

ABSTRACT
Piwi-interacting RNAs (piRNAs/piRs) are small non-coding RNAs that play important roles in stablizing genome through silencing transposable genetic elements. The piR-651 was reported to be dysregulated in several human solid cancer tissues, such as gastric and lung cancers. However, the role of piRNA-651 in carcinogenesis of breast cancer has not been defined. We found that piR-651 was highly expressed in breast cancer tissues and cell lines. Overexpression of piR-651 facilitated cell proliferation and invasion, restrained cell apoptosis and the percentage of arrested cells in G0/G1 phase, accompanied by upregulated expression of oncogenes (MDM2, CDK4 and Cyclin D1), whereas piR-651 downregulation showed the opposite effects. Additionally, piR-651 could promote phosphatase and tensin homolog (PTEN) methylation and its downregulated expression by recruiting DNA methyltransferase 1 (DNMT1) to the PTEN promoter region through complex formation with PIWIL2. PTEN overexpression reversed the effects of upregulated piR-651 on cell functions. This study reveals that piR-651 promotes proliferation and migration and induces apoptosis of breast cancer cells by facilitating DNMT1-mediated PTEN promoter methylation, which may provide a potential therapeutic mechanism for breast cancer.
KEYWORDS: Breast cancer, piR-651, PTEN, DNMT1, methylation
Introduction
Breast cancer is the most common malignant tumor in women all over the world. The incidence of breast cancer in China is 42.55/10 million, and by 2021, breast cancer patients in China will be more than 2.5 million [1]. Although breast cancer is regarded as a curable cancer at early stage and the 5-year survival rate has been increased to about 90% in advanced countries, 5-year survival rate of breast cancer in China is just about 80% and the patients tend to be younger [2]. What’s worse, only 22% of Chinese patients are treated with breast conserving surgery, which is far less than 50% of the advanced countries [3]. It is urgent for the Chinese researchers making progresses in molecular oncology and new therapies to extend the survival of breast cancer patients and improve the patients’ quality of life. For the moment, gene therapy against neoplasms has been a novel research focus in cancer treatment. Therefore, a better understanding of such molecular mechanisms is required to facilitate the development of more accurate prognostic markers and effective therapeutic strategies.
piRNAs are a type of novel non-coding small RNA with a length of 25–33 nucleotides. They have biological effects through specific combinations with PIWI proteins [4]. Abundant expression of PIWI proteins was found in breast, gastrointestinal, gastric and endometrial tissues from patients with cancer, whereas no expression has been found in normal tissues. PIWIL2 has been reported to be widely expressed in breast and cervical cancer cells, suggesting that PIWIL2 may be involved in tumorigenesis [5]. In addition to PIWI proteins, piRNAs have also been found to be abnormally expressed in cancer cells. Recent studies have shown that abnormal expression of piRNA is a hallmark feature of multiple cancer types. Circulating PIWI-interacting RNAs piR-5937 and piR-28,876 have been reported to be upregulated in colon cancer and are promising diagnostic biomarkers for cancer therapy [6]. Zhou et al found that piR-1245 was up-regulated in gastric cancer tissues and was associated with tumor node metastasis (TNM) stage. The authors concluded that piR-1245 is involved in the development of gastric cancer and may be a potential marker for diagnosis [7]. Notably, increasing evidence on the function of piRNAs is involved in the regulation of epigenetic mechanisms in tumorigenesis. PIWIL4/piR-31,470 was able to recruit DNA methyltransferase 1 or DNA methyltransferase 3B and methyl-CpG binding domain protein 2 to initiate and maintain the hypermethylation and inactivation of GSTP1, which in turn suppressed GSTP1 levels and increased susceptibility to oxidative stress and DNA damage in human prostate epithelial cancer cells [8]. PIWI-interacting RNA 021285 participates in breast tumorigenesis by remodeling the cancer epigenome [9]. PIWI/PiR-651 was reported to be up-regulated in several cancer tissues and cell lines, including NSCLC and gastric cancer, and may be an oncogene in carcinogenesis [10,11]. However, the regulatory mechanism of PIWI/piR-651 in breast cancer carcinogenesis remains to be elucidated.
In the present study, we aimed to investigate whether piR-651 is involved in breast cancer progression and its underlying mechanisms, contributing to the clinical treatment for breast cancer.
Materials and methods
Tissue sample collection
Breast cancer tissue and adjacent tissue samples were collected from 16 patients (aged 37.93 ± 10.05 years) at The Second Affiliated Hospital, Medical School of Xi’an Jiaotong University (Xi’an, China) between January 2018 and February 2019. And all patients had not received radiotherapy or chemotherapy. They don’t have other gynecological diseases and tumors. All breast cancer patients gave the written informed consent and conducted in accordance with the Declaration of Helsinki. This study was performed with the approval of the ethics committee of Xi’an Jiaotong University. The ethical number is XAJTDX-2018-31.
Cell culture and treatment
Human breast cancer cell lines (MDA-MB-231, MCF-7, AU565 and HCC38) and human breast epithelial cells (Hs-578Bst) were obtained from Chinese Academy of Sciences (Shanghai, China). The cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Rockville, MD) supplemented with 10% fetal bovine serum (FBS) (HyClone, Salt Lake City, UT) and 1% penicillin‐streptomycin (Sigma, St. Louis, MO, USA) at 37°C in a controlled humidified atmosphere with 5% CO2. 2 × 105 cells were plated in 6-well plates and incubated for 24 h. The culture medium was replaced once every 2 days, and the cells were passaged by 1% trypsin when they reached 80%~90% confluence.
Cell transfection
Overexpression plasmid of piR-651 (pcDNA-piR-651) and PTEN (pcDNA-PTEN), short hairpin RNA against piR-651 (piR-651 shRNA) and its negative control (sh-NC) were purchased from RiboBio Co., Ltd (Guangzhou, China). Cells were transfected by using 1 μg and 2 μg of piR-651 shRNA, 0.5 μg and 1 ug of pcDNA-piR-651 mixed with Lipofectamine®3000 (Thermo, Waltham, MA, USA), respectively. All transfection was performed by using Lipofectamine®3000 according to manufacturer’s instructions. In brief, transfection was performed when cells were about 70% confluent. Lipofectamine®3000 reagent was diluted by using Opti-MEM medium (2 tubes) and mix well sufficiently. DNA master mix was then prepared by diluting the DNA by using Opti-MEM medium, followed by the addition of p3000™ reagent. Lipofectamine®3000 that had been diluted in each tube reagent with diluted DNA (1:1 ratio). DNA-liposome complexes were added to the cells after 5 min incubation at room temperature. Cells were collected after transfection for 48 h for further experiments.
Cell proliferation assay
Cell proliferation was determined by Cell Counting Kit-8 (CCK-8) assay (Dojindo, Kumamoto, Japan). The MDA-MB-231 and MCF-7 cells were seeded in a 96-well plate at 1.5 × 104 cells/well. After 24, 48 and 72 h of transfection, the supernatant was discarded and 10 μL of CCK-8 solution was added to each well. After incubating at 37°C for 2 h, the absorbance at 450 nm wavelength was measured by an automatic microplate reader (Bio-Rad, Hercules, CA, USA).
Cell apoptosis assay
Annexin V-FITC/PI double staining was used to analyze apoptosis on the flow cytometer. After 48 h of transfection, the cells were detached with EDTA-free trypsin and collected afterward. The cells were centrifuged for 5 min at 4°C at 1000 rpm, and the supernatant was discarded. Apoptosis was detected by an Annexin V-FITC/PI Apoptosis Detection Kit (CA1020; Beijing Solarbio Science & Technology, Beijing, China). The cells were suspended in a mixture of Annexin V-FITC and binding buffer (1:40) and incubated at room temperature for 30 min. A mixture of PI and binding buffer (1:40) was then added and shaken, followed by incubation at room temperature for 15 min. Fluorescence was detected by the flow cytometer (BD FACSVerse 6 color Flow Cytometer, BD Bioscience, San Jose, CA, USA), and the apoptosis rate was calculated and determined.
Transwell invasion assay
We use 8.0 μm chamber plates to determinate cells invasiveness. The upper surface of the Transwell filter we used was coated with Matrigel (BD, New Jersey, USA). Firstly, cells were planted into the 8.0 μm chamber plates, then 300 μL of serum-free DMEM medium was added to the upper compartment of the chamber, and 500 μL of DMEM medium supplemented with 10% FBS was added to the lower chamber for 48 h incubations. Then, the noninvasive cells on the upper side of the chamber were suspended with a cotton swab, and then the invasive cells were fixed in 4% paraformaldehyde and stained with a crystal violet solution. We stained infiltrating cells using an Olympus IX70 inverted microscope (Olympus Corp, Tokyo, Japan) and randomly selected the best six fields of view, and each experiment was repeated three times.
Cell cycle assay
MDA-MB-231 and MCF-7 cells were fixed with 70% ethanol overnight. After resuspending the cells with 50 µg/ml propidium iodide, the cells were treated with 0.5 µg/ml Annexin V-PE and 0.5 µg/ml 7-aminoactinomycin D (7-AAD) (BD Biosciences, Franklin Lakes, NJ, USA) at room temperature for 15 min and assayed by using a FACSCanto flow cytometer. The signals of Annexin V, 7-AAD, or both were detected and cell death was analyzed by using CellQuestPro software (BD Biosciences).
Quantitative real-time polymerase chain reaction (RT-qPCR)
Total RNA from clinical samples or cultured cells was extracted by using Trizol reagent (Invitrogen, Carlsbad, CA, USA) with the manufacturer’s instructions. RNA (1 µg) was used to synthesize first-strand cDNA. To perform RT-qPCR of piRNA, the miScript SYBR-Green PCR kit provided by Qiagen (Hilden, Germany) was used for reverse transcription, and the PrimeScript™ II 1st Strand cDNA Synthesis kit from Takara (Dalian, China) was used for gene expression detection. Real-time PCR analyses were conducted with the SYBR Premix Ex Taq II (Takara, Dalian, China) on an ABI 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA) under the following conditions: 95°C for 1 min, and then 95°C for 20 s, 56°C for 10 s and 72°C for 15 s for 35 cycles. The PCR conditions are as follows: 2 µL of cDNA was added to 10 µL of the 2× SYBR green PCR master mix with 0.4 µL of Taq polymerase enzyme (RiboBio Co., Ltd, Guangzhou, China), 0.8 µL of each primer and 6 µL of ddH2O to a final volume of 20 µL. The sequences of primers were as follows: piR-651 forward, 5ʹ-AGA GAG GGG CCC GTG CCT TG-3ʹ; and reverse, 5ʹ-CCA TCC AGC TCT TTC ACC AT-3ʹ; phosphatase and tensin homolog (PTEN) forward, 5ʹ-ACA CCG CCA AAT TTA ACT GC-3ʹ; and reverse, 5ʹ-TAC ACC AGT CCG TCC TTT CC-3ʹ; DNMT1 forward: 5ʹ-CCA CCA CCA AGC TGG TCT AT-3ʹ; and reverse, 5ʹ-TAC GGC CAA GTT AGG ACA CC-3ʹ; GAPDH forward, 5ʹ-TGC AGT GGC AAA GTG GAG ATT-3ʹ; and reverse, 5ʹ-TCG CTC CTG GAA GAT GGT GAT-3ʹ; U6 forward, 5ʹ- GCT TCG GCA GCA CAT ATA CTA AAA T-3ʹ; reverse: 5ʹ-CGC TTC ACG AAT TTG CGT GTC AT-3ʹ. We used GAPDH and U6 (only against piR-651) as endogenous control, respectively. The relative expression levels were calculated by using 2−ΔΔCT method.
Western blotting
The cells were washed twice with ice-cold PBS and lysed by using RIPA lysis buffer (CW Biotech, Beijing, China) supplemented with protease inhibitor (Roche Diagnostics, Basel, Switzerland). Then the protein concentration was measured by BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, USA). Equal amount of protein was subjected to 10% SDS-PAGE at 70 V for 30 min then 120 V for 90 min. And the protein bands were transferred to PVDF membranes at 300 mA for 2 h. The membranes were blocked with 5% skim milk for 2 h at room temperature, then incubated with the following primary antibodies: rabbit monoclonal GAPDH antibody (1:2500 dilution, Abcam, ab9485, Cambridge, UK), rabbit monoclonal Cyclin D1 antibody (1:200 dilution, ab16663, Abcam, Cambridge, UK), rabbit monoclonal CDK4 antibody (1:1000 dilution, ab108357, Abcam, Cambridge, UK), rabbit monoclonal MDM2 antibody (1:1000 dilution, ab259265, Abcam, Cambridge, UK), rabbit monoclonal PTEN antibody (1:1000 dilution, ab170941, Abcam, Cambridge, UK), DNMT1 (1:1000 dilution, ab188453, Abcam, Cambridge, UK), PIWIL4/PIWI (1:500 dilution, ab111714, Abcam, Cambridge, UK). After washing 3 times with TBS-T, the membranes were incubated with the horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (1:5000, ab6721, Abcam, Cambridge, London, UK) for 1 h at room temperature. GAPDH was used as endogenous controls. Protein bands were visualized by the ECL system and the quantification was performed by using Image J software.
Chromatin immunoprecipitation (ChIP)
ChIP assay was conducted to verify the effects of DNMT1 on the PTEN promoter region. Briefly, MDA-MB-231 cells (4 × 106 cells/well) were seeded into 10 cm flat petri dishes for 24 h, and then cells were subjected to standard cross-linking. DNA-protein complexes were cross-linked with 1% formaldehyde for 10 min at 37°C. Then, cells were lysed in the lysate, and the chromatin DNA was sonicated into fragments. Immunoprecipitation was performed with DNMT1 antibody. IgG as an isotype control antibody. The immunoprecipitated DNA was treated with RNase A and proteinase K and purified with phenol-chloroform extraction followed by ethanol precipitation. Input DNA was purified with phenol chloroform extraction and ethanol precipitation. Purified DNA and input genomic DNA were analyzed by real-time PCR. The PTEN promoter primer sequences used in this study to amplify purified DNA were as follows: Forward primer: 5ʹ- AAG CTG CAG CCA TGA TGG AA −3ʹ, Reverse primer: 5ʹ- CTC CAG GAG CGG GAG GTG −3ʹ. Fold enrichment at the promoter was calculated with the standard method according to the manufacturer’s protocol.
Co-immunoprecipitation (Co-IP)
MDA-MB-231 cells were lysed by using RIPA buffer (Beyotime, Shanghai, China), and the supernatant was collected and incubated with or DNMT1 antibody at 4°C overnight. Then, the mixture was incubated with 100 μL of protein A/G agarose beads (Takara Biotechnology, Dalian, China) overnight at 4°C. Subsequently, the agarose beads-antigen-antibody complex was collected by instantaneous centrifugation and washed with PBS for three times. Next, the complex was boiled with protein loading buffer for 5 min. The supernatant was collected by centrifugation and analyzed by using Western blotting to detect the expression of interaction proteins.
Methylation-specific PCR
Analysis of CpG island of PTEN promoter region was performed by using MethPrimer software (https://www.urogene.org/methprimer/). The primers specific for the methylated PTEN gene promoter were 5ʹ -TTG TTA TTA TTT TTA GGG TTG GGA A − 3ʹ and 5ʹ – CTA AAC CTA CTT CTC CTC AAC AAC C − 3ʹ. MSP-PCR was used to detect the methylation status of the PTEN promoter. Cell DNA extraction was conducted with the genomic DNA extraction kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The DNA concentration was determined by using a UV spectrophotometer. The extracted DNA (10 μg) was added with 5.5 μL of 3 M NaOH to denature at 37°C for 10 min. Next, DNA was added with 20 μL of 10 mM hydroquinone and 520 μL of 40.5% sodium hydrogen sulfite, and then covered by 200 μL mineral oil. Then, the modified DNA was purified by wizard DNA column. The purified solution was added with 5.5 μL of 3 mol/L NaOH to denature for 10 min, and then 5.5 μL of 3 M sodium acetate and 120 μL of cold absolute ethanol were added to precipitate and recycle DNA. DNA was then modified with CpGenome DNA Modification Kit (S7820; Millipore Sigma) and underwent MSP followed by PCR reaction on a Thermocycler (Biometra, Dublin, Ireland). The amplified products were subjected to 18 g/L agarose gel electrophoresis, imaged by UV imager gel electrophoresis imaging, and analyzed by using a gel imager (FireReader, UVItec, Cambridge, United Kingdom).
Northern blotting
Total RNA (30 µg) was separated in a 15% denaturing polyacrylamide gel and then transferred onto a GeneScreen Plus Hybridization Transfer membrane (NEN Life Science Products, Inc., Boston, MA, USA) and blotted by using a 32P-labeled oligonucleotide. The membrane was then stripped in 0.1X SSPE and hybridized with the probe (piR-651, 5ʹ-GAC GCU UUC CAA GGC ACG GGC CCC UCU CU-3ʹ) overnight at 40°C. The antisense oligonucleotide (5ʹ-AAA ATA TGG AAC GCT TCA CGA-3ʹ) of U6 snRNA was used as a loading control. After washing with 2X SSPE containing 1% sodium dodecyl sulfate (SDS), the membrane was exposed to a PhosphorImager screen (Molecular Dynamics, Inc., Sunnyvale, CA, USA) for 20 min.
Enzyme-linked immunosorbent assay (ELISA)
The activity of DNMT1 in MD-MB-231 cells was determined by ELISA kit (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions.
RNA Immunoprecipitation (RIP)
RIP assay was performed to validate the binding relationship between piR-651 and PIWIL2. MDA-MB-231 cells were lysed by using RIP lysis buffer, and 100 μL of the lysate was incubated with RIP buffer containing magnetic beads, which were conjugated with PIWI2 antibodies (Abcam, 1:500, ab181340, Cambridge, UK) or control normal mouse IgG (Millipore, Billerica, MA, USA). Among the antibodies, IgG was considered as a negative control. Proteinase K buffer was then added to the samples to digest protein. Finally, the bounded RNA was isolated from the beads by using Trizol reagent and purified piR-651 was assayed by using RT-qPCR.
Statistical analysis
All the experimental data were expressed as mean ± standard error of mean (SEM), and SPSS Statistics, version 23.0 (IBM Corporation, NY, USA) was used for statistical analysis. The Shapiro–Wilk test was used to verify whether the data was normally distributed. Levene’s test was used to verify the homogeneity of variances. A one-way analysis of variance (ANOVA) or Kruskal–Wallis non-parametric test was used, depending on data distribution and variance homogeneity. The Kruskal–Wallis test followed by the post hoc analysis (Mann–Whitney U-test) was used to evaluate pairwise differences among the adjusted means. Values of P < 0.05 were considered statistically significant.
Results
piR-651 was up-regulated in breast cancer tissues and cell lines
The mRNA expression of piR-651 in breast cancer tissues or cell lines was determined by Northern blotting and RT-qPCR analysis, respectively. The results showed that piR-651 expression in the cancer lesions of breast cancer patients was prominently higher than that in the para-carcinoma tissues (Figure 1(a), p < 0.01). Similarly, piR-651 was overexpressed in breast cancer cell lines (AU565, HCC38, MCF-7 and MDA-MB-231) compared with that of normal breast epithelial cells (Hs-578Bst), especially in MDA-MB-231 cells (Figure 1(b), p < 0.01).
Figure 1.
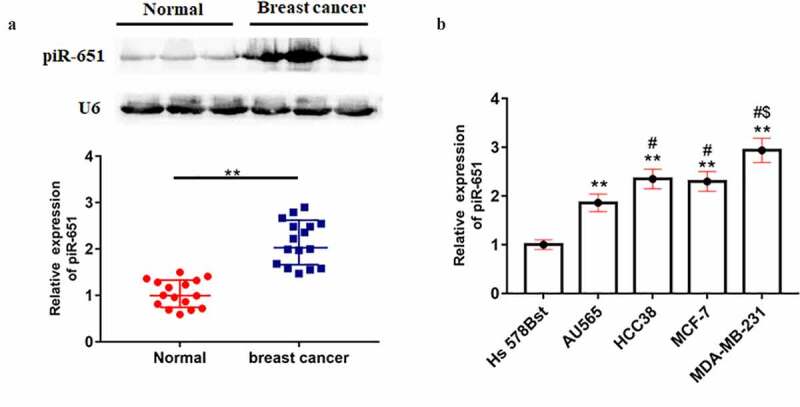
piR-651 was up-regulated in breast cancer tissues and cell lines. (a) The expression of piR-651 in breast cancer tissue and adjacent tissue samples collected from 16 patients (37.93 ± 10.05 years) was detected by Northern blotting. ** P < 0.01, unpaired t test. (b) Relative expression of piR-651 in breast cancer cells and normal breast epithelial cells was detected by RT-qPCR. N = 4, **P < 0.01 compared with Hs-578Bst. #P < 0.05 compared with AU565. $P < 0.05 compared with HCC38 or MCF-7. The difference of the samples between the two groups were analyzed with independent sample t test. The analysis of variance (ANOVA) test was used to evaluate multigroup comparisons of the means
Overexpression of piR-651 promotes cell proliferation and invasion and inhibits apoptosis
We then analyzed the effects of piR-651 upregulation in breast cancer cell functions. Different doses of pcDNA-piR-651 and its negative control were transfected into MDA-MB-231 and MCF-7 cells. Figure 2(a) and Figure 2(b) displayed that the piR-651 expression was elevated in a dose-dependent manner in pcDNA-piR-651 transfected cells (P < 0.01). Overexpression of piR-651 could observably enhance the cell proliferation and invasion and inhibit cell apoptosis, especially in the high-dose transfection group (Figure 2(c-j), P < 0.01). Additionally, the cell cycle results illustrated that upregulation of piR-651 markedly decreased the percentage of G0/G1 phase cells and increased the percentage of G2/M phase cells, indicating that overexpression of piR-651 could promote cell cycle progression and arrest cells in G2/M phase, especially in the high-dose transfection group (Figure 2(k-m), P < 0.01).
Figure 2.
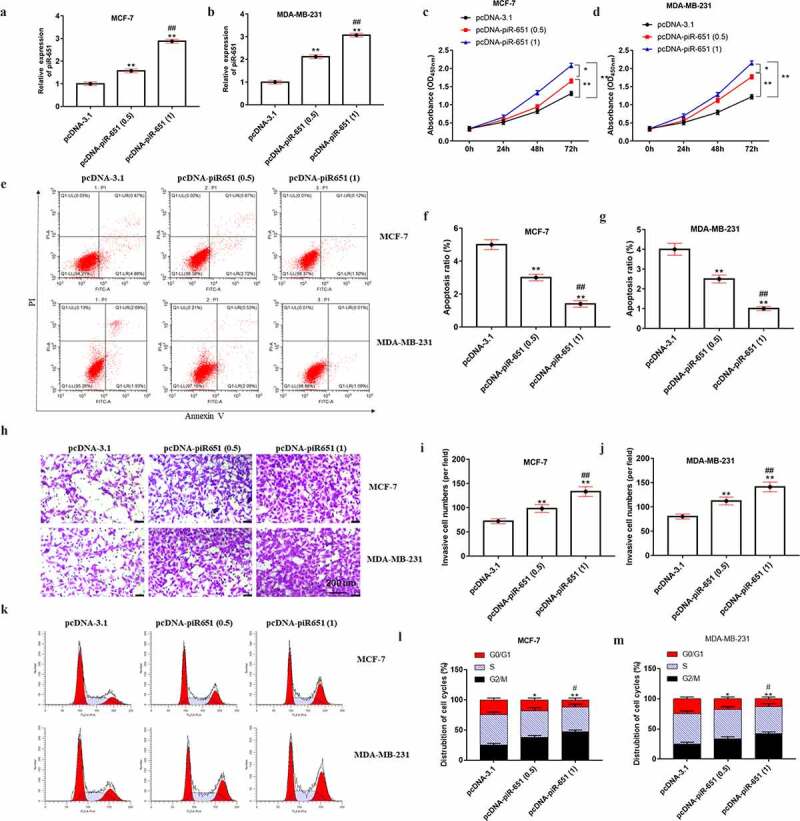
Overexpression of piR-651 promotes cell proliferation and invasion and inhibits apoptosis. Different doses of pcDNA-piR-651 and its negative control were transfected into MDA-MB-231 and MCF-7 cells for 24 h. (a) RT-qPCR was used to perform the expression of piR-651 in MCF-7 cells. (b) RT-qPCR was used to perform the expression of piR-651 in MDA-MB-231 cells. (c) The cell viability of MCF-7 cells was detected by CCK-8 assay. (d) The cell viability of MDA-MB-231 cells was detected by CCK-8 assay. (e-g) The cell apoptosis ability of MCF-7 cells and MDA-MB-231 was determined by flow cytometry. (h-j) The cell invasion ability of MCF-7 and MDA-MB-231 cells was determined by Transwell assay. (k-m) The percentage of MCF-7 and MDA-MB-231 cells in each phase of the cell cycle was conducted by flow cytometry analysis. N = 4, **P < 0.01 compared with pcDNA-3.1 group. ##P < 0.01 compared with pcDNA-piR-651 (0.5) group. Data were presented as mean ± SEM, (N = 4). The difference of the samples between the two groups were analyzed with independent sample t test. The analysis of variance (ANOVA) test was used to evaluate multigroup comparisons of the means
Interference piR-651 expression inhibits cell proliferation and invasion and promotes apoptosis
To further explore the effect of piR-651 interference on cell proliferation, invasion, apoptosis and cell cycle of breast cancer cells, MB-231 and MCF-7 cells were transfected with 1 μg or 2 μg piR-651 shRNA and its negative control for 24 h, respectively. The results displayed that the piR-651 expression was decreased in a dose-dependent manner in piR-651 shRNA transfected cells (Figure 3(a) and Figure 3(b), P < 0.01). Downregulation of piR-651 inhibited cell proliferation and invasion and facilitated cell apoptosis, in which the high-dose group of piR-651 shRNA had prominently higher inhibitory effects on cell proliferation and invasion and promotion effect on apoptosis than its low-dose transfection group (Figure 3(c-j), P < 0.01). Furthermore, silencing of piR-651 expression showed a notably higher percentage of cells in the G0/G1 phase and markedly lower percentage of cells in the G2/M phase, indicating that piR-651 interference promoted cell cycle progression and arrests cells in G0/G1 phase (Figure 3(k-m), P < 0.01).
Figure 3.
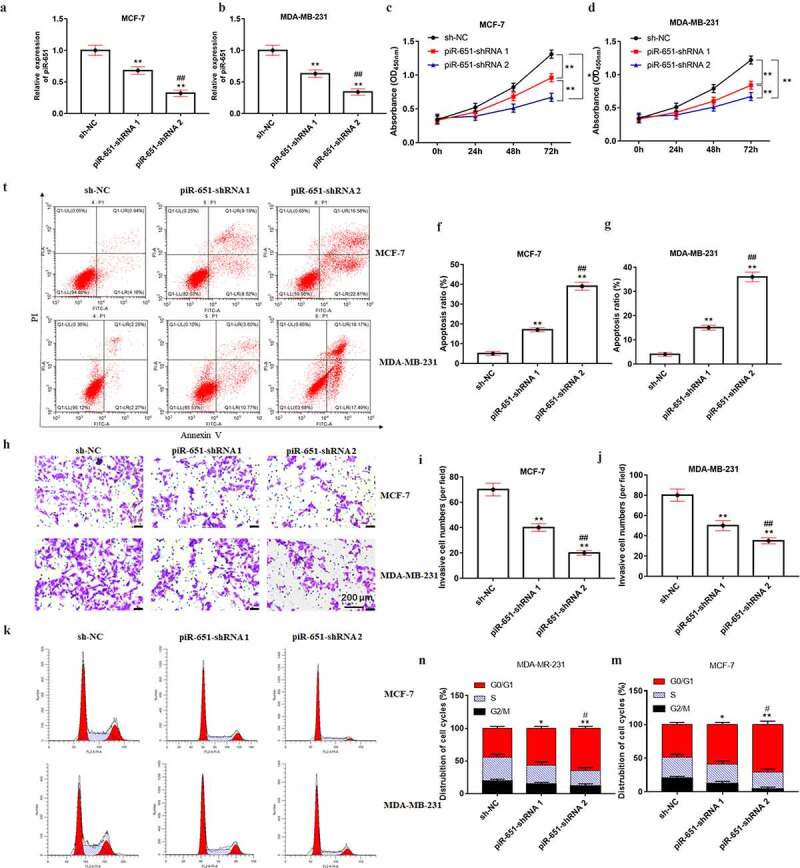
Interference piR-651 expression inhibits cell proliferation and invasion and promotes apoptosis. Different doses of piR-651 shRNA and its negative control were transfected into MDA-MB-231 and MCF-7 cells for 24 h. (a) RT-qPCR was used to perform the expression of piR-651 in MCF-7 cells. (b) RT-qPCR was used to perform the expression of piR-651 in MDA-MB-231 cells. (c) The cell viability of MCF-7 cells was detected by CCK-8 assay. (d) The cell viability of MDA-MB-231 cells was detected by CCK-8 assay. (e-g) The cell apoptosis ability of MCF-7 cells and MDA-MB-231 was determined by flow cytometry. (h-j) The cell invasion ability of MCF-7 and MDA-MB-231 cells was determined by Transwell assay. (k-m) The percentage of MCF-7 and MDA-MB-231 cells in each phase of the cell cycle was conducted by flow cytometry analysis. N = 4, **P < 0.01 compared with sh-NC group. ##P < 0.01 compared with piR-651 shRNA 1 group. Data were presented as mean ± SEM, (N = 4). The difference of the samples between the two groups were analyzed with independent sample t test. The analysis of variance (ANOVA) test was used to evaluate multigroup comparisons of the means
piR-651 overexpression promotes the expression of oncogenes
Western blotting results displayed that the protein levels of MDM2, CKD4 and CyclinD1 were significantly increased after overexpression of piR-651, whereas inhibition of piR-651 prominently decreased the protein levels of MDM2, CKD4 and CyclinD1 (Figure 4(a-d), P < 0.01).
Figure 4.
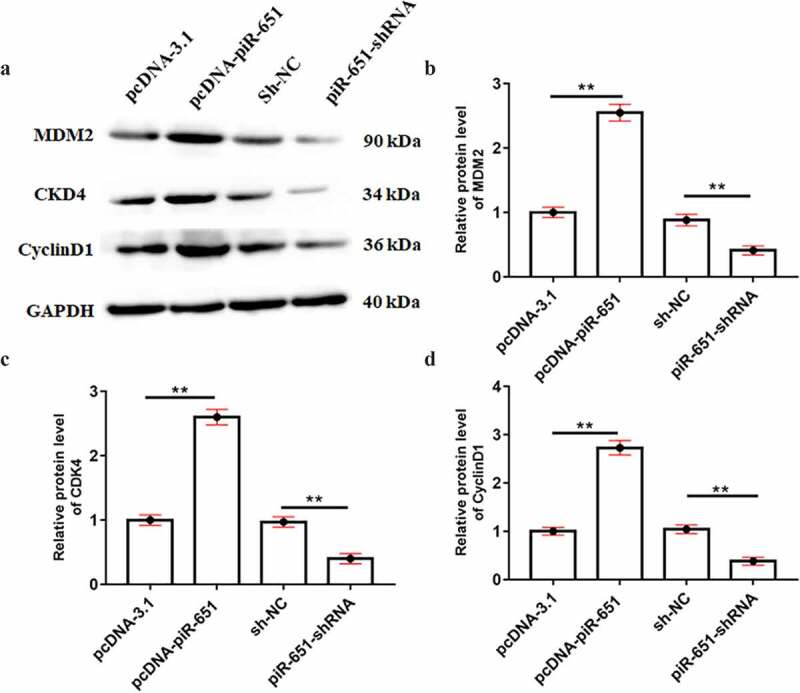
piR-651 overexpression promotes the expression of oncogenes. PcDNA-piR-651 and piR-651 shRNA were transfected into MDA-MB-231 cells for 24 h. (a) Western blotting was used to detect the protein levels of MDM2, CKD4 and CyclinD1 in MDA-MB-231 cells. (b) The quantitative analysis of MDM2 protein level. (c) The quantitative analysis of CKD4 protein level. (d) The quantitative analysis of CyclinD1 protein level. Data were presented as mean ± SEM, (N = 4). The difference of the samples between the two groups were analyzed with independent sample t test. The analysis of variance (ANOVA) test was used to evaluate multigroup comparisons of the means
PIWI/piR-651 facilitates DNMT1-mediated PTEN promoter methylation
To further investigate the underlying mechanism of piR-651 in the regulation of breast cancer cell behaviors, we focused on the piR-651 downstream pathway. RIP results showed that PIWIL2 could combine with piR-651 to form a complex (Figure 5(a), p < 0.01). Co-IP results displayed that DNMT1 was an interacting protein of PIWI/piR-651 (Figure 5(b), p < 0.01). Furthermore, upregulated piR-651 resulted in elevated enzymatic activity of DNMT1, which was reduced when piR-651 was interfered (Figure 5(c), p < 0.01). We conducted ChIP assay and the results showed that DNMT1 could bind to promoter region of PTEN, and silencing of piR-651 prominently attenuated DNMT1 enrichment (Figure 5(d), p < 0.01). PiR-651 overexpression increased PTEN promoter region methylation and restrained PTEN expression, whereas piR-651 knockdown decreased PTEN promoter region methylation and facilitated PTEN expression. Furthermore, overexpression of piR-651 significantly reversed the inhibitory effect of DNMT1 inhibitor 5-Aza-Dc on PTEN promoter methylation rate as well as the promoting effect on PTEN protein level (Figure 5(e) and Figure 5(f), P < 0.01).
Figure 5.
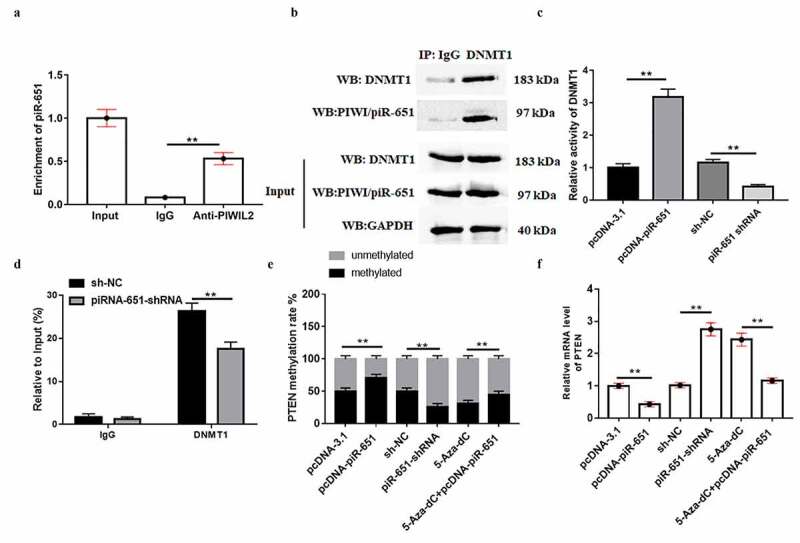
PIWI/piR-651 facilitates DNMT1-mediated PTEN promoter methylation. (a) RIP assay was used to determine the binding relationship between PIWIL2 and piR-651. (b) Co-IP was used to determine the interaction between PIWI/piR-651 and DNMT1. (c) The enzyme activity of DNMT1 was detected by ELISA. (d) The enrichment of DNMT1 in the PTEN promoter region was detected by ChIP assay. (e) MSP-PCR assay was used to detect the methylation level of PTEN promoter region in MDA-MB-231 cells. (f) RT-qPCR was used to detect the mRNA level of PTEN in MDA-MB-231 cells. 5-Aza-dC: DNMT1 inhibitor. N = 4, **P < 0.01. The difference of the samples between the two groups were analyzed with independent sample t test. The analysis of variance (ANOVA) test was used to evaluate multigroup comparisons of the means
Overexpression of PTEN antagonizes the promotive effects of piR-651 upregulation on breast cancer cell functions
The expression of PTEN in breast cancer tissues or cell lines was determined by Western blotting and RT-qPCR analysis, respectively. The results displayed that PTEN expression in breast cancer cell lines (AU565, HCC38, MCF-7 and MDA-MB-231) and the cancer lesions of breast cancer patients was prominently lower than that in normal breast epithelial cells (Hs-578Bst) and the para-carcinoma tissues (Figure 6(a-c), P < 0.01). Additionally, survival analysis of breast cancer patients showed that patients with low expression of PTEN had poor overall survival (Figure 6(d), p < 0.01). To further explore the effect of PTEN on piR-651 regulation of breast cancer cell proliferation, invasion, apoptosis and cell cycle, pcDNA-piR-651, pcDNA-PTEN and its corresponding negative control were transfected into MDA-MB-231 cells. Figure 6(e-Figure 6g) displayed that overexpression of PTEN had no significant effect on piR-651 expression, whereas the PTEN expression was observably decreased (P < 0.01). PTEN overexpression could reverse the facilitative effects of piR-651 overexpression on cell proliferation and invasion, as well as the inhibitory effect on cell apoptosis (Figure 6(h-l), P < 0.01). Flow cytometry analysis showed that PTEN overexpression reversed the regulation of the cell cycle by piR-651 upregulation and arrested cells in G0/G1 phase (Figure 6(m) and Figure 6(n), P < 0.01). Furthermore, Western blot assay demonstrated that PTEN overexpression reversed the promoting effects of piR-651 upregulated on the protein levels of MDM2, CDK4 and CyclinD1 (Figure 6(o), p < 0.01).
Figure 6.
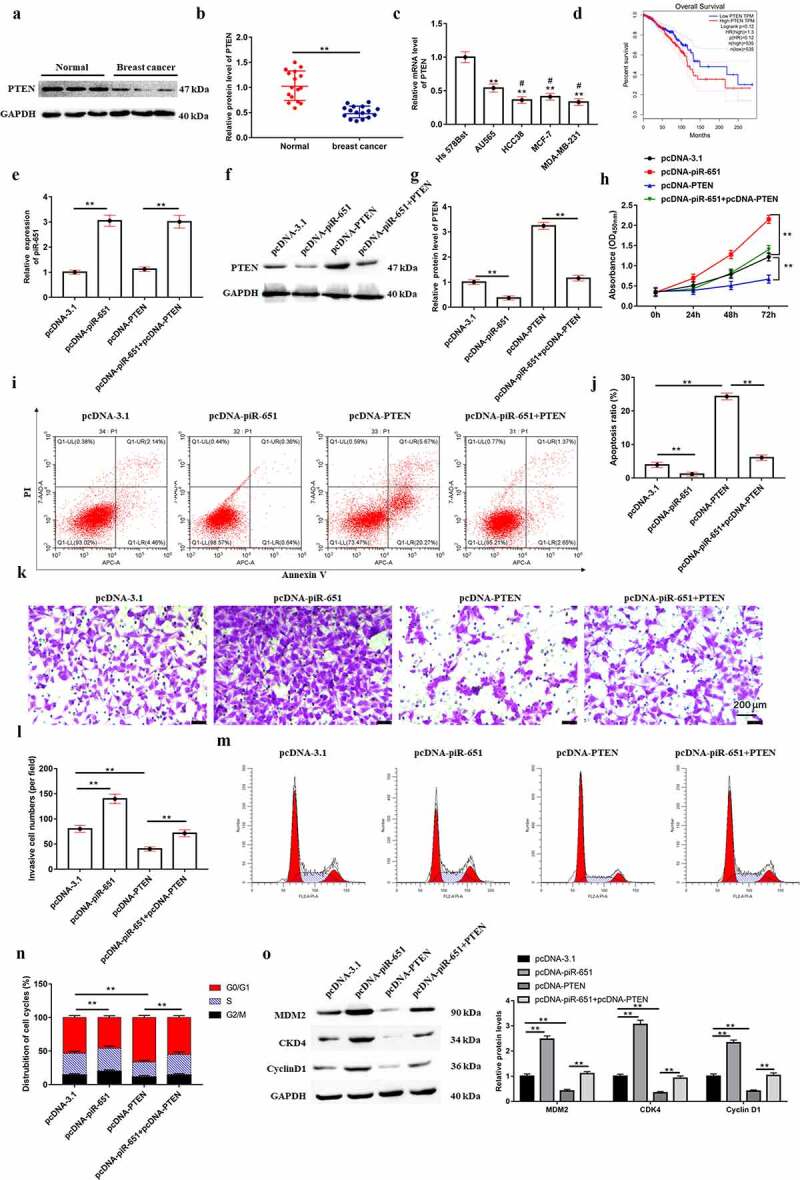
Overexpression of PTEN antagonizes the promotive effects of piR-651 upregulation on proliferation and invasion in breast cancer cells. (a-b) The expression of PTEN in breast cancer tissue and adjacent tissue samples collected from 16 patients (37.93 ± 10.05 years) was detected by Western blotting. ** P < 0.01, unpaired t test. (c) Relative expression of PTEN in breast cancer cells and normal breast epithelial cells was detected by RT-qPCR. (d) The survival analysis of breast cancer patients by PTEN expression. pcDNA-piR-651, pcDNA-PTEN and its corresponding negative control were transfected into MDA-MB-231 cells for 24 h. (e) RT-qPCR was used to perform the expression of piR-651 in MDA-MB-231 cells. (f-g) Western blotting was used to perform the expression of PTEN in MDA-MB-231 cells. (h) The cell viability of MDA-MB-231 cells was detected by CCK-8 assay. (i-j) The cell apoptosis ability of MDA-MB-231 cells was determined by flow cytometry. (k-l) The cell invasion ability of MDA-MB-231 cells was determined by Transwell assay. (m-n) The percentage of MDA-MB-231 cells in each phase of the cell cycle was conducted by flow cytometry analysis. (o) Western blotting was used to detect the protein levels of MDM2, CKD4 and CyclinD1 in MDA-MB-231 cells. N = 4, **P < 0.01. The difference of the samples between the two groups were analyzed with independent sample t test. The analysis of variance (ANOVA) test was used to evaluate multigroup comparisons of the means
Discussion
Increasing evidence indicates that abnormal piRNAs expression is detected in many human cancers and cell lines, including breast cancer, and their various functions are demonstrated by regulating several biological features. For example, PIWIL2/piR-932 is a positive regulator in the process of breast cancer stem cells by promoting the methylation of Latexin [12]. The expression of piR-651 was reported to be higher in gastric, colon, lung and breast cancer tissues than in paired non-cancerous tissues [13]. PiR-651 regulated lung tumorigenesis by promoting the highly metastatic properties of human lung cancer cell line 95-D [10]. In addition, a study found that upregulation of piR-651 in non‑small cell lung carcinoma cells resulted in a significant increase in cell viability and metastasis, as well as with a decrease in the percentage of cells arrested in G0/G1 phase [14]. Consistent with previous findings, our results verified that piR-651 was upregulated in breast cancer cell lines, including MDA-MB-231 and MCF-7, and piR-651 overexpression significantly promoted cell proliferation and migration of breast cancer cells. The flow cytometry assays confirmed that the piR-651 overexpression markedly induced cell apoptosis and arrested cells in G2/M phase by regulating cell cycle, while piR-651 interference showed the opposite results. So, we inferred that the biological function of piR-651 was closely related to the proliferation, invasion, apoptosis and cell cycle of breast cancer cells. Subsequently, piR-651 regulates the expression of tumor-related genes, which further confirms this hypothesis.
Epigenetics is the study of inheritable modifications of gene expression and regulation, including DNA methylation, histone modification, genomic imprinting, and small molecule RNA-related gene silencing [15]. DNA methylation has critical roles in the control of gene activity. Hypermethylation of CpG islands in the promoter region of tumor suppressor genes is a major factor in the development of many cancers. Hypermethylation and inactivation of these genes could affect several cellular processes, such as cell cycle, DNA repair, cell–cell interactions, carcinogen metabolism, angiogenesis and apoptosis, all of which may be involved in the development of cancer [16]. Methylation of genomic DNA is catalyzed by DNA methyltransferases (DNMTs), which are essential for cytosine methylation and maintenance of methylation throughout cell replication [17]. Enhanced expression of DNMTs is associated with hypermethylation of tumor suppressor genes in breast cancer [18]. Additionally, PIWIL4/piR-31,470 was able to recruit DNA methyltransferase 1 or DNA methyltransferase 3B and methyl-CpG binding domain protein 2 to initiate and maintain the hypermethylation and inactivation of GSTP1, which in turn suppressed GSTP1 levels and increased susceptibility to oxidative stress and DNA damage in human prostate epithelial cancer cells [8]. Our results demonstrated that the methylation level of PTEN gene promoter is regulated by piR-651, which could promote PTEN methylation by recruiting methyltransferase DNMT 1 to the PTEN promoter region in MDA-MB-231 cells, thereby inhibiting PTEN expression.
PTEN, a tumor suppressor gene, exists in various tumor cell chromosomes and is located at 10q23.3. In the nucleus, PTEN promotes chromosome stability and DNA repair. Conversely, loss of PTEN function increases genomic instability [19]. PTEN is mutated in variety of cancers. For example, PTEN is inhibited in prostate cancer cells, and Notch can participate in the development of epithelial tumors by regulating PTEN/PI3K/Akt and other pathways [20]. Furthermore, Methylation of the PTEN promoter is directly associated with gastric cancer, hematotoxicity and other diseases. CagA promotes the methylation of PTEN and reduces its expression in human gastric cancer [21]. PTEN methylation is involved in benzene-induced hematotoxicity through suppressing PTEN mRNA expression [22]. Similarly, loss of PTEN expression has been observed during the progression of breast cancer, in which aberrant promoter methylation is one of the silencing mechanisms of PTEN. Recent studies have reported that PTEN hypermethylation is one of the possible mechanisms leading to the levels of PTEN decreased in human breast cancer [23]. Tamoxifen promotes PTEN promoter methylation by recruiting DNMT1, thereby alleviating drug resistance in breast cancer cells [24]. Consistent with this, our results illustrate that PTEN expression in breast cancer cell lines and the cancer lesions of breast cancer patients was prominently lower than that in normal breast epithelial cells and the para-carcinoma tissues. Furthermore, PTEN overexpression reversed the promotive effects of piR-651 upregulated on proliferation and invasion and inhibitory effects on apoptotic ability in MDA-MB-231 cells, and arrested cells in G0/G1 phase by regulating cell cycle. Previous reports have reported that a range of oncogenes, including MDM2, CDK4 and Cyclin D1, are amplified and overexpressed in a variety of tumor types, including breast cancer [25–27]. Our results displayed that upregulation of piR-651 restrained the expression of MDM2, CDK4 and CyclinD1, while PTEN overexpression reversed these effects.
In conclusion, the present study demonstrated that piR-651 promotes proliferation and migration and inhibits apoptosis of breast cancer cells by facilitating DNMT1-mediated PTEN promoter methylation, which may have applications as a potential diagnostic indicator and therapeutic target in the management of breast cancer.
Supplementary Material
Funding Statement
This study was supported by the National Natural Science Foundation of China (Grant No. 81871366)
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Wang L, Zhang S, Wang X . The metabolic mechanisms of breast cancer metastasis. Front Oncol. 2020;10:602416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wei W, Zeng H, Zheng R, et al. Cancer registration in China and its role in cancer prevention and control. Lancet Oncol. 2020;21:e342–e349. [DOI] [PubMed] [Google Scholar]
- [3].Chen W, Zheng R, Zeng H, et al. Annual report on status of cancer in China, 2011. Chin J Cancer Res. 2015;27:2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Liu Y, Dou M, Song X, et al. The emerging role of the piRNA/piwi complex in cancer. Mol Cancer. 2019;18:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Erber R, Meyer J, Taubert H, et al. PIWI-Like 1 and PIWI-like 2 expression in breast cancer. Cancers (Basel). 2020;12:2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Vychytilova-Faltejskova P, Stitkovcova K, Radova L, et al. Circulating PIWI-interacting RNAs piR-5937 and piR-28876 are promising diagnostic biomarkers of colon cancer. Cancer Epidemiol Biomarkers Prev. 2018;27:1019–1028. [DOI] [PubMed] [Google Scholar]
- [7].Zhou X, Liu J, Meng A, et al. Gastric juice piR-1245: a promising prognostic biomarker for gastric cancer. J Clin Lab Anal. 2020;34:e23131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang L, Meng X, Pan C, et al. piR-31470 epigenetically suppresses the expression of glutathione S-transferase pi 1 in prostate cancer via DNA methylation. Cell Signal. 2020;67:109501. [DOI] [PubMed] [Google Scholar]
- [9].Fu A, Jacobs DI, Hoffman AE, et al. PIWI-interacting RNA 021285 is involved in breast tumorigenesis possibly by remodeling the cancer epigenome. Carcinogenesis. 2015;36:1094–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yao J, Wang YW, Fang BB, et al. piR-651 and its function in 95-D lung cancer cells. Biomed Rep. 2016;4:546–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li PF, Chen SC, Xia T, et al. Non-coding RNAs and gastric cancer. World J Gastroenterol. 2014;20:5411–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang H, Ren Y, Xu H, et al. The expression of stem cell protein Piwil2 and piR-932 in breast cancer. Surg Oncol. 2013;22:217–223. [DOI] [PubMed] [Google Scholar]
- [13].Cheng J, Guo JM, Xiao BX, et al. piRNA, the new non-coding RNA, is aberrantly expressed in human cancer cells. Clin Chim Acta. 2011;412:1621–1625. [DOI] [PubMed] [Google Scholar]
- [14].Li D, Luo Y, Gao Y, et al. piR-651 promotes tumor formation in non-small cell lung carcinoma through the upregulation of cyclin D1 and CDK4. Int J Mol Med. 2016;38(3):927–936. [DOI] [PubMed] [Google Scholar]
- [15].Nebbioso A, Tambaro FP, Dell’Aversana C, et al. Cancer epigenetics: moving forward. PLoS Genet. 2018;14:e1007362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang YP, Lei QY.. Metabolic recoding of epigenetics in cancer. Cancer Commun (Lond). 2018;38:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19:81–92. [DOI] [PubMed] [Google Scholar]
- [18].Leppert S, Matarazzo MR. De novo DNMTs and DNA methylation: novel insights into disease pathogenesis and therapy from epigenomics. Curr Pharm Des. 2014;20:1812–1818. [DOI] [PubMed] [Google Scholar]
- [19].Álvarez-Garcia V, Tawil Y, Wise HM, et al. Mechanisms of PTEN loss in cancer: it’s all about diversity. Semin Cancer Biol. 2019;59:66–79. [DOI] [PubMed] [Google Scholar]
- [20].Bertrand FE, McCubrey JA, Angus CW, et al. NOTCH and PTEN in prostate cancer. Adv Biol Regul. 2014;56:51–65. [DOI] [PubMed] [Google Scholar]
- [21].Zhang B, Zhang X, Jin M, et al. CagA increases DNA methylation and decreases PTEN expression in human gastric cancer. Mol Med Rep. 2019;19:309–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yang J, Zuo X, Bai W, et al. PTEN methylation involved in benzene-induced hematotoxicity. Exp Mol Pathol. 2014;96:300–306. [DOI] [PubMed] [Google Scholar]
- [23].Zhao D, Lu X, Wang G, et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature. 2017;542:484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Phuong NT, Kim SK, Lim SC, et al. Role of PTEN promoter methylation in tamoxifen-resistant breast cancer cells. Breast Cancer Res Treat. 2011;130:73–83. [DOI] [PubMed] [Google Scholar]
- [25].Haupt S, Vijayakumaran R, Miranda PJ, et al. The role of MDM2 and MDM4 in breast cancer development and prevention. J Mol Cell Biol. 2017;9:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Maeda A, Irie K, Hashimoto N, et al. Serum concentration of the CKD4/6 inhibitor abemaciclib, but not of creatinine, strongly predicts hematological adverse events in patients with breast cancer: a preliminary report. Invest New Drugs. 2021;39:272–277. [DOI] [PubMed] [Google Scholar]
- [27].Holah NS, Hemida AS. Cyclin D1 and PSA act as good prognostic and clinicopathological indicators for breast cancer. J Immunoassay Immunochem. 2020;41:28–44. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.