

Abstract
Neutrophil gelatinase associated lipocalin (NGAL, Lcn2) is the most widely studied biomarker of acute kidney injury (AKI). Previous studies have demonstrated that NGAL is produced by the kidney and released into the urine and plasma. Consequently, NGAL is currently considered a tubule specific injury marker of AKI. However, the utility of NGAL to predict AKI has been variable suggesting that other mechanisms of production are present. IL-6 is a proinflammatory cytokine increased in plasma by two hours of AKI and mediates distant organ effects. Herein, we investigated the role of IL-6 in renal and extra-renal NGAL production. Wild type mice with ischemic AKI had increased plasma IL-6, increased hepatic NGAL mRNA, increased plasma NGAL, and increased urine NGAL; all reduced in IL-6 knockout mice. Intravenous IL-6 in normal mice increased hepatic NGAL mRNA, plasma NGAL and urine NGAL. In mice with hepatocyte specific NGAL deletion (Lcn2hep−/−) and ischemic AKI, hepatic NGAL mRNA was absent, and plasma and urine NGAL were reduced. Since urine NGAL levels appear to be dependent on plasma levels, the renal handling of circulating NGAL was examined using recombinant human NGAL. After intravenous recombinant human NGAL administration to mice, human NGAL in mouse urine was detected by ELISA during proximal tubular dysfunction, but not in pre-renal azotemia. Thus, during AKI, IL-6 mediates hepatic NGAL production, hepatocytes are the primary source of plasma and urine NGAL, and plasma NGAL appears in the urine during proximal tubule dysfunction. Hence, our data change the paradigm by which NGAL should be interpreted as a biomarker of AKI.
Keywords: Acute kidney injury, cytokines, IL-6, ischemia reperfusion, nephrotoxicity, biomarkers
Introduction
Acute kidney injury (AKI) is a common complication in hospitalized patients occurring in up to 20% of hospital admissions and 30 to 50% of intensive care unit (ICU) admissions. AKI is an independent risk factor for death, with mortality rates as high as 50% in the ICU. 1 The development of biomarkers that can identify AKI early and more reliably than serum creatinine rise has been a translational research priority for over a decade with the premise that early recognition may lead to improved care and the development of new therapies. 2
Neutrophil gelatinase-associated lipocalin (NGAL) (also known as lipocalin 2) is one of the most widely studied AKI biomarkers having been tested in thousands of patients. 3–6 NGAL has been considered an advantageous AKI biomarker 7 because it is reported to be a kidney specific marker of AKI that produced by the kidney during tubular injury, 8 not influenced by systemic diseases, and appears in the plasma and urine within 2 hours of injury after being released by injured tubules. 9 The performance of NGAL to predict AKI has been variable, however, possibly because it is produced by cell types outside of the kidney or in response to other stimuli such as systemic inflammation and circulating cytokines. 10
IL-6 is a proinflammatory cytokine that increases by 2 hours in the plasma in patients and mice after AKI 11, 12 and thus might play a role in NGAL production. In the present study, we investigated the role of IL-6 in extra-renal NGAL production during AKI.
Results
IL-6 deficient (IL-6−/−) mice have reduced plasma and urine NGAL after sham (surgery alone), ischemic AKI, and bilateral nephrectomy (BNx).
To determine the effect of IL-6 on plasma and urine NGAL levels during AKI, wild type (WT) or IL-6−/− mice were subjected to sham (surgery/laparotomy), ischemic AKI (laparotomy with bilateral renal pedicle clamping) or BNx (laparotomy with removal of both kidneys). Normal mice were studied as a control for all three procedures.
No kidney injury occurred after sham in either WT or IL-6−/−; kidney injury was similar after ischemic AKI in WT and IL-6−/− (as previously reported 12); BUN and plasma creatinine were similar in WT and IL-6−/− with BNx (Figure 1). Plasma IL-6 was increased in WT 4 hours after sham, AKI and bilateral nephrectomy, and absent in IL-6−/−; plasma NGAL was increased in WT and reduced in IL-6−/− versus WT at both 4 and 24 hours after sham, AKI, and BNx; urine NGAL was increased 4 and 24 hours after sham and AKI in WT and reduced in IL-6−/− versus WT at both time points and in both conditions in (Figure 2).
Figure 1. IL-6 deficient (IL-6−/−) mice have similar kidney function and injury after sham (surgery alone), ischemic acute kidney injury (AKI) or bilateral nephrectomy (BNx).
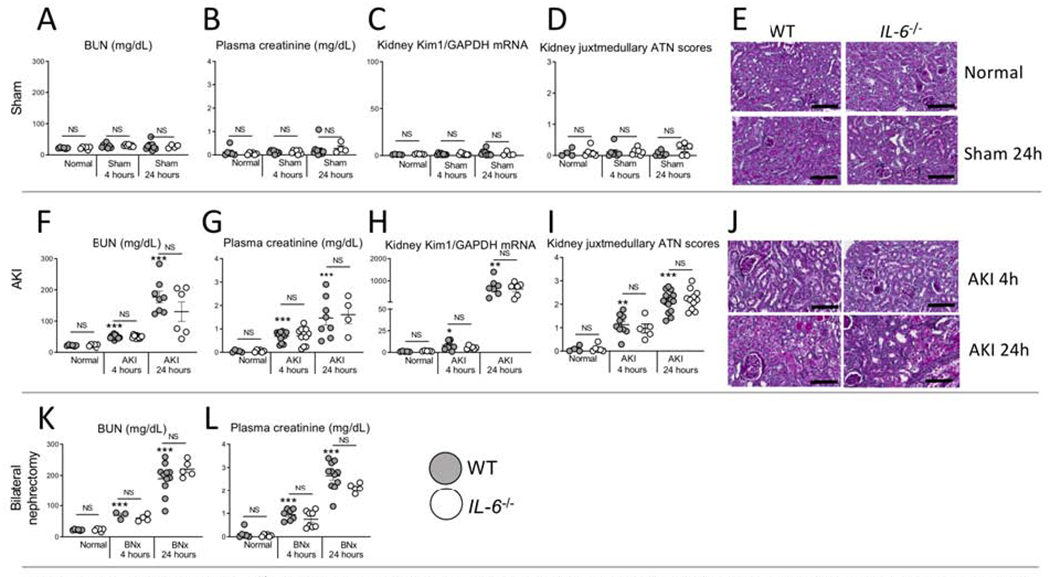
Kidney injury was determined in normal (unmanipulated) mice, and after sham, AKI or BNx at 4 and 24 hours in wild type (WT) and IL-6−/− mice. No kidney injury occurred after sham (surgery alone) in either WT or IL-6−/− as judged by (A) BUN, (B) plasma creatinine, (C) kidney KIM-1 mRNA, and (D) ATN score with (E) representative PAS images. Kidney injury was similar at 4 and 24 hours after AKI between WT and IL-6−/− mice as judged by (F) BUN, (G) plasma creatinine, (H) kidney mRNA KIM-1, and (I) ATN scores with (J) representative PAS images. Wild type and IL-6−/− mice with bilateral nephrectomy had similar levels of (K) BUN and (L) plasma creatinine. N=3 to 12 mice per group from 3 experiments. Results are expressed as mean ±SEM and analyzed by t test: WT versus IL-6−/− at the same time point (indicated above the bar) and versus normal WT (indicated below the bar: *P<0.05; **P<0.01, ***P < 0.0001). Scale bar: 100 μm.
Figure 2. Plasma NGAL is reduced in IL-6 deficient IL-6−/− mice after sham (surgery alone), ischemic acute kidney injury (AKI) or bilateral nephrectomy (BNx) at 4 and 24 hours post-procedure.
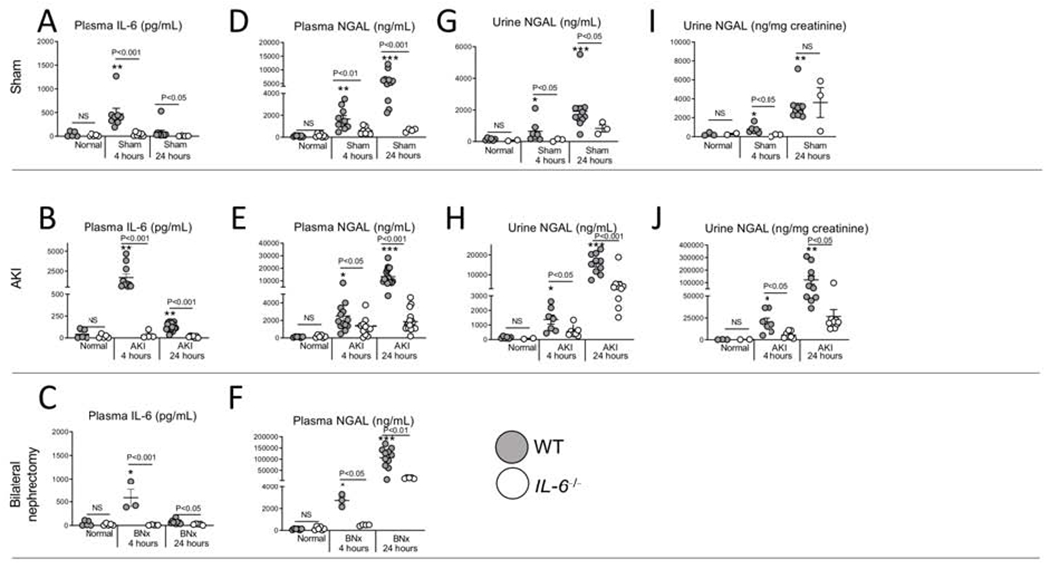
Plasma IL-6, plasma NGAL, and urine NGAL were determined in normal (unmanipulated) mice, and after sham, ischemic AKI or BNx at 4 and 24 hours in wild type (WT) and IL-6−/− mice. Plasma IL-6 was increased in WT 4 and 24 hours after (A) sham, (B) AKI, and (C) BNx. Plasma NGAL was significantly reduced in IL-6−/− at 4 and 24 hours after (D) sham, (E) AKI and (F) BNx. Urine NGAL was reduced after sham and ischemic AKI in IL-6−/− (G-J). N=3 to 12 mice per group from 3 experiments. Results are expressed as mean ±SEM and analyzed by t test: WT versus IL-6−/− at the same time point (indicated above the bar) and versus normal WT (indicated below the bar: *P<0.05; **P<0.01, ***P < 0.0001).
Together, these data demonstrate that IL-6 plays a key role in NGAL production after sham, ischemic AKI and bilateral nephrectomy.
NGAL production occurs predominantly in the liver after sham, ischemic AKI, and bilateral nephrectomy (BNx), and is significantly reduced in IL-6−/−.
To determine the sources of IL-6-mediated NGAL production after sham, AKI and Bnx, NGAL mRNA levels were determined in the liver, kidney, lung and spleen in WT and IL-6−/− (Figure 3)
Figure 3. NGAL production (mRNA) occurs predominantly in the liver after sham (surgery alone}, ischemic acute kidney injury (AKI) and bilateral nephrectomy (BNx), and liver NGAL mRNA is reduced in IL-6−/− mice.

mRNA NGAL levels were determined in the (A-C) liver, (D, E) kidney, (F-H) lung, and (I-K) spleen in normal (unmanipulated) mice, and 4 and 24 hours after sham, AKI or BNx in wild type (WT) and IL-6−/− mice. N=3 to 12 mice per group from 3 experiments. Results are expressed as mean ±SEM and analyzed by t test: WT versus IL-6−/− at the same time point (indicated above the bar) and versus normal WT (indicated below the bar: *P<0.05; **P<0.01, ***P < 0.0001).
In the liver, NGAL mRNA levels were increased 4 and 24 hours after sham, AKI and BNx, and were significantly reduced at both time points and in all three conditions in IL-6−/−. In the kidney, NGAL mRNA increased at 4 and 24 hours post AKI, and was not affected in IL-6−/−. In the lung and spleen, NGAL mRNA were minimally affected.
NGAL protein accumulates in the liver, kidney, lung, and spleen after sham, ischemic AKI and bilateral nephrectomy.
To determine the sites of NGAL accumulation in organs, protein levels of NGAL were measured in the liver, kidney, lung, and spleen 4 and 24 hours after sham, AKI and bilateral nephrectomy and immunofluorescence was examined 24 hours after ischemic AKI.
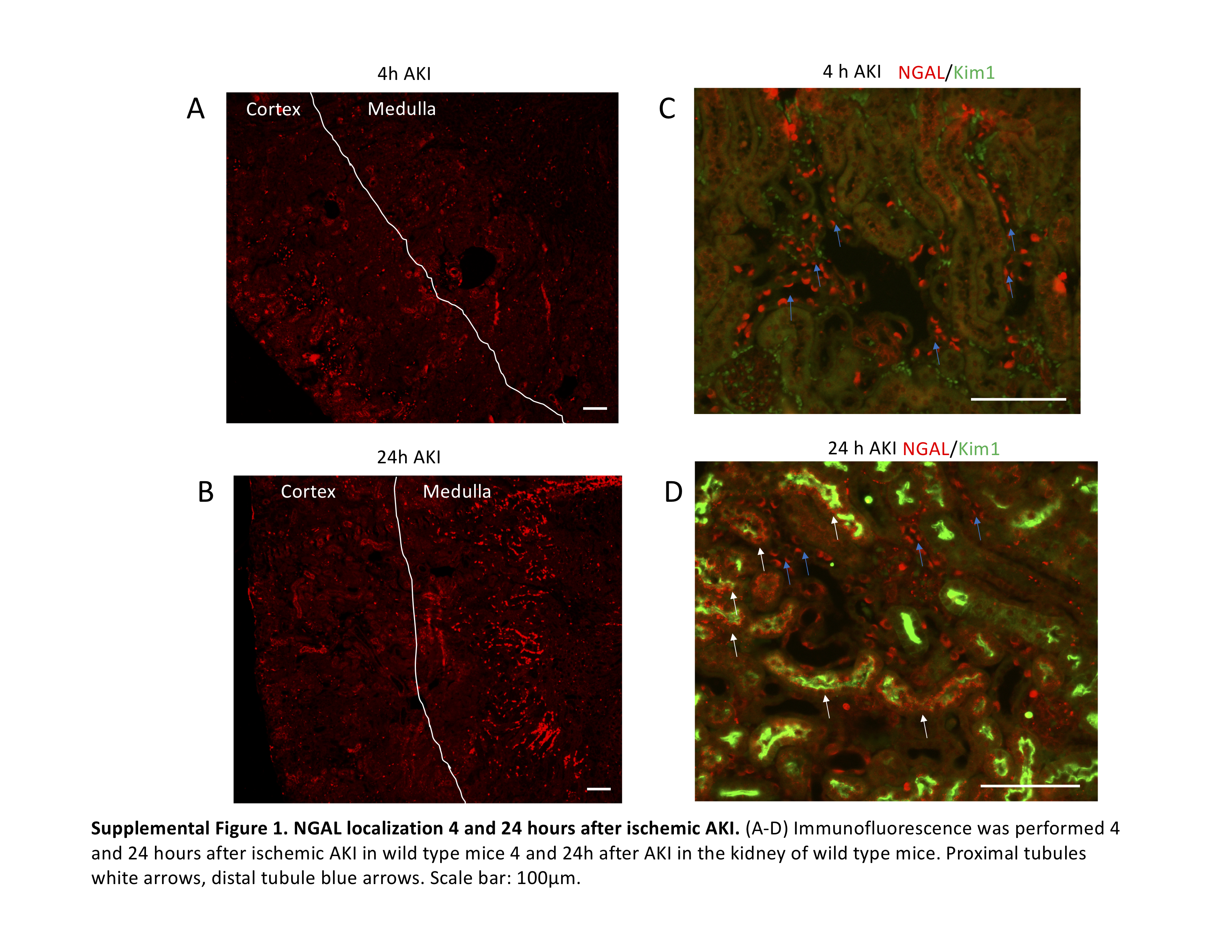
Unlike mRNA, NGAL protein levels were elevated in all the organs after sham, AKI and bilateral nephrectomy and was reduced in IL-6−/− (Figure 4). NGAL localizes to both the proximal and distal tubule after AKI (Supplemental Figure 1).
Figure 4. NGAL protein accumulates in the liver, lung, spleen, and kidney at 4 and 24 hours after sham (surgery alone), ischemic acute kidney injury (AKI) or bilateral nephrectomy (BNx).
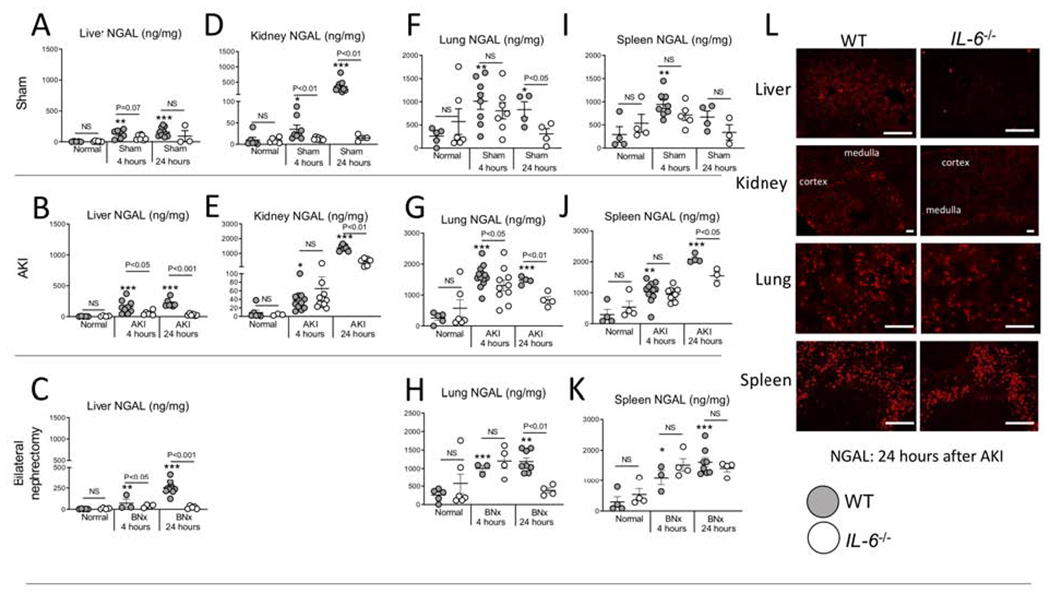
NGAL protein levels (ELISA) were determined in normal (unmanipulated) mice, and 4 and 24 hours after sham, AKI and BNx in wild type (WT) and IL-6−/− mice in the (A-C) liver, (D-E) kidney, (F-H) lung, and (I-K) spleen. (L) Immunofluorescence staining for NGAL was performed 24 hours after AKI in WT and IL-6−/− in the liver, kidney, lung, and spleen (representative images). N=3 to 12 mice per group from 3 experiments. Results are expressed as mean ±SEM and analyzed by t test: WT versus IL-6−/− at the same time point (indicated above the bar) and versus normal WT (indicated below the bar: *P<0.05; **P<0.01, ***P < 0.0001). Scale bar: 100 μm.
The relative contribution of organ production (mRNA) and accumulation (protein) of NGAL after ischemic AKI is graphically represented in Figure 5.
Figure 5. Relative NGAL production (mRNA) and accumulation (protein) after ischemic AKI in wild type (WT) and IL-6−/− mice.
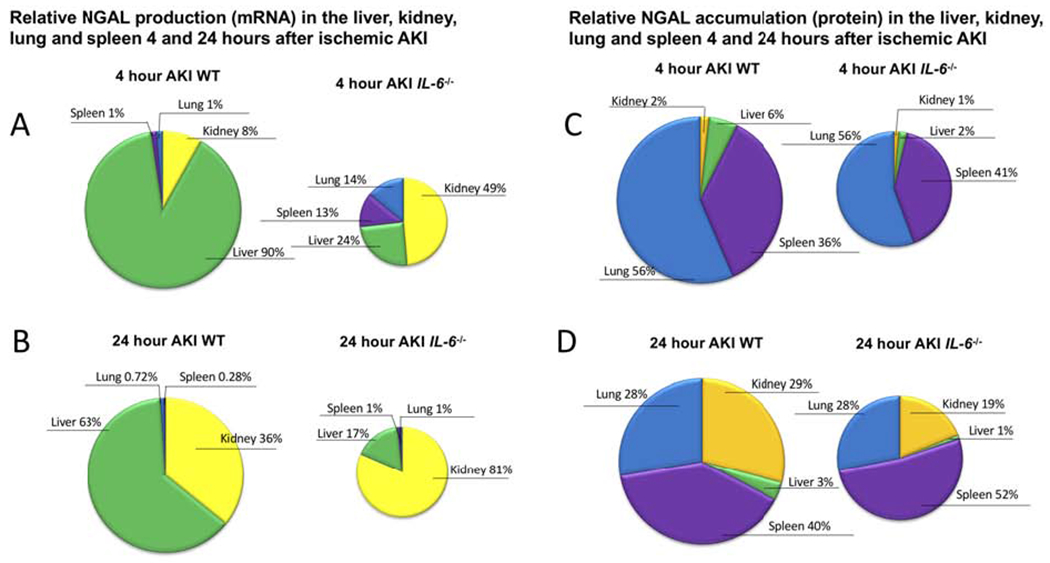
(A,B) NGAL production (mRNA) in liver, kidney, lung, spleen is expressed as % organ production and demonstrates that the liver is the major organ source of NGAL production 4 and 24 hours post-AKI in WT mice after ischemic AKI; in IL-6−/−, the kidney is the major source of NGAL production. Thus, IL-6 is a mediator of hepatic, but not renal, NGAL production. (C, D) NGAL protein levels in the liver, kidney, lung, and spleen are expressed as % organ levels and demonstrate that after AKI, NGAL accumulates predominantly within the kidney, lung, and spleen (total protein was determined within organs, thus, organ accumulation does not distinguish free NGAL from NGAL contained within neutrophils). A similar distribution of NGAL accumulation was observed in IL-6−/− mice. (The size of the pie chart were adjusted to indicate relative production and accumulation between WT and IL-6−/−.) The differences in mRNA and protein highlight that while the liver is the primary site of organ production, NGAL accumulation is widely distributed.
Altogether, the data in Figures 1 to 5 demonstrate that IL-6 contributes to hepatic NGAL production which results in an increase in plasma NGAL levels in all three conditions tested: (sham) surgery, ischemic AKI and bilateral nephrectomy.
Intravenous IL-6 administration induces hepatic NGAL production.
Since the increase in plasma IL-6 corresponded to an increase in hepatic NGAL production after sham, ischemic AKI, and bilateral nephrectomy, we hypothesized that circulating IL-6 was acting at the liver to induce NGAL production in hepatocytes. Since hepatocytes contain the IL-6 receptor, a direct effect of IL-6 on hepatocytes is plausible. Therefore, to test whether circulating IL-6 mediates hepatic NGAL production, IL-6 was administered IV every hour for 3 hours to WT mice and endpoints were determined 1 hour after the last IV injection (Figure 6). After IL-6 injection, hepatic NGAL production (mRNA) increased as did plasma and urine NGAL protein levels. NGAL mRNA levels did not increase in the kidney, lung, or spleen. IL-6 did not affect kidney function; plasma IL-6 was increased in the IL-6-treated mice, as expected.
Figure 6. Intravenous (IV) administration of IL-6 to normal WT mice induces NGAL production (mRNA) in the liver and increases plasma NGAL.
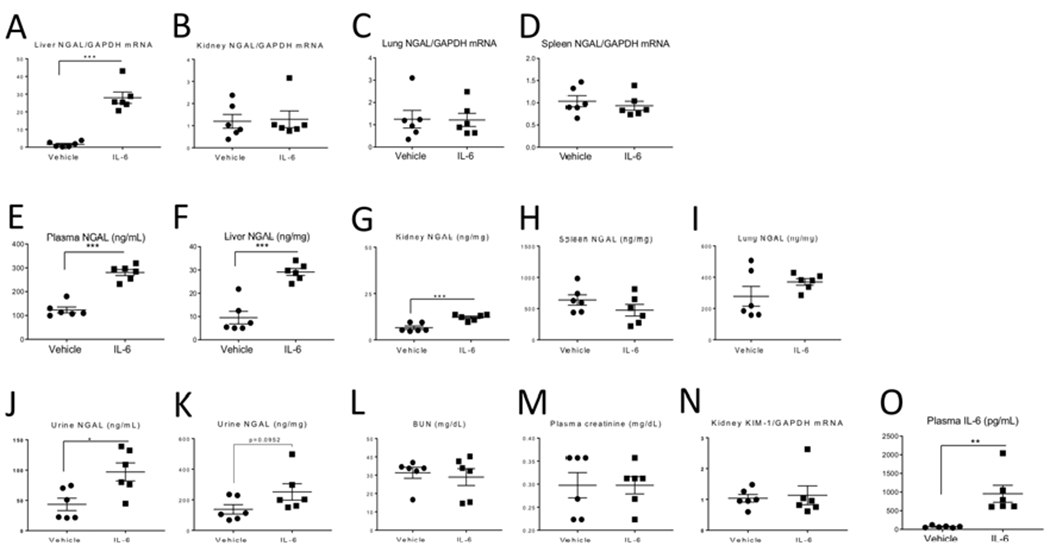
200 ng of recombinant murine IL-6 or 0.1% BSA was administered IV every hour for 3 hours to healthy WT mice; endpoints were determined 1 hour after the last IV injection. mRNA NGAL expression was significantly increased in the (A) liver, but not the (B) kidney, (C) lung, or (D) spleen in IL-6-treated. Protein NGAL levels were significantly increased in the (E) plasma, (F) liver, and (G) kidney but not the (H) spleen, or (I) lung in IL-6-treated. Urine NGAL was significantly increased (J), but not when corrected for urine creatinine (K) in IL-6-treated. Kidney injury did not occur after IL-6 administration as judged by similar levels of (L) BUN, (M) plasma creatinine and (N) kidney KIM-1 mRNA expression. (O) Plasma levels of IL-6 were increased in mice receiving IV IL-6, as expected. Data were analyzed by t test, *P < 0.05, **P < 0.01 and ***P < 0.001, Vehicle vs IL-6. Results are expressed as mean ±SEM of expression values for 6 mice per group from 1 experiment, n=6).
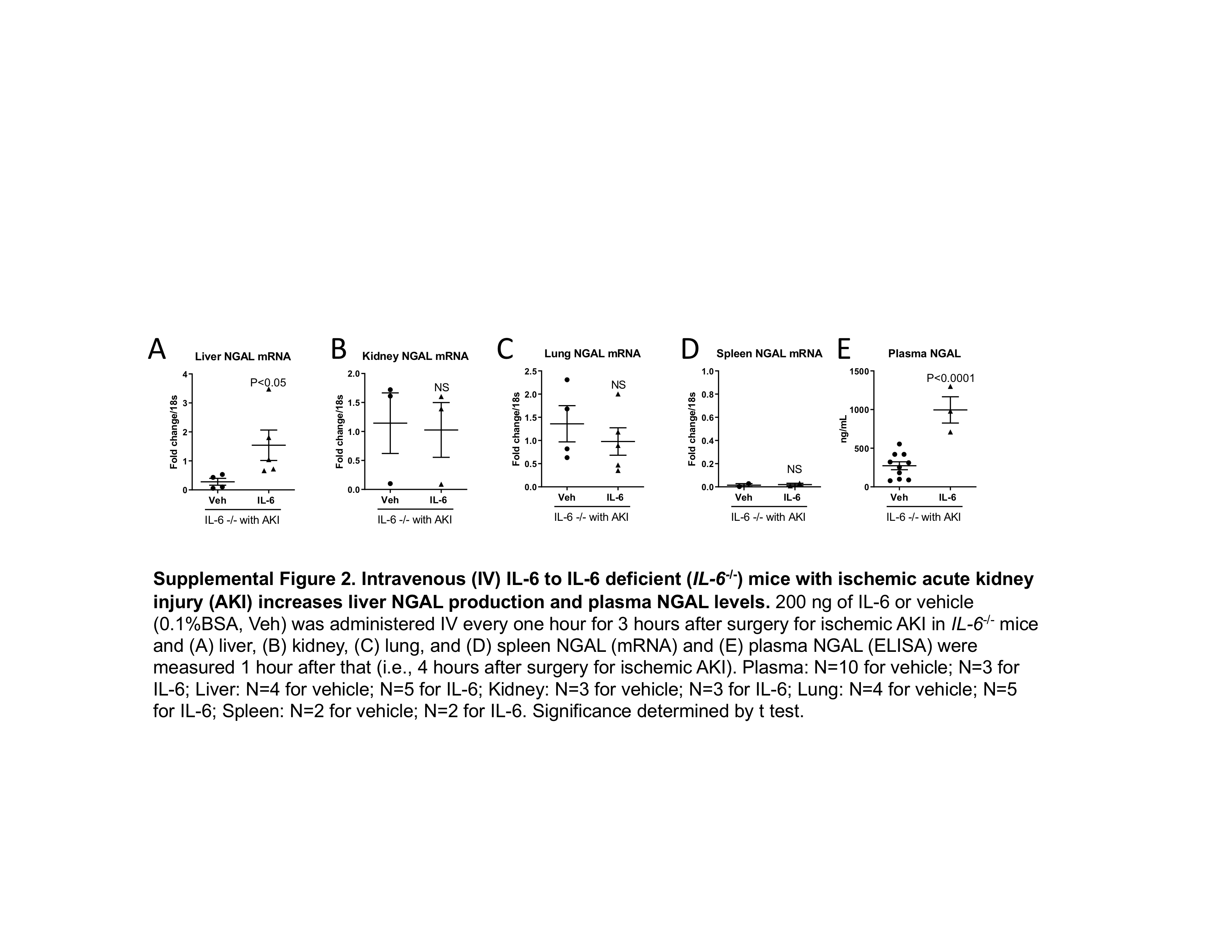
Similarly, IV IL-6 to IL-6−/− with AKI increased liver NGAL mRNA levels and plasma NGAL, but not kidney, lung, or spleen NGAL mRNA levels (supplemental Figure 2).
Since the only source of IL-6 after IV injection is the plasma, these data demonstrate that circulating IL-6 mediates hepatic NGAL production that leads to increased plasma NGAL levels.
IL-6 mediates hepatic NGAL production within 2 hours, in vitro.
To determine the onset of IL-6 mediated hepatic NGAL production, recombinant human IL-6 was added to human (HepG2) hepatocytes and NGAL mRNA levels were determined in cells, and NGAL protein levels were determined in the supernatant at 1, 2, and 4 hours (Figure 7). NGAL mRNA significantly increased in the cells by 1 hour, and in the supernatant by 2 hours.
Figure 7. IL-6 mediates NGAL production by 1 hour in hepatocytes, in vitro.
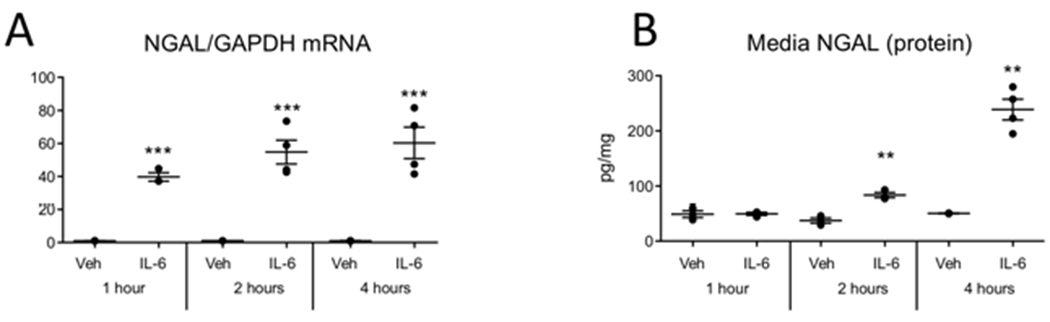
Vehicle (Veh) or 50 ng/mL of human recombinant IL-6 was added to human (HepG2) hepatocytes. (A) NGAL mRNA was increased in hepatocytes at 1, 2, and 4 hours after IL-6 addition. (B) NGAL protein levels in the media increased at 2 and 4, hours after IL-6 addition **P < 0.01 and ***P < 0.001 vs vehicle treated at the same time point by t test. n=2-5 for each condition.
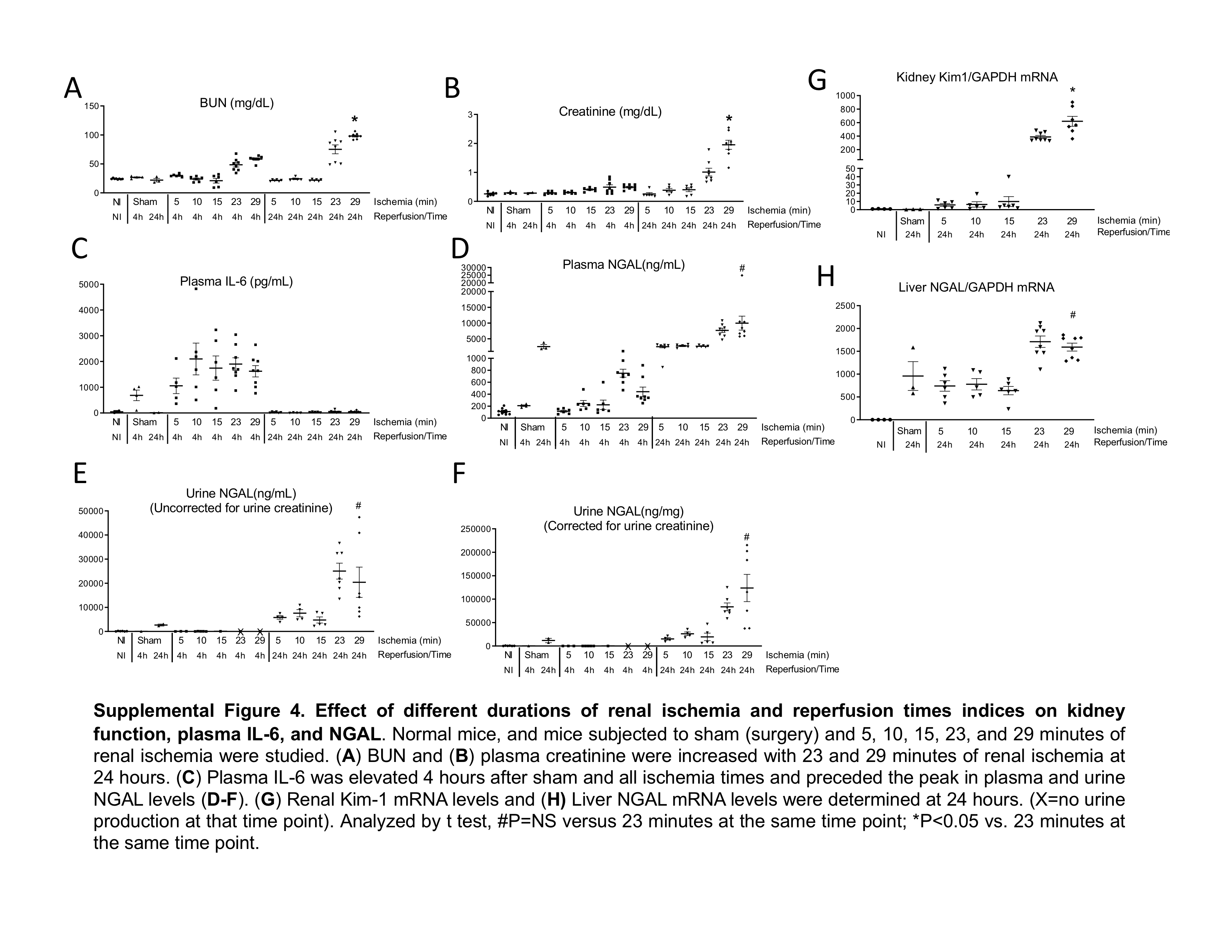
The effect of dose and time on NGAL levels in response to IL-6 was assessed, in vitro, and duration of IL-6 exposure (24 hours) had a much greater effect on supernatant NGAL levels than dose (Supplemental Figure 3). In vivo, the effect of various renal ischemia times on plasma IL-6 and NGAL levels was assessed and demonstrated that plasma IL-6 levels peaked at 4 hours while plasma and urine NGAL levels peaked at 24 hours (Supplemental Figure 4). These data demonstrate that NGAL production persists up to 24 hours after IL-6 exposure.
IL-6 mediates NGAL production in the liver and hepatocytes via phosphorylation of the transcription factor STAT3, in vivo and in vitro.
Many cytokines, including IL-6, mediate their effects through the phosphorylation and activation of the transcription factor STAT3. To assess the role of IL-6-mediated STAT3 activation after ischemic AKI, pSTAT3 was determined in nuclear and cytosolic fractions of the liver and kidney 4 and 24 hours after AKI in WT and IL-6−/− (Figure 8A–D). In the liver, but not the kidney, IL-6−/− had reduced pSTAT3 and absent translocation to the nucleus; in the liver in WT, translocation peaked at 24 hours, correlating with the peak of NGAL production.
Figure 8. IL-6 mediated NGAL production in the liver and hepatocytes is dependent on phosphorylation of STAT3.
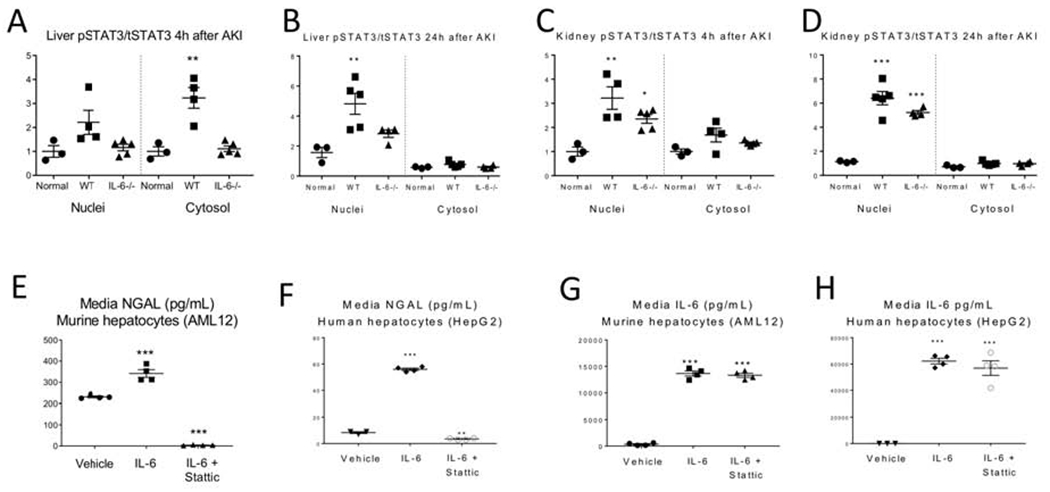
Nuclear and cytosolic fractions were isolated from the liver and kidney from normal wild type (WT) mice and at 4 and 24 hours after AKI in WT and IL-6−/− mice and total and phosphorylated STAT3 (pSTAT3) was determined by ELISA. (A, B) In the liver. pSTAT3 increased and completely translocated to the nuclei at 24 hours in WT, but not IL-6−/− mice, with AKI. (C, D) In the kidney, pSTAT3 increased and translocated to the nucleus in both WT and IL-6−/− mice at 4 and 24 hours. IL-6 and an inhibitor of STAT3 phosphorylation (STATTIC) was added to murine (AML12) and human (HepG2) hepatocyte cell lines in vitro. IL-6 was added 1 hour after addition of Stattic; media NGAL was determined 4 hours after adding IL-6. (E-H) NGAL was increased with addition of IL-6 and reduced with IL-6+STATTIC in AML12 and HepG2. IL-6 was unchanged by STATTIC in AML12 and HepG2. Data represent the expression values for 3 to 5 mice per group from 1 experiment. Results are expressed as mean ±SEM. Data were analyzed by t test, *P < 0.05, **P < 0.01 and ***P < 0.001 vs. WT normal (uninjured) mice or vs. vehicle in cell culture experiments.
In vitro, vehicle, IL-6, or IL-6 plus Stattic (an inhibitor of STAT3 phosphorylation) was added to murine (AML12) or human (HepG2) hepatocytes. The increase in NGAL in the supernatant after IL-6 treatment was completely suppressed in the Stattic-treated group; IL-6 levels were not affected (Figure 8E–H).
Together these data demonstrate that STAT3 activation is required for IL-6 mediated NGAL production in hepatocytes.
Plasma and urine NGAL levels are reduced in mice with deficient hepatocyte NGAL producing capacity after ischemic AKI.
To confirm that the liver is the major source of NGAL production after AKI, ischemic AKI was performed in WT and Lcn2Hep−/− mice (Figure 9). Hepatocytes are specifically unable to produce NGAL in the Lcn2Hep−/− mice, while NGAL production capacity in all other cells remains intact. 13 24 hours post AKI in Lcn2Hep−/−, plasma and urine NGAL levels were significantly reduced versus WT. NGAL mRNA was absent in the liver and not affected in the kidney, lung or spleen. NGAL protein levels were significantly decreased in the liver, but not the kidney, lung or spleen; kidney function/injury and plasma IL-6 levels were similar to WT.
Figure 9. Hepatocyte-specific Lcn2 (NGAL) deficient mice have reduced plasma and urine NGAL after ischemic AKI.
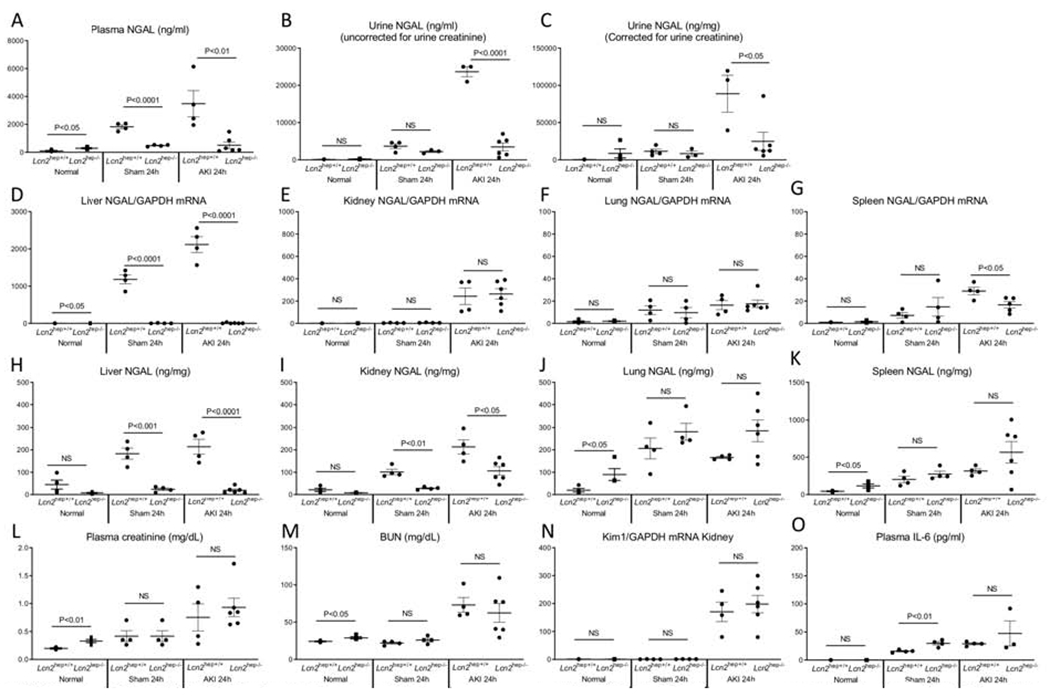
Normal mice (no procedure), sham (surgery alone) and ischemic AKI were studied in hepatocyte specific Lcn2 deficient (Lcn2hep−/−) mice, Cre-negative floxed littermates were used as controls (Lcn2hep+/+). (A, B, C) Plasma and urine NGAL were dramatically reduced in Lcn2Hep−/− versus Lcn2Hep+/+. As expected, Liver mRNA NGAL expression was decreased in Lcn2hep−/− versus Lcn2Hep+/+; NGAL mRNA was similar in the (B) kidney and (C) lung in Lcn2Hep−/− versus Lcn2Hep+/+; (D) spleen mRNA NGAL was reduced after AKI in Lcn2Hep−/− versus Lcn2Hep+/+. NGAL protein levels were reduced in Lcn2hep−/− Lcn2Hep−/− versus Lcn2Hep+/+ in the (E) liver, and (K) kidney, but not the lung and spleen in AKI. Kidney function with AKI was similar as judged by (L) BUN, (M) plasma creatinine and (N) kidney mRNA KIM-1 expression. (O) Plasma 1L-6 levels were similar in AKI. Results are expressed as mean ±SEM; N = 4-6 mice per group from 3 exDeriments. Analyzed by t test. *P < 0.05. **P < 0.01 and ***P < 0.001, Lcn2hep+/+ vs. Lcn2hep−/−.
Neutrophils are not a source of plasma or urine NGAL levels after ischemic AKI.
Since NGAL is constitutively expressed in neutrophils, 7 we tested whether IL-6−/− mice might have also had lower plasma and urine NGAL levels due to an effect on neutrophils by subjecting neutrophil depleted mice to ischemic AKI (Figure 10). Neutrophil depleted mice with AKI had a similar level of kidney function/injury. Neutrophil depletion did not affect plasma NGAL, urine NGAL, or NGAL mRNA levels in the liver, kidney, lung or spleen. Organ accumulation of NGAL was similar in the liver and kidney, but significantly reduced in the spleen (24 hours) and lung (4 and 24 hours). Since lung neutrophil accumulation occurs after AKI,12, 14, 15 the reduction in lung NGAL in the neutrophil depleted mice likely reflects a reduction in lung neutrophil recruitment and a similar mechanism may be at play within the spleen.
Figure 10. Ly6G+ cells are not the source of plasma or urine NGAL after ischemic acute kidney injury (AKI).
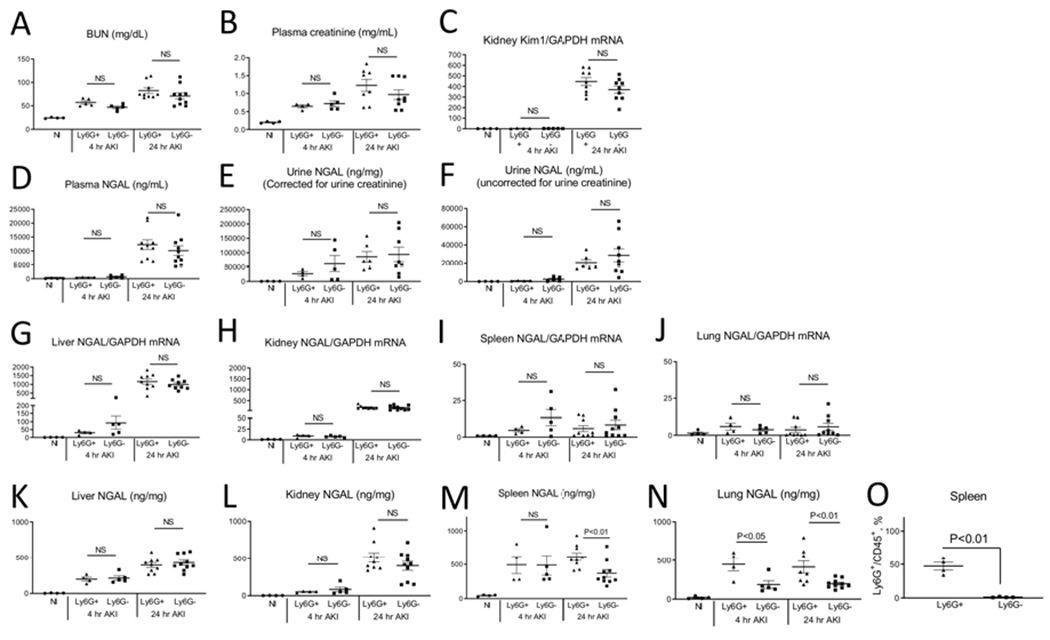
Normal mice (Nl), and mice with and without depletion of Ly6G+ cells were studied 4 and 24 hours after ischemic AKI. Ly6G is predominantly expressed on neutrophils. There was no change in kidney function/injury after AKI with Ly6G+ cell depletion as judged by (A) BUN, (B) plasma creatinine and (C) kidney Kim1 mRNA. Neither (D) plasma nor (E,F) urine NGAL were affected by Ly6G+ cell depletion. (G) Liver, (H) kidney, (I) spleen, and (J) lung NGAL production was not affected by Ly6G+ depletion. NGAL protein levels in the liver and kidney (K and L) were not affected by Ly6G+ cell deletion. NGAL protein was reduced in the spleen at 24 hours in the lung at 4 and 24 hours after AKI with Ly6G+ cell depletion (N). Neutrophil depletion was confirmed by flow cytometry of the spleen (O). Results are expressed as mean ±SEM; 2 separate experiments. Analyzed by t test, Ly6G+ versus Ly6G− at the same time point. (N=4-10)
Renal handling of circulating NGAL.
Since our data suggest that urine NGAL is predominantly derived from the plasma, we assessed the renal handling of circulating NGAL by determining the fate of intravenously administered recombinant human NGAL (hNGAL) in six different conditions with varying degrees of renal impairment or tubular injury in which glomerular filtration rate (GFR) was known (Figure 11). hNGAL is homologous to murine NGAL, and is handled similarly by the kidney, but shows no cross reactivity to murine NGAL by ELISA and does not affect endogenous murine NGAL levels (data not shown).
Figure 11. Models of AKI.
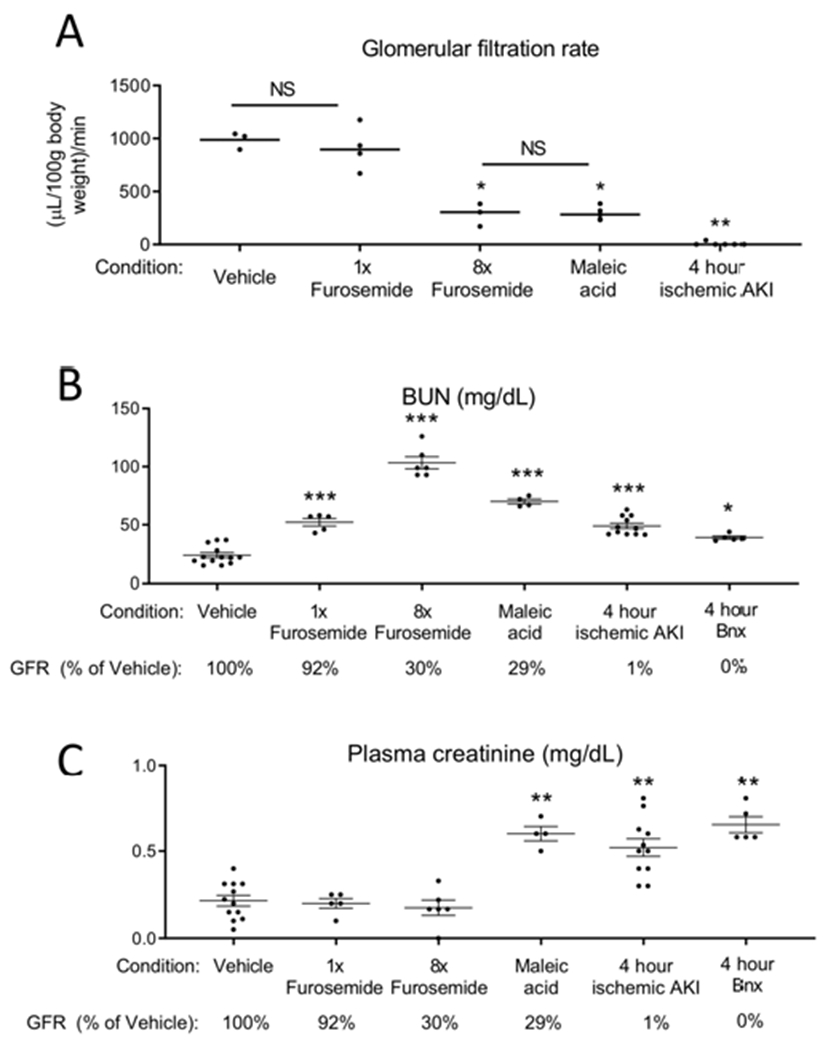
(A) Transcutaneous GFR was measured in 5 groups: 1) mice treated with vehicle (saline), 2) mice treated with 0.5 mg furosemide (1x furosemide) to induce mild prerenal azotemia, 3) mice treated with 4 mg furosemide for 2 doses (8x furosemide) to induce severe prerenal azotemia, 4) mice treated with maleic acid to induce proximal tubular injury and Fanconi syndrome, and 5) mice subjected to ischemic AKI. GFR was determined 6 hours after vehicle, furosemide, and maleic acid treatment, and 4 hours after ischemic AKI. GFR was analyzed by one way ANOVA, comparing all groups. *P < 0.01 versus Vehicle and 1x furosemide, **P < 0.001 versus all other groups. Results are expressed as mean ±SEM. n=3-6. (B) BUN and (C) plasma creatinine were determined 6 hours after vehicle, 6 hours after 1x furosemide, 6 hours after 8x furosemide, 6 hours after maleic acid, 4 hours after ischemic AKI, and 4 hours after bilateral nephrectomy. GFR as a percentage of vehicle treated is indicated for reference (GFR was not measured in the bilateral nephrectomy group, but is assumed to be “0” since both kidneys are removed). BUN and plasma creatinine were analyzed by t test versus vehicle: *P<0.05; **P<0.001, ***P<0.0001. Results are expressed as mean ±SEM. n=5-12 (groups include mice that directly had GFR measured, as well as additional mice).
GFRs were as follows (percent of vehicle): 1) vehicle treatment: 100%, 2) mild prerenal azotemia: 92%, 3) severe pre-renal azotemia: 30%, 4) PT injury due to maleic acid: 29%, 5) ischemic AKI: 1%, and 6) bilateral nephrectomy: 0% (not measured). Maleic acid is a toxin specifically affects the proximal tubule and induces Fanconi syndrome. 16 Plasma and urine hNGAL were determined 1 hour after IV injection (Figure 12).
Figure 12. Intravenously administered recombinant human (h) NGAL increases in the urine of mice with maleic acid administration and ischemic AKI to a greater extant than during pre-renal azotemia (furosemide administration).
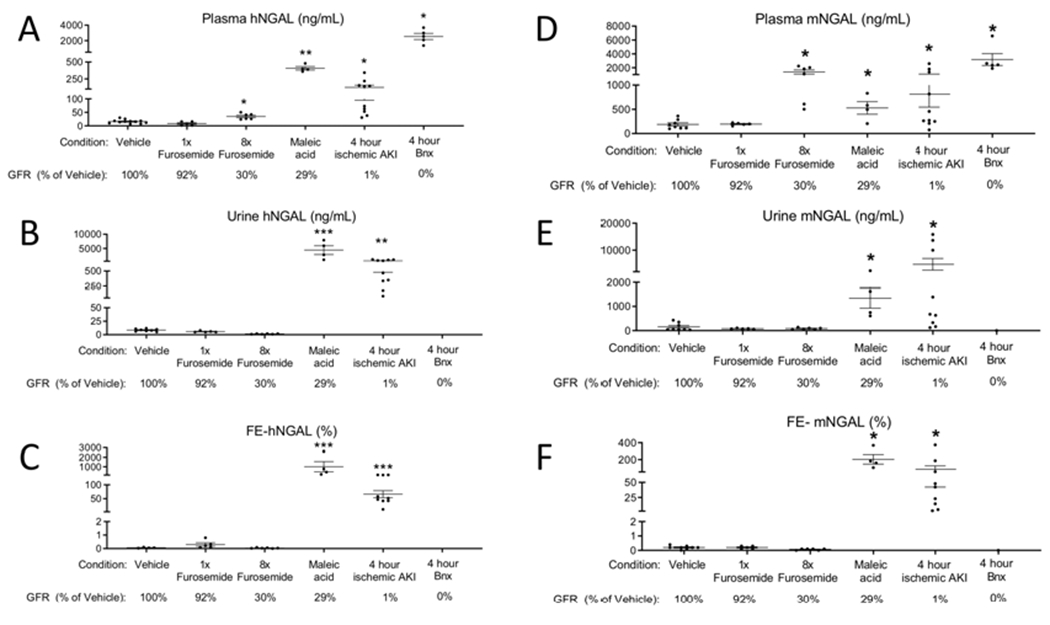
rhNGAL was administered intravenously(IV) 6h after vehicle, furosemide, and maleic acid; and 4 hours ischemic AKI and bilateral nephrectomy (BNx). Endpoints were determined 1 after IV hNGAL administration. (A) hNGAL was significantly increased in the plasma after severe prerenal azotemia (8x furosemide), maleic acid administration, ischemic AKI, and bilateral nephrectomy versus vehicle. (B) Urine hNGAL was significantly increased after maleic acid treatment and ischemic AKI versus vehicle. (C) Fractional excretion (FE) of hNGAL was less than 1% in vehicle, mild prerenal azotemia (1x furosemide), and severe prerenal azotemia (8x furosemide), and was greater than 10% after maleic acid treatment and ischemic AKI. (D-F) Endogenous murine (m) NGAL was determined in the plasma and urine, and FE-mNGAL was calculated. Analyzed by t test versus vehicle, *P<0.05, **P<0.01, ***P<0.001. n=5-12 (groups include mice that directly had GFR measured, as well as additional mice; GFR for each model relative to vehicle is indicated below for reference).
Plasma and urine levels of hNGAL were low in mice with normal kidney function. Plasma and urine hNGAL were similar to normal in mild pre-renal azotemia; plasma hNGAL was slightly, but significantly, higher in severe prerenal azotemia, although urine hNGAL levels were similar to normal. In contrast, both plasma and urine hNGAL were elevated in PT injury and ischemic AKI and plasma levels were increased in mice with bilateral nephrectomy.
In order to account for plasma levels and PT function in the assessment of urine excretion of NGAL, we calculated the fractional excretion of hNGAL (FE-hNGAL) as follows: FE-hNGAL = ((urine [hNGAL] x plasma creatinine)/(plasma h[NGAL] x urine creatinine)) x 100. FE-hNGAL was less than 1% in normal mice, mild pre-renal azotemia, and severe pre-renal azotemia, and was greater than 20% in maleic acid-treated and ischemic AKI. Endogenous (murine (m)) levels of plasma and urine NGAL, and FE-mNGAL were also determined, with similar results to hNGAL.
Taken together, these data indicate that circulating NGAL is normally filtered, and then resorbed and metabolized/degraded by the proximal tubule. Excretion of the intact protein is not a normal mechanism of NGAL elimination since urine levels were very low in mice with normal kidney function. With PT dysfunction, filtered NGAL is not resorbed and metabolized, and appears in the urine. FE-NGAL distinguishes between severe pre-renal azotemia and proximal tubular injury (maleic acid treated) in mice with similar GFRs.
Discussion.
NGAL is the most widely studied AKI biomarker, having been tested in thousands of patients for its potential to diagnose AKI early. Based on early studies demonstrating that NGAL is produced by the injured kidney and is then released into the urine and plasma,8 NGAL has been classified and studied specifically as a tubular injury biomarker.17 In the present study, we systematically examined renal and extra-renal production of NGAL, the influence of the proinflammatory cytokine IL-6, and the renal handling of NGAL. The major novel findings of our report are that 1) IL-6 mediates hepatic NGAL production during AKI, 2) the liver – not the kidney – is the primary source of both plasma and urine NGAL levels during AKI, and 3) circulating NGAL appears in the urine during AKI associated with proximal tubular dysfunction. These data change the paradigm by which NGAL should be interpreted as a biomarker of AKI and extend the known role of IL-6 in the systemic response after AKI.
To date, pre-clinical studies that have examined NGAL during AKI have focused on its role as an AKI biomarker 8, 18 and in kidney function recovery.19, 20 Independent of AKI, ongoing studies have clarified the systemic functions of NGAL in a variety of settings. In particular, it is now recognized that NGAL is part of the hepatic acute phase response (APR).13 The hepatic APR is characterized by the hepatic production of acute-phase proteins (APPs) that collectively function to prepare the body to battle infection, minimize tissue injury, and initiate wound repair.21 NGAL participates in the hepatic APR as a bactericidal agent via the sequestration of iron, and works in conjunction with other iron regulatory hepatic APPs such as hepcidin, 22 haptoglobin, 23 and ferritin.24 Since bacteria require iron for growth, iron sequestration is a key defense against infection. The important role of NGAL during infection is highlighted by studies demonstrating that NGAL knock-out mice that have increased susceptibility to infection.25, 26
IL-6 is a major mediator of the hepatic APR and induces hepatocyte production of virtually all acute phase proteins. 27 IL-6 production is triggered within minutes by numerous systemic insults such as surgery, trauma, and sepsis as part of the systemic inflammatory response syndrome (SIRS) – which also occurs during AKI. In mice and patients with AKI, plasma IL-6 increases by 2 hours.28, 29 The production and renal handling of IL-6 during AKI is well characterized. Notably the kidney is not a major source of IL-6 production as evidenced by the fact that plasma IL-6 increases after bilateral nephrectomy. 28, 30 In contrast, the spleen and liver are the major organ sources of IL-6 in AKI 31 and the major cell source is the macrophage. 32 Clearance of IL-6 is decreased during AKI, largely as a result of decreased proximal tubular resorption and metabolism.11 Since normal kidney function is necessary to eliminate plasma IL-6, conditions associated with both SIRS and AKI would be expected to have increased levels of plasma IL-6, as has been shown after cardiopulmonary bypass surgery.29, 33
Although IL-6 is a known mediator of the hepatic APR, and IL-6 levels are increased during AKI, the role of IL-6 mediated NGAL production during AKI has not been previously examined. With multiple lines of evidence, we demonstrate that circulating IL-6 mediates hepatic NGAL production during AKI. Notably, IL-6−/− mice with ischemic AKI had significantly reduced plasma NGAL levels that were associated with a reduction in hepatic, but not renal, mRNA NGAL. Plasma NGAL and hepatic NGAL levels were also reduced IL-6−/− mice with bilateral nephrectomy; data in the bilateral nephrectomy model also highlight the fact that the kidney is not necessary for NGAL production since plasma NGAL levels were very high in wild type mice. We are aware of one other study that has examined the role of IL-6 in hepatic NGAL production, in vivo; in this study, IL-6 receptor deficient mice were subjected to bacterial infection and partial hepatectomy and had reduced plasma NGAL.13 As with our study, IL-6 hepatic NGAL production was mediated by phosphorylation of STAT3.13
An important finding of our study is the rapidity by which IL-6 can induce hepatic NGAL production. In vivo, our novel data with IV administration of IL-6 to normal mice demonstrated that both hepatic NGAL production (mRNA) and plasma NGAL were increased within 4 hours. In vitro, addition of IL-6 to hepatocytes increased NGAL levels by 2 hours, while previous studies have examined much later time points (8 and 24 hours).13 The rapidity of this response is biologically plausible, since NGAL production is part of the hepatic APR, which occurs rapidly after diverse injuries. Whether other APPs are similarly affected during AKI by has not been investigated, but our data would predict that IL-6 mediated production of other APPs such as ferritin and hepcidin would also be affected during AKI. Our results also predict that most conditions associated with SIRS or an increased in plasma IL-6 would also be associated with a rise in plasma NGAL – independent of AKI - as we observed after sham (surgery) in the present study. Indeed, there are multiple examples of clinical conditions in which both plasma IL-6 and NGAL are known to be elevated and include sepsis,34–37 pancreatitis,38, 39 myocardial infarction, and cardiopulmonary bypass surgery.29, 33
The rapidity of the effect of IL-6 on the liver during AKI aligns with our previous studies of systemic effects of IL-6 during AKI. For example, circulating IL-6 mediates lung inflammation and neutrophil accumulation that is observed 4 hours after the onset of ischemic AKI. 40, 41 IL-6 also mediates IL-10 production in the spleen within 4 hours which functions to limit sustained inflammation. 14 Together, these data highlight the diverse systemic effects of IL-6 which may be viewed as a coordinated innate immune response that primes the host to battle infection. It is also important to note that while IL-6 initiates many responses early after AKI, plasma IL-6 levels are diminished by 24 hours post-AKI. With regard to NGAL, our data demonstrate that IL-6 initiates hepatic NGAL production within 4 hours but it is unknown whether other factors contribute to sustained NGAL production since hepatic NGAL production and plasma levels are dramatically increased 24 hours post AKI, when plasma IL-6 levels are low. In this regard, our data demonstrate that STAT3 activation is required for IL-6 mediated NGAL production in hepatocytes and suggest that the delay in complete translocation of STAT3 to the nucleus (24 hours) may explain the observation that NGAL production continues to increase 24 hours after IL-6 exposure. Together, these observations highlight the importance of examining early and later time points during AKI, and that AKI characteristics can change substantially over time.
Our data in the Lcn2Hep−/− mice with ischemic AKI definitively demonstrated the role of hepatocyte NGAL production during AKI and demonstrated that hepatocyte NGAL production accounts for the majority of plasma NGAL levels, which – in turn – accounts for the majority of urine NGAL levels at 24 hours. Previous studies examining the source of urine NGAL during AKI demonstrated that NGAL is produced by the kidney and then released into the urine and plasma from damaged tubules; in these studies, however, the contribution of plasma NGAL to urine NGAL levels during AKI was not assessed, plasma NGAL during AKI was not determined, and extra-renal production of NGAL via mRNA and protein levels during AKI were not systematically examined.8, 18 Our report then builds on this foundation of previous work demonstrating that urine NGAL appearance in AKI is also due to impaired PT resorption and degradation of circulating NGAL – thus explaining the utility of urine NGAL as a biomarker of AKI, despite being predominantly produced by the liver.
Since a key novel finding our study is that a major source of urine NGAL is the plasma, a major implication is that plasma levels of NGAL should be considered when interpreting urine NGAL levels as a biomarker of AKI. To account for plasma NGAL levels, we calculated the fractional excretion of NGAL (FE-NGAL). In clinical practice, this formula is an estimate of the % of a filtered substance that is excreted in the urine and may be used to indirectly infer tubular function, with a low percent corresponding to a high level of tubular resorption and a high percent corresponding to a low level of tubular resorption. Since our data demonstrate that circulating NGAL appears in the urine as a result of impaired proximal tubule resorption and degradation of circulating NGAL, a method to assess for tubular function and resorption would be predicted to be useful in the interpretation of urine NGAL levels. Indeed, in mice with normal proximal tubular function – including severe pre-renal azotemia - FE-NGAL was less than 1 % in all examples measured; in mice with proximal tubular dysfunction from maleic acid or ischemic AKI, FE-NGAL was greater than 10% in all examples tested. Of note, after IV injection, the FE of hNGAL was greater than 100% in maleic-acid treated; since the only source of hNGAL is that which was administered intravenously, renal production or secretion cannot be the explanation for this high percent, rather we interpret this result to indicate that during PT dysfunction, NGAL may transiently accumulate in the PT but then back-leak into the urine due to a failure of degradation. This explanation is in line with previous reports demonstrating that intravenous NGAL normally localizes to the proximal tubule, 20,19 with uptake thought to be due to endocytic delivery of NGAL via megalin and degradation due to lysosomes (LAMP) within proximal tubules19. We anticipate that there are many caveats to consider with the potential use and interpretation of this formula in clinical (and pre-clinical) practice, for example, since some urine NGAL is released into the urine during AKI, this would “falsely” increase the FE-NGAL level - yet the increase would still favor the diagnosis of tubular injury. In sum, calculation of FE-NGAL demonstrates that a method that accounts for plasma NGAL levels and PT function has the potential improve the utility of urine NGAL as a biomarker of AKI – especially to distinguish pre-renal azotemia from acute tubular injury, which is a longstanding clinical conundrum.
Since previous studies have interpreted urine NGAL as a kidney tubule specific injury marker versus a marker of systemic inflammation and proximal tubule dysfunction, we suggest that studies that have utilized NGAL as a tubular specific marker of AKI may need to be reinterpreted and/or reanalyzed. There are a number of cases where this is especially true; for example, studies that had identified sub-clinical AKI (tubular injury without a rise in serum creatinine) based on urine NGAL may not have been detecting tubular injury, but causes of systemic inflammation and increased plasma IL-6 – as would be associated with sepsis and other causes of SIRS. Conversely, NGAL levels should be interpreted with caution in situations where inflammation is suppressed, as evidenced by our studies in IL-6−/− mice which plasma and urine NGAL were markedly reduced without a true change in kidney injury/function.
Although our study has many strengths, there are a number of important limitations and unanswered questions. First, it is important to comment that AKI is an extremely complex systemic disorder and that marked variability on the systemic response may occur depending on the cause of AKI and the timing at which measurements are assessed. While we studied multiple models of AKI, the majority of our studies were during ischemic AKI, and at 4 and 24 hours. The fate of NGAL and its metabolism by PT or other nephron segments was not specifically examined. Better characterization of NGAL production and handling may be warranted during other causes of AKI – particularly sepsis – and at additional time points. The renal handling of NGAL during proximal tubule recovery was not investigated. Although this is the first study to identify a specific mediator of NGAL production during AKI, other mediators of hepatic NGAL production besides IL-6 are present since the reduction in plasma NGAL was incomplete; thus IL-6 dependent and independent effects on the hepatic production of NGAL – and other APPs - during AKI merits investigation.
Conclusions.
The major findings or our report are that hepatic NGAL production is the primary source of plasma and urine NGAL during AKI, and that IL-6 is a major mediator of hepatic NGAL production. Our data extend the known role of IL-6 in the systemic response after AKI and change the paradigm by which NGAL should be interpreted as a biomarker of AKI: specifically, NGAL is a marker of tubular function, and plasma levels should be accounted for in its interpretation. With this approach, the diagnostic utility on NGAL may be greatly improved, particularly to distinguish prerenal azotemia from acute tubular injury.
Brief Methods (Additional details for all methods are in Supplemental Methods)
Mice.
10-12 week old male wild type C57BI/6J (WT), IL-6 deficient IL6tm1Kopf (IL-6−/−)42, hepatocyte specific Lcn2 deficient (Lcn2hep−/−), and cre-negative floxed littermates mice were used as previously described.13 WT and IL-6−/− were purchased from The Jackson Laboratories; the IL-6−/− mice are on a C56Bl/6J background (JAX stock 002650). Lcn2hep−/− breeding pairs were provided by Dr. Bin Gao, M.D., Ph.D. NIH, Bethesda, MD.
Surgical procedures.
Ischemic AKI was induced by bilateral clamping of renal pedicles for 5, 10, 15, 23, 27 or 29 minutes on a bath heated platform 38°C via abdominal approach. Sham and bilateral nephrectomy were performed as previously described.12
Kidney function, histology, immunofluorescence staining, RNA Isolation and Quantitative RT-PCR, IL-6 injection, neutrophil depletion, and STAT3 (Phospho/Total) expression.
See supplemental methods.
In vitro studies.
AML12 (hepatocyte mouse cell line) and HepG2 (hepatocyte human cell line) were purchased from ATCC® (CRL-2254, HB-8065). See supplemental methods.
Flow cytometry to assess neutrophil depletion was performed as previously described 15 using the following strategy to identify neutrophils by cell surface markers: CD45+, CD11b+, CD11c−, MHCII−, CD24+, and Ly6g+.
Transcutaneous glomerular filtration rate (GFR).
Transcutaneous GFR (tGFR) was measured and performed as we have previously reported. 43 GFR measurements were obtained at baseline, and then after the pre-specified time point in mice treated with vehicle, furosemide or maleic acid or after ischemic AKI. The NIC-Kidney device (MediBeacon Inc, Amtsgericht, Germany) was utilized for tGFR measurements per manufacturer’s instructions. 44
Study approval.
All animal experiments were conducted with adherence to the NIH Guide for the Care and Use of Laboratory Animals. The animal protocol was approved by the Animal Care and Use Committee of the University of Colorado at Denver.
Supplementary Material
Supplemental Figure 1. NGAL localization 4 and 24 hours after ischemic AKI. (A-D) Immunofluorescence was performed 4 and 24 hours after ischemic AKI in wild type mice 4 and 24h after AKI in the kidney of wild type mice. Proximal tubules white arrows, distal tubule blue arrows. Scale bar: 100μm.
{kind=link}
Supplemental Figure 2. Intravenous (IV) IL-6 to IL-6 deficient (IL-6−/−) mice with ischemic acute kidney injury (AKI) increases liver NGAL production and plasma NGAL levels. 200 ng of IL-6 or vehicle (0.1%BSA, Veh) was administered IV every one hour for 3 hours after surgery for ischemic AKI in IL-6−/− mice and (A) liver, (B) kidney, (C) lung, and (D) spleen NGAL (mRNA) and (E) plasma NGAL (ELISA) were measured 1 hour after that (i.e., 4 hours after surgery for ischemic AKI). Plasma: N=10 for vehicle; N=3 for IL-6; Liver: N=4 for vehicle; N=5 for IL-6; Kidney: N=3 for vehicle; N=3 for IL-6; Lung: N=4 for vehicle; N=5 for IL-6; Spleen: N=2 for vehicle; N=2 for IL-6. Significance determined by t test.
{kind=link}
Supplemental Figure 3. Effect of dose and time on IL-6 dependent hepatic NGAL production. Various doses of recombinant human NGAL were added to human (HepG2) hepatocytes and NGAL protein levels were determined in the media. Analysis by one way ANOVA. N=3, 2 separate experiments.
{kind=link}
Supplemental Figure 4. Effect of different durations of renal ischemia and reperfusion times indices on kidney function, plasma IL-6, and NGAL. Normal mice, and mice subjected to sham (surgery) and 5, 10, 15, 23, and 29 minutes of renal ischemia were studied. (A) BUN and (B) plasma creatinine were increased with 23 and 29 minutes of renal ischemia at 24 hours. (C) Plasma IL-6 was elevated 4 hours after sham and all ischemia times and preceded the peak in plasma and urine NGAL levels (D-F). (G) Renal Kim-1 mRNA levels and (H) Liver NGAL mRNA levels were determined at 24 hours. (X=no urine production at that time point). Analyzed by t test, #P=NS versus 23 minutes at the same time point; *P<0.05 vs. 23 minutes at the same time point.
{kind=link}
Translational statement.
Acute kidney injury is the most common inpatient consultation to nephrologists, and is associated with increased morbidity and mortality. Current tools to identify and classify AKI are inadequate. Herein, we extensively characterize the production and renal handling of one of the most widely studied AKI biomarkers – NGAL– and demonstrate several new insights into its production and renal handling that may improve its accuracy to identify AKI and distinguish between prerenal azotemia and acute tubular injury, which have major differences in the approach to clinical care. Since we demonstrate that the major source of urine NGAL is the plasma and increases during proximal tubule dysfunction, future studies should incorporate methods to account for plasma NGAL levels and proximal tubule function in patients with adjudicated pre-renal azotemia and acute tubular injury.
Acknowledgements
This work was supported by VA Merit 1 I01 BX001498 and NHLBI R01 HL 63012693 to SF and VA BLR&D 1 IK2 BX003839-01A1 to JM and NIDDK K08DK109226-01A1 to DS. We would like to thank Joel Topf, MD for assistance with the Graphical Abstract.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Palevsky PM, Zhang JH, O’Connor TZ, et al. Intensity of renal support in critically ill patients with acute kidney injury. The New England journal of medicine 2008; 359: 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.American Society of Nephrology Renal Research Report. J Am Soc Nephrol 2005; 16: 1886–1903. [DOI] [PubMed] [Google Scholar]
- 3.Nickolas TL, Schmidt-Ott KM, Canetta P, et al. Diagnostic and prognostic stratification in the emergency department using urinary biomarkers of nephron damage: a multicenter prospective cohort study. J Am Coll Cardiol 2012; 59: 246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parikh CR, Devarajan P, Zappitelli M, et al. Postoperative biomarkers predict acute kidney injury and poor outcomes after pediatric cardiac surgery. J Am Soc Nephrol 22: 1737–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parikh CR, Coca SG, Thiessen-Philbrook H, et al. Postoperative Biomarkers Predict Acute Kidney Injury and Poor Outcomes after Adult Cardiac Surgery. J Am Soc Nephrol 2011; 22: 1748–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim S, Kim HJ, Ahn HS, et al. Is plasma neutrophil gelatinase-associated lipocalin a predictive biomarker for acute kidney injury in sepsis patients? A systematic review and meta-analysis. J Crit Care 2016; 33: 213–223. [DOI] [PubMed] [Google Scholar]
- 7.Schmidt-Ott KM. Neutrophil gelatinase-associated lipocalin as a biomarker of acute kidney injury--where do we stand today? Nephrol Dial Transplant 2011; 26: 762–764. [DOI] [PubMed] [Google Scholar]
- 8.Paragas N, Qiu A, Zhang Q, et al. The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat Med 2011; 17: 216–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mishra J, Dent C, Tarabishi R, et al. Neutrophil gelatinase-associated lipocalin (NGAL) as a biomarker for acute renal injury after cardiac surgery. Lancet 2005; 365: 1231–1238. [DOI] [PubMed] [Google Scholar]
- 10.Xiao X, Yeoh BS, Vijay-Kumar M. Lipocalin 2: An Emerging Player in Iron Homeostasis and Inflammation. Annu Rev Nutr 2017; 37: 103–130. [DOI] [PubMed] [Google Scholar]
- 11.Dennen P, Altmann C, Kaufman J, et al. Urine interleukin-6 is an early biomarker of acute kidney injury in children undergoing cardiac surgery. Crit Care 2010; 14: R181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klein CL, Hoke TS, Fang WF, et al. Interleukin-6 mediates lung injury following ischemic acute kidney injury or bilateral nephrectomy. Kidney Int 2008; 74: 901–909. [DOI] [PubMed] [Google Scholar]
- 13.Xu MJ, Feng D, Wu H, et al. Liver is the major source of elevated serum lipocalin-2 levels after bacterial infection or partial hepatectomy: a critical role for IL-6/STAT3. Hepatology 2015; 61: 692–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andres-Hernando A, Okamura K, Bhargava R, et al. Circulating IL-6 upregulates IL-10 production in splenic CD4+ T cells and limits acute kidney injury-induced lung inflammation. Kidney Int 2017. [DOI] [PubMed] [Google Scholar]
- 15.Altmann C, Ahuja N, Kiekhaefer CM, et al. Early peritoneal dialysis reduces lung inflammation in mice with ischemic acute kidney injury. Kidney Int 2017; 92: 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reynolds R, McNamara PD, Segal S. On the maleic acid induced Fanconi syndrome: effects on transport by isolated rat kidney brushborder membrane vesicles. Life Sci 1978; 22: 39–43. [DOI] [PubMed] [Google Scholar]
- 17.Moledina DG, Parikh CR. Phenotyping of Acute Kidney Injury: Beyond Serum Creatinine. Semin Nephrol 2018; 38: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mishra J, Ma Q, Prada A, et al. Identification of neutrophil gelatinase-associated lipocalin as a novel early urinary biomarker for ischemic renal injury. J Am Soc Nephrol 2003; 14: 2534–2543. [DOI] [PubMed] [Google Scholar]
- 19.Mori K, Lee HT, Rapoport D, et al. Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J Clin Invest 2005; 115: 610–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mishra J, Mori K, Ma Q, et al. Amelioration of ischemic acute renal injury by neutrophil gelatinase-associated lipocalin. J Am Soc Nephrol 2004; 15: 3073–3082. [DOI] [PubMed] [Google Scholar]
- 21.Jain S, Gautam V, Naseem S. Acute-phase proteins: As diagnostic tool. J Pharm Bioallied Sci 2011; 3: 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hepcidin Ganz T. and iron regulation, 10 years later. Blood 2011; 117: 4425–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boe A, Canosi U, Donini S, et al. Determination of haptoglobin expression in IL-6 treated HepG2 cells by ELISA and by RNA hybridization--evaluation of a quantitative method to measure IL-6. J Immunol Methods 1994; 171: 157–167. [DOI] [PubMed] [Google Scholar]
- 24.Kobune M, Kohgo Y, Kato J, et al. Interleukin-6 enhances hepatic transferrin uptake and ferritin expression in rats. Hepatology 1994; 19: 1468–1475. [PubMed] [Google Scholar]
- 25.Berger T, Togawa A, Duncan GS, et al. Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc Natl Acad Sci U S A 2006; 103: 1834–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan YR, Liu JS, Pociask DA, et al. Lipocalin 2 is required for pulmonary host defense against Klebsiella infection. J Immunol 2009; 182: 4947–4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gruys E, Toussaint MJ, Niewold TA, et al. Acute phase reaction and acute phase proteins. J Zhejiang Univ Sci B 2005; 6: 1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hoke TS, Douglas IS, Klein CL, et al. Acute renal failure after bilateral nephrectomy is associated with cytokine-mediated pulmonary injury. J Am Soc Nephrol 2007; 18: 155–164. [DOI] [PubMed] [Google Scholar]
- 29.Liu KD, Altmann C, Smits G, et al. Serum interleukin-6 and interleukin-8 are early biomarkers of acute kidney injury and predict prolonged mechanical ventilation in children undergoing cardiac surgery: a case-control study. Crit Care 2009; 13: R104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klein CL, Hoke TS, Fang WF, et al. Interleukin-6 mediates lung injury following ischemic acute kidney injury or bilateral nephrectomy. Kidney Int 2008. [DOI] [PubMed] [Google Scholar]
- 31.Andres-Hernando A, Altmann C, Ahuja N, et al. Splenectomy exacerbates lung injury after ischemic acute kidney injury in mice. Am J Physiol Renal Physiol 2011; 301: F907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altmann C, Andres-Hernando A, McMahan RH, et al. Macrophages mediate lung inflammation in a mouse model of ischemic acute kidney injury. Am J Physiol Renal Physiol 2012; 302: F421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang WR, Garg AX, Coca SG, et al. Plasma IL-6 and IL-10 Concentrations Predict AKI and Long-Term Mortality in Adults after Cardiac Surgery. J Am Soc Nephrol 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hou T, Huang D, Zeng R, et al. Accuracy of serum interleukin (IL)-6 in sepsis diagnosis: a systematic review and meta-analysis. Int J Clin Exp Med 2015; 8: 15238–15245. [PMC free article] [PubMed] [Google Scholar]
- 35.Matsumoto H, Ogura H, Shimizu K, et al. The clinical importance of a cytokine network in the acute phase of sepsis. Sci Rep 2018; 8: 13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martensson J, Bell M, Xu S, et al. Association of plasma neutrophil gelatinase-associated lipocalin (NGAL) with sepsis and acute kidney dysfunction. Biomarkers 2013; 18: 349–356. [DOI] [PubMed] [Google Scholar]
- 37.Aydogdu M, Gursel G, Sancak B, et al. The use of plasma and urine neutrophil gelatinase associated lipocalin (NGAL) and Cystatin C in early diagnosis of septic acute kidney injury in critically ill patients. Dis Markers 2013; 34: 237–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fisic E, Poropat G, Bilic-Zulle L, et al. The Role of IL-6, 8, and 10, sTNFr, CRP, and Pancreatic Elastase in the Prediction of Systemic Complications in Patients with Acute Pancreatitis. Gastroenterol Res Pract 2013; 2013: 282645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sporek M, Dumnicka P, Gala-Bladzinska A, et al. Determination of serum neutrophil gelatinase-associated lipocalin at the early stage of acute pancreatitis. Folia Med Cracov 2016; 56: 5–16. [PubMed] [Google Scholar]
- 40.Ahuja N, Andres-Hernando A, Altmann C, et al. Circulating IL-6 mediates lung injury via CXCL1 production after acute kidney injury in mice. Am J Physiol Renal Physiol 2012; 303: F864–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhargava R, Janssen W, Altmann C, et al. Intratracheal IL-6 protects against lung inflammation in direct, but not indirect, causes of acute lung injury in mice. PLoS ONE 2013; 8:e61405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kopf M, Baumann H, Freer G, et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 1994; 368: 339–342. [DOI] [PubMed] [Google Scholar]
- 43.Soranno DE, Gil HW, Kirkbride-Romeo L, et al. Matching Human Unilateral AKI, a Reverse Translational Approach to Investigate Kidney Recovery after Ischemia. J Am Soc Nephrol 2019; 30: 990–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schreiber A, Shulhevich Y, Geraci S, et al. Transcutaneous measurement of renal function in conscious mice. Am J Physiol Renal Physiol 2012; 303: F783–788. [DOI] [PubMed] [Google Scholar]
- 45.Chiba T, Skrypnyk NI, Skvarca LB, et al. Retinoic Acid Signaling Coordinates Macrophage-Dependent Injury and Repair after AKI. J Am Soc Nephrol 2016; 27: 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C-T method. Nat Protoc 2008; 3: 1101–1108. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. NGAL localization 4 and 24 hours after ischemic AKI. (A-D) Immunofluorescence was performed 4 and 24 hours after ischemic AKI in wild type mice 4 and 24h after AKI in the kidney of wild type mice. Proximal tubules white arrows, distal tubule blue arrows. Scale bar: 100μm.
Supplemental Figure 2. Intravenous (IV) IL-6 to IL-6 deficient (IL-6−/−) mice with ischemic acute kidney injury (AKI) increases liver NGAL production and plasma NGAL levels. 200 ng of IL-6 or vehicle (0.1%BSA, Veh) was administered IV every one hour for 3 hours after surgery for ischemic AKI in IL-6−/− mice and (A) liver, (B) kidney, (C) lung, and (D) spleen NGAL (mRNA) and (E) plasma NGAL (ELISA) were measured 1 hour after that (i.e., 4 hours after surgery for ischemic AKI). Plasma: N=10 for vehicle; N=3 for IL-6; Liver: N=4 for vehicle; N=5 for IL-6; Kidney: N=3 for vehicle; N=3 for IL-6; Lung: N=4 for vehicle; N=5 for IL-6; Spleen: N=2 for vehicle; N=2 for IL-6. Significance determined by t test.
Supplemental Figure 3. Effect of dose and time on IL-6 dependent hepatic NGAL production. Various doses of recombinant human NGAL were added to human (HepG2) hepatocytes and NGAL protein levels were determined in the media. Analysis by one way ANOVA. N=3, 2 separate experiments.
Supplemental Figure 4. Effect of different durations of renal ischemia and reperfusion times indices on kidney function, plasma IL-6, and NGAL. Normal mice, and mice subjected to sham (surgery) and 5, 10, 15, 23, and 29 minutes of renal ischemia were studied. (A) BUN and (B) plasma creatinine were increased with 23 and 29 minutes of renal ischemia at 24 hours. (C) Plasma IL-6 was elevated 4 hours after sham and all ischemia times and preceded the peak in plasma and urine NGAL levels (D-F). (G) Renal Kim-1 mRNA levels and (H) Liver NGAL mRNA levels were determined at 24 hours. (X=no urine production at that time point). Analyzed by t test, #P=NS versus 23 minutes at the same time point; *P<0.05 vs. 23 minutes at the same time point.