

ABSTRACT
Clinical trials involving anti-programmed cell death protein-1 (anti-PD-1) failed to demonstrate improved overall survival in glioblastoma (GBM) patients. This may be due to the expression of alternative checkpoints such as B- and T- lymphocyte attenuator (BTLA) on several immune cell types including regulatory T cells. Murine GBM models indicate that there is significant upregulation of BTLA in the tumor microenvironment (TME) with associated T cell exhaustion. We investigate the use of antibodies against BTLA and PD-1 on reversing immunosuppression and increasing long-term survival in a murine GBM model. C57BL/6 J mice were implanted with the murine glioma cell line GL261 and randomized into 4 arms: (i) control, (ii) anti-PD-1, (iii) anti-BTLA, and (iv) anti-PD-1 + anti-BTLA. Kaplan–Meier curves were generated for all arms. Flow cytometric analysis of blood and brains were done on days 11 and 16 post-tumor implantation. Tumor-bearing mice treated with a combination of anti-PD-1 and anti-BTLA therapy experienced improved overall long-term survival (60%) compared to anti-PD-1 (20%) or anti-BTLA (0%) alone (P = .003). Compared to monotherapy with anti-PD-1, mice treated with combination therapy also demonstrated increased expression of CD4+ IFN-γ (P < .0001) and CD8+ IFN-γ (P = .0365), as well as decreased levels of CD4+ FoxP3+ regulatory T cells on day 16 in the brain (P = .0136). This is the first preclinical investigation into the effects of combination checkpoint blockade with anti-PD-1 and anti-BTLA treatment in GBM. We also show a direct effect on activated immune cell populations such as CD4+ and CD8 + T cells and immunosuppressive regulatory T cells through this combination therapy.
KEYWORDS: glioblastoma immunotherapy, anti-PD-1, anti-BTLA, B and T lymphocyte attenuator, immune checkpoint inhibitor therapy, glioblastoma
Introduction
Glioblastoma (GBM) is a highly aggressive primary brain tumor with a dismal prognosis.1 Despite current standards of care involving maximal surgical resection, chemotherapy, and radiation therapy, median overall survival (mOS) is approximately 19 months.2,3 Recently, the advent of immune checkpoint blockade (ICB) with antibodies against programmed cell death protein-1 (anti-PD-1) served as a novel immunotherapy treatment strategy that has shown promise in many solid tumors including melanoma and non-small cell lung cancer (NSCLC).4,5
Given the success of ICB in other cancers, there has been avid interest in applying anti-PD-1 therapy to GBM. However, the recent phase III CheckMate 143 clinical trial evaluating anti-PD-1 in patients with GBM failed to improve mOS compared to current standard of care.6,7 The lack of significant clinical benefit from anti-PD-1 therapy is thought to be in part due to both intrinsic resistance to the immune system due to poor antigen presentation/priming, low-quality neoantigens, and relatively low mutational burden, as well as extrinsic resistance from an immunosuppressive tumor microenvironment (TME).8,9 Moreover, regulatory T cells (Tregs) – an immunosuppressive T cell population usually not seen in the brain – have significantly increased representation within high-grade gliomas such as GBM.10
With anti-PD-1 monotherapy failing to show significant clinical benefit, several studies have examined the efficacy of combining multiple checkpoint inhibitors in order to target different pathways of immunosuppression in GBM.11,12 B and T lymphocyte attenuator (BTLA) is a co-inhibitory checkpoint molecule that is structurally related to PD-1.13 Previous studies have shown that BTLA is highly expressed on tumor-specific T cells in cancer patients14 and is upregulated in several cancers including gastric cancer,15 hepatocellular carcinoma,16 and GBM.17 Furthermore, it has been shown that increased BTLA expression correlates with resistance to anti-PD-1 immunotherapy and poor clinical outcomes.15,16
In the context of GBM, the ligand for BTLA – Herpes virus entry mediator (HVEM) – is overexpressed on high-grade gliomas and is associated with the suppression of T cell-mediated anti-tumor activity in the GBM TME.18 Given the likely immunosuppressive effect of BTLA in the GBM TME, we sought to evaluate the survival efficacy of anti-BTLA therapy in combination with anti-PD-1 in a murine GBM model and assess the immunological effects of this dual immune checkpoint blockade therapy.
Materials and methods
Murine glioma model and cell lines
6–8 week-old C57BL/6 J wild-type female mice were maintained at the Johns Hopkins University Animal Facility per the Institutional Animal Care and Use Committee (IACUC) protocol. For all experiments, mice were anesthetized with Ketathesia (100 mg/kg)/xylazine (10 mg/kg) by intra-peritoneal (i.p.) injection and had topical eye gel for lubrication. After every procedure, mice were placed on a heating pad and observed until fully recovered. Orthotopic murine glioma models utilized GL261-Luc2 (RRID:CVCL_X986) cells grown in DMEM (Life Technologies) + 10% FBS (Sigma-Aldrich) + 1% penicillin-streptomycin (Life Technologies) as described in previous studies (Zeng, 2013). GL261-Luc2 cells were obtained from Reardon Lab, Dana Farber Cancer Institute. All experiments were performed with mycoplasma-free cells. 1.3e5 GL261-Luc2 cells in a volume of 2 μl were stereotactically injected 2 mm posterior to the coronal suture, 2 mm lateral to the sagittal suture, and 3 mm deep to the cortical surface in the area of the left striatum. After implantation of tumor cells, mice were assessed for tumor growth on post-implantation day 7 using bioluminescent IVIS imaging (PerkinElmer).
Mice with tumor burden at day 7 were then randomly separated into control (non-treated) and treatment arms. Survival experiments were repeated in triplicate with 8–10 mice in each control or treatment arm. Animals were euthanized according to humane endpoints, including central nervous system disturbances, hunched posture, lethargy, weight loss, and inability to ambulate per our IACUC protocol. Long-term survivors were rechallenged with re-implantation of GL261-Luc2 in the right striatum on post-implantation day 60. Additionally, 8 naïve control mice were implanted with tumor the day of rechallenge to serve as controls.
Therapeutic antibodies
G4 hybridomas were cultured and used to develop hamster monoclonal antibodies (mAbs) against murine PD-1, as described in previous studies (Hirano F, 2005). Individual treatment dose was 200 μg per animal on post-implantation days 10, 12, and 14 for anti-PD-1 monotherapy and combination therapy arms. Anti-murine BTLA antibody was received from Bristol Myers Squibb (BMS) and stored at −80°C in 1 mg/mL aliquots. Per the manufacturer, anti-BTLA is a non-depleting, blocking antibody with a defective Fc region preventing antibody-dependent cellular cytotoxicity. This antibody was administered in three 400 μg doses on post-implantation days 7, 10, and 14.
Immune cell harvest and isolation
Mice were deeply anesthetized or euthanized before harvesting blood (120 μl, retro-orbital) and brains for immunological assays. 120 μl blood was collected retro-orbitally from mice under deep anesthesia on days 11 and 16 according to our IACUC protocol. Red blood cells were lysed using ACK lysis buffer (ThermoFisher) and resuspended in PBS for further cytometric analysis. Brains were removed, tissue was mechanically dissociated through a 70 μm filter, and homogenates were centrifuged (ThermoFisher Sorvall Legend X1R centrifuge) in a 30%/70% Percoll® (Sigma-Aldrich) gradient at 2200 rpm for 20 minutes without brakes to separate out brain myeloid cells and lymphocytes from tumor cells and myelin. Brain immune cells were extracted at the 30%/70% interface and resuspended in phosphate-buffered saline (PBS) buffer (Sigma-Aldrich) for further cytometric analysis.
Flow cytometric analysis of murine immune cells
Mouse immune cells were stained for Live/Dead (L/D) (Invitrogen), CD45, CD3, CD4, CD8, PD-1, IFN-γ, FoxP3, and BTLA (Supplementary Figure S1 for antibody fluorophores). To stain for the intracellular marker IFN-γ and intranuclear marker FoxP3, samples were fixed in 1:3 fixation/permeabilization concentrate:diluent mixture (eBioscence) for 30 minutes and subsequently stained in permeabilization buffer (eBioscience). Fluorescence minus one (FMO) was used to control for data spread due to multiple fluorochromes and nonbinary expression of markers such as IFN-γ. Flow data was acquired using a FACSCelesta flow cytometer (BD) and analyzed using FlowJo (BD). Nonviable cells and doublets were excluded by forward versus side scatter gating, forward scatter height versus forward scatter area gating, and L/D staining.
Co-culture and ELISA
For IFN-γ assays, 5e3 CD45.2+ CD3+ CD8 + T cells were sorted from each mouse brain and co-cultured with 100 μg/ml GL261-Luc2 tumor cell lysate and 25e3 dendritic cells isolated from CD45.1 mouse spleen (isolated with a pan-dendritic cell isolation kit) (Miltenyi Biotec) in a 96-well round bottomed plate with T cell media (RPMI 1640 + 10% FBS + 1% NEAA + 1% 2-Mercaptoethanol + 1% Penicillin/Streptomycin). Co-cultured cells were incubated at 37°C for 48 hours. Supernatant was collected for subsequent ELISA for IFN-γ and run on a plate reader (PerkinElmer Victor3 1420 Multilabel plate reader).
Depletion study
GL261-luc2 tumor was confirmed in mice on post-implantation day 7. After randomization, tumor bearing mice received IP injections of either anti-CD4 (clone GK1.5, Bio X Cell, catalog#: BE0003-1) at 200 μg/dose or anti-CD8 (clone 2.43, Bio X Cell, catalog#: BE0004-1) at 500 μg/dose. Control mice received 200 μL of PBS IP. All depletion antibodies were administered 48 hours and 24 hours prior to the administration of the first dose of anti-BTLA treatment. Depletion antibodies were administered every seven days thereafter until conclusion of the study. T cell depletion was confirmed via flow cytometry of blood in each treatment arm on day 14 post-implantation.
Statistical analysis
All replicates were biological replicates. Survival was analyzed via Kaplan-Meier method and compared by log-rank (Mantel-Cox) test. Calculated variables were treated as continuous variables under the assumption that data follow Student T-distribution. Mouse experimental data were analyzed using a 2-tailed Student’s T-test for experiments containing 2 groups. All data were analyzed using GraphPad Prism 8 and values of P < .05 were considered statistically significant.
Results
BTLA expression increases over time on tumor-infiltrating non-Treg CD4 + T cells
To evaluate the expression of BTLA over time on immune cells, a time point experiment was conducted in mice on post-implantation day 7, 16, and 21 (Figure 1). 1.3e5 GL261-Luc+ cells were implanted in the left striatum on day 0. Blood and brain were harvested at each of the three time points for 5 mice after confirming presence of tumor on IVIS. Single cell suspensions were prepared and stained for flow cytometric analysis of BTLA. In the blood, BTLA expression significantly increased on non-Treg CD4 + T cells from 6.1% on day 7 to 55.1% on day 16 (P < .0001) and 67.8% on day 21 (P < .0001) (Figure 1b). In the brain, BTLA expression on tumor-infiltrating Treg (CD3+ CD4+ FoxP3+) cells did not significantly change from an average of 13.4% on day 7 to 16.2% on day 16 (P = .003) and 20.0% on day 21 (P = .003) (Figure 1c). However, BTLA expression on tumor-infiltrating non-Treg (CD3+ CD4+ FoxP3-) T cells significantly increased from a mean of 10.1% on day 7 to 37.7% on day 16 (P < . 0001) and 37.8% on day 21 (P < .0001) (Figure 1c). On CD3+ CD8 + T cells in the blood, BTLA expression increased from 8.2% on day 7 to 35.3% on day 21 (P < .001) (Figure 1b). In the brain, BTLA expression on CD3+ CD8 + T cells did not significantly change from 12.9% on day 7 to 17.1% on day 21 (P = .49) (Figure 1c).
Figure 1.

Timing of BTLA expression in tumor-bearing mice on days 7, 16, and 21. (a) Representative flow cytometry plots of TILs demonstrating BTLA expression on day 7, 16, and 21 on CD4+ FoxP3 + T cells, CD4+ FoxP3- T cells, and CD8 + T cells (b)Aggregate (n = 5) flow cytometry data of BTLA expression in PBMCs from untreated tumor-bearing mice on days 7, 16, and 21. (c) Aggregate (n = 5) flow cytometry data of BTLA expression in TILs of untreated tumor-bearing mice on days 7, 16, and 21. All plots represent mean with standard deviation. Comparison between two groups is made via the Student’s t test. (* p < .05)
Next, expression of BTLA and its ligand HVEM was studied in the glioma setting on various tumor-infiltrating immune cell populations including CD45+ CD3 + T cells, CD45+ CD11b+ cells (commonly denoting macrophages), CD45+ CD11 c+ cells (commonly denoting macrophages and dendritic cells) and CD45+ CD19 + B cells (Supplemental Figure S1). There was significantly lower HVEM expression on CD3+ TILs compared to tumor-infiltrating CD11b+ cells (P < .0001), CD11 c+ cells (P < .0001), or CD19 + B cells (P < .0001) (Supplemental Figure S1b). There was significantly lower BTLA expression on CD11 c+ cells compared to CD3 + T cells (P = .0405) and tumor-infiltrating CD11b+ cells (P = .0062) (Supplemental Figure S1b). HVEM expression was also analyzed after administration of control (no treatment), anti-PD-1 alone, anti-BTLA alone or combination therapy. There was no significant difference in HVEM expression on tumor-infiltrating CD11b+ cells, CD11 c+ cells, or CD19 + B cells with the four treatment arms. There was significantly higher HVEM expression on CD3 + T cells with anti-BTLA (P = .0135) or combination therapy (P = .0285) compared to control (Supplemental Figure S1c).
Combination anti-BTLA and anti-PD-1 treatment increases activation of CD4+ and CD8 + T cells and modulates presence of Tregs in the brain and blood
In order to assess the immunologic effects of anti-PD-1 and anti-BTLA in murine GBM models, blood was harvested on days 11 and 16 with brains also harvested on day 16. T cells were gated on forward versus side scatter for lymphocyte populations with exclusion of doublets, which were then gated for live CD45+ CD3 + T cells and separated into CD4+ and CD8+ populations (Supplemental Figure S2).
On day 16 in the brain, IFN-γ expression of CD4+ and CD8+ populations were compared between experimental arms, with 32.9% of CD4+ tumor-infiltrating lymphocytes (TILs) expressing IFN-γ in the combination anti-PD-1 and anti-BTLA arm compared to 11.7% in the non-treated (control) arm, 16.7% in the anti-PD-1 monotherapy arm (P < .0001), and 19.1% in the anti-BTLA arm (P = .0033) (Figure 2a, b). Similarly, 76.3% of CD8+ TILs expressed IFN-γ in the combination anti-PD-1 and anti-BTLA arm compared to 43.8%, 59.9%, and 44.8% in control, anti-PD-1 monotherapy (P = .0365), and anti-BTLA (P = .0004) monotherapy arms, respectively (Figure 2b).
Figure 2.
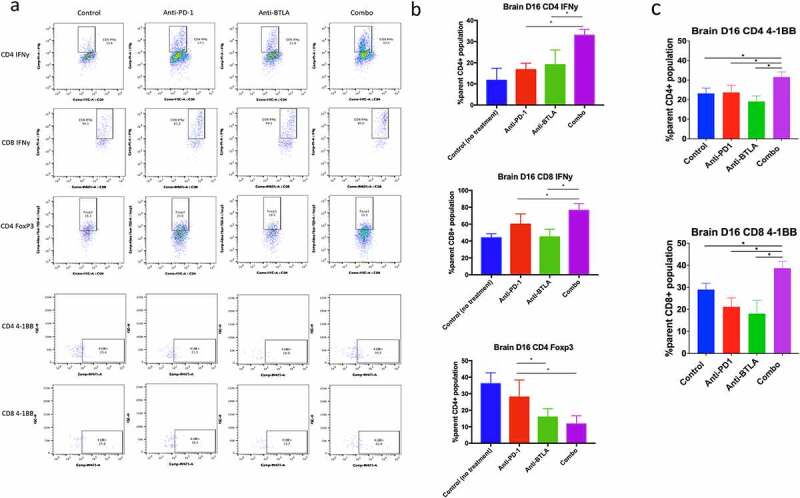
Flow cytometric analysis of T cell populations in mouse brain at day 16 post-tumor implantation. (a) Flow plots demonstrating CD4 and CD8 IFN-γ expression, 4–1BB expression, and CD4 Foxp3 presence in the brain. (b) Aggregate (n = 5 per group) data of control (no treatment), anti-PD-1, anti-BTLA, and combination therapy groups for CD4 and CD8 IFN-γ and CD4 Foxp3 presence in the brain. Differences between two treatment arms were analyzed via Student’s t test. (**** P < .0001, *** P < .001, ** P < .01, * P < .05)
Of note, in the combination therapy arm, there was a marked decrease in the proportion of CD4+ FoxP3 + T regs of the parent CD4+ TIL population in the brain compared to monotherapy and control arms, with 11.7% of CD4+ TILs expressing FoxP3 in mice treated with both anti-PD-1 and anti-BTLA compared to 36.0% of CD4 + T cells in non-treated mice expressing FoxP3 and 27.9% of CD4+ TILs in mice treated with anti-PD-1 monotherapy expressing FoxP3 (P = .0136) (Figure 2b). However, there was no statistically significant difference between combination and anti-BTLA monotherapy arms for FoxP3 expression in the brain, with 15.8% of CD4+ TILs in mice with anti-BTLA monotherapy expressing FoxP3 (P = .2349). However, there was a statistically significant difference in FoxP3 expression between anti-PD-1 and anti-BTLA monotherapy arms (P = .0483) (Figure 2a, b).
Next, we evaluated for the expression of additional co-stimulatory molecules such as 4–1BB and co-inhibitory molecules such as TIM-3 and LAG-3 on TILs. 4–1BB expression on CD4+ TILs was significantly higher with combination therapy (31.6%) compared to control (23. 2%, P = .0098), anti-PD-1 monotherapy (23.7%, P = .0135), anti-BTLA monotherapy (19.1%, P = .0007) (Figure 2c). Likewise, 4–1BB expression on CD8+ TILs was significantly higher with combination therapy (38.7%) compared to control (29.0%, P = .016), anti-PD-1 monotherapy (21.2%, P = .0016), anti-BTLA monotherapy (18.1%, P = .0063) (Figure 2c). There was no significant difference in LAG-3 or TIM-3 expression between control, monotherapy, or combination therapy.
We also studied if anti-PD-1 and anti-BTLA combination therapy was affecting tumor-infiltrating myeloid or B cell populations. After treating mice with control, anti-PD-1 only, anti-BTLA only, or combination therapy, we found no significant change in tumor infiltration of CD45+ CD11b+ macrophages between the four treatment arms. Of note, there was a trend toward significantly greater CD45+ CD11c+ macrophage and dendritic cell infiltration of the tumor in the combination therapy arm compared to control (P = .09). Likewise, there was a trend toward significantly greater CD45+ CD19 + B cell tumor infiltration with combination therapy compared to control (P = .06) (Supplemental Figure S3).
To assess the characteristics of possible homing and peripheral proliferation of lymphocytes, blood was analyzed on day 11, one day after exposure to both anti-PD-1 and anti-BTLA, as well as on day 16 – the day of brain harvest. Indications of T cell homing were present across all arms for CD8 + T cells, with marked decrease in percentage of CD8+ IFN-γ-secreting T cells on day 16 compared to day 11. Furthermore, combination treatment with anti-PD-1 and anti-BTLA showed the greatest levels of CD4+ and CD8+ IFN-γ expression on day 11 with 21.5% of CD4+ cells and 37.9% of CD8+ cells expressing IFN-γ (Supplemental Figure S4). Notably, tumor bearing mice treated with combination therapy did not observe a similar trend in peripheral regulatory T cell distribution on days 11 and 16; while other monotherapy arms showed a marked decrease in Tregs between days 11 and 16, mice treated with combination therapy showed similar proportions of Tregs in the blood on both days (1.52% and 1.56% on days 11 and 16, respectively) (P = .8271) (Figure S3).
Treatment with anti-BTLA and anti-PD-1 correlate with increased IFN-γ secretion
To further determine if combination therapy with anti-PD-1 and anti-BTLA resulted in decreased immunosuppression, TILs belonging to mice ± treatment with anti-PD-1 monotherapy, anti-BTLA monotherapy, or combination therapy were sorted for live CD45+ CD3+ CD8 + T cells and co-incubated with dendritic cells and GL261-Luc2 tumor cell lysate (Figure 3a). Resultant supernatant with secreted IFN-γ was then analyzed by ELISA. IFN-γ levels directly correlated with treatment, with significantly increased secretion of IFN-γ in combination treatment compared to no treatment (P = .0033) and anti-BTLA monotherapy (P = .0032). There is a trend toward increased secretion of IFN-γ in combination treatment compared to anti-PD-1 monotherapy (P = .0776) (Figure 3b).
Figure 3.

In vitro co-culture assay for IFN-γ secretion with ELISA. (a) CD8 + T cells were isolated via FACS microfluidic sorting from untreated mice and mice treated in vivo with anti-PD-1 monotherapy, anti-BTLA monotherapy, and combination anti-PD-1 and anti-BTLA. Cells were co-cultured with GL261 tumor cell lysate and CD11c+ dendritic cells. (b) Plot of concentration of IFN-γ in supernatant of co-culture wells. All plots represent mean with standard error. Differences between two treatment arms were analyzed via Student’s t test. (**** P < .0001, *** P < .001, ** P < .01, * P < .05)
Combination treatment with anti-PD-1 and anti-BTLA results in improved overall long-term survival
Having confirmed BTLA expression on Tregs in GBM, we hypothesized that dual blockade with anti-PD-1 and anti-BTLA antibodies would result in a synergistic survival benefit due to targeted reversal of immunosuppression of CD8+ and CD4 + T cells. After ensuring tumor burden with GL261-Luc2 on post-implantation day 7, mice were treated with three doses of BTLA on days 7, 10, and 14 as well as anti-PD-1 on days 10, 12, and 14 per previous protocols19 (Figure 4a, b). Compared to anti-PD-1 monotherapy, combination treatment with dual checkpoint blockade resulted in increased overall survival (60% vs. 20%, P = .0003). There were no long-term survivors with anti-BTLA monotherapy as well as no statistically significant difference between control and anti-BTLA monotherapy for median overall survival (P = .0954). On post-implantation day 60, long-term survivors and naïve control mice were contralaterally rechallenged/implanted with intracranial GL261-Luc2. Long-term survivors rechallenged with glioma demonstrated significantly better survival than control mice (P = .0031) (Supplemental Figure S5).
Figure 4.

Survival schema and Kaplan-Meier curve. (a) Mice were implanted with 1.3e5 GL261-Luc2 cells with a stereotactic frame. On post-implantation day 7, mice were assessed for presence of tumor by injecting them with 200 μl of 1 mg/ml D-luciferin and imaged with IVIS (In Vivo Imaging Systems), and subsequently randomly assorted into four groups: control (no treatment), anti-BTLA monotherapy, anti-PD-1 monotherapy, and combination anti-BTLA and anti-PD-1 therapy. 5 mice in each arm were saved for blood and brain harvest to undergo flow cytometric analysis. Remaining 8 mice in each arm underwent a survival study and were rechallenged on day 60. (b) Treatment with anti-BTLA was on days 7, 10, and 14. Treatment with anti-PD-1 was on days 10, 12, and 14. (c) Kaplan-Meier curve demonstrating differences in overall long-term survival with combination therapy. (**** P < .0001, *** P < .001, ** P < .01, * P < .05)
Depletion of CD4 + T cells or CD8 + T cells diminishes the efficacy of anti-PD-1 and anti-BTLA combination therapy
The results thus far suggest that anti-PD-1 and anti-BTLA combination therapy provide a synergistic survival benefit in murine glioma (Figure 4) potentially mediated through CD4+ and CD8 + T cells (Figures 2, Figures 3). To determine whether CD4+ and CD8 + T cells are necessary for the observed survival benefit with combination therapy, we performed a depletion study (Figure 5). After confirming tumor burden on post-implantation day 7, mice were depleted of either CD4 + T cells or CD8 + T cells. After depletion of these T cell populations, mice were treated with anti-PD-1 plus anti-BTLA combination therapy. Depletion of CD4+ or CD8 + T cells was confirmed on Day 14 (Supplemental Figure S6). There was no significant difference in survival between the control arm, CD4 + T cell depletion plus combination therapy arm, and the CD8 + T cell depletion plus combination therapy arm. Combination therapy without any T cell depletion had significantly greater median overall survival compared to control (P = .004), CD4 + T cell depletion plus combination therapy (P = .036), and CD8 + T cell depletion plus combination therapy (P = .046).
Figure 5.
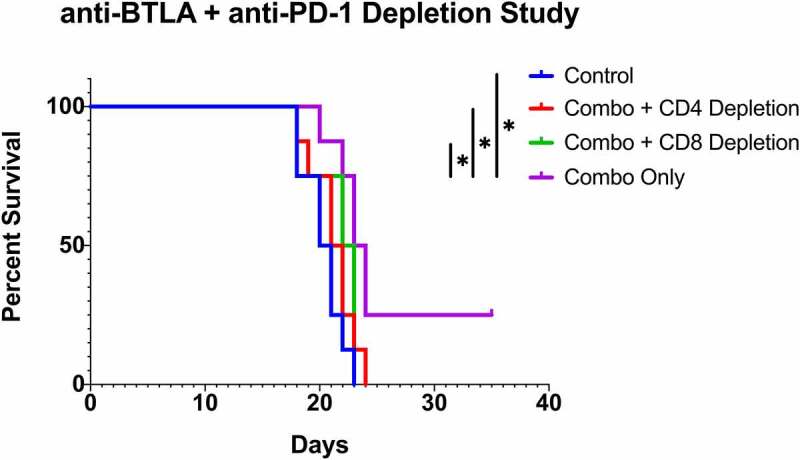
CD4+ and CD8 + T cell depletion Kaplan-Meier curve. Mice in the depletion arms underwent depletion of either CD4 + T cells or CD8 + T cells prior to treatment with anti-BTLA and anti-PD-1 therapy. There was no significant difference in survival between the control arm, CD4 + T cell depletion arm, and CD8 + T cell depletion arm. Combination therapy without any T cell depletion had significantly greater median overall survival compared to control (P = .004), CD4 + T cell depletion plus combination therapy (P = .036), and CD8 + T cell depletion plus combination therapy (P = .046)
Discussion
Despite advances in immune checkpoint blockade for solid tumors, GBM has demonstrated resistance to anti-PD-1 therapy with multiple mechanisms of immunosuppression.9 Of note, beyond PD-1, TILs in GBM demonstrate upregulation of additional inhibitory immune checkpoint molecules such as Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), Indoleamine 2,3-dioxygenase 1 (IDO1), T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), and Lymphocyte-activation gene-3 (LAG-3).17 As such, the utilization of combination checkpoint therapy involving additional checkpoint blockade such as antibodies against CTLA-4, LAG-3, TIM-3, and IDO1 have been explored in both pre-clinical and clinical investigations.
In the clinical setting, the precedent for success with combination checkpoint therapy for brain tumors is most salient in treatment for metastatic melanoma, in which anti-CTLA-4 and anti-PD-1 combination therapy was approved by the FDA in 2016 as a first-line therapy and showed promising results of improved progression-free survival (PFS) compared with anti-PD-1 therapy alone.20 There is currently an ongoing trial that is studying antibodies against PD-1 and LAG-3 (NCT02658981), with several other combination studies bolstered by promising preclinical findings.11,12,21In preclinical GBM murine models, combination therapy involving antibodies against TIM-3 (anti-TIM-3), anti-PD-1, and stereotactic radiation resulted in 100% overall survival.11 As the success of this mechanism is thought to be in part from targeting both myeloid and lymphocyte populations with anti-TIM-3 and anti-PD-1, respectively, these preclinical studies show proof of principle of the synergistic survival benefit that can result from targeting multiple pathways of immunosuppression.
In this study, we found a synergistic survival benefit accompanying combination therapy with anti-PD-1 and anti-BTLA in a mouse glioma model, with increased 4–1BB expression and IFN-γ secretion from CD4+ and CD8 + T cells. There was also a decreased proportion of infiltrating Tregs of the CD4+ TILs (Figures 2, Figures 4c). In tumor-bearing mice, combination therapy demonstrated T cells with significantly increased IFN-γ expression in both CD4+ and CD8 + T cell populations that correlated with survival outcomes. This reversal of immunosuppression when compared to control and monotherapy arms in conjunction with a decreased proportion of Treg infiltration demonstrates that BTLA therapy may possibly involve a CD4-based mechanism, likely involving decreased polarization of CD4+ TILs toward the Treg phenotype in the TME, leading to a greater proportion of non-Treg effector CD4+ TILs (Figure 2a, b, 3b). Furthermore, our depletion study (Figure 4) suggests that both CD4 + T cells and CD8 + T cells are necessary for the synergistic survival benefit observed with anti-PD-1 and anti-BTLA combination therapy (Figure 5).
CD4+ FoxP3+ Treg cells in the TME of GBM are thought to play an important role in immunosuppression. Both preclinical and clinical studies have shown increased infiltration of Tregs within GBM,22–24 with data showing that the presence of these increased Treg cell fractions in patients correlates with decreased effector T cell proliferation and responsiveness.25 Several studies have since explored the potential survival impact of Treg based therapy in GBM,26 with anti-CD25 or anti-CTLA demonstrating preclinical efficacy by targeting Tregs.22,27,28 However, these therapies have not yet successfully been translated into the clinical setting.29
Interestingly, our BTLA timepoint experiment (Figure 1) demonstrated differential expression of BTLA on tumor-infiltrating CD4+ FoxP3+ Treg cells and CD4+ FoxP3- non-Treg cells over time. BTLA expression remained constant over time on tumor-infiltrating Treg cells, however BTLA expression significantly increased over time on tumor-infiltrating non-Treg cells. Indeed, there is a similar percentage of BTLA expression on both tumor-infiltrating Treg cells and tumor-infiltrating non-Treg cells on day 7 (13.4% vs. 10.1% respectively). However, there is significantly higher BTLA expression on tumor-infiltrating non-Treg cells compared to tumor-infiltrating Treg cells by day 21 (37.8% vs 20.0%). This data parallels data in human GBM patients showing PD-1 expression on TILs is increased in patients with GBM compared to control.30
Given that BTLA is an immune checkpoint, we posit that tumor-infiltrating non-Treg cells become more suppressed over time due to increased BTLA expression over the course of tumor progression. We believe there may not be BTLA-mediated suppression of tumor-infiltrating Treg cells given constant low expression of the BTLA immune checkpoint on this cell population over tumor progression from day 7 to day 21. Our data also showed that there was not a statistically significant difference in the infiltration of Tregs between anti-BTLA and combination therapy arms. This has two possible implications: (1) the immune phenomenon affecting Treg infiltrating with combination therapy is likely due to the inhibition of BTLA rather than PD-1 and (2) despite a similar decrease in Treg infiltration, CD8+ IFN-γ expression as well as survival outcomes with anti-BTLA monotherapy failed to reach the same level as anti-PD-1 monotherapy or combination therapy (Figures 2b, Figures 4). Interestingly, while the immunosuppressive role of Tregs have been well explored in both preclinical models and in human GBM tissue, the impact of Tregs on overall survival and prognosis are less clear. An immunohistochemical analysis of glioma tissue by Heimberger et al. with multivariate analysis for the presence of Tregs on overall survival found that when accounting for confounding factors such as age and Karnofsky performance score, the presence of FoxP3+ Tregs did not have a significant prognostic impact.10 This may in part offer insights as to why anti-BTLA monotherapy may not confer as great a survival benefit, but in turn is an excellent candidate for combination immunotherapy as targeting Tregs has been shown to impact immunosuppression in GBM.10
Additional lines of exploration that would be essential in characterizing this avenue of treatment would be to delve into further mechanisms involving BTLA, including understanding of involved essential trafficking molecules since our data suggests anti-BTLA decreases Treg infiltration into the brain. For example, a notable target that has already been shown to be highly associated with Treg trafficking in GBM is C-C chemokine receptor type 4 (CCR4), which is highly expressed on Tregs with its cognate antigen being produced by GBM.31 Furthermore, our supplemental data suggests BTLA and HVEM are also expressed on myeloid and B cells. In our study, we did not find a significant change in tumor-infiltrating CD11b+ cells, CD11c+ cells, or CD19 + B cells with combination therapy compared to control. Despite this result, It may be interesting to characterize in future studies whether anti-BTLA therapy affects the interaction between myeloid or B cells and TILs.
With any animal model research, consideration must be given to the degree to which the animal model accurately mimics human disease. GBM is notorious for being difficult to replicate in vivo. Indeed, many therapeutic combinations that work well in murine glioma models fail to translate to clinical trials in human GBM.6,19,32 In this project, all experiments were conducted using the GL261-Luc2 cell line. The GL261 cell line is one of the most well-studied glioma models in the field of GBM immunotherapy.33 The primary benefit of this cell line is that it is well characterized, can be easily implanted into immunocompetent C57BL/6 mice given that it is syngeneic, and effectively recapitulates many of the characteristics of human GBM.34,35 The Luc2 tag allows for tumor visualization on IVIS, and indeed many recent studies evaluating checkpoint inhibition therapy in GBM have used the GL261-Luc2 model.11,19,32,36 The primary disadvantage of this model is that it does not fully recapitulate the significant intra-tumoral and inter-tumoral heterogeneity of GBM. Furthermore, the GL261-Luc2 model is a moderately immunogenic model whereas GBM is significantly immunoresistant.35 It is important to bear in mind the strengths and limitations of the preclinical model utilized to study GBM in the preclinical setting, and additional experiments should be conducted in other GBM cell lines to improve generalizability of these results.
The appeal of BTLA as a target for immunotherapy in GBM comes from its multimodal effect on regulatory T cells and CD8 + T cells. The dramatic changes in Treg representation as well as the synergistic survival benefit seen in murine in vivo models demonstrate a treatment schema that can target multiple immunosuppressive cells when used in combination with antibodies involved in the well-established PD-1 pathway. With immune checkpoint therapy continuing to gain traction in the clinical setting, new targets that synergize survival outcomes are of interest. Our preclinical study involving antibodies against PD-1 and BTLA offer proof of principle for the efficacy of this combination therapy and offers the first exploration of targeting BTLA in GBM in a mouse model.
Supplementary Material
Funding Statement
This research was funded from a gift fund by private donors Conflict of Interest: Michael Lim (Funding from Arbor Pharmaceuticals, Accuray, BMS, Novartis; Consultant: BMS, Merck, SQZ Biotechnologies, Tocagen, VBI; Patents: Combining Focused Radiation and Immunotherapy, Combining Local Chemotherapy and Immunotherapy)
Abbreviations
Band T lymphocyte attenuator (BTLA); Bristol Myers Squibb (BMS); C-C chemokine receptor type 4 (CCR4); Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4); Fluorescence minus one (FMO); Glioblastoma (GBM); Herpes virus entry mediator (HVEM); immune checkpoint blockade (ICB); Indoleamine 2,3-dioxygenase 1 (IDO1); Institutional Animal Care and Use Committee (IACUC); intra-peritoneal (i.p.); IVIS (In Vivo Imaging System); Lymphocyte-activation gene-3 (LAG-3); median overall survival (mOS); monoclonal antibodies (mAbs); non-small cell lung cancer (NSCLC); phosphate-buffered saline (PBS); programmed cell death protein-1 (anti-PD-1); progression-free survival (PFS); regulatory T cells (Tregs); T-cell immunoglobulin and mucin-domain containing-3 (TIM-3); tumor infiltrating lymphocytes (TILs); tumor microenvironment (TME);
Novelty and Impact:
BTLA is an immune checkpoint thought to downregulate the immune system. BTLA has been shown to be upregulated in the GBM tumor microenvironment. In this study, we study synergy between anti-BTLA and anti-PD-1 immunotherapy and show that combination therapy significantly improves survival in a murine GBM model. Furthermore, we demonstrate a direct effect of combination therapy on several immune cell populations including CD4+, CD8+, and regulatory T cells.
Disclosure statement
The authors do not have any relevant conflicts of interest associated with this manuscript.
Data availability statement:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics statement:
This study protocol was approved by the Johns Hopkins Institutional Animal Care and Use Committee. This study did not involve any human patients.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Ostrom QT, Gittleman H, Liao P, Vecchione-Koval T, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS.. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2010-2014. Neuro Oncol. 2017Nov;19(5):v1–9. doi: 10.1093/neuonc/nox158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005Mar;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Di Carlo DT, Cagnazzo F, Benedetto N, Morganti R, Perrini P. Multiple high-grade gliomas: epidemiology, management, and outcome. A systematic review and meta-analysis. Vol. 42, Neurosurgical Review. Springer Verlag; 2019. p. 263–275. [DOI] [PubMed] [Google Scholar]
- 4.Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, et al. Pembrolizumab plus chemotherapy for squamous non–small-cell lung cancer. N Engl J Med. 2018Nov;379(21):2040–2051. doi: 10.1056/NEJMoa1810865. [DOI] [PubMed] [Google Scholar]
- 5.Paz-Ares L, Horn L, Borghaei H, Spigel DR, Steins M, Ready N, et al. Phase III, randomized trial (CheckMate 057) of nivolumab (NIVO) versus docetaxel (DOC) in advanced non-squamous cell (non-SQ) non-small cell lung cancer (NSCLC). J Clin Oncol. 2015Jun;33(18_suppl):LBA109–LBA109. doi: 10.1200/jco.2015.33.18_suppl.lba109. [DOI] [Google Scholar]
- 6.Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T, Voloschin A, Ramkissoon SH, Ligon KL, Latek R, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase i cohorts of CheckMate 143. Neuro Oncol. 2018;20(5):674–686. doi: 10.1093/neuonc/nox208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Vol. 15, nature reviews clinical oncology. Nature Publishing Group; 2018. p. 422–442. [DOI] [PubMed] [Google Scholar]
- 8.Razavi SM, Lee KE, Jin BE, Aujla PS, Gholamin S, Li G. Immune evasion strategies of glioblastoma. Vol. 3, frontiers in surgery. Frontiers Media S.A.; 2016. p. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Vol. 20, nature immunology. Nature Publishing Group; 2019. p. 1100–1109. [DOI] [PubMed] [Google Scholar]
- 10.Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W, Hiraoka N, Fuller GN. Incidence and prognostic impact of FoxP3+regulatory T cells in human gliomas. Clin Cancer Res. 2008Aug;14(16):5166–5172. doi: 10.1158/1078-0432.CCR-08-0320. [DOI] [PubMed] [Google Scholar]
- 11.Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, Liu A, Sankey EW, Tam A, Xu H, et al. Combination therapy with anti-PD-1, anti-TIM-3, and focal radiation results in regression of murine gliomas. Clin Cancer Res. 2017Jan;23(1):124–136. doi: 10.1158/1078-0432.CCR-15-1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris-Bookman S, Mathios D, Martin AM, Xia Y, Kim E, Xu H, Belcaid Z, Polanczyk M, Barberi T, Theodros D, et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int J Cancer. 2018Dec;143(12):3201–3208. doi: 10.1002/ijc.31661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003Jul;4(7):670–679. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- 14.Derré L, Rivals JP, Jandus C, Pastor S, Rimoldi D, Romero P, Michielin O, Olive D, Speiser DE. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest. 2010Jan;120(1):157–167. doi: 10.1172/JCI40070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lan X, Li S, Gao H, Nanding A, Quan L, Yang C, Ding S, Xue Y. Increased BTLA and HVEM in gastric cancer are associated with progression and poor prognosis. Onco Targets Ther. 2017Feb;10:919–926. doi: 10.2147/OTT.S128825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J, Li J, He M, Zhang GL, Zhao Q. Distinct changes of BTLA and HVEM expressions in circulating CD4+ and CD8+ T cells in hepatocellular carcinoma patients. J Immunol Res. 2018;2018: 4561571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, Elsamadicy AA, Cui X, Koyama S, Jackson C. et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res. Internet]. 2018Sep1;24(17):4175LP–4186. Available from. ;:. 10.1158/1078-0432.CCR-17-1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han MZ, Wang S, Zhao WB, Ni SL, Yang N, Kong Y, Huang B, Chen A-J, Li X-G, Wang J, et al. Immune checkpoint molecule herpes virus entry mediator is overexpressed and associated with poor prognosis in human glioblastoma. EBioMedicine. 2019May;43:159–170. doi: 10.1016/j.ebiom.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013Jun;86(2):343–349. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL, Lao CD, Schadendorf D, Wagstaff J, Dummer R, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018Nov;19(11):1480–1492. doi: 10.1016/S1470-2045(18)30700-9. [DOI] [PubMed] [Google Scholar]
- 21.Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, Cheng Y, Kim JW, Qiao J, Zhang L, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. 2014Oct;20(20):5290–5301. doi: 10.1158/1078-0432.CCR-14-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El AA, Lesniak MS. An increase in CD4+CD25+FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme1. Neuro Oncol. 2006Jul;8(3):234–243. doi: 10.1215/15228517-2006-006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sugihara AQ, Rolle CE, Lesniak MS. Regulatory T cells actively infiltrate metastatic brain tumors. Int J Oncol. 2009;34:1533–1540. [DOI] [PubMed] [Google Scholar]
- 24.Kennedy BC, Maier LM, D’Amico R, Mandigo CE, Fontana EJ, Waziri A, Assanah MC, Canoll P, Anderson RC, Anderson DE, et al. Dynamics of central and peripheral immunomodulation in a murine glioma model. BMC Immunol. 2009Feb;10(1):11. doi: 10.1186/1471-2172-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, Herndon JE, Bigner DD, Dranoff G, Sampson JH, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006Mar;66(6):3294–3302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- 26.Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, Grau S, Hiraoka N, Eckstein V, Ecker RC, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-β. Clin Cancer Res. 2011Jul;17(13):4296–4308. doi: 10.1158/1078-0432.CCR-10-2557. [DOI] [PubMed] [Google Scholar]
- 27.Fecci PE, Sweeney AE, Grossi PM, Nair SK, Learn CA, Mitchell DA, Cui X, Cummings TJ, Bigner DD, Gilboa E, et al. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin Cancer Res. 2006;12(14 Pt 1):4294–4305. doi: 10.1158/1078-0432.CCR-06-0053. [DOI] [PubMed] [Google Scholar]
- 28.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013Jul;1(1):32–42. doi: 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 29.Lim M;, Weller M;, Chiocca EA. Current State of Immune-Based Therapies for Glioblastoma. 2016; [DOI] [PubMed]
- 30.Davidson TB, Lee A, Hsu M, Sedighim S, Orpilla J, Treger J, Mastall M, Roesch S, Rapp C, Galvez M. et al. Expression of PD-1 by T Cells in Malignant Glioma Patients Reflects Exhaustion and Activation. Clin Cancer Res. Internet]. 2019Mar15;25(6):1913 LP– 1922. Available from. ;:. 10.1158/1078-0432.CCR-18-1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, Heimberger AB. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol Immunother. 2008May;57(1):123–131. doi: 10.1007/s00262-007-0336-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reardon DA, Gokhale PC, Klein SR, Ligon KL, Rodig SJ, Ramkissoon SH, Jones KL, Conway AS, Liao X, Zhou J, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2016Feb;4(2):124–135. doi: 10.1158/2326-6066.CIR-15-0151. [DOI] [PubMed] [Google Scholar]
- 33.Maes W, Van Gool SW. Experimental immunotherapy for malignant glioma: lessons from two decades of research in the GL261 model. Cancer Immunol Immunother. 2011Feb;60(2):153–160. doi: 10.1007/s00262-010-0946-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lenting K, Verhaak R, Ter Laan M, Wesseling P, Leenders W. Glioma: experimental models and reality. Acta Neuropathol. 2017Feb;133(2):263–282. doi: 10.1007/s00401-017-1671-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh T, Fakurnejad S, Sayegh ET, Clark AJ, Ivan ME, Sun MZ et al. Immunocompetent murine models for the study of glioblastoma immunotherapy J Transl Med 2014;12:107Internet]. ;(1):. Available from 10.1186/1479-5876-12-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mathios D, Kim JE, Mangraviti A, Phallen J, Park C-K, Jackson CM. et al. Anti-PD-1 antitumor immunity is enhanced by local and abrogated by systemic chemotherapy in GBM. Sci Transl Med. Internet]. 2016Dec21;8(370):370ra180–370ra180. Available from. ;:. 10.1126/scitranslmed.aag2942 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.