

ABSTRACT
Avelumab is an IgG1 anti–programmed death ligand 1 (anti–PD-L1) monoclonal antibody that has been approved as a monotherapy for metastatic Merkel cell carcinoma and advanced urothelial carcinoma, and in combination with axitinib for advanced renal cell carcinoma. Avelumab is cleared faster and has a shorter half-life than other anti–PD-L1 antibodies, such as atezolizumab and durvalumab, but the mechanisms underlying these differences are unknown. IgG antibodies can be cleared through receptor-mediated endocytosis after binding of the antibody Fab region to target proteins, or via Fcγ receptor (FcγR)-mediated endocytosis. Unlike other approved anti–PD-L1 antibodies, avelumab has a native Fc region that retains FcγR binding capability. We hypothesized that the rapid clearance of avelumab might be due to the synergistic effect of both FcγR-mediated and PD-L1 target–mediated internalization. To investigate this, we performed in vitro and in vivo studies that compared engineered variants of avelumab and atezolizumab to determine mechanisms of cellular internalization. We found that both FcγR and PD-L1 binding contribute to avelumab internalization. While FcγR binding was the dominant mechanism of avelumab internalization in vitro, with CD64 acting as the most important FcγR, studies in mice and cynomolgus monkeys showed that both FcγR and PD-L1 contribute to avelumab elimination, with PD-L1 binding playing a greater role. These studies suggest that the rapid internalization of avelumab might be due to simultaneous binding of both PD-L1 and FcγR in trans. Our findings also provide a basis to alter the clearance and half-life of monoclonal antibodies in therapeutic development.
KEYWORDS: Avelumab, internalization, FCγr binding, PD-L1 binding, clearance
Introduction
Tumors employ various strategies to evade immunosurveillance, including the inactivation of tumor-infiltrating immune effector cells.1,2 Of particular importance is the interaction between the programmed death 1 (PD-1) receptor expressed on the surface of activated T cells and its ligand, PD-L1, which is expressed on the surface of various immune cells and other host cells, in addition to some cancer cells, and which acts to suppress antitumor T cell responses.1–6 Blocking the PD-1/PD-L1 interaction using immune checkpoint inhibitor (ICI) monoclonal antibodies is an established therapeutic strategy in multiple tumor types.7
All approved monoclonal antibodies developed to date are IgG, and their elimination from the body mostly occurs via intracellular catabolism after nonspecific fluid-phase pinocytosis or receptor-mediated endocytosis.8 Following the binding of the antibody Fab region to cell surface proteins, receptor-mediated endocytosis results in antibody recycling via endosomes or degradation within lysosomes. Thus, target binding can significantly contribute to the kinetics of antibody elimination. IgG antibodies can also trigger elimination through binding of their Fc region to Fcγ receptors (FcγRs).8 These receptors are expressed on various types of immune cells that can be recruited to tumors through FcγR-Fc interactions with antibody-bound targets.9 Recruited immune cells can then elicit or mediate immunologic activities, including endocytosis and phagocytosis.8,9 FcγR-mediated elimination might dominate when an antibody can form soluble immune complexes with three or more IgGs, or in cases where the antibody binds cells suspended in blood or body fluid.8
During therapeutic development, effector functions of monoclonal antibodies are often modulated. For example, FcγR binding capability has been enhanced for some targeted therapies.10–12 In contrast, most approved ICIs have either been engineered to remove FcγR binding capability (eg, via Fc sequence changes as in atezolizumab and durvalumab) or have been developed using an IgG subclass with limited FcγR-binding functionality (eg, IgG4 subclass of nivolumab and pembrolizumab).10,11,13–16 Clearance of atezolizumab has been shown to involve both specific (target-mediated) and nonspecific clearance mechanisms, with the former providing a greater contribution at lower doses.14 It has also been shown that IgG antibodies with a low isoelectric point (pI) have a longer half-life and reduced elimination.17 Thus, the intrinsic properties of therapeutic antibodies can have profound effects on their potency and pharmacokinetics (PK).
Avelumab is an anti–PD-L1 inhibitor that has been approved in several indications, including advanced urothelial carcinoma (first-line maintenance and second-line therapy), metastatic Merkel cell carcinoma, and advanced renal cell carcinoma (first-line treatment in combination with axitinib).18–23 Unlike other approved anti–PD-L1 antibodies, avelumab has a native IgG1 Fc region that retains FcγR binding.7,24,25 Preclinical studies have shown that in addition to stimulating T cell–mediated immune responses by binding to PD-L1 and inhibiting PD-1/PD-L1 interactions, the native Fc region of avelumab induces innate effector functions against tumor cells (eg, antibody-dependent cell-mediated cytotoxicity [ADCC]).10,25–28 This extra mode of action for avelumab compared with other anti–PD-L1 antibodies might provide additional therapeutic potential. Although the contribution of ADCC to therapeutic benefit has not been demonstrated in clinical trials, it has been demonstrated in nonclinical monotherapy studies.29
Avelumab is cleared significantly faster than some other anti–PD-L1 antibodies, resulting in a shorter half-life.30 We speculated that both PD-L1 binding and FcγR-mediated mechanisms might contribute to the rapid clearance of avelumab. To elucidate mechanisms responsible for clearance and to better understand FcγR-mediated internalization vs PD-L1 target–mediated internalization, in vitro and in vivo studies were performed using variants of avelumab and atezolizumab.
Materials and methods
Antibody variants
Versions of avelumab studied comprised the wild-type (WT) version with full FcγR binding capability, an FcγR binding–deficient variant (containing an N297A substitution), a PD-L1 binding–deficient variant (R99K substitution), and a lower pI variant containing several substitutions (Q16E, S17R, T72D, and Q81E), which has a calculated pI of 8.7 compared with 9.1 for WT avelumab (Table 1). Additionally, an anti–PD-L1 antibody with an amino acid sequence identical to atezolizumab, including its N297A substitution (FcγR binding–deficient), was assessed, along with a modified version of atezolizumab with a restored WT Fc region and intact glycosylation site (FcγR binding). All antibody variants were generated internally. Initial in vitro studies included assessment of different antibody batches to confirm that observations were not due to batch-specific effects.
Table 1.
Characteristics and affinity of anti–PD-L1 antibody variants analyzed. The binding affinities of anti–PD-L1 antibodies were determined using surface plasmon resonance. Human PD-L1 analyte was bound at 0, 1.25, 2.5, 5, 10, and 20 nM. Mouse PD-L1 analyte was bound at 0, 2.5, 5, 10, 20, and 40 nM. Binding affinities were determined from the measured association and dissociation rate constants
| Anti-PD-L1 antibody (amino acid substitutions) | Outcome of mutation(s) |
Human PD-L1 affinity, nM |
Mouse PD-L1 affinity, nM |
|---|---|---|---|
| WT avelumab | NA | 0.4 | 0.7 |
| Avelumab (N297A) | FcγR binding–deficient | 0.3 | 0.8 |
| Avelumab (R99K) | PD-L1 binding–deficient | NB | NB |
| Avelumab (Q16E, S17R, T72D, Q81E) | Lower pI* | 0.8 | 1.2 |
| WT atezolizumab | NA | 0.2 | 2.5 |
| Atezolizumab (N297A) | FcγR binding–deficient | 0.2 | 2.4 |
FcγR, Fcγ receptor; NA, not applicable; NB, no binding; PD-L1, programmed death-ligand 1; pI, isoelectric point; WT, wild-type.
*Calculated pI of 8.7 compared with 9.1 for WT avelumab.
Antibody affinity determination
The binding affinity of anti–PD-L1 antibodies for histidine-tagged PD-L1 (PD-L1–His; human and mouse) was determined using a Biacore 4000 instrument (GE Healthcare). The Fc region of anti–PD-L1 antibodies was captured using goat anti-human IgG immobilized to a CM5 sensor chip. Anti–PD-L1 antibodies were captured at 0.5 and 1.0 µg/mL for 120 sec at 30 μL/mL. Human PD-L1–His analyte was bound at 0, 1.25, 2.5, 5, 10, and 20 nM. Mouse PD-L1–His analyte was bound at 0, 2.5, 5, 10, 20, and 40 nM. During the association phase, samples were injected at 30 μL/min for 180 sec. For the dissociation phase, wash buffer was injected at 30 μL/min for 600 sec. Binding affinities (KD) were determined from the measured association (ka) and dissociation (kd) rate constants, where KD = kd/ka, using the Biacore 4000 Evaluation software.
Internalization assay
Antibody internalization in healthy donor blood was measured using a commercially available flow, cytometry–based assay in which pHrodo (ThermoFisher Scientific), a pH-sensitive fluorescent dye, was used to directly monitor antibody endocytosis and lysosomal degradation.31,32 Antibodies were labeled with pHrodo iFL Red Microscale Labeling kits (ThermoFisher Scientific), following the manufacturer’s instructions. Briefly, 100 µL of 1 mg/mL antibodies were purified from primary amines using Zeba Spin Desalting Columns (Thermo Fisher Scientific). Antibodies were then mixed with 1 M sodium bicarbonate and 2 mM pHrodo iFL Red labeling solution and incubated in the dark at room temperature for 15 min. Finally, the reaction mixture was purified using a purification spin column, according to the manufacturer’s instructions. The DOL (degree of labeling) and absorbance maximum of the dye were determined from the absorbance at 280 nm and 560 nm, respectively, using a Nanodrop ND-1000 (Thermo Scientific). Labeled antibodies were measured with the same DOL during investigation.
Blood samples were collected from the different donors using sodium heparin tubes. Internalization was measured using 100 µL of whole blood. Each tube was treated with 30 µg/mL of selected pHRodo-labeled antibody and incubated at 37°C with 5% CO2 for different times. For negative controls, whole blood was incubated with pHrodo-labeled antibody at 4°C. After treatment, each tube was incubated for 20 min on ice with external staining antibodies. Cells were stained with CD45-PE Cy5, CD3-FITC, CD16-PE Cy7, CD14-BV421, and CD56-APC (Becton Dickinson). Internalization in natural killer (NK) cells (CD56+CD16+), lymphocytes (CD3+), total monocytes (CD14+CD16−, CD14+CD16+, and CD14−CD16+), and granulocytes was assessed.
Antibody internalization studies were also performed in the presence of various soluble Fc receptors.33 Blood samples were collected from the different donors using CPT tubes (Becton Dickinson). Peripheral blood mononuclear cells (PBMCs) were isolated and counted using a Nucleocounter NC-200 instrument (Chemometec), following manufacturer’s instructions. PBMCs were then resuspended in Gibco AIM V medium (ThermoFisher Scientific) and aliquoted at 5 × 105 cells/tube. Each tube was first treated with 30 µg/mL of selected recombinant human receptor proteins (FcγRI/CD64, FcγRIIA/CD32a, FcγRIIB/C [CD32b/c], FcγRIIIA/CD16a, FcγRIIIB/CD16b, FcRn [neonatal Fc receptor]; R&D Systems) or with WT avelumab. PBMCs were then treated with 30 µg/mL of selected pHrodo-labeled antibodies and incubated at 37°C with 5% CO2 for 2 h. For negative controls, PBMCs were incubated with pHrodo-labeled antibody at 4°C in parallel. After treatment, each tube was incubated for 20 min on ice with various staining antibodies (CD3, CD14, CD16, CD45, and CD56; Becton Dickinson). Internalization was assessed on NK cells (CD56+CD16+), lymphocytes (CD3+), and total monocytes (CD14+CD16−, CD14+CD16+, and CD14−CD16+).
PK studies in mice
C57BL/6 male and female mice were dosed with 200 µg of antibody variants in 0.3 mL phosphate-buffered saline at pH 7.4 and blood samples of 0.1 mL were drawn to obtain Li-heparin plasma at six time points: 1, 2, 4, 6, 8, and 10 days post dosing (three alternating time points for subgroups of three mice; six mice per antibody). Plasma concentrations were measured by immunoassay, as reported previously.30 The initial concentration (C0) was fixed at 200 µg/mL assuming a plasma volume of 1 mL (40 mL/kg). When plotting the plasma concentration vs time profiles, concentrations at the first time point below the lower limit of quantitation (LLOQ) of the assay (0.2 µg/mL) were set at 0.5 × LLOQ.
PK studies in cynomolgus monkeys
Cynomolgus monkeys (n = 3 per group) received a single intravenous (IV) bolus injection of 5 mg/kg of each antibody variant. WT avelumab was investigated in an earlier study and was dosed at 4 mg/kg and serum concentrations were normalized to 5 mg/kg. Serum concentrations of antibodies were measured by immunoassay, as reported previously30; profiles affected by anti-drug antibodies were excluded. Internalization of antibody variants in circulating immune cells obtained from cynomolgus monkey whole blood was assessed using the internalization assay described above.
Ethical approval
Studies using human blood were approved by the local ethics committee where studies were performed (Hesse, Germany). Healthy donors provided written informed consent. All studies in animals were conducted in accordance with national guidelines and with formal approval by relevant animal care committees. Studies in mice were performed in an institute fully authorized by the national Ministry of Health (Italy), all parts of the study plan were reviewed by the institute’s designated veterinarian and animal welfare officer, and animal protection, housing, and welfare were consistent with national laws. Studies in cynomolgus monkeys complied with all relevant national regulations (United States), and the protocol and procedures involving use of animals were reviewed and approved by the institutional committee before studies were initiated.
Results
Development of the internalization assay
An internalization assay was developed using antibodies labeled with pHrodo dyes targeting human immune cells in whole blood or PBMCs. The fluorescence intensity of the dyes increases as the pH decreases from neutral in the early endosome to acidic in the late lysosome, enabling measurement of antibody endocytosis and internalization.31,32 The main immune cells that internalize labeled antibodies are monocytes and granulocytes (Supplementary Figure S1). NK cells can also internalize antibodies, whereas T and B cells do not contribute to internalization (data not shown). Antibody internalization and kinetics were observable after 1 h and the signal plateaued after 6 h. The gating strategy is shown (Supplementary Figure S1).
Internalization of WT avelumab and its FcγR binding–deficient variant in human monocytes and granulocytes in vitro
To determine the role of FcγR binding in the internalization of avelumab by human immune cells, internalization of WT avelumab in human blood was compared with an FcγR binding–deficient variant (N297A). Compared with WT avelumab, the N297A variant had significantly reduced internalization in FcγR-expressing cells (CD14+ CD16− monocytes; Figure 1a). Up to 80% of monocytes internalized avelumab, with peak internalization reached in 4–6 hours. Similarly, N297A variants were also internalized significantly less than WT avelumab in granulocytes (Figure 1b). Up to 25% of granulocytes internalized avelumab, with peak internalization reached in 4–6 hours. Thus, the avelumab FcγR binding–deficient variant (N297A) showed diminished uptake in monocytes and granulocytes compared with WT avelumab.
Figure 1.
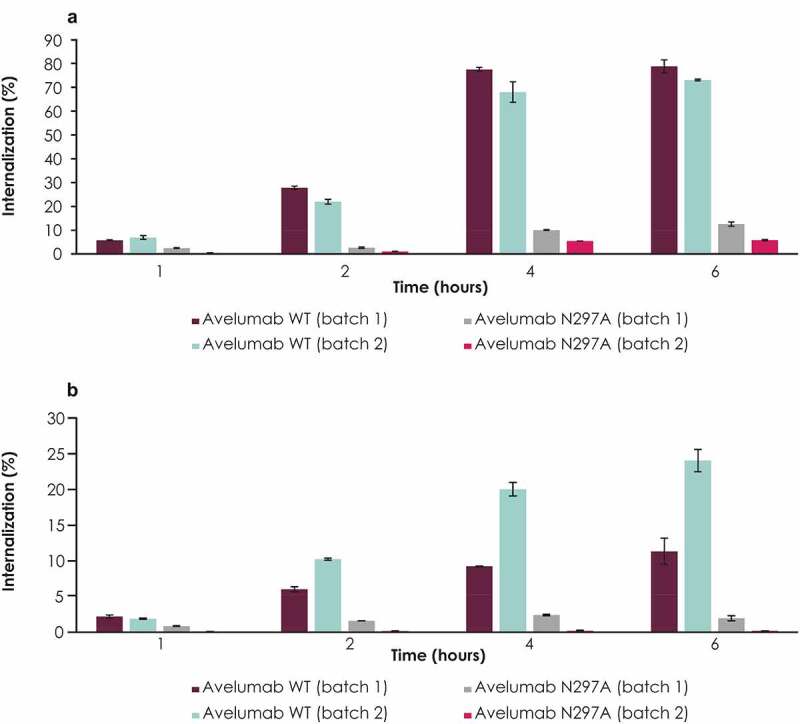
Internalization of WT avelumab compared with its FcγR binding–deficient variant (N297A) in human monocytes and granulocytes. Internalization in (a) CD14+ CD16− monocytes and (b) granulocytes. A flow cytometry–based antibody internalization assay with pH-sensitive fluorescent dye, pHrodo, was used to directly monitor antibody internalization in healthy donor blood. Blood samples were treated with pHrodo-labeled antibodies, and immune cells were detected with external staining antibodies. Error bars represent standard deviations. FcγR, Fcγ receptor; WT, wild type
Internalization of avelumab anti–PD-L1 binding–deficient variant in human monocytes and granulocytes in vitro
Although the FcγR binding–deficient variant of avelumab was internalized less than WT avelumab, internalization of the FcγR binding–deficient variant was still observed at later time points (Figure 1a, b). This observation suggested a role for PD-L1–mediated internalization. To test this, internalization of WT avelumab and the FcγR binding–deficient variant (N297A) was compared with a PD-L1 binding–deficient variant (R99K). In both human monocytes and granulocytes, the R99K avelumab variant had reduced internalization compared with WT avelumab but some internalization was observed, particularly at late time points (Figure 2a, b). In contrast to the R99K variant, and consistent with prior experiments, the N297A variant showed substantially reduced internalization at early time points (Figure 2a, b).
Figure 2.
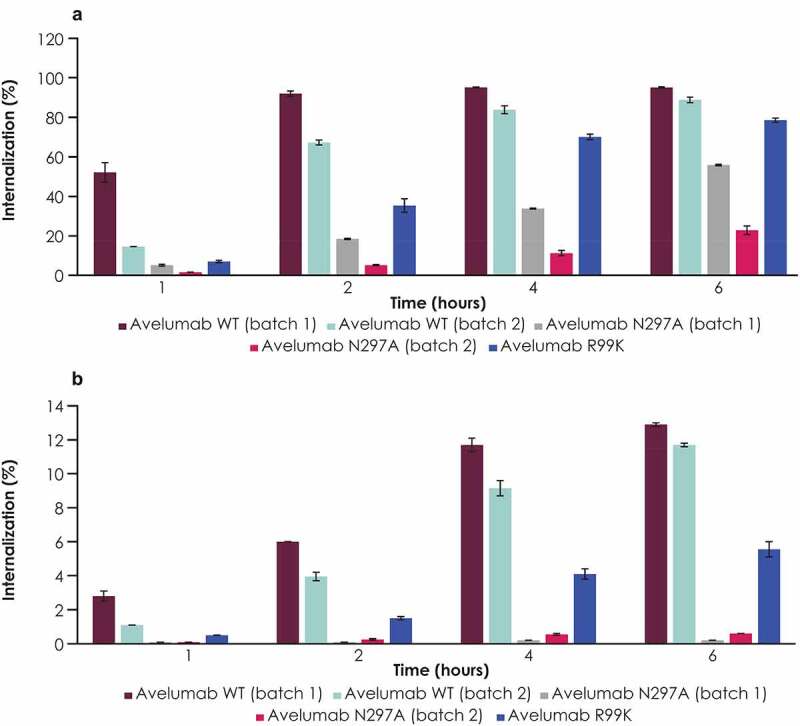
Internalization of WT avelumab compared with its anti–PD-L1 binding–deficient variant (R99K) and FcγR binding–deficient variant (N297A) in total human monocytes and granulocytes. Internalization in (a) total monocytes and (b) granulocytes from a representative donor is shown. Results were similar in other donors (data not shown). Error bars represent standard deviations of replicate assays from the same donor. Anti–PD-L1, anti–programmed death ligand 1; WT, wild-type
To confirm that both FcγR and PD-L1 binding contributed to avelumab internalization, the internalization of WT avelumab was compared with internally generated antibodies that had the same Fab domain as atezolizumab linked either to an FcγR binding–deficient (N297A) Fc domain or its WT IgG1 (FcγR binding–restored) variant (Figure 3a, 3b, Supplementary Figure 1b, 1c). The atezolizumab and avelumab WT IgG1 antibodies had starkly different levels of internalization, with WT atezolizumab internalized at a much lower level than WT avelumab, indicating that FcγR binding alone does not explain the difference in internalization between avelumab and atezolizumab. However, the FcγR binding–deficient (N297A) variant of atezolizumab also had very low levels of internalization, lower than WT atezolizumab, indicating the occurrence of FcγR-mediated internalization (Figure 3a, 3b).
Figure 3.
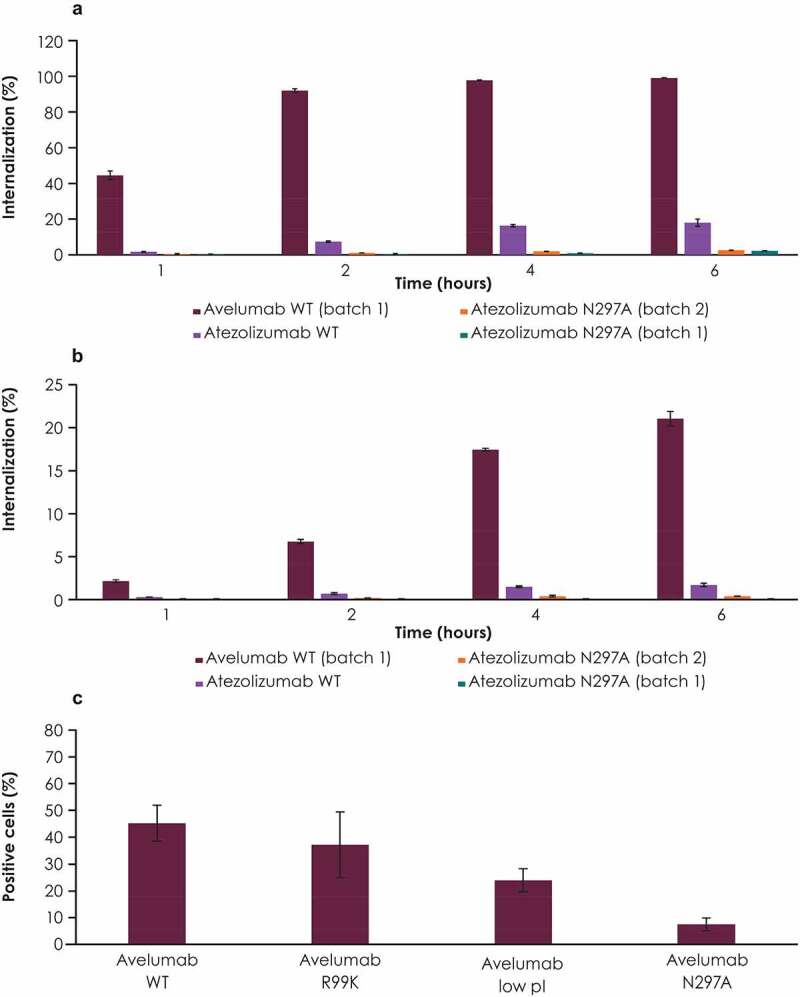
Internalization of anti–PD-L1 antibodies compared with their variants. Internalization of WT avelumab was compared with antibodies containing the atezolizumab Fab domain attached to either an FcγR binding–deficient (N297A) or a WT IgG1 Fc domain (FcγR binding–restored) in (a) CD14+ CD16− monocytes and (b) granulocytes. (c) Internalization of WT avelumab compared with a low pI variant, as well as FcγR binding–deficient (N297A) and PD-L1 binding–deficient (R99K) variants, in human PBMCs. Error bars represent standard deviations. FcγR, Fcγ receptor; PBMC, peripheral blood mononuclear cell; PD-L1, programmed death ligand 1; pI, isoelectric point; WT, wild-type
Internalization of a low pI variant of avelumab in human PBMCs in vitro
To confirm that PD-L1 binding characteristics are an important factor that contributes to internalization, studies were performed to compare an additional variant of avelumab that had a lower pI (containing several amino acid substitutions) with WT avelumab, the PD-L1 binding–deficient variant (R99K), and the FcγR binding–deficient variant (N297A). Differences in affinity for PD-L1 for each variant are shown (Table 1). In human PMBCs, the N297A variant had the lowest internalization of the variants assessed; in comparison, WT avelumab had the highest internalization, followed by the PD-L1 binding–deficient variant (R99K) and the lower pI variant (Figure 3c).
PK studies in mice
The PK of the PD-L1 antibodies and variants was investigated in mice (Figure 4). Following IV dosing, WT avelumab was cleared faster than each of the variants assessed. This difference in clearance was observed only in the terminal phase, 8–10 days after dosing. In the terminal phase, elimination was fastest for the WT avelumab and slowest for the PD-L1 binding–deficient variant (R99K) (Figure 4a, 4b). The elimination rates of the FcγR binding–deficient (N297A) and low pI variants were between that of WT avelumab and the PD-L1 binding–deficient variant (R99K). Similarly, WT atezolizumab was eliminated slightly faster than its FcγR binding–deficient variant (N297A) in mice (Figure 4c).
Figure 4.
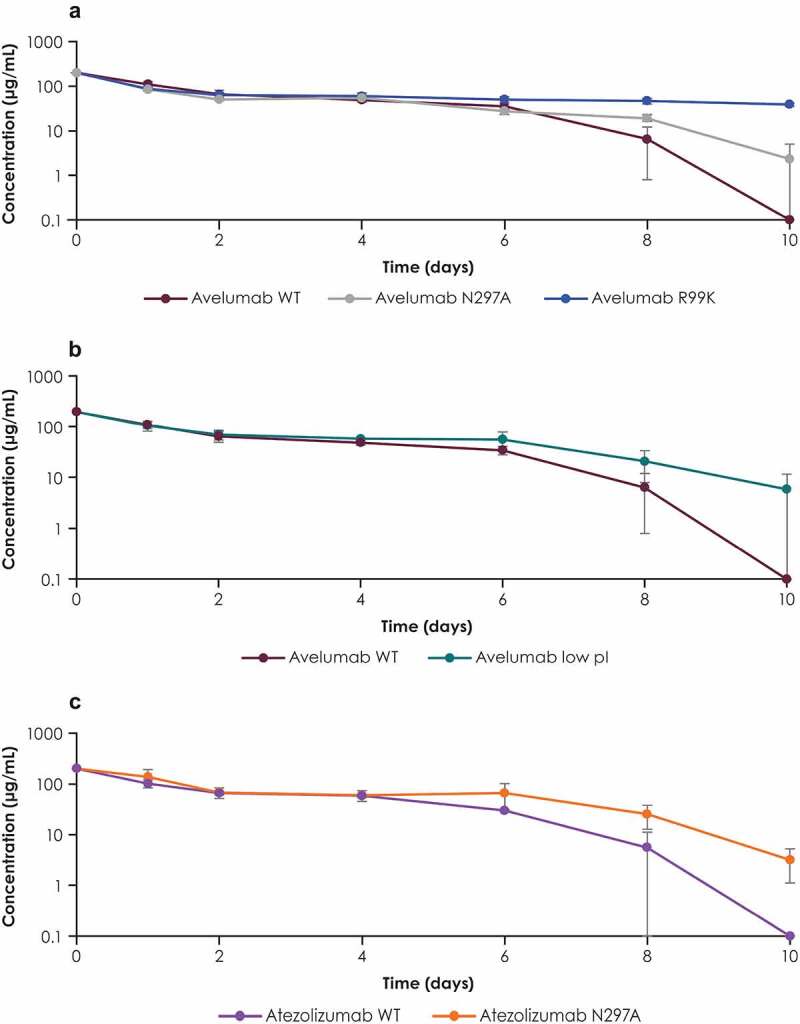
Serum concentration profiles of PD-L1 antibodies and their variants in mice. Serum concentration profiles of (a) WT avelumab and its FcγR binding–deficient (N297A) and PD-L1 binding–deficient (R99K) variants, (b) WT avelumab and its low pI variant, and (c) WT atezolizumab and its FcγR binding–deficient variant (N297A). C57BL/6 mice were dosed with 200 µg of antibodies. Blood samples were collected at various time points, and serum concentrations were measured by immunoassay. Error bars represent standard deviations. FcγR, Fcγ receptor; PD-L1, programmed death ligand 1; pI, isoelectric point; WT, wild-type
PK studies in cynomolgus monkeys
Internalization of different antibody variants was assessed in vitro in whole blood obtained from cynomolgus monkeys, and results were similar to in vitro studies in human blood. Compared with WT avelumab, the FcγR binding–deficient variant (N297A) was internalized less in monocytes and granulocytes, similar to WT atezolizumab (Figure 5a). The PD-L1 binding–deficient variant (R99K) of avelumab was internalized less than WT avelumab but more than the FcγR binding–deficient variant (N297A). PK studies with different antibody variants were performed in cynomolgus monkeys to correlate in vitro findings with in vivo studies. Following IV dosing, WT avelumab was eliminated faster than each of the variants assessed (Figure 5b). In the terminal phase, clearance was fastest for WT avelumab and slowest for PD-L1 binding deficient variant (R99K); clearance rates for the FcγR binding–deficient variant (N297A) and the low pI variant were very similar and were between clearance rates for WT avelumab PD-L1 binding–deficient variant (Figure 5b, Supplementary Table S1). In studies of atezolizumab variants in vivo, the FcγR binding–deficient variant (N297A) showed a minimal reduction in elimination compared with WT atezolizumab (Figure 5c). Consequently, WT avelumab had a shorter half-life (53.6 h) and faster clearance (1.18 mL/h/kg) than the other antibody variants (Supplementary Table 1).
Figure 5.
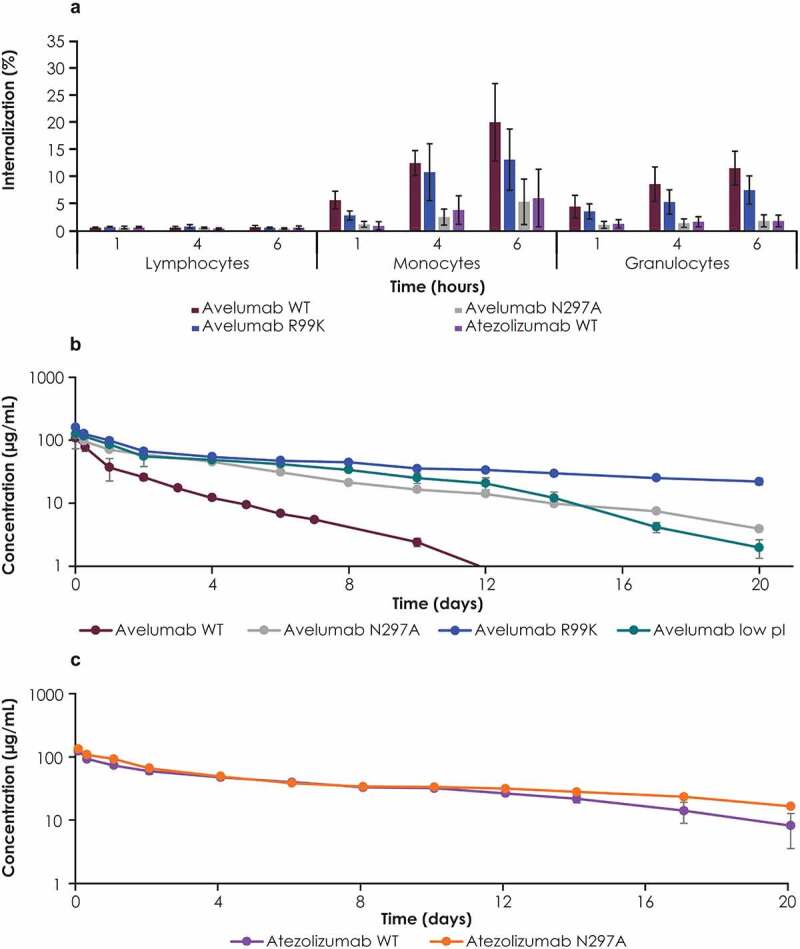
Studies with PD-L1 antibodies and their variants in cynomolgus monkeys. (a) Internalization of avelumab and its FcγR binding–deficient (N297A) and PD-L1 binding–deficient (R99K) variants was assessed in whole blood samples from cynomolgus monkeys. Serum concentration profiles of (b) WT avelumab, n = 2; avelumab FcγR binding–deficient (N297A), n = 1; avelumab PD-L1 binding–deficient (R99K), n = 3; avelumab low pI: n = 3; and (c) WT atezolizumab with, n = 2 and atezolizumab N297A (FcγR binding-deficient), n = 1. Cynomolgus monkeys were dosed with 5 mg/kg of antibody variants (WT avelumab was dosed at 4 mg/kg and normalized to 5 mg/kg). Serum concentrations were measured by immunoassay; profiles affected by antidrug antibodies were excluded. Error bars represent standard deviations. FcγR, Fcγ receptor; pI, isoelectric point; WT, wild-type
Internalization of avelumab and variants in the presence of competing soluble receptors
To assess the contribution of different FcγRs to the internalization of avelumab, further internalization studies were performed in the presence of various competing soluble FcγRs (Figure 6). The presence of soluble CD64, which is an FcγR with high affinity for IgG1, significantly reduced the internalization of WT avelumab and the PD-L1 binding–deficient variant (R99K). Other soluble FcγRs had minimal effects on the internalization of avelumab. Free avelumab had no impact on the internalization of labeled avelumab.
Figure 6.
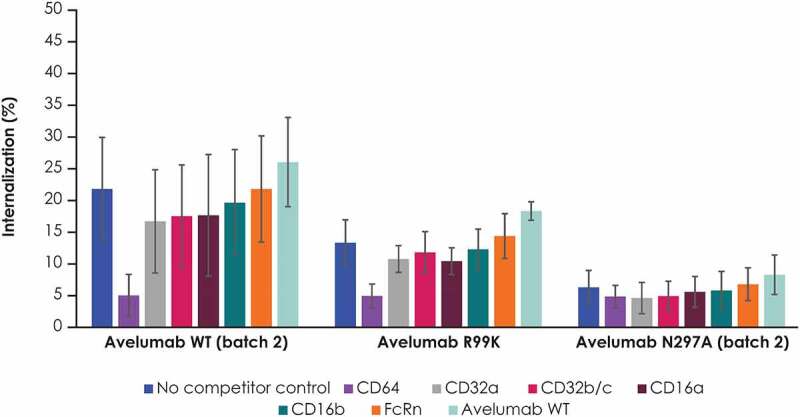
Internalization of avelumab and its FcγR binding–deficient (N297K) and PD-L1 binding–deficient (R99K) variants in the presence of competing soluble receptors in humans blood samples. PBMCs were incubated with selected recombinant human receptor proteins or with WT avelumab, followed by selected fluorescently labeled antibodies. Internalization was assessed using external staining antibodies. Error bars represent standard deviations. PBMC, peripheral blood mononuclear cell; FcRN; neonatal Fc receptor; WT, wild-type
Discussion
Avelumab has a shorter half-life than some other anti–PD-L1 antibodies, namely atezolizumab and durvalumab (≈4 days for avelumab vs ≈21 days for atezolizumab and durvalumab)30; however, the reasons for this observation had not previously been investigated. Mechanisms of antibody clearance from circulation include target-mediated and FcγR-mediated endocytosis, and FcγR-mediated phagocytosis.8 In addition, IgGs with a higher pI are cleared efficiently via nonspecific pinocytosis.17 The half-life and clearance of therapeutic antibodies can be fine-tuned by modulating their biophysical properties, which influences internalization by circulating immune cells.
Previously, avelumab, durvalumab, and atezolizumab were shown to bind PD-L1 with different binding characteristics, including use of distinct surfaces and with different binding orientations.34 Furthermore, avelumab was reported to bind to PD-L1 with a higher affinity than durvalumab and atezolizumab, which have similar affinities (KD values: avelumab, 0.0467 nmol/L; durvalumab, 0.667 nmol/L; atezolizumab, 1.75 nmol/L).34 In our studies, however, WT avelumab showed a binding affinity of 0.4 nmol/L, similar to other anti–PD-L1 antibodies. Despite their similar affinities, these anti–PD-L1 antibodies demonstrated unique binding kinetics that might contribute to differences in PK.34 Furthermore, unlike other approved anti–PD-L1 antibodies, avelumab has a native IgG1 Fc region with FcγR binding capability.7,24,25 We hypothesized that FcγR-mediated clearance, in addition to PD-L1 binding-mediated internalization, might underlie the faster clearance and shorter half-life of avelumab compared with other anti–PD-L1 antibodies.
To investigate this, we performed studies with variants of avelumab engineered to remove its capability to bind to either FcγR or PD-L1, or to reduce its pI.17 The lower pI variant was found to have a slightly lower affinity for PD-L1 than WT avelumab (KD values for human PD-L1: low PI variant, 0.8 nM; WT avelumab, 0.4 nM). For comparison studies, engineered antibodies containing the Fab region of atezolizumab joined to FcγR-binding or non–FcγR-binding Fc regions were also assessed. The avelumab FcγR binding–deficient variant showed reduced internalization in human monocytes and granulocytes in vitro; however, internalization still occurred at later time points. The avelumab PD-L1 binding–deficient variant also showed reduced internalization, albeit mostly at early time points and to a lesser extent than the FcγR binding–deficient variant. These data suggested that both FcγR-mediated and PD-L1–mediated mechanisms contribute to avelumab internalization, with FcγR binding playing the major role in vitro. Compared with monocytes, FcγR-mediated internalization in granulocytes was severely impacted at all the time points, indicating that granulocytes internalize avelumab efficiently. The involvement of both monocytes and granulocytes might increase avelumab clearance compared with other ICIs. Such clearance mechanisms might include FcγR-mediated endocytosis or opsonization followed by phagocytosis.8 The FcγR-binding (WT) variant of avelumab was internalized more than the FcγR-binding variant of atezolizumab, indicating that FcγR binding alone is not solely responsible for differences in PK characteristics between avelumab and atezolizumab, and suggesting that differences in PD-L1 binding affinity, structural conformation, and/or pI might influence either internalization or fate after internalization. The combination of FcγR and PD-L1 binding might increase internalization of avelumab, providing a primary elimination pathway. Subsequent studies showed that a low pI variant of avelumab had lower levels of internalization in vitro than WT avelumab and PD-L1 binding–deficient and FcγR binding–deficient variants. Thus, the distinct PD-L1 binding characteristics of avelumab, including its unique binding kinetics and conformation, might increase specific or nonspecific internalization, leading to faster clearance.30,34
While FcγR largely determined internalization in vitro, serum concentration profiles in mice and cynomolgus monkeys showed an observable decrease in avelumab elimination in the terminal phase associated with loss of either FcγR or PD-L1 binding capability. Loss of PD-L1 binding had the greatest effect, suggesting that binding to antigens in trans strongly influences FcγR-mediated antibody internalization. Consistent with these observations, the FcγR and PD-L1 binding–deficient variants had longer half-lives and slower clearance than WT avelumab, with the PD-L1 binding–deficient variant showing the greatest differences. The avelumab low pI variant also showed reduced elimination; given that internalization is strongly influenced by antigen binding, the presence of a surface charge patch that lowers affinity might slow the internalization rate and decrease clearance.
In assays performed in the presence of soluble competitor FcγR proteins, avelumab internalization was strongly inhibited by CD64, a high-affinity receptor for human IgG1.33 These data show that CD64 is an important receptor for FcγR-mediated internalization of avelumab, although it does not rule out a lesser role for other FcγRs. Furthermore, free avelumab did not impact the internalization of labeled avelumab, indicating the occurrence of FcγR-mediated endosome recycling and sorting into lysosomes. This suggests that the machinery and/or pathways mediating internalization and recycling are always present and cannot be saturated by the presence of free avelumab or other antibodies in whole blood. Therefore, internalization might be more efficient when antibodies bind simultaneously to the antigen and FcγR in trans; as such, the affinity for the antigen determines the efficiency of internalization by FcγR and vice versa.
In patients, avelumab clearance might also be influenced by tissue-specific Fc receptors.35 For example, liver endothelial and Kupffer cells, which express FcγRs, can phagocytose and degrade IgG.35,36 Thus, in addition to circulating immune cells, avelumab could also be removed from circulation by other cell types.
In conclusion, our findings suggest that the faster clearance of avelumab compared with some other anti–PD-L1 antibodies is due to multiple mechanisms, with FcγR binding acting in synergy with the characteristics of PD-L1 binding (eg, pI) to determine the elimination rate. These results provide guidance for altering the clearance and half-life of monoclonal antibodies in future therapeutic antibody development. The avelumab clinical dosing schedule involving administration every 2 weeks was selected based on the short half-life of avelumab compared with other PD-L1 inhibitors. The results described provide an explanation for the need to administer avelumab with this schedule, which has been associated with clinical efficacy in patients with Merkel cell carcinoma, renal cell carcinoma, and urothelial cancer, and why longer dosing windows are not advisable from a PK perspective.
Supplementary Material
Acknowledgments
Medical writing support was provided by Hiba Al-Ashtal of ClinicalThinking and funded by the healthcare business of Merck KGaA and Pfizer. Our work was supported by help from many scientists including Maria Soloviev, John S. Wesolowski, Bijan Zakeri, Vera Sellers, Mike Lavallee, Selena Li, Jessica Dawson, and Nadine Barron.
Funding Statement
This research was sponsored by the healthcare business of Merck KGaA, Darmstadt, Germany (CrossRef Funder ID: 10.13039/100009945), as part of an alliance between the healthcare business of Merck KGaA, Darmstadt, Germany and Pfizer.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Darvin P, Toor SM, Sasidharan Nair V, Elkord E.. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hamilton G, Rath B. Avelumab: combining immune checkpoint inhibition and antibody-dependent cytotoxicity. Expert Opin Biol Ther. 2017;17:515–523. [DOI] [PubMed] [Google Scholar]
- 3.Tang H, Liang Y, Anders RA, Taube JM, Qiu X, Mulgaonkar A, Liu X, Harrington SM, Guo J, Xin Y, et al. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J Clin Invest. 2018;128(2):580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, Szeliga W, Herbst R, Harms PW, Fecher LA, et al. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest. 2018;128(4):1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu B, Sun X, Gupta HB, Yuan B, Li J, Ge F, Chiang H-C, Zhang X, Zhang C, Zhang D, et al. Adipose PD-L1 modulates PD-1/PD-L1 checkpoint blockade immunotherapy efficacy in breast cancer. Oncoimmunology. 2018;7(11):e1500107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marciscano AE, Gulley JL, Kaufman HL. Avelumab: is it time to get excited? Expert Rev Anticancer Ther. 2018;18:815–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84:548–558. [DOI] [PubMed] [Google Scholar]
- 9.Nimmerjahn F, Ravetch JV. Fcγ receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. [DOI] [PubMed] [Google Scholar]
- 10.Chen X, Song X, Li K, Zhang T. FcγR-binding is an important functional attribute for immune checkpoint antibodies in cancer immunotherapy. Front Immunol. 2019;10:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang TH, Jung ST. Boosting therapeutic potency of antibodies by taming Fc domain functions. Exp Mol Med. 2019;51:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A. 2006;103(11):4005–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolt S, Routledge E, Lloyd I, Chatenoud L, Pope H, Gorman SD, Clark M, Waldmann H. The generation of a humanized, non-mitogenic CD3 monoclonal antibody which retains in vitro immunosuppressive properties. Eur J Immunol. 1993;23(2):403–411. [DOI] [PubMed] [Google Scholar]
- 14.Deng R, Bumbaca D, Pastuskovas CV, Boswell CA, West D, Cowan KJ, Chiu H, McBride J, Johnson C, Xin Y, et al. Preclinical pharmacokinetics, pharmacodynamics, tissue distribution, and tumor penetration of anti-PD-L1 monoclonal antibody, an immune checkpoint inhibitor. MAbs. 2016;8(3):593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li M, Zhao R, Chen J, Tian W, Xia C, Liu X, Li Y, Li S, Sun H, Shen T, et al. Next generation of anti-PD-L1 atezolizumab with enhanced anti-tumor efficacy in vivo. Sci Rep. 2021;11(1):5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barnhart BC, Quigley M. Role of Fc-FcγR interactions in the antitumor activity of therapeutic antibodies. Immunol Cell Biol. 2017;95:340–346. [DOI] [PubMed] [Google Scholar]
- 17.Igawa T, Tsunoda H, Tachibana T, Maeda A, Mimoto F, Moriyama C, Nanami M, Sekimori Y, Nabuchi Y, Aso Y, Hattori K, et al. Reduced elimination of IgG antibodies by engineering the variable region. Protein Eng Des Sel. 2010;23(5):385–392. [DOI] [PubMed] [Google Scholar]
- 18.Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D'Angelo SP, Shih KC, Lebbé C, Linette GP, Milella M, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17(10):1374–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel MR, Ellerton J, Infante JR, Agrawal M, Gordon M, Aljumaily R, Britten CD, Dirix L, Lee K-W, Taylor M, et al. Avelumab in metastatic urothelial carcinoma after platinum failure (JAVELIN Solid Tumor): pooled results from two expansion cohorts of an open-label, phase 1 trial. Lancet Oncol. 2018;19(1):51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Powles T, Park SH, Voog E, Caserta C, Valderrama BP, Gurney H, Kalofonos H, Radulović S, Demey W, Ullén A, et al. Avelumab maintenance therapy for advanced or metastatic urothelial carcinoma. N Engl J Med. 2020;383(13):1218–1230. [DOI] [PubMed] [Google Scholar]
- 21.Choueiri TK, Larkin J, Oya M, Thistlethwaite F, Martignoni M, Nathan P, Powles T, McDermott D, Robbins PB, Chism DD, et al. Preliminary results for avelumab plus axitinib as first-line therapy in patients with advanced clear-cell renal-cell carcinoma (JAVELIN Renal 100): an open-label, dose-finding and dose-expansion, phase 1b trial. Lancet Oncol. 2018;19(4):451–460. [DOI] [PubMed] [Google Scholar]
- 22.Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, Venugopal B, Kollmannsberger C, Negrier S, Uemura M, et al. Avelumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380(12):1103–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bavencio (avelumab) prescribing information. EMD Serono ; 2020.
- 24.Boyerinas B, Jochems C, Fantini M, Heery CR, Gulley JL, Tsang KY, Schlom J. Antibody-dependent cellular cytotoxicity activity of a novel anti-pd-l1 antibody avelumab (msb0010718c) on human tumor cells. Cancer Immunol Res. 2015;3(10):1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Julia EP, Amante A, Pampena MB, Mordoh J, Levy EM. Avelumab, an IgG1 anti-PD-L1 immune checkpoint inhibitor, triggers NK cell-mediated cytotoxicity and cytokine production against triple negative breast cancer cells. Front Immunol. 2018;9:2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khanna S, Thomas A, Abate-Daga D, Zhang J, Morrow B, Steinberg SM, Orlandi A, Ferroni P, Schlom J, Guadagni F, Hassan R. Malignant mesothelioma effusions are infiltrated by CD3(+) T cells highly expressing PD-L1 and the PD-L1(+) tumor cells within these effusions are susceptible to ADCC by the anti-PD-L1 antibody avelumab. J Thorac Oncol. 2016;11(11):1993–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujii R, Friedman ER, Richards J, Tsang KY, Heery CR, Schlom J, Hodge JW. Enhanced killing of chordoma cells by antibody-dependent cell-mediated cytotoxicity employing the novel anti-PD-L1 antibody avelumab. Oncotarget. 2016;7(23):33498–33511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Donahue RN, Lepone LM, Grenga I, Jochems C, Fantini M, Madan RA, Heery CR, Gulley JL, Schlom J, et al. Analyses of the peripheral immunome following multiple administrations of avelumab, a human IgG1 anti-PD-L1 monoclonal antibody. J Immunother Cancer. 2017;5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV. FcγRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 axis. Cancer Cell. 2015;28:543. [DOI] [PubMed] [Google Scholar]
- 30.Heery CR, O’Sullivan-Coyne G, Madan RA, Cordes L, Rajan A, Rauckhorst M, ELamping E, Oyelakin I, Marté JL, Lepone LM, et al. Avelumab for metastatic or locally advanced previously treated solid tumours (JAVELIN Solid Tumor): a phase 1a, multicohort, dose-escalation trial. Lancet Oncol. 2017;18(5):587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Topper MB, Tonra JR, Pytowski B, Eastman SW. Differentiation between the EGFR antibodies necitumumab, cetuximab, and panitumumab: antibody internalization and EGFR degradation. J Clin Oncol. 2011;29:e13022. [Google Scholar]
- 32.Langsdorf C, Mandavilli B, Hu Y-Z, Chen A, Marcy W. Evaluating antibody-mediated cellular cytotoxicity and potency of antibody-drug conjugates within three- dimensional tumor models. J Immunother Cancer. 2018;6:3.29307306 [Google Scholar]
- 33.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daëron M. Specificity and affinity of human Fcγ receptors and their polymorphic variants for human IgG subclasses. Blood. 2009;113(16):3716–3725. [DOI] [PubMed] [Google Scholar]
- 34.Tan S, Liu K, Chai Y, Zhang CW-H, Gao S, Gao GF, Qi J. Distinct PD-L1 binding characteristics of therapeutic monoclonal antibody durvalumab. Protein Cell. 2018;9(1):135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johansson AG, Lovdal T, Magnusson KE, Berg T, Skogh T. Liver cell uptake and degradation of soluble immunoglobulin G immune complexes in vivo and in vitro in rats. Hepatology. 1996;24:169–175. [DOI] [PubMed] [Google Scholar]
- 36.Bruggeman CW, Houtzager J, Dierdorp B, Kers J, Pals ST, Lutter R, van Gulik T, den Haan JMM, van den Berg TK, van Bruggen R, Kuijpers TW. Tissue-specific expression of IgG receptors by human macrophages ex vivo. PLoS One. 2019;14(10):e0223264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.