescence analysis.
At the experimental endpoint, animals were subdivided into 1 of 2 possible tissue preservation and harvesting modalities. Approximately 30% of endpoint animals were anesthetized and subjected to cardiac perfusion with 4% formaldehyde in PBS. The brain was removed and post-fixed overnight. The tissue was then cryoprotected with sequential treatments first in a solution of 30% sucrose in PBS and then in a 1:1 mixture of 30% sucrose (in PBS)/OCT. Finally, the tissue was embedded in OCT, and 20 μm coronal sections were prepared for subsequent immunofluorescence analysis using standard protocols. The specific antibodies used can be found in Table 1. In all cases, primary antibodies were coupled with appropriate Alexa Fluor 488– or Alexa Fluor 555–conjugated secondary antibodies. Nuclei were visualized using either DAPI or Hoechst 33342. The stained sections were mounted onto slides, coverslipped, and examined using an inverted Leica SP8 confocal microscope.
To quantify the tumor microenvironment, tissue was examined from 3 separate animals per diet. For a given animal, 15 representative images were captured (5 images × 3 tumor-bearing sections). From the 45 representative images captured per diet, one necrotic site was identified within the HFD group as compared with 39 sites identified under control conditions. For a given animal, 12 representative images were captured (4 images × 3 tumor-bearing sections).
The remaining 70% of endpoint animals were anesthetized and subjected to cardiac perfusion with PBS. The brain was dissected away from the calvarium and bisected along the midline, generating a pair of matched samples: the tumor-bearing hemisphere and the contralateral, healthy control hemisphere. Labeled samples were immediately flash frozen in liquid nitrogen and stored at –20°C until downstream tissue analysis.
Untargeted mass spectrometry-based lipidomic analysis.
To characterize the lipid landscape of the HFD- versus chow-fed brain and tumor, we employed a mass spectrometry-based shotgun lipidomic analysis method that enables identification of multiple structurally distinct lipid species. This method, which we adopted without alteration, was originally published by Gromovsky et al. (58) and later optimized by Neumann et al. (59).
Preparation of oleic and linoleic acid.
The free fatty acids oleic and linoleic acid were delivered to cultured cells bound to BSA. Therefore, we initially generated a 3% (mass/vol) fatty acid-free BSA solution using D-PBS at room temperature. Oleic acid and linoleic acid were solubilized separately in 90% aqueous ethanol, producing a 50 mM free fatty acid (FFA) solution. These solutions were then diluted to 50 nM stock solutions in the 3% BSA solution, aliquoted, and stored at −20°C until needed.
Tumor cell proliferation assessment.
Proliferation was assessed in the contexts of excess oleic and linoleic acid, in the presence of PAG, and in response to the exogenous H2S donors NaHS and GYY4137 using the CellTiter Glo viability assay following the protocol established by the manufacturer. A total of 2000 cells/well were plated onto Geltrex-coated 96-well plates. BSA-bound oleic and linoleic acids were added to the media at the following concentrations: vehicle (3% BSA alone), 100 nM, 1 μM, and 10 μM. All conditions were plated in 5 technical replicates. Readings were taken on the day of plating (Day 0) as well as on Days 1, 3, and 5. Cell viability was normalized to Day 0 to account for any plating irregularity. Each of these experiments were repeated in triplicate. Points represent the mean ± SEM taking all experimental replication into account.
Cellular proliferation was validated under conditions of H2S inhibition through direct cell counting. In this case, 20,000 cells/well were plated onto Geltrex-coated 12-well plates. Vehicle (DMSO), PAG (solubilized in growth media), or temozolomide (TMZ, solubilized in DMSO) was added at the time of plating. All conditions were plated in 4 technical replicates. Cells were dissociated, collected, and counted with an automated hemocytometer 3 days after plating.
Proliferation was monitored and quantified in real-time using the Incucyte Live-Cell Analysis System. For these experiments, 5 technical replicates of 25,000 cells were plated per condition. Cells were plated in standard growth media supplemented with 5 μM OA, fatty acid-free BSA vehicle control, 500 μM NaHS (solubilized in growth media), or 5 μM OA + 500 μM NaHS. Data were captured every 8 hours over a period of 4 days. Complete media, including supplements, was exchanged every 48 hours.
In vitro limiting-dilution analysis (LDA).
Extreme LDA was performed to assess self-renewal in the context of excess mono- and di-unsaturated lipids. Cells were diluted progressively throughout the plate beginning with 100 cells/well across the first row. Subsequent dilutions resulted in the delivery of 50, 25, 13, 6, 3, 1, and 0 cells/well. Separate plates were prepared containing vehicle (3% BSA alone) and 10 μM, 50 μM, and 100 μM oleic or linoleic acid bound to 3% BSA. Cells were maintained in culture for 10 to 14 days. Fifty microliters of media plus lipids or media plus vehicle was added to the appropriate wells every 5 to 7 days. Each well was scored as either positive or negative for sphere outgrowth. The Walter and Eliza Hall Institute Bioinformatics Division ELDA analyzer (60) was used to analyze data and calculate stem cell frequency. Each of these experiments were repeated in duplicate. Points represent the mean ± SEM, taking all experimental replication into account.
IC50 assessment of the H2S donors NaHS and GYY 4137.
The half maximal inhibitory concentration (IC50) was established for the H2S donors NaHS and GYY 4137 across a variety of cellular models. Fresh NaHS was solubilized in D-PBS at the time of experimental setup. GYY 4137 was solubilized in 100% ethanol and stored at –20°C. A total of 1000 cells/well was plated in white-walled, Geltrex-coated 96-well plates. Five technical replicates were plated per H2S donor concentration. Wells were then supplemented with either NaHS or GYY 4137 at the following concentrations: 10 μM, 25 μM, 50 μM, 75 μM, 100 μM, and 500 μM, along with appropriate vehicle controls. Following the manufacturer’s instructions, cellular viability was determined using the CellTiter Glo assay after 5 days of incubation. Each of these experiments was repeated in triplicate. Points represent the mean ± SEM, taking all experimental replication into account.
Seahorse metabolic analysis.
Prior to analysis, either the CGL-selective inhibitor DL-PAG or vehicle was applied to the syngeneic GBM models CT2A or KR158. After 3 consecutive passages in the presence of PAG or vehicle, we evaluated metabolic output using the Agilent Seahorse Analyzer before and after the secondary administration of 5 μM oleic acid, incorporating 10 technical replicates per condition per experiment. Standard protocols were followed, including cell plating strategy, which included 4 blank wells omitted for background normalization. Each of these experiments was repeated in triplicate. Points represent the mean ± SEM, taking all experimental replication into account.
Protein analysis.
Denatured protein (20 μg) was loaded on 12% SDS-PAGE gels, electrophoresed, and transferred onto PVDF membranes. Blots were blocked with 5% BSA in TBS-T, probed with primary antibodies (Table 1) and appropriate horseradish peroxidase–conjugated secondary antibodies, and developed using the Pierce ECL Plus kit (Thermo Fisher Scientific) on the Bio-Rad ChemiDoc Imaging System.
Lead acetate/lead sulfide H2S production assay.
The endogenous H2S production capacity of tissues (and pelleted cells) was measured using the lead acetate/lead sulfide method. This method, which we adopted without alteration, was originally published by Hine and colleagues (56). H2S production was assessed from tumor-containing specimens collected from HFD- versus control-fed mice, LepOB versus C57BL/6J mice, and human GBM versus noncancerous control specimens. In brief, prior to assay setup, the protein content of each sample was measured using the BCA protocol, enabling an equivalent amount of protein to be added to each well of the assay. Chemical reaction between the lead acetate embedded within the assay filter paper and the H2S produced by the tissue lysate generates a pigmented substrate that can be quantified based on densitometry using the Integrated Density function (IntDen) of the ImageJ software package.
Protein S-Sulfhydration landscape analysis.
To characterize the S-sulfhydration landscape of human GBM tumors and noncancerous control tissues, we adopted a multistage biotin thiol–based procedure to isolate S-sulfhydrated proteins. Isolated proteins were then identified and quantified using mass spectrometry in collaboration with The Cleveland Clinic Proteomics Core Facility. The complete method, which we employed without alteration, was recently described by Bithi and colleagues (61). S-sulfhydration profiling data have been uploaded to the ProteomeXchange Consortium via the PRIDE Partner Repository. Human specimen data (Figure 5) were deposited under accession number PXD025902. HFD versus control diet–fed mouse specimen data (Supplemental Figure 5F) were deposited under accession number PXD023685.
Statistics for S-Sulfhydration profiling.
Microsoft Excel, GraphPad Prism, and OriginPro were used for data analysis and statistics. Microsoft Excel and GraphPad Prism data are presented here as mean ± SEM. The difference between 2 groups was analyzed by unpaired t-test, with the significance level set to P less than 0.05. OriginPro was used to generate volcano plots in which the x axis reported the fold change between GBM and noncancerous control groups represented in log2 scale. The y axis reported negative log10 of the P values from the comparative t tests.
Statistics for in vitro and in vivo experimentation.
All experiments were performed in triplicate. Results are reported as mean ± SEM. Unless otherwise stated, 1-way ANOVA was used to calculate statistical significance; P values and sample size are detailed in the text and figure legends. In vivo survival analysis was calculated by log-rank analysis.
Interrogation of The Cancer Genome Atlas database.
Gene expression data were obtained from the University of California Santa Cruz Xena Functional Genomics Explorer (62), leveraging data from The Cancer Genome Atlas Low-Grade Glioma and Glioblastoma (LGGGBM) study. Expression levels were categorized into 2 groups based on median gene expression. Log-rank survival analysis was performed according to the overall survival times and statuses for patients in both groups.
Study approvals.
All animal procedures were evaluated and approved prior to initiation by the IACUC of the Cleveland Clinic Lerner Research Institute. The Rose Ella Burkhardt Brain Tumor Center collected, catalogued, and distributed all human brain tumor and control brain specimens according to policies and procedures specified by the IRB of the Cleveland Clinic.
Author contributions
DJS conceived, designed, and performed experiments, analyzed and interpreted data, wrote and revised the manuscript, and provided financial support for the project. GAR and NB designed and performed experiments, analyzed data, and contributed to the writing of the manuscript. SZW, KMT, CKAN, and GKA performed experiments and provided critical feedback on the revision of the manuscript. OR, JMB, CH, and JDL helped guide overall strategic planning as well as the experimental design process, provided critical feedback on the revision of the manuscript, and provided financial support for the project.
Supplementary Material
Acknowledgments
We thank the members of the Lathia, Hine, and Brown laboratories for insightful conversations and constructive criticism. We are grateful to Judith Drazba of the Lerner Digital Imaging Microscopy Core, who provided assistance with confocal microscopy. The Leica SP8 confocal microscope used in this study was purchased with support from the National Institutes of Health (1S10OD019972-01). We appreciate Belinda Willard of the Lerner Proteomics and Metabolomics Core, who provided support with mass spectrometry. This investigation employed the Orbitrap Elite and Fusion Lumos mass spectrometers. Both instruments were purchased with funding from the National Institutes of Health (1S10RR031537-01, 1S10OD023436-01). We thank Amanda Mendelsohn from the Cleveland Clinic Center for Medical Art & Photography for illustration assistance; Loic Deleyrolle, Erik Sulman, and Jeremy Rich for providing syngeneic and patient-derived GBM models; and Gene Barnett and Mary McGraw of the Rose Ella Burkhardt Brain Tumor Center for assistance with patient tissue acquisition. The Lathia Laboratory is currently supported by the National Institutes of Health (R01 NS089641, R01 NS109742, R01 NS083629, and P01 CA245705) and the VeloSano Bike Race and was previously supported by a Sontag Foundation Distinguished Scientist Award. The Hine Laboratory is supported by the National Institutes of Health (R01 HL148352 and R00 AG050777). The Brown Laboratory receives funding from the National Institutes of Health (R01 DK120679 and P50 AA024333). DJS was supported by the Case Comprehensive Cancer Center Cancer Biology Training Grant (T32 CA059366) and an NIH Kirschstein NRSA Fellowship (F32 CA213727).
Version 1. 07/13/2021
In-Press Preview
Version 2. 09/01/2021
Electronic publication
Funding Statement
Award issued to J Lathia. Supports work in the Lathia Lab.
Awards issued to J Lathia. Support work in the Lathia Lab.
Award issued to J Lathia. Supports work in the Lathia Lab.
Award issued to J Lathia, C Hine, and D Silver in direct support of these efforts.
Awards issued to C Hine.
Awards issued to JM Brown.
Kirschstein NRSA Postdoctoral Fellowship awarded to D Silver
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2021, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2021;131(17):e138276.https://doi.org/10.1172/JCI138276.
Contributor Information
Daniel J. Silver, Email: silverd2@ccf.org.
Gustavo A. Roversi, Email: roversig@ccf.org.
Nazmin Bithi, Email: bithin@ccf.org.
Sabrina Z. Wang, Email: szw7@case.edu.
Katie M. Troike, Email: troikek@ccf.org.
Chase K.A. Neumann, Email: chase.neumann@biochem.utah.edu.
Grace K. Ahuja, Email: gka11@case.edu.
Ofer Reizes, Email: reizeso@ccf.org.
J. Mark Brown, Email: brownm5@ccf.org.
Christopher Hine, Email: hinec@ccf.org.
Justin D. Lathia, Email: lathiaj@ccf.org.
References
- 1.Tran B, Rosenthal MA. Survival comparison between glioblastoma multiforme and other incurable cancers. J Clin Neurosci. 2010;17(4):417–421. doi: 10.1016/j.jocn.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Weller M, et al. Glioma. Nat Rev Dis Primers. 2015;39:15017. doi: 10.1038/nrdp.2015.17. [DOI] [PubMed] [Google Scholar]
- 3.Sottoriva A, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci U S A. 2013;110(10):4009–4014. doi: 10.1073/pnas.1219747110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel AP, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reinartz R, et al. Functional subclone profiling for prediction of treatment-induced intratumor population shifts and discovery of rational drug combinations in human glioblastoma. Clin Cancer Res. 2016;23(2):562–574. doi: 10.1158/1078-0432.CCR-15-2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siebzehnrubl FA, et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med. 2013;5(8):1196–1212. doi: 10.1002/emmm.201302827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lathia JD, et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell. 2010;6(5):421–432. doi: 10.1016/j.stem.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haas TL, et al. Integrin α7 is a functional marker and potential therapeutic target in glioblastoma. Stem Cell. 2017;21(1):35–50. doi: 10.1016/j.stem.2017.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 10.Badr CE, et al. Metabolic heterogeneity and adaptability in brain tumors. Cell Mol Life Sci. 2020;77(24):5101–5119. doi: 10.1007/s00018-020-03569-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitchell K, et al. The evolution of the cancer stem cell state in glioblastoma: emerging insights into the next-generation of functional interactions. Neuro Oncol. 2021;23(2):199–213. doi: 10.1093/neuonc/noaa259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hale JS, et al. Cancer stem cell-specific scavenger receptor CD36 drives glioblastoma progression. Stem Cells. 2014;32(7):1746–1758. doi: 10.1002/stem.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X, et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat Neurosci. 2017;20(5):661–673. doi: 10.1038/nn.4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung J, et al. Nicotinamide metabolism regulates glioblastoma stem cell maintenance. JCI Insight. 2017;2(10):766–724. doi: 10.1172/jci.insight.90019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warburg O, Minami S. Versuche an Überlebendem Carcinom-gewebe. Klin Wochensch. 1923;2:776–777. doi: 10.1007/BF01712130. [DOI] [Google Scholar]
- 16.Hoang Minh LB, et al. Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018;37(23):e98772. doi: 10.15252/embj.201798772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chianese R, et al. Impact of dietary fats on brain functions. Curr Neuropharmacol. 2018;16(7):1059–1085. doi: 10.2174/1570159X15666171017102547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He C, et al. High-fat diet induces dysbiosis of gastric microbiota prior to gut microbiota in association with metabolic disorders in mice. Front Microbiol. 2018;9:979–979. doi: 10.3389/fmicb.2018.00979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mehrian-Shai R, et al. The gut-brain axis, paving the way to brain cancer. Trends Cancer. 2019;5(4):1–207. doi: 10.1016/j.trecan.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crevel RW, et al. High-fat diets and the immune response of C57Bl mice. Br J Nutr. 1992;67(1):17–26. doi: 10.1079/BJN19920005. [DOI] [PubMed] [Google Scholar]
- 21.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92(2):791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 22.Peh MT, et al. Effect of feeding a high fat diet on hydrogen sulfide (H2S) metabolism in the mouse. Nitric Oxide. 2014;41:138–145. doi: 10.1016/j.niox.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Paul BD, Snyder SH. H2S: A novel gasotransmitter that signals by sulfhydration. Trends Biochem Sci. 2015;40(11):687–700. doi: 10.1016/j.tibs.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang D, et al. H2S-induced sulfhydration: biological function and detection methodology. Front Pharmacol. 2017;8:608. doi: 10.3389/fphar.2017.00608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szabo C, et al. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci U S A. 2013;110(30):12474–12479. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hellmich MR, Szabo C. Hydrogen sulfide and cancer. Handb Exp Pharmacol. 2015;230:233–241. doi: 10.1007/978-3-319-18144-8_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu D, et al. Hydrogen sulfide in cancer: friend or foe? Nitric Oxide. 2015;50:38–45. doi: 10.1016/j.niox.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 28.Takano N, et al. Decreased expression of cystathionine β-synthase promotes glioma tumorigenesis. Mol Cancer Res. 2014;12(10):1398–1406. doi: 10.1158/1541-7786.MCR-14-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wróbel M, et al. Is development of high-grade gliomas sulfur-dependent? Molecules. 2014;19(12):21350–21362. doi: 10.3390/molecules191221350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Villa GR, et al. An LXR-cholesterol axis creates a metabolic co-dependency for brain cancers. Cancer Cell. 2016;30(5):683–693. doi: 10.1016/j.ccell.2016.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martuscello RT, et al. A supplemented high-fat low-carbohydrate diet for the treatment of glioblastoma. Clin Cancer Res. 2016;22(10):2482–2495. doi: 10.1158/1078-0432.CCR-15-0916. [DOI] [PubMed] [Google Scholar]
- 32.Bi J, et al. Altered cellular metabolism in gliomas — an emerging landscape of actionable co-dependency targets. Nat Rev Cancer. 2019;20(1):57–70. doi: 10.1038/s41568-019-0226-5. [DOI] [PubMed] [Google Scholar]
- 33.Calle EE, et al. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348(17):1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 34.Lauby-Secretan B, et al. Body fatness and cancer--viewpoint of the IARC working group. N Engl J Med. 2016;375(8):794–798. doi: 10.1056/NEJMsr1606602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts MN, et al. A ketogenic diet extends longevity and healthspan in adult mice. Cell Metab. 2017;26(3):539–546. doi: 10.1016/j.cmet.2017.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newman JC, et al. Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab. 2017;26(3):547–557. doi: 10.1016/j.cmet.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Obeid LM, et al. Programmed cell death induced by ceramide. Science. 1993;259(5102):1769–1771. doi: 10.1126/science.8456305. [DOI] [PubMed] [Google Scholar]
- 38.Yabu T, et al. Stress-induced ceramide generation and apoptosis via the phosphorylation and activation of nSMase1 by JNK signaling. Cell Death Differ. 2015;22(2):258–273. doi: 10.1038/cdd.2014.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alvarado AG, et al. Glioblastoma cancer stem cells evade innate immune suppression of self-renewal through reduced TLR4 expression. Cell Stem Cell. 2016;20(4):450–461. doi: 10.1016/j.stem.2016.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silver DJ, Lathia JD. Revealing the glioma cancer stem cell interactome, one niche at a time. J Pathol. 2018;244(3):260–264. doi: 10.1002/path.5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stoll EA, et al. Neural stem cells in the adult subventricular zone oxidize fatty acids to produce energy and support neurogenic activity. Stem Cells. 2015;33(7):2306–2319. doi: 10.1002/stem.2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshimoto S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499(7456):97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 43.Aboody KS, et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc Nat Acad Sci U S A. 2000;97(23):12846–12851. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sergentanis TN, et al. Obesity and risk for brain/CNS tumors, gliomas and meningiomas: a meta-analysis. PLoS One. 2015;10(9):e0136974. doi: 10.1371/journal.pone.0136974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Disney-Hogg L, et al. Influence of obesity-related risk factors in the aetiology of glioma. Br J Cancer. 2018;118(7):1020–1027. doi: 10.1038/s41416-018-0009-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuan AS, et al. Diet and risk of glioma: combined analysis of 3 large prospective studies in the UK and USA. Neuro Oncol. 2019;21(7):944–952. doi: 10.1093/neuonc/noz013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Derr RL, et al. Association between hyperglycemia and survival in patients with newly diagnosed glioblastoma. J Clin Oncol. 2009;27(7):1082–1086. doi: 10.1200/JCO.2008.19.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grunberger G, et al. Insulin receptor of human cerebral gliomas. Structure and function. J Clin Invest. 1986;77(3):997–1005. doi: 10.1172/JCI112402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hambardzumyan D, et al. The probable cell of origin of NF1- and PDGF-driven glioblastomas. PLoS One. 2011;6(9):e24454. doi: 10.1371/journal.pone.0024454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee JH, et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature. 2018;560(7717):243–247. doi: 10.1038/s41586-018-0389-3. [DOI] [PubMed] [Google Scholar]
- 51.Khosh N, et al. Contact and encirclement of glioma cells in vitro is an intrinsic behavior of a clonal human neural stem cell line. PLoS One. 2012;7(12):e51859. doi: 10.1371/journal.pone.0051859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arvidsson A, et al. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8(9):963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- 53.Kokaia Z. Neurogenesis after ischaemic brain insults. Curr Opin Neurobiol. 2003;13(1):127–132. doi: 10.1016/S0959-4388(03)00017-5. [DOI] [PubMed] [Google Scholar]
- 54.Mukherjee P, et al. Therapeutic benefit of combining calorie-restricted ketogenic diet and glutamine targeting in late-stage experimental glioblastoma. Commun Biol. 2019;2:200. doi: 10.1038/s42003-019-0455-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang X, et al. Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells. Sci Transl Med. 2019;11(504):4972. doi: 10.1126/scitranslmed.aau4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hine C, et al. Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell. 2015;160(1–2):132–144. doi: 10.1016/j.cell.2014.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hine C, et al. Dietary and endocrine regulation of endogenous hydrogen sulfide production: implications for longevity. Antioxid Redox Signal. 2018;28(16):1483–1502. doi: 10.1089/ars.2017.7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gromovsky AD, et al. Δ-5 fatty acid desaturase FADS1 impacts metabolic disease by balancing proinflammatory and proresolving lipid mediators. Arterioscler Thromb Vasc Biol. 2018;38(1):218–231. doi: 10.1161/ATVBAHA.117.309660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Neumann CKA, et al. MBOAT7-driven phosphatidylinositol remodeling promotes the progression of clear cell renal carcinoma. Mol Metab. 2020;34:136–145. doi: 10.1016/j.molmet.2020.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1–2):70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 61.Bithi N, et al. Dietary restriction transforms the mammalian protein persulfidome in a tissue-specific and cystathionine γ-lyase-dependent manner. Nat Commun. 2021;12(1):1745. doi: 10.1038/s41467-021-22001-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. doi: 10.1101/326470. Goldman M, et al. The UCSC Xena platform for cancer genomics data visualization and interpretation [preprint]. Posted on bioRxiv September 26, 2019. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.