

ABSTRACT
Oncolytic viruses (OVs) can eradicate tumor cells and elicit antitumor immunity. VSV-GP, a chimeric vesicular stomatitis virus (VSV) with the glycoprotein (GP) of the lymphocytic choriomeningitis virus, is a promising new OV candidate. However, the interaction of VSV-GP with host immune cells is not fully understood. Dendritic cells (DCs) are essential for inducing efficient antitumor immunity. Thus, we aimed to investigate the interaction of VSV-GP with different murine and human DCs subsets in direct comparison to the less cytopathic variant VSV-dM51-GP and wild type VSV. Immature murine bone marrow-derived DCs (BMDCs) were equally infected and killed by VSV and VSV-GP. Human monocyte-derived DCs (moDCs) were more permissive to VSV. Interestingly, VSV-dM51-GP induced maturation instead of killing in both BMDCs and moDCs as well as a pronounced release of pro-inflammatory cytokines. Importantly, matured BMDCs and moDCs were no longer susceptible to VSV-GP infection. Mouse splenic conventional DC type 1 (cDC1) could be infected ex vivo by VSV and VSV-GP to a higher extent than cDC2. Systemic infection of mice with VSV-GP and VSV-dM51-GP resulted in strong activation of cDCs despite low infection rates in spleen and tumor tissue. Human blood cDC1 were equally infected by VSV and VSV-GP, whereas cDC2 showed preferential infection with VSV. Our study demonstrated differential DC infection, activation, and cytokine production after the treatment with VSV and VSV-GP variants among species and subsets, which should be taken into account when investigating immunological mechanisms of oncolytic virotherapy in mouse models and human clinical trials.
KEYWORDS: Oncolytic viruses, dendritic cells, vesicular stomatitis virus, infection
Introduction
The critical importance of the immune system for the treatment of cancer has become evident over the last decades and several immunotherapies have been shown to induce a durable immune response against the tumor.1 As one of the most potent antigen-presenting cell (APC) populations, dendritic cells (DCs) are crucial for the initiation of an efficient antitumor immune response in cancer patients due to their unique capacity to elicit T cell responses and activate innate immune cells.2 Furthermore, DCs orchestrate lymphocyte infiltration into tumor lesions and contribute to the efficacy of immunotherapies such as checkpoint inhibitors (CPIs) or adoptive T cell therapy.3–5 Moreover, it has been reported that high numbers of conventional DC subsets with a mature phenotype in tumor lesions correlate with better clinical outcomes.6–9 DCs are classified based on their ontogeny and function. Conventional DCs (cDC), subdivided in cDC type 1 (cDC1) and type 2 (cDC2), are the most potent CD8+ and CD4+ T cell activators, respectively.10,11 Plasmacytoid DCs (pDC) are type I interferon (IFN) producers, specifically involved in antiviral responses but can also modulate tumor immunity.12 Monocyte-derived DCs (moDCs) are functionally very heterogeneous and the generation of moDCs requires inflammatory stimuli.13 Human moDCs and murine bone marrow-derived DCs (BMDCs) are often used for in vitro studies.10 Moreover, due to their pivotal function in cancer immunology, DCs are also harnessed as cell-based vaccines for immunotherapy.2 In sum, due to their highly important functions, DCs are widely evaluated for cancer treatment and the increasing knowledge on DC heterogeneity helps to constantly improve therapies.14,15
Oncolytic virotherapy represents a new promising approach for cancer treatment. Oncolytic viruses (OVs) not only target and eradicate tumor cells but initiate also antitumor immunity and modulate the tumor microenvironment (TME). Thereby, the immune tolerance of the tumor can be overridden.16 The vesicular stomatitis virus (VSV) belongs to the family of Rhabdoviridae and is an oncolytic agent currently under pre-clinical and clinical testing.17 However, the neurotropism of VSV presents a safety concern. The exchange of the glycoprotein G of VSV with the glycoprotein GP of the lymphocytic choriomeningitis virus (LCMV) abrogates VSV’s neurotoxicity while maintaining the oncolytic activity.18 This chimeric virus called VSV-GP is safe, induces strong antigen-specific immune responses but low vector-neutralizing antibodies, and is applicable as OV19–22 or vaccine vector.23,24
Regarding oncolytic virotherapy, little is known how OVs interact or affect APCs. Previous studies suggest that virotherapy promotes DC functionality by enhancing tumor-antigen presentation. Importantly, OV-DC interactions can impact adaptive immune responses not only against the tumor but also against the oncolytic agent itself.25,26 It was demonstrated that VSV infects DCs in vitro.27–29 However, the chimeric VSV-GP might behave differently compared to wild-type VSV in terms of host immune cell interaction. Thus, we aimed to investigate the infection of DCs by VSV-GP in detail. We examined infection of in vitro generated murine BMDCs and human moDCs as well as various mouse and human DC subsets ex vivo and in vivo. Additionally, the less cytopathic variant VSV-dM51-GP was included in our study. The M protein of VSV is responsible for counteracting antiviral responses by inhibiting host-directed gene expression. Virus variants bearing a mutation in the M protein are less pathogenic and have been favored as safer OV candidates.30–32 This study offers a head to head comparison of DC susceptibility to three vesicular stomatitis virus variants in the murine and human systems. Here, we demonstrated that infection levels, activation, and cytokine profiles of DCs treated with VSV, VSV-GP, and VSV-dM51-GP differ between subsets and species.
Methods
Cell lines and viruses
The hamster cell line BHK21 (clone 13, ECACC) was cultured in GMEM (Lonza, Basel, Switzerland) supplemented with 10% FCS (Life Technologies), 5% tryptose phosphate broth (Gibco, Carlsbad, CA, USA), and 2% L-glutamine. This cell line was used for virus production, titration, and control infection experiments. The melanoma B16-F10 interferon-alpha receptor (IFNAR) knock out cells (B16 IFNAR−/-) (kindly provided by Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany) were cultured in DMEM (Lonza) with 10% FCS (Life Technologies, Carlsbad, CA, USA) and 2% L-glutamine (Gibco).
VSV (Indiana serotype), VSV pseudotyped with the glycoprotein of LCMV (VSV-GP), and VSV-GP with a deletion of methionine at position 51 in the viral M protein (VSV-dM51-GP) were used for infection experiments. The three aforementioned viruses encode the fusion protein eGFP-ovalbumin at the fifth position in the viral genome and have been described elsewhere.23 VSV-GP encoding the Photius pyralis luciferase (VSV-GP-Luc) has been described previously.19 All viruses were propagated and titrated on BHK21 cells via the plaque and the TCID50 assay as reported previously.33
Isolation of peripheral blood mononuclear cells and generation of human monocyte-derived DCs
For the isolation of monocytes or peripheral blood mononuclear cells (PBMCs), whole blood (in citrate-phosphate-dextrose (CPD) bags (Fresenius Kabi, Graz, Austria) or EDTA monovettes (Sarstedt, Nümbrecht, Germany)) of healthy blood donors was obtained from the Central Institute of Blood Transfusion and Immunology (University Hospital of Innsbruck, Austria). Whole blood was centrifuged and the plasma was removed. The remaining blood cells were mixed 1:2 with phosphate-buffered saline (PBS) (Lonza) and placed onto a Pancoll (PAN Biotech, Aidenbach, Germany) gradient. After centrifugation, the interface was collected and washed two times with RPMI-1640 (Sigma-Aldrich, St. Louis, MO, USA) plus PBS (1:2). PBMCs were counted and 4 × 106 cells were seeded into six-well plates in RPMI supplemented with 10% FCS (Gibco), 2% L-glutamine (Gibco) for infection experiments. To generate human moDCs, monocytes were isolated from PBMCs by adherence to gelatin-coated plates as described previously.34 Isolated monocytes were cultured with human recombinant GM-CSF and IL-4 in the absence or presence of LPS as described previously to obtain immature and mature moDCs, respectively.34 At day 7, moDCs were harvested and the phenotype was routinely tested via flow cytometry (propidium iodide (PI) for discrimination of dead cells, CD11c, CD11b, and CD83). Cells were washed and 5 × 105 cells were seeded in 24-well plates for infection experiments.
Mice and ethics statement
Six to eight-week-old female C57BL/6JRj or C57BL/6JRj albino mice were purchased from Janvier (Le Genest St Isle, France) and housed under specific pathogen-free conditions at the animal facilities of the Medical University of Innsbruck. Animal experiments were approved by the animal ethics committees of the Medical University of Innsbruck and the Austrian Federal Ministry of Science and Research (BMWF-66.011/0092-WF/V/3b/2016; BMWF-66.011/0156-V/3b/2019). No animals were excluded from the described experiments. The exact animal numbers are given for each experiment.
Generation of bone marrow-derived DCs
The generation of BMDCs from C57BL/6Rj mice was performed as described.35 Shortly, isolated bone marrow cells were cultured for 8 days in presence of recombinant mouse (rm)GM-CSF (4 ng/ml, BD Pharmingen, San Jose, CA, USA). On the eighth day, loosely adherent cells were harvested and washed with RPMI and PBS (1:2). The phenotype of BMDCs was routinely tested via flow cytometry (PI for discrimination of dead cells, CD11c, CD11b, and CD86). For infection experiments, 5 × 105 cells were seeded in 24-well plates in RPMI-1640 supplemented with 10% FCS (Gibco), 2 mM L-glutamine (Gibco), and rmGM-CSF.
Virus infection
PBMCs, human moDCs, and mouse BMDCs were infected with either VSV, VSV-GP, or VSV-dM51-GP at a multiplicity of infection (MOI) of 1 and 10 according to plaque-forming unit (PFU) titer and were incubated for 6 to 48 h at 37°C. As a control for infection, BHK21 cells were treated with the virus variants as described above. Infection was monitored by determining GFP positive (GFP+) cells as all viruses encode GFP as a reporter gene. BHK21 cells were analyzed simultaneously to BMDCs and human moDCs at 6 to 24 h post infection (hpi). For the analysis of productive viral infection, input virus was removed by washing 3 times with PBS 1 h after infection. Supernatants were collected at 1, 24, and 48 hpi, and infectious viral particles were determined by TCID50 assays. The titers obtained from 1 hpi samples were subtracted from the respective 24 hpi and 48 hpi samples to exclude that determined titers originate from infectious input virus.
Mouse splenocyte infection
To generate single cell suspension, spleens were cut into small pieces and incubated in RMPI-1640 containing 0.4 mg/ml Collagenase P (Roche, Basel, Switzerland) and 0.1 mg/ml DNase I (Roche) for 30 min at 37°C in a water bath. After incubation, the whole suspension was smashed through a Falcon® 70 µm cell strainer (BD). Cells were washed with cold PBS. After the lysis of erythrocytes with BD Pharm Lyse™ (BD) according to the manufacture’s protocol followed by two washing steps, cell numbers were determined. For infection experiments, 5 × 106–1 × 107 splenocytes were cultured in RMPI-1640 supplemented with 10% FCS (Life Technologies), 2% L-glutamine (Gibco) in six-well plates. Cells were infected immediately with VSV, VSV-GP, and VSV-dM51-GP at an MOI of 1, 10, and 100 according to the PFU titer. Infection was analyzed after 6 h via flow cytometry.
In vivo studies
To analyze the effects of VSV-GP variants on splenic DCs, C57BL/6JRj mice were injected intravenously (i.v.) with either PBS (200 µl) or with VSV-GP and VSV-dM51-GP (108 TCID50 in 200 µl PBS). Spleens were harvested 6 or 12 hpi to determine viral RNA in splenocytes and to assess the DC phenotype via flow cytometry, respectively.
B16 IFNAR-/- cells (1 × 106) were implanted subcutaneously (s.c.) in 100 µl PBS into the right flank of albino and normal C57BL/6JRj mice. Tumor size was measured three times a week and tumor volume was calculated using the formula: length × width2× 0.4. Treatments were performed when tumors reached a size of 0.05 to 0.07 cm3. To investigate virus replication in the tumors, VSV-GP-Luc (108 TCID50 in 50 µl PBS) was injected intratumorally (i.t.). Luciferase imaging was performed up to 7 d post treatment as described previously using the IVIS Lumina II (Perkin Elmer, Waltham, MA, US).36 To assess infection of tumor-associated DCs (TADCs), tumors were treated i.t. with PBS, VSV-GP, or VSV-dM51-GP (108 TCID50 in 50 µl PBS). After 12 h, tumors were resected and minced in a digestion enzyme mix (RPMI-1640 supplemented with 0.4 mg/ml Collagenase P (Roche) and 0.1 mg/ml DNase I (Roche)). After incubation for 30 min at 37°C, the tumor cell suspension was filtered through a 70 µm cell strainer and washed two times with PBS supplemented with 2% FCS and 5 mM EDTA. Single cell suspensions were stained for flow cytometric analysis to assess virus infection of TADCs.
Conventional CD8+ DC isolation
To determine viral RNA after immunization in splenic DCs, cDC1 were isolated from spleens of PBS, VSV-GP or VSV-dM51-GP immunized mice using the CD8+ Dendritic Cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Single cell suspensions were generated as described above. CD8+ DCs were isolated according to the manufacturer’s protocol. The purity of isolated CD8+ DCs was assessed via flow cytometry and was always over 97%.
RNA extraction and RT-qPCR
Viral RNA was isolated from the supernatant of infected BMDCs and BHK21 cells at 1 h, 24 h, and 48 h post infection using the NucliSENS® easyMAG® system (BioMérieux, Marcy-l’Etoile, France) as recommended by the manufacturer.
RNA from isolated CD8+ cDC1 was extracted using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Viral RNA was quantified using reverse transcriptase (RT) quantitative PCR (qPCR) with the forward primer 5ʹ-AGT ACC GGA GGA TTG ACG ACT AAT-3ʹ, the reverse primer 5ʹ-TCA AAC CAT CCG AGC CAT TC-3ʹ, and the probe 5ʹ-FAM-ACC GCC ACA AGG CAG AGA TGT GGT-BH Q-3ʹ (all from Sigma Aldrich). RT-qPCRs were performed with the iTaq™ Universal Probes one-step kit (Bio-Rad, Hercules, CA, USA) in a 10 µl reaction mix. The reaction was run in duplicates on an iCycler iQ (Bio-Rad) with the following settings: 10 min at 50°C, 2 min at 95°C, followed by 40 cycles of at 95°C for 15 s and 60°C for 30 s. To normalize viral RNA to cell numbers RT-qPCRs for actin were performed using the forward primer 5ʹ-GTC CCT CAC CCT CCC AAA AG-3ʹ and the reverse primer 5ʹ-GCT GCC TCA ACA CCT CAA CCC-3ʹ (Sigma-Aldrich). Reactions were performed with iTaq™ Universal SYBR® Green One-Step kit (Bio-Rad) and run in duplicates with following settings: 10 min at 50°C, 1 min at 95°C, followed by 40 cycles of at 95°C for 10 s, 55°C for 20 s and 72°C for 30 s. The data were analyzed using an iCycler iQ data analysis module. Fold changes of viral RNA in the supernatant of infected BMDCs and BHK21 cells were calculated using the samples 2ΔCt method based on the respective 1 hpi samples.37
Viral genome copies from isolated CD8+ cDC1 were determined using a standard for VSV-N. The viral genome copies were normalized to actin gene expression of the PBS controls and virus treated samples.
Flow cytometry
To determine cell death, PI (Sigma-Aldrich) was used for the quality controls and infection experiments with BMDCs and human moDCs. The LIVE/DEADTM Fixable Dead Cell Stain Kit (L/D) (Thermo Fischer Scientific, Waltham, MA, USA) was used for the splenocyte infection experiments, for samples from in vivo experiments, and human PBMC samples according to the manufacturer’s instruction. PBS supplemented with 2% FCS and 5 mM EDTA was used for washing and staining the samples. In murine-derived samples, Fc-receptors were blocked by incubating cells for 10 min at 4°C with anti-mouse CD16/32 (BioXCell, Lebanon, New Hampshire, US). For intracellular VSV-N staining, the Transcription Factor Buffer Set (BD Pharmingen) was used as recommended. Samples for surface and intracellular staining were incubated with the antibodies at 4°C for 30 and 50 min, respectively. The anti-mouse antibodies CD8a (53–6.7), CD11b (M1/70), CD11c (HL3), CD45 (30-F11), CD86 (GL1), and MHCII (I-Ad/I-Ed) (2G9) were purchased from BD Biosciences. Additionally, CD11c (REA754) and XCR1 (REA707) were purchased from Miltenyi Biotec. The viral VSV-N protein was detected using the primary anti-VSV-N (10G4) from Kerafast (Boston, MA, USA) and the secondary IgG2a/IgG2b (R2-40, BD). An anti-IgG2 (TIB-109, in-house) was applied as an isotype control. The anti-α-dystroglycan antibody (D-3) was purchased by Santa Cruz Biotechnology (Dallas, TX, USA). The following antibodies against human CD3 (OKT3), CD11b (ICRF44), CD11c (B-ly6), CD14 (M5E2), CD19 (HIB19), CD20 (H1), CD45 (HI30), CD64 (10.1), CD83 (HB15e) and HLA-DR (G46-6) were purchased from BD. Additionally, anti-human CD1c (AD5-8E7), CD16 (REA423), CD123 (AC145) and CD141 (AD5-14H12) antibodies were purchased from Miltenyi Biotec. Stained samples were measured using the FACS Canto IITM cytometer (BD Biosciences) and data analysis was performed using the FlowJo software (BD).
Quantification of cytokines and chemokine
Murine BMDCs and human moDCs were infected with the virus variants at an MOI of 10 as described. As a positive control, BMDCs and moDCs were treated with 100 ng/µl and 0.5 µg/µl LPS (Enzo Life Sciences, New York, USA), respectively. Supernatants were collected at 6 and 24 hpi. Cytokines from murine BMDCs were tested using the bead-based immunoassay Legendplex™ (Biolegend, San Diego, CA, USA) in the mouse inflammation panel (IL-23, IL-1α, IFN-γ, TNF-α, MCP-1, IL-12p70, IL-1β, IL-10, IL-6, IL-27, IL-17A, and IFN-β) according to the manufacturer’s instructions. Supernatants of human moDCs were run in the Legendplex™ human inflammation panel (IL-1β, IFN-α2, IFN-γ, TNF-α, MCP-1, IL-6, IL-8, IL-10, IL-12p70, IL-17A, IL-18, IL-23, and IL-33) and in the Legendplex™ human virus panel to assess IFN-λ1, IFN-λ2/3, and IP-10. Of note, GM-CSF was excluded from the analysis due to exogenous supplementation in BMDC and moDC culture medium.
Statistics
GraphPad Prism software (version 9, GraphPad Software, La Jolla, CA, USA) was used for statistical analysis. Kruskal-Wallis non-parametric analysis with Dunn’s post hoc test was applied to assess significance levels. Data are presented as mean ± SD or SEM as noted. Statistically significant differences were encoded as follows: *p < .05; **p < .01; ***p < .001; ****p < .0001, not significant (ns).
Results
VSV-GP infects mouse bone marrow-derived DCs as efficiently as VSV, but cells are less susceptible to VSV-dM51-GP
To test the susceptibility of DCs in vitro, first BMDCs were infected with VSV, VSV-GP, and VSV-dM51-GP at an MOI of 1 and 10. As all viruses encoded GFP as a reporter, BMDC infection was monitored by determining GFP+ infected cells at different time points. We found that VSV-GP infected BMDCs at similar levels as VSV, reaching about 25% and 35% GFP+ cells after 6 h at an MOI of 1 and 10, respectively (Figure 1(a)). VSV-dM51-GP showed significantly lower infection of BMDCs at 6 hpi and 24 hpi. Of note, simultaneous infections of BHK21 cells revealed similar infection levels for all 3 viruses at both time points (Supplementary Figure 1). After 24 h, low levels of GFP+ cells were only detectable after VSV-dM51-GP infection. To further investigate BMDC infection, intracellular staining against the viral N protein was performed. Whereas anti-VSV-N and GFP signals were similar after 6 h (Figure 1(b)), an increase in VSV-N positive cells (VSV-N+) was observed at 24 hpi. This indicates that GFP may be degraded within 24 h while the N protein or potential N degradation products can still be detected by the anti-VSV-N antibody. Another explanation could be quenching of the GFP signal in acidic cellular compartments or the acidic environment in dying cells.38,39 When high virus concentrations (MOI 10) were applied, around 50–60% of BMDCs after VSV and VSV-GP and 35% after VSV-dM51-GP application were infected (VSV-N+). Conclusively, both detection methods corroborate that VSV-GP variants can infect BMDCs and infection was not significantly different for VSV-GP compared to the wild type. The detection of VSV-N protein in BMDCs required active virus replication as UV light-inactivated virus did not result in detectable VSV-N+ cells in the culture (Supplementary Figure 2a).
Figure 1.
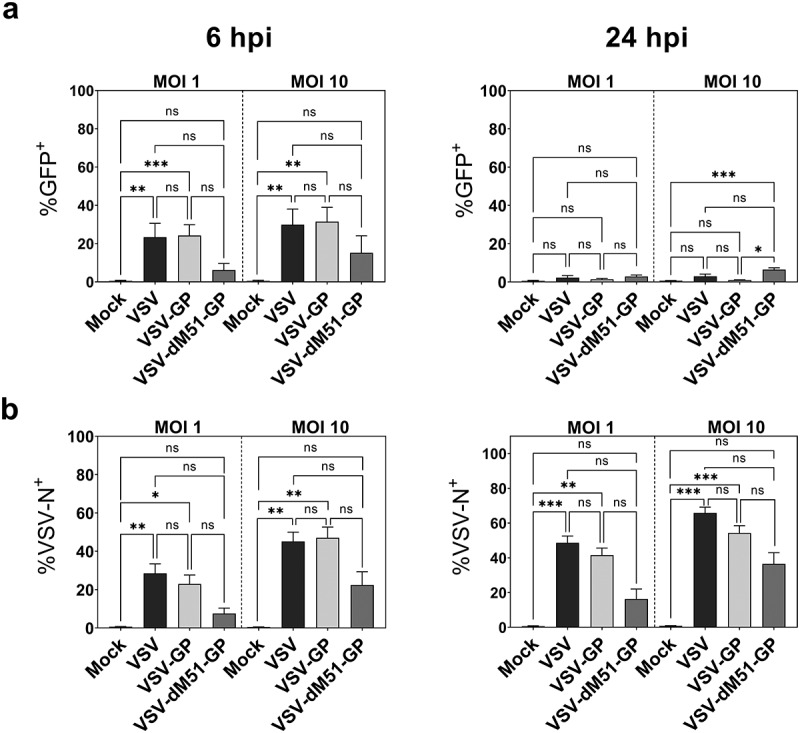
Infection of mouse BMDCs with VSV, VSV-GP, and VSV-dM51-GP
a GM-CSF bone marrow-derived DCs (BMDCs) were infected with the different virus variants at an MOI of 1 and MOI 10 according to PFU titers. GFP encoded by the viruses was measured via flow cytometry 6 and 24 h post infection (hpi). b Intracellular staining for the viral N protein was performed and analyzed via flow cytometry. Data represent at least five independent experiments. Mean ± standard deviations (SD) are depicted *p < .05; **p < .01; ***p < .001; not significant (ns); non-parametric Kruskal–Wallis test, followed Dunn’s multiple comparisons post-hoc test.
VSV-GP preferentially infects and kills immature BMDCs while VSV-dM51-GP induces maturation of BMDC cultures
On day 8, murine BMDCs generated in the presence of GM-CSF contained DCs in different maturation states as shown in the representative dot plot in Figure 2(a). Cells expressing CD11c represented about 85–90% in BMDC cultures including 60–70% immature DCs (iDCs, CD11c+CD86−) and 15–25% mature DCs (mDCs, CD11c+CD86+). Of note, CD11b+ cells that neither express CD11c nor CD86 remained in the culture (nonDCs, 10–15%). As they differ in their phenotype and function, we next examined the infection of iDCs and mDCs by VSV, VSV-GP, and VSV-dM51-GP. All three virus variants preferentially infected immature DCs as shown in Figure 2(a). GFP+ cells made up about 30% within the iDC population at 6 hpi if infected with VSV or VSV-GP, whereas VSV-dM51-GP application resulted in 10% infected iDCs. The frequency of GFP+ mDCs was under 15% for VSV and under 5% for VSV-GP variants. Representative dot plots of infected iDCs and mDCs are given in Supplementary Figure 2b. Of note, the preferential iDC infection by VSV-GP variants could not be explained by a differential expression of the entry receptor, as α-dystroglycan (α-DG) was expressed to similar levels on iDCs and mDCs (Supplementary Figure 2c). We also investigated the productive infection of DCs. An increase of viral RNA could be detected in the supernatants for all three viruses after 24 and 48 h (Supplementary Figure 3a). Infectious virus particles were analyzed via a TCID50 assay. Low levels of virus progeny were produced by BMDCs for all virus variants (Supplementary Figure 3b). Generally, the levels of viral RNA and infectious virus particles derived from BMDCs were much lower compared to highly susceptible BHK21 cells.
Figure 2.
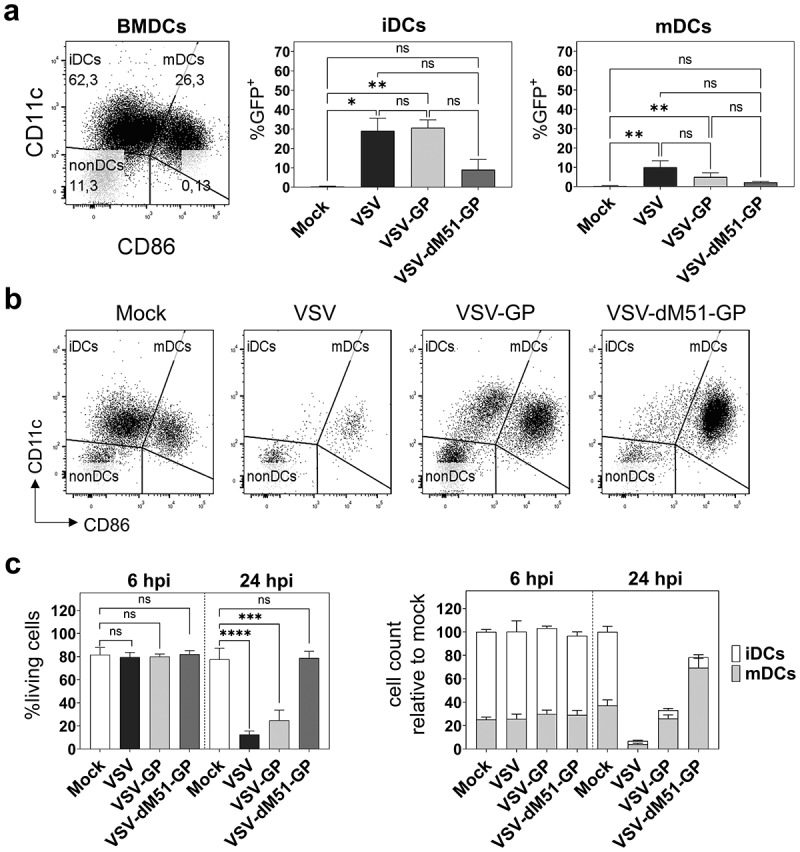
VSV-GP infects and kills immature BMDCs while VSV-dM51-GP leads to maturation of BMDC cultures
BMDCs were infected with VSV, VSV-GP, and VSV-dM51-GP at an MOI of 1. a A representative flow cytometry dot plot of a BMDC culture is depicted (top, left). GFP encoded by the virus was measured via flow cytometry at 6 hpi post. Combined data of GFP+ infected iDCs and mDCs are given. b Representative dot plots of either mock or virus infected living BMDCs (PI−) after 24 h are shown. c BMDCs were infected as mentioned above and cell viability was analyzed via flow cytometry after 6 and 24 h. Combined graphical data are shown (left). Cell counts of iDCs and mDCs were calculated relative to the cell counts of the respective mock infected controls (right). Data are shown as mean ± SD and represent five independent experiments. *p < .05; **p < .01; ***p < .001; ****p < .0001; not significant (ns); non-parametric Kruskal–Wallis test, followed Dunn’s multiple comparisons post-hoc test.
As VSV has been reported to kill BMDCs,40 cell viability was determined via flow cytometry. No differences in viability were observed at 6 hpi compared to non-infected cells (mock). However, after 24 h only up to 10–15% of VSV infected DCs were alive. In contrast, VSV-GP killed fewer cells (Figure 2(b,c)). Counting of living iDCs and mDCs and calculating the ratio to non-infected controls revealed that the living mDCs remained stable and mainly iDCs were killed by VSV-GP (Figure 2(c)). Although a substantial proportion of BMDCs was infected by VSV-dM51-GP, cells were hardly killed by this virus variant. Instead, an almost complete maturation was observed and total cell counts remained stable (Figure 2(b,c)). The prolonged survival may be responsible for the remaining GFP signal after 24 h in VSV-dM51-GP infected BMDCs (Figure 1(a)).
Splenic conventional DCs type 1 are preferentially infected by VSV-GP variants ex vivo
Our data gave clear evidence that BMDCs can be infected by VSV and VSV-GP variants. However, in vitro generated BMDCs remain only a model system and do not fully reflect the diversity of DC subsets in vivo. Upon intravenous delivery of VSV-GP, one of the main target organs is the spleen representing an important secondary lymphoid organ where the adaptive immune response is generated. Infection of splenic DC subsets could potentially influence the adaptive immune response against the tumor and the OV. Thus, we investigated VSV-GP infection of splenic DC subsets ex vivo. Single cell suspensions of splenocytes were generated and cells were either mock infected or infected with an MOI of 1, 10, or 100 of VSV, VSV-GP, and VSV-dM51-GP. The infection of total resident DCs (resDCs; MHCII+CD11chigh), and the subsets cDC1 (XCR1+CD8+CD11b−), and cDC2 (XCR1−CD11b+) was analyzed via flow cytometry after 6 h. The gating strategy for the splenic resDCs and the cDC subsets is presented in Supplementary Figure 4a. Generally, infection levels of resDCs were found to be relatively low, only reaching significant levels when higher MOIs of VSV and VSV-GP were applied. The treatment with VSV-dM51-GP did not result in a statistically significant infection level (Figure 3). Six hours after VSV and VSV-GP application, GFP+ infected cells were detectable among cDCs. Up to 10% of cDC1 and around 5% of cDC2 were GFP positive after application of VSV-GP and VSV, respectively (MOI 100). In contrast, , cDC1 infection by VSV-dM51-GP was only significant when the highest MOI was applied and cDC2 infection was negligible. Representative dot plots of infected cells among resDCs, cDC1 and cDC2 are given in Supplementary Figure 4b. Taken together, VSV and VSV-GP equally infected murine BMDCs as well as splenic cDC1 and cDC2 in vitro. VSV-dM51-GP showed generally lower infections.
Figure 3.

VSV-GP variants and VSV infect splenic conventional DCs ex vivo
Single cell suspensions of naïve mouse spleens were prepared via enzymatic digestion. Total cells were mock infected or infected with VSV, VSV-GP, and VSV-dM51-GP at an MOI of 1, 10, or 100 according to PFU titers. Infection levels among resident DCs (resDCs), conventional type 1 DCs (cDC1), and conventional type 2 DCs (cDC2) were determined by GFP positivity as all viruses encode GFP as a reporter after 6 h via flow cytometry. The data are shown as mean ± SD and are obtained from five independent experiments. *p < .05; **p < .01; ***p < .001; not significant (ns); non-parametric Kruskal–Wallis test, followed Dunn’s multiple comparisons post-hoc test compared to mock control.
Low infection of splenic and tumor-associated DCs after systemic VSV-GP and VSV-dM51-GP application
Next, we aimed to investigate infection of splenic DCs in vivo upon systemic virus treatment. The high virus concentrations required for significant ex vivo infection already indicated a low permissiveness of splenic DCs. Accordingly, we were not able to detect GFP+ splenocytes via flow cytometry after applying either VSV-GP or VSV-dM51-GP intravenously (i.v.), which might be due to technical limitations when only low numbers of DCs are infected. To increase detection sensitivity, susceptibility was assessed by analyzing viral genome copies in isolated splenic cDC1. Mice were treated with PBS or infected with VSV-GP or VSV-dM51-GP (108 TCID50, i.v.), spleens were harvested after 6 h. CD8+ cDC1 were purified via magnetic bead separation. As a positive control, we isolated cDC1 from ex vivo VSV-GP infected splenocytes (MOI 100, 6 hpi), where we observed around 10% GFP+ infected cDC1 (Figure 3). An RT-qPCR for VSV-N was performed and viral genome copies were calculated based on a standard curve. The viral genome copies were normalized to actin. Similar numbers of viral genome copies were detected in cDC1 of VSV-GP and VSV-dM51-GP infected mice (Figure 4(a)). However, the number of viral genomes was 2-fold lower compared to the ex vivo infected cDC1. These results show that splenic cDC1 are susceptible to VSV-GP variants after i.v administration but direct infection may be a rare event in vivo.
Figure 4.

Infection of splenic DCs and tumor-associated DC is low in vivo but VSV-GP and VSV-dM51-GP activated the DC subsets
a CD8+ cDC1 from either ex vivo infected splenocytes (VSV-GP, MOI 100) or spleens of infected mice were isolated. Viral genome copies were determined via RT-qPCR in isolated CD8+ cDC1 and normalized to actin. Data are shown are means ± SD obtained from three independent experiments. b C57BL/6Rj mice were treated i.v. with PBS or infected with VSV-GP or VSV-dM51-GP (108 TCID50). Spleens were harvested after 12 h. Activation of conventional DCs type 1 and type 2 (cDC1, cDC2) was determined via the geometric mean fluorescent intensity (gMFI) of MHCII and CD86. Data are shown as mean (n = 5). *p < .05; not significant (ns); non-parametric Kruskal–Wallis test, followed Dunn’s multiple comparisons post-hoc test. c B16 IFNAR-/- mouse melanoma tumors were implanted in syngeneic albino C57BL/6Rj mice. Tumors were treated i.t. with either PBS or with VSV-GP expressing the firefly luciferase (VSV-GP-Luc, 108 TCID50). Virus replication was analyzed using the in vivo imaging system (IVIS). Representative pictures from PBS and VSV-GP-Luc treated mice after 12, 24, and 48 h are shown. The average radiances in the tumor area over time are presented (n = 5; mean ± SEM). d B16 IFNAR-/- mouse melanoma tumors were treated i.t. with PBS, VSV-GP, or VSV-dM51-GP (108 TCID50) and resected after 12 h. Frequencies of GFP+ infected XCR1+ tumor-associated DCs (XCR1+ TADCs) are summarized graphically (left). The gMFI of MHCII on XCR1+ TADCs is shown (right). Data are shown as mean (n = 5 to 7 mice per group). *p < .05; **p < .01; not significant (ns); non-parametric Kruskal–Wallis test, followed Dunn’s multiple comparisons post-hoc test.
As we observed a strong maturation of BMDCs by VSV-dM51-GP, we next analyzed if DC maturation could be induced in vivo. Thus, mice were either treated with PBS or infected with the VSV-GP and VSV-dM51-GP (i.v., 108 TCID50), and spleens were harvested after 12 h. An upregulation of MHCII on splenic resDCs was observed in virus infected mice compared to control animals (PBS) (Supplementary Figure 5a). The increased geometric mean fluorescent intensity (gMFI) of CD86 and MHCII on cDC1 and cDC2 in infected animals indicated that VSV-GP and VSV-dM51-GP application activated and matured both cDC subsets (Figure 4(b)).
DC infection in lymphoid organs might be limited as VSV-GP is assumed to be suppressed rapidly after administration due to type I interferon (IFN) responses.19 However, cancer cells have often limited potential to respond to viral infection due to defects in the IFN pathway. Thus, VSV-GP replication retains in tumor cells. Therefore, we next investigated whether tumor-associated DCs (TADCs; MHC-II+CD11c+XCR1+CD11b−, referred to as XCR1+ TADCs) can be infected when the virus is present locally nearby in the tumor. Even if certain tumor cell lines are defective in intrinsic IFN pathways, some, such as the B16 mouse melanoma tumors, can respond to exogenous IFN.41 Hence, we used the B16 IFNAR-/- cells as it was shown that IFNAR deficiency prolongs virus replication.22 Virus replication in the tumor was confirmed by in vivo bioluminescence imaging (BLI) using a VSV-GP variant expressing the firefly luciferase (VSV-GP-Luc). In comparison to the PBS treated group, i.t. injection of VSV-GP-Luc (108 TCID50) resulted in measurable bioluminescence signal up to 5 d (Figure 4(c)). The strongest virus activity was determined between 12 and 24 h post treatment. Next, XCR1+ TADC infection was analyzed in this model. For this, established B16 IFNAR-/- tumors were treated i.t. with either PBS, VSV-GP, or VSV-dM51-GP (108 TCID50), and tumors were resected after 12 h. In general, XCR1+ TADCs form a minor population of MHCII+CD11c+ myeloid cells in B16 IFNAR−-/- tumors (Supplementary Figure 5b). In comparison to PBS treated tumors, GFP+ cells could be detected among XCR1+ TADCs of around 6.3% and 4.6% after VSV-GP and VSV-dM51-GP application, respectively (Figure 4(d), left). Of note, we cannot completely exclude that GFP+ infected tumor cells incorporated by DCs contribute to the GFP signal. Interestingly, XCR1+ TADCs of virus treated tumors showed higher gMFI of MHCII than PBS treated tumors (Figure 4(d), right). This may indicate that both virus variants mature XCR1+ TADCs, although it was not statistically significant for VSV-dM51-GP.
Infection of human monocyte-derived DCs is lower for VSV-GP variants
VSV-GP infected mouse DCs to a similar extent as wild type VSV. Therefore, we addressed the question of whether this also applies to human DCs. Human monocyte-derived DCs (moDCs) were generated from peripheral monocytes of healthy blood donors. The DC phenotype (CD11c+CD11b+) and maturation status (CD83+/-) were routinely analyzed before each experiment. Whereas unstimulated moDCs showed a complete immature (iDCs) phenotype, the application of lipopolysaccharide (LPS) matured moDCs (mDCs) (Supplementary Figure 6a). Frequencies of GFP+ infected cells and moDC viability were determined 6 and 24 h after infection with VSV, VSV-GP, and VSV-dM51-GP. In contrast to murine BMDC, which were equally infected by VSV and VSV-GP, human moDCs were less susceptible to VSV-GP variants compared to wild type VSV. Even with high virus amounts (MOI 10) of VSV-GP and VSV-dM51-GP, cells were not infected to a significant extent after 6 h (Figure 5(a)). Conversely, VSV infected immature moDCs very efficiently, reaching up to 45% GFP+ cells at 6 hpi. After 24 h, frequencies of VSV infected cells elevated to 65%, while VSV-GP infection of moDCs (25%) was still lower (Figure 5(a)). The viability of VSV-GP infected moDCs did not change up to 48 hpi in comparison to mock treated cells. VSV infected cells started to die already 24 hpi and at 48 hpi no more than 40% of living cells could be detected in the culture (Supplementary Figure 6b). Infection rates of moDCs did not significantly differ between VSV-dM51-GP and VSV-GP. Notably, frequencies of VSV-N+ cells were similar to GFP+ cells in moDCs for all tested virus variants (Supplementary Figure 6c). Although only 15% of immature moDCs were infected with VSV-dM51-GP, up to 90% of the cells showed a matured phenotype expressing CD83 after 24 h (Figure 5(a), right). Interestingly, also VSV-GP (20–30%) and to a much lesser extent, VSV (<10%) led to a partial maturation of moDCs, which was not observed in mouse BMDCs. No GFP+ cells could be detected among LPS-matured moDCs, indicating that mature moDCs are neither permissive for VSV-GP variants nor VSV. In line with this, the viability of mDCs was not affected by virus application (Figure 5(b)). In summary, human moDCs were less permissive to VSV-GP variants compared to VSV. Furthermore, VSV-GP did not kill the cells in the analyzed time range, which was the opposite for mouse BMDCs. Interestingly, VSV-dM51-GP infection led to a strong maturation in both murine and human DCs.
Figure 5.

VSV-GP variants infect human monocyte-derived immature DCs less than VSV
a Human monocyte-derived immature DCs (iDCs) were either mock infected or infected with the different virus variants at an MOI of 10 according to PFU titers. Frequencies of GFP+ infected cells, living cells (PI−), and CD83 expression were measured by flow cytometry after 6 and 24 h post infection (3 donors; 2 technical replicates). b LPS-matured moDCs (mDCs) were infected and flow cytometric analysis was performed 24 h post infection as described above (2 donors, 2 technical replicates). Data are shown as mean ± SD. *p < .05; **p < .01; ****p < .0001; not significant (ns); non-parametric Kruskal–Wallis test, followed Dunn’s multiple comparisons post-hoc test.
VSV-GP infects human cDC1s and cDC2s but classical monocytes are the main target population
As moDC cultures do not reflect the different DC subsets present in vivo, we investigated human DC subset infection ex vivo.10,13 Therefore, peripheral blood mononuclear cells (PBMCs) were isolated from whole blood from healthy blood donors and were immediately infected with VSV, VSV-GP, or VSV-dM51-GP. Initially, infection levels among total PBMCs were determined by measuring GFP+ cells. VSV infected around 10% of whole living PBMCs (MOI 10, 6hpi). VSV-GP and VSV-dM51-GP infections were first detectable after 24 h. Around 1–5% of total PBMCs were infected by VSV-GP. VSV-dM51-GP infection was under 2% for each tested donor (Supplementary Figure 7a). Next, we analyzed infection of human blood DCs in more detail. Among PBMCs three different DC subsets namely pDCs (HLA-DR+CD11c−CD123+), cDC1 (HLA-DR+CD11c+CD141+), and cDC2 (HLA-DR+CD11c+CD1c+) can be found.42 In Figure 6(a), the gating strategy for the DC subsets based on CD45+ single cells is depicted. Infection was monitored via flow cytometry at 6, 24, and 48 h post virus application. Data obtained from 5 donors are summarized in Figure 6(b). Neither VSV nor VSV-GP variants were able to infect pDCs. In contrast, after 6 h around 7% and 16% of cDC1 and cDC2, respectively, were infected by VSV. Infection of the cDC2 subset declined, most likely because infected cDC2 died over time. VSV-GP infection could be detected after 24 h and levels of GFP+ cells increased over time. Interestingly, higher frequencies of infected cDC1 (up to 27%, 48 hpi) than infected cDC2 (12%, 48 hpi) were observed for VSV-GP. VSV-dM51-GP infection was with 5% GFP+ cells among cDCs lower compared to the other viruses. We observed up to 25% and 5% infected cells among total PBMCs after VSV and VSV-GP application, respectively, suggesting that next to DCs other cells are infected by the virus variants (Supplementary Figure 7a). It is known, that viruses can infect myeloid cells like monocyte.43 Thus, we evaluated whether monocytes are susceptible to VSV-GP variants. We analyzed classical monocytes (CD14+CD16−), intermediate monocytes (CD14+CD16+), and non-classical monocytes (CD14-CD16+) which all express CD11c and except for the latter also CD64 (Supplementary Figure 7b). Non-classical monocytes were not infected by the viruses and intermediated monocytes showed only few GFP+ cells at 6 hpi. The main target of all three virus variants were CD14+ classical monocytes (Supplementary Figure 7c). Around 50% of classical monocytes were infected by VSV after 6 h. Already at this time point, the number of CD14−GFP+ cells increased in the culture. These cells are referred to as ‘CD14− subset’. Within the first 24 h, VSV infection peaked and CD14+ classical monocyte population declined drastically. Reduction of classical monocytes and the increase of the CD14− subset are depicted in representative plots in Supplementary Figure 7d. These observations indicated that classical monocytes may downregulate CD14 after virus infection. The same phenomenon was observed after VSV-GP and to a very low extent also after VSV-dM51-GP infection. As mentioned above, infection for those virus variants could be first detected after 24 h. At this time point, CD14 was already downregulated and around 40% and 10% among the CD14− subset were infected by VSV-GP and VSV-dM51-GP, respectively.
Figure 6.

Human blood conventional DCs can be infected by VSV-GP variants and VSV while plasmacytoid DCs are resistant to infection
a Peripheral blood mononuclear cells (PBMCs) were isolated from healthy donor blood and DC populations were analyzed via flow cytometry. The gating strategy among living CD45+, lineage negative (CD3−CD19−CD20−) cells for conventional DC type 1 (cDC1; HLA-DR+CD11c+CD141+), conventional DC type 2 (cDC2; HLA-DR+CD11c+CD1c+) and plasmacytoid DCs (pDC; HLA-DR+CD11c−CD123+) is shown. b PBMCs were infected with VSV, VSV-GP, and VSV-dM51-GP at an MOI of 10 according to PFU titer. GFP+ cells among pDC, cDC1, and cDC2 were analyzed via flow cytometry to determine infected cells after 6, 24, and 48 h. Combined data from five donors are shown as mean ± SEM.
Taken together, cDC1 were stronger infected by VSV-GP than other blood DC populations. Nevertheless, classical monocytes were the main target population of all virus variants. This experiment underlined that human PBMC infection differs significantly between VSV and the chimeric GP virus variants.
VSV-dM51-GP induces the strongest cytokine release by murine and human DCs
Next to infection and activation, we analyzed cytokine and chemokine secretion of DCs to further investigate their functionality upon virus treatment. Therefore, supernatants of infected murine BMDCs were collected (6 and 24 hpi), and cytokines were quantified using the Legendplex™ assay. Cytokines with significant or pronounced changes are presented in Figure 7(a). In Supplementary Figure 8, cytokines with slight or no changes are shown. We could observe strong differences in the cytokine profile between the virus variants. Generally, VSV-dM51-GP induced a pronounced increase of cytokine secretion after 6 and 24 h. The amount of IL-1α, IL-6, IL-27, and IFN-β was significantly higher compared to mock control (24 hpi). Additionally, TNF-α secretion was already 2 logs higher at 6 hpi. A slight increase was also found for the release of other tested cytokines. VSV-GP induced more than 1 log increase of IL-6 and IFN-β amounts after 24 h. Furthermore, slightly higher levels of IL-1α and IL-1β were detectable. VSV infection led to a minimal increase of IL-1α, IL-1β, and IFN-β. Interestingly, the amount of MCP-1 and TNF-α decreased after 6 h and 24 h of VSV treatment compared to mock control. This was also observed for VSV-GP, although not as distinctive as for wild type VSV.
Figure 7.
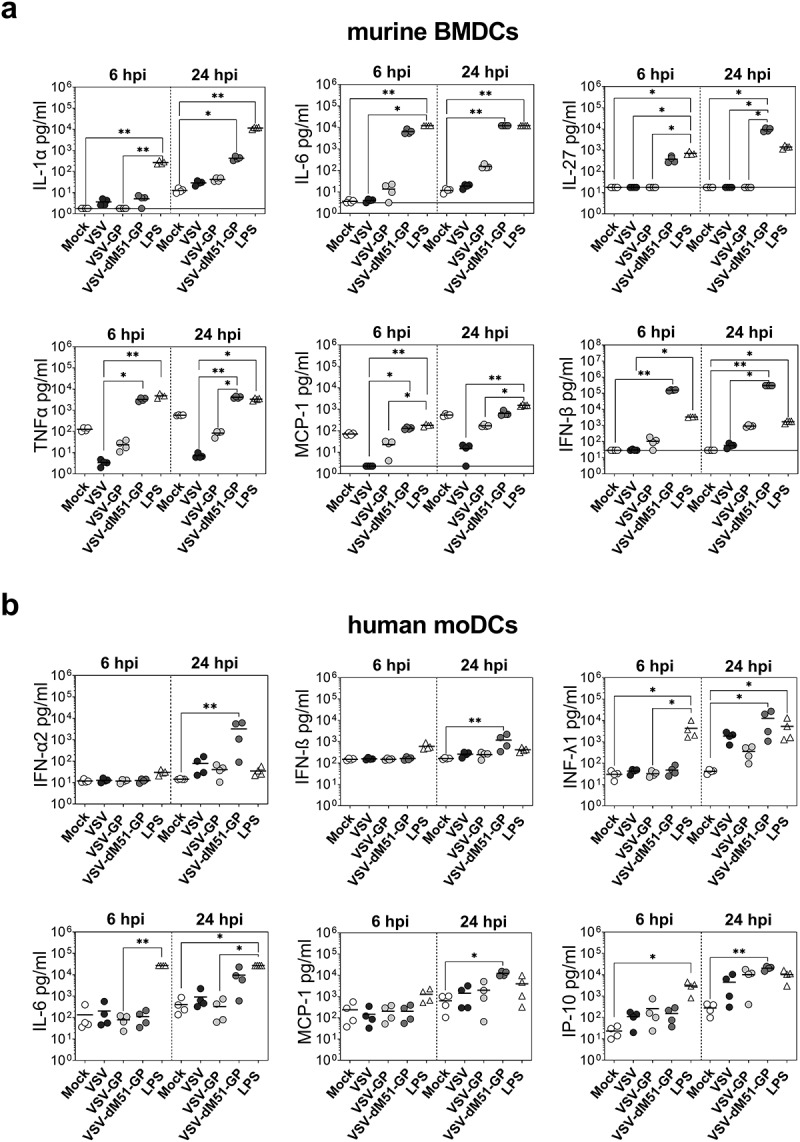
Different cytokine profiles are induced in murine and human DCs in response to VSV variants
a Murine bone marrow-derived DCs (BMDCs) were infected with VSV, VSV-GP, and VSV-dM51-GP at an MOI of 10. As a positive control, BMDCs were treated with LPS (100 ng/µl). Cytokines and chemokines were determined in the supernatant at 6 and 24 h post infection (hpi). The horizontal lines indicate the lower detection of the assay for each cytokine. Data are shown as mean (n = 4). b Human monocyte-derived DCs (moDCs) were infected with VSV, VSV-GP, and VSV-dM51-GP at an MOI of 10. As a positive control, moDCs were treated with LPS (0.5 µg/µl). Cytokines and chemokines were determined in the supernatant at 6 and 24 h post infection (hpi). Data are shown as mean and are obtained from four independent experiments. Significant changes are indicated. *p < .05; **p < .01; non-parametric Kruskal–Wallis test, followed Dunn’s multiple comparisons post-hoc test.
Human moDCs were also analyzed for cytokine secretion upon virus application after 6 and 24 h. Again, VSV-dM51-GP treatment showed the most prominent changes in cytokine levels compared to VSV and VSV-GP, although direct infection was the lowest. A significant elevation of pro-inflammatory INF-α, IFN-β, IFN-λ1, MCP-1, and IP-10 as well as a strong increase of IL-6 was measured after 24 h (Figure 7(b)). In contrast, cytokine profiles of VSV-GP and VSV changed less compared to mock control. These viruses induced only significantly higher release of IP-10. Additionally, slightly higher amounts of INF-α and IFN-λ1 were detected. No changes for other investigated cytokines/chemokines were found after virus infection at the tested time points (Supplementary Figure 9).
Discussion
In this study, we explored the permissiveness of murine and human DCs for VSV-GP and the attenuated VSV-dM51-GP in comparison to wild type VSV. We could confirm that VSV infected BMDCs as already shown in previous studies.28,40,44,45 Although, VSV-GP uses α-DG46 and not the low-density lipoprotein (LDL) receptor as VSV,47 murine BMDCs were equally infected by both viruses. Interestingly, VSV and VSV-GP infected preferentially immature BMDCs. VSV infection resulted in infection and killing of iDCs and, to a lesser extent, of mDCs. In contrast, mature BMDCs were completely resistant to VSV-GP infection and the cell viability of mDCs was not affected by VSV-GP application. In our hands, no maturation of BMDCs was observed neither upon VSV nor VSV-GP infections. This is in contrast to previously published data showing a VSV-induced maturation of BMDCs,28 which might be caused by the proportional switch of iDCs to mDCs due to the preferential infection and killing of iDCs in VSV-infected cultures. As we counted both iDCs and mDCs in BMDC cultures in the presence of live/dead staining, we excluded this possibility. The reduced production of TNF-α and MCP-1 by VSV and VSV-GP after infection seen in the cytokine analysis may be a reason for the impaired maturation of the DCs.48–50
Despite infection of iDCs by VSV-dM51-GP, no marked reduction of viable cell counts was observed. More importantly, VSV-dM51-GP treated BMDC cultures underwent complete maturation most likely due to the strong induction of IFN-β and TNF-α.49,51,52 The incomplete host-gene shut down due to the mutated M protein of this virus seems to allow the generation of an antiviral state of BMDCs resulting in survival, activation, and pronounced cytokine production.40,45 The maturation and prolonged survival led to a significantly higher expression of pro-inflammatory cytokines (IL-1α, IL-6, IL-27). Together with IFN-β and IFN-γ, these cytokines may generate a prime environment for efficient T cell activation by VSV-dM51-GP infected DCs. Moreover, only VSV-dM51-GP treatment induced strong IL-27 secretion which is a key initiator for Th1 CD4+ T cell differentiation.53 In contrast, we observed that VSV-GP treated BMDCs secreted pro-inflammatory cytokines to much lower levels compared to VSV-dM51-GP, while VSV did barely lead to an elevation of the analyzed cytokines.
By infecting splenocytes ex vivo, we observed only a low permissiveness of resDCs for all three virus variants even when high MOIs were applied. These results are in line with previous data showing that BMDCs differentiated in the presence of FLT3 ligand, a culture model for splenic DCs,10 are resistant to VSV infection.28 VSV and VSV-GP could infect both cDC1 and cDC2 subsets ex vivo, showing comparability of both viruses in the murine system. After systemic application of VSV-GP and VSV-dM51-GP, we detected low levels of viral RNA in cDC1. This indicates that VSV-GP variants can enter murine cDC1 in vivo. However, we hypothesize that direct infection and replication may be a rather rare event. In line with this, also others showed only marginal infection of CD11c+ cells in the spleen within 36 h post VSV application.54,55 A further explanation for the low in vivo susceptibility of splenic DCs to VSV-GP infection may relate to the used GP variant. The glycoprotein of VSV-GP is derived from the WE (HPI) strain of LCMV binding with a low affinity to α-DG as published recently.56,57 While low-affinity LCMV variants do not infect splenic DCs significantly58 due to a binding competition of GP with tissue laminin to α-DG,59 high-affinity LCMV can displace laminin and bind to α-DG resulting in DC infection.60 Additionally, previous studies demonstrated that the marginal zone or CD169+ macrophages located in the marginal zone of the spleen and the subcapsular sinus of lymph nodes provide a replication niche for type I IFN sensitive viruses, such as VSV and LCMV.54,61,62 Thus, CD169+ macrophages may well be an immune cell population preferentially infected by VSV-GP variants in vivo. Although direct splenic and tumor-associated DC infection by VSV-GP variants might be a rather rare event, DCs in both tissues were highly activated after virus application. Immature DCs in the tumor are often anergic and possess tolerogenic traits.63 Thus, the observed DC activation induced by VSV-GP and VSV-dM51-GP might be critical for antitumor T cell priming. This should be investigated in future experiments as Leveille et al. postulate that VSV-dM15 abolishes antigen presentation due to the killing of TADCs.64
Using mouse models for pre-clinical testing of OVs opens the question of how far animal models are appropriable to predict therapeutic success in humans. With this respect, we investigated VSV and VSV-GP infection of different human DC subsets. While murine DCs infection was similar, we indeed observed significant differences for the infection of human moDCs and blood cDCs. Human moDCs were significantly less susceptible to VSV-GP variants compared to VSV. Lower susceptibility of moDCs to VSV-GP and VSV-dM51-GP resulted in better viability of cells relative to VSV infection. Interestingly, next to VSV-dM51-GP causing a complete maturation of moDCs also VSV-GP induced partial maturation.
Similar to murine BMDCs, moDCs showed a more pronounced pro-inflammatory cytokine profile upon VSV-dM51-GP treatment. Especially, higher amounts of the IL-6, MCP-1, and IFN-α2 were released in response to this virus variant. Interestingly, no statistically significant induction of IFN-β was observed after VSV and VSV-GP in human moDCs. However, next to a slight increase of IFN-α2, type III IFN-λ1 production was induced by all three tested virus variants. It was shown that both IFNs promote maturation and IFN-λ1 can additionally suppress IL-12p70 release,65 which was indeed not detectable upon virus treatment in our experiments. Next to IFN production, VSV and VSV-GP variants induced IP-10/CXCL-10 secretion, which is important for effector T cell recruitment into the tumor.4 In sum, we could show that a stronger infection does not necessarily lead to a higher pro-inflammatory cytokine release and that VSV-GP and VSV-dM51-GP induce a considerably different cytokine profile.
Plasmacytoid DCs are not permissive to viral infection,66,67 which we could demonstrate for VSV and VSV-GP variants in human blood pDCs. Interestingly, human blood cDC1 seem to be slightly more susceptible to VSV-GP infection than cDC2. However, we could show that classical monocytes are the major target of VSV and VSV-GP variants, while intermediate and non-classical monocytes were weakly infected. Corresponding to the infection level, both VSV and VSV-GP led to a downregulation of CD14 on classical monocytes. The CD14 downregulation was described recently for VSV and the data suggest that VSV infection triggers differentiation of monocytes into cDCs or immature DCs.68,69 However, we could not confirm differentiation of classical monocytes to cDCs (or pDCs) as none of the key markers (CD123, CD141, or CD1c) were detected neither on the remaining classical monocytes nor on the CD14−subset that appeared upon infection. Thus, we assume that infected monocytes might gain an inflamed phenotype rather than differentiating into DCs. Here, more functional assays and transcriptional profile analysis will be necessary to thoroughly characterize this population to define functional consequences of VSV-GP infection, as monocytes and monocyte-derived inflammatory DCs can acquire APC functions and may thereby influence the adaptive immune response.70 Hence, these results highlight the need for more sophisticated studies to understand the influence of novel OV candidates such as VSV-GP on DCs and other myeloid cells. In summary, we showed that VSV and VSV-GP variants interact differently with mouse and human DC subsets as well as human blood monocytes. We demonstrated substantial divergences in infection levels, cell killing, and cytokine release between murine and human DCs. Our results indicate that mouse models are valuable to answer certain questions but results should be carefully considered when translating pre-clinical data to clinical trials. Future studies should investigate how the effects of VSV-GP variants on DCs and entailed phenotypical and functional changes shape the adaptive immune system. The understanding of these mechanisms might help to improve the activation of antitumor immunity by OVs and cancer vaccine vectors.
Supplementary Material
Acknowledgments
We thank B. Müllauer and V. Rinnofner for excellent technical assistance. We thank ViraTherapeutics (Boerhinger Ingelheim) for kindly providing the B16 IFNAR-/- cell line. This research was funded by the Austrian Science Fund (FWF) through a research project (FWF, P 25499-B13 to D.v.L.). D.v.L. is the inventor of VSV-GP and advisor to the company ViraTherapeutics GmbH, but competing financial interests do not exist. The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
Funding Statement
This work was supported by the Austrian Science Fund (FWF) [P 25499-B13].
Disclosure statement
No potential conflicts of interest were disclosed.
Author contributions
L.P.: conception and design of the study, performed experiments, acquisition, and analysis of data, interpretation of data, and wrote the manuscript; L.R.: conduction of experiments; A.S.: organization of blood samples; J.K.: contributed with valuable scientific discussions and critical revision of the manuscript; P.S.: contributed with valuable scientific discussions and critical revision of the manuscript; Z.B.: conception and design of the study, interpretation of data, and critical revision of the manuscript; D.v.L.: conception and design of the study, interpretation of data, and critical revision of the manuscript.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, Miller WH.. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol. 2020;27(12):87–15. doi: 10.3747/co.27.5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20(1):7–24. doi: 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
- 3.Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, et al. Successful anti-PD-1 cancer immunotherapy requires T Cell-Dendritic cell crosstalk involving the cytokines IFN gamma and IL-12. Immunity. 2018;49(6):1148–1161. doi: 10.1016/j.immuni.2018.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31(5):711–723. doi: 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, Casanova-Acebes M, Khudoynazarova M, Agudo J, Tung N, et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF Inhibition. Immunity. 2016;44(4):924–938. doi: 10.1016/j.immuni.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Movassagh M, Spatz A, Davoust J, Lebecque S, Romero P, Pittet M, Rimoldi D, Liénard D, Gugerli O, Ferradini L, et al. Selective accumulation of mature DC-Lamp+ dendritic cells in tumor sites is associated with efficient T-Cell-Mediated antitumor response and control of metastatic dissemination in melanoma. Cancer Res. 2004;64(6):2192–2198. doi: 10.1158/0008-5472.CAN-03-2969. [DOI] [PubMed] [Google Scholar]
- 7.Truxova I, Kasikova L, Hensler M, Skapa P, Laco J, Pecen L, Belicova L, Praznovec I, Halaska MJ, Brtnicky T, et al. Mature dendritic cells correlate with favorable immune infiltrate and improved prognosis in ovarian carcinoma patients. J Immunother Cancer. 2018;6(1):139. doi: 10.1186/s40425-018-0446-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Michea P, Noël F, Zakine E, Czerwinska U, Sirven P, Abouzid O, Goudot C, Scholer-Dahirel A, Vincent-Salomon A, Reyal F, et al. Adjustment of dendritic cells to the breast-cancer microenvironment is subset specific. Nat Immunol. 2018;19(8):885–897. doi: 10.1038/s41590-018-0145-8. [DOI] [PubMed] [Google Scholar]
- 9.Verneau J, Sautés-Fridman C, Sun CM. Dendritic cells in the tumor microenvironment: prognostic and theranostic impact. Semin Immunol. 2020;48:101410. doi: 10.1016/j.smim.2020.101410. [DOI] [PubMed] [Google Scholar]
- 10.Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31(1):1–48. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eisenbarth SC. Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol. 2019;19(2):89–103. doi: 10.1038/s41577-018-0088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitchell D, Chintala S, Dey M. Plasmacytoid dendritic cell in immunity and cancer. J Neuroimmunol. 2018;322:63–73. doi: 10.1016/j.jneuroim.2018.06.012. [DOI] [PubMed] [Google Scholar]
- 13.Domínguez PM, Ardavín C. Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol Rev. 2010;234(1):90–104. doi: 10.1111/j.0105-2896.2009.00876.x. [DOI] [PubMed] [Google Scholar]
- 14.Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol. 2016;37(12):855–865. doi: 10.1016/j.it.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber A, Dammeijer F, Aerts JGJV, Vroman H. Current state of dendritic cell-Based immunotherapy: opportunities for in vitro antigen loading of different DC subsets? Front Immunol. 2018;9:2804. doi: 10.3389/fimmu.2018.02804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Russell L, Peng KW, Russell SJ, Diaz RM. Oncolytic viruses: priming time for cancer immunotherapy. BioDrugs. 2019;33(5):485–501. doi: 10.1007/s40259-019-00367-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felt SA, Grdzelishvili VZ. Ecent advances in vesicular stomatitis virus-based oncolytic virotherapy: a 5-year update. J Gen Virol. 2017;98(12):2895–2911. doi: 10.1099/jgv.0.000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muik A, Stubbert LJ, Jahedi RZ, Geib Y, Kimpel J, Dold C, Tober R, Volk A, Klein S, Dietrich U, et al. Re-engineering vesicular stomatitis virus to abrogate neurotoxicity, circumvent humoral immunity, and enhance oncolytic potency. Cancer Res. 2014;74(13):3567–3578. doi: 10.1158/0008-5472.CAN-13-3306. [DOI] [PubMed] [Google Scholar]
- 19.Dold C, Rodriguez Urbiola C, Wollmann G, Egerer L, Muik A, Bellmann L, Fiegl H, Marth C, Kimpel J, von Laer D, et al. Application of interferon modulators to overcome partial resistance of human ovarian cancers to VSV-GP oncolytic viral therapy. Mol Ther — Oncolytics. 2016;3:16021. doi: 10.1038/mto.2016.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kimpel J, Urbiola C, Koske I, Tober R, Banki Z, Wollmann G, von Laer D. The oncolytic virus VSV-GP is effective against malignant melanoma. Viruses. 2018;10(3):1–16. doi: 10.3390/v10030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Urbiola C, Santer FR, Petersson M, van der Pluijm G, Horninger W, Erlmann P, Wollmann G, Kimpel J, Culig Z, von Laer D, et al. Oncolytic activity of the rhabdovirus VSV-GP against prostate cancer. Int J Cancer. 2018;143(7):1786–1796. doi: 10.1002/ijc.31556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schreiber LM, Urbiola C, Das K, Spiesschaert B, Kimpel J, Heinemann F, Stierstorfer B, Müller P, Petersson M, Erlmann P, et al. The lytic activity of VSV-GP treatment dominates the therapeutic effects in a syngeneic model of lung cancer. Br J Cancer. 2019;121(8):647–658. doi: 10.1038/s41416-019-0574-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tober R, Banki Z, Egerer L, Muik A, Behmuller S, Kreppel F, Greczmiel U, Oxenius A, von Laer D, Kimpel J, et al. VSV-GP: a potent viral vaccine vector that boosts the immune response upon repeated applications. J Virol. 2014;88(9):4897–4907. doi: 10.1128/JVI.03276-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilmschen S, Schneider S, Peters F, Bayer L, Issmail L, Bánki Z, Laer V. RSV vaccine based on rhabdoviral vector protects after single immunization. Vaccines. 2019;7(3):59. doi: 10.3390/vaccines7030059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gujar SA, Lee PWK. Oncolytic virus-mediated reversal of impaired tumor antigen presentation. Front Oncol. 2014;4:1–7. doi: 10.3389/fonc.2014.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim Y, Clements DR, Sterea AM, Jang HW, Gujar SA, Lee PWK. Dendritic cells in oncolytic virus-based anti-cancer therapy. Viruses. 2015;7(12):6506–6525. doi: 10.3390/v7122953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahmed M, Mckenzie MO, Puckett S, Hojnacki M, Poliquin L, Lyles DS. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and Protein Synthesis Ability of the Matrix Protein of Vesicular Stomatitis Virus To Suppress. J Virol. 2003;77(8):4646. doi: 10.1128/JVI.77.8.4646-4657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Westcott MM, Ahmed M, Smedberg JR, Rajani KR, Hiltbold EM, Lyles DS. Preservation of dendritic cell function during vesicular stomatitis virus infection reflects both intrinsic and acquired mechanisms of resistance to suppression of host gene expression by viral M protein. J Virol. 2013;87(21):11730–11740. doi: 10.1128/JVI.00680-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frenz T, Graalmann L, Detje CN, Döring M, Grabski E, Scheu S, Kalinke U. Independent of Plasmacytoid Dendritic Cell (pDC) infection, pDC triggered by virus-infected cells mount enhanced type I IFN responses of different composition as opposed to pDC stimulated with free virus. J Immunol. 2014;193(5):2496–2503. doi: 10.4049/jimmunol.1400215. [DOI] [PubMed] [Google Scholar]
- 30.Jayakar HR, Whitt MA. Identification of two additional translation products from the matrix (M) gene that contribute to vesicular stomatitis virus cytopathology. J Virol. 2002;76(16):8011–8018. doi: 10.1128/JVI.76.16.8011-8018.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stojdl DF, Lichty BD, TenOever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003;4(4):263–275. doi: 10.1016/S1535-6108(03)00241-1. [DOI] [PubMed] [Google Scholar]
- 32.Wollmann G, Rogulin V, Simon I, Rose JK, van den Pol AN. van den Pol AN. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J Virol. 2010;84(3):1563–1573. doi: 10.1128/JVI.02040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muik A, Kneiske I, Werbizki M, Wilflingseder D, Giroglou T, Ebert O, Kraft A, Dietrich U, Zimmer G, Momma S, et al. Pseudotyping vesicular stomatitis virus with lymphocytic choriomeningitis virus glycoproteins enhances infectivity for glioma cells and minimizes neurotropism. J Virol. 2011;85(11):5679–5684. doi: 10.1128/JVI.02511-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bánki Z, Kacani L, Müllauer B, Wilflingseder D, Obermoser G, Niederegger H, Schennach H, Sprinzl GM, Sepp N, Erdei A, et al. Cross-Linking of CD32 induces maturation of human monocyte-derived dendritic cells via NF-κB signaling pathway. J Immunol. 2003;170(8):3963–3970. doi: 10.4049/jimmunol.170.8.3963. [DOI] [PubMed] [Google Scholar]
- 35.Lutz MB, Kukutsch N, Ogilvie ALJ, Rößner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223(1):77–92. doi: 10.1016/S0022-1759(98)00204-X. [DOI] [PubMed] [Google Scholar]
- 36.Schreiber L-M, Urbiola C, Erlmann P, Wollmann G. In Vivo Bioimaging for Monitoring Intratumoral Virus Activity. In: Engeland C. (eds) Oncolytic Viruses. Methods in Molecular Biology, Chapter 15, vol 2058. New York: Humana. 2020. p. 237–248. [DOI] [PubMed] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Lagadic-Gossmann D, Huc L, Lecureur V. Alterations of intracellular pH homeostasis in apoptosis: origins and roles. Cell Death Differ. 2004;11(9):953–961. doi: 10.1038/sj.cdd.4401466. [DOI] [PubMed] [Google Scholar]
- 39.Shinoda H, Shannon M, Nagai T. Fluorescent proteins for investigating biological events in acidic environments. Int J Mol Sci. 2018;19(6):1548. doi: 10.3390/ijms19061548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahmed M, Brzoza KL, Hiltbold EM. Matrix protein mutant of vesicular stomatitis virus stimulates maturation of myeloid dendritic cells. J Virol. 2006;80(5):2194–2205. doi: 10.1128/JVI.80.5.2194-2205.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bart RS, Porzio NR, Kopf AW, Cheng EH, Farcet Y, Vilcek JT. Inhibition of growth of b16 murine malignant melanoma by exogenous interferon. Cancer Res. 1980;40:614–619. [PubMed] [Google Scholar]
- 42.Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. 2018;154(1):3–20. doi: 10.1111/imm.12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nikitina E, Larionova I, Choinzonov E, Kzhyshkowska J. Monocytes and macrophages as viral targets and reservoirs. Int J Mol Sci. 2018;19(9):2821. doi: 10.3390/ijms19092821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahmed M, Mitchell LM, Puckett S, Brzoza-Lewis KL, Lyles DS, Hiltbold EM. Vesicular stomatitis virus M protein mutant stimulates maturation of toll-Like receptor 7 (TLR7)-Positive dendritic cells through TLR-Dependent and -Independent mechanisms. J Virol. 2009;83(7):2962–2975. doi: 10.1128/JVI.02030-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boudreau JE, Bridle BW, Stephenson KB, Jenkins KM, Brunellière J, Bramson JL, Lichty BD, Wan Y. Recombinant vesicular stomatitis virus transduction of dendritic cells enhances their ability to prime innate and adaptive antitumor immunity. Mol Ther. 2009;17(8):1465–1472. doi: 10.1038/mt.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao W, Henry MD, Borrow P, Yamada H, Elder JH, Ravkov EV, Henry MD, Borrow P, Yamada H, Elder JH, et al. Identification of α-Dystroglycan as a receptor for lymphocytic choriomeningitis virus and lassa fever virus. Science. 1998;282(5396):2079–2081. (80-). doi: 10.1126/science.282.5396.2079. [DOI] [PubMed] [Google Scholar]
- 47.Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci. 2013;110(18):7306–7311. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jonuleit H, Kiihn U, Miiller G, Steinbrink K, Paragnik L, Schmitp E, Knop J, Enk AH. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions European Journal of immunology wiley online library. Eur J Immunol. 1997;27(12):3135–3142. doi: 10.1002/eji.1830271209. [DOI] [PubMed] [Google Scholar]
- 49.Maney NJ, Reynolds G, Krippner-heidenreich A, Catharien MU. Dendritic cell maturation and survival are differentially regulated by TNF receptors 1 and 21. J Immunol. 2016;193(10):4914–4923. doi: 10.4049/jimmunol.1302929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gschwandtner M, Derler R, Midwood KS. More than just attractive: how CCL2 influences myeloid cell behavior beyond chemotaxis. Front Immunol. 2019;10:1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Honda K, Sakaguchi S, Nakajima C, Watanabe A, Yanai H, Matsumoto M, Ohteki T, Kaisho T, Takaoka A, Akira S, et al. Selective contribution of IFN-α/β signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc Natl Acad Sci U S A. 2003;100(19):10872–10877. doi: 10.1073/pnas.1934678100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP. Direct Type I IFN but Not MDA5/TLR3 Activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol. 2014;12(1):e1001759. doi: 10.1371/journal.pbio.1001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J, Hibbert L, Churakova T, Travis M, Vaisberg E, et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+T cells. Immunity. 2002;16(6):779–790. doi: 10.1016/S1074-7613(02)00324-2. [DOI] [PubMed] [Google Scholar]
- 54.Honke N, Shaabani N, Cadeddu G, Sorg UR, Zhang DE, Trilling M, Klingel K, Sauter M, Kandolf R, Gailus N, et al. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat Immunol. 2012;13(1):51–57. doi: 10.1038/ni.2169. [DOI] [PubMed] [Google Scholar]
- 55.Naumenko V, Van S, Dastidar H, Kim DS, Kim SJ, Zeng Z, Deniset J, Lau A, Zhang C, Macia N, et al. Visualizing oncolytic Virus-Host interactions in live mice using intravital microscopy. Mol Ther - Oncolytics. 2018;10:14–27. doi: 10.1016/j.omto.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beyer WR, Miletic H, Ostertag W, von Laer D. Recombinant expression of lymphocytic choriomeningitis virus strain WE glycoproteins: a single amino acid makes the difference. J Virol. 2001;75(2):1061–1064. doi: 10.1128/JVI.75.2.1061-1064.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hastie KM, Igonet S, Sullivan BM, Legrand P, Zandonatti MA, Robinson JE, Garry RF, Rey FA, Oldstone MB, Saphire EO, et al. Crystal structure of the prefusion surface glycoprotein of the prototypic arenavirus LCMV. Nat Struct Mol Biol. 2016;23(6):513–521. doi: 10.1038/nsmb.3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sevilla N, Kunz S, Holz A, Lewicki H, Homann D, Yamada H, Campbell KP, de la Torre JC, Oldstone MBA. Immunosuppression and resultant viral persistence by specific viral targeting of dendritic cells. J Exp Med. 2000;192(9):1249–1260. doi: 10.1084/jem.192.9.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kunz S, Sevilla N, McGavern DB, Campbell KP, Oldstone MBA. Molecular analysis of the interaction of LCMV with its cellular receptor [alpha]-dystroglycan. J Cell Biol. 2001;155(2):301–310. doi: 10.1083/jcb.200104103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oldstone MBA, Campbell KP. Decoding arenavirus pathogenesis: essential roles for alpha-dystroglycan-virus interactions and the immune response. Virology. 2011;411(2):170–179. doi: 10.1016/j.virol.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iannacone M, Moseman EA, Tonti E, Bosurgi L, Junt T, Henrickson SE, Whelan SP, Guidotti LG, von Andrian UH. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature. 2010;465(7301):1079–1083. doi: 10.1038/nature09118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shaabani N, Duhan V, Khairnar V, Gassa A, Ferrer-Tur R, Häussinger D, Recher M, Zelinskyy G, Liu J, Dittmer U, et al. CD169+ macrophages regulate PD-L1 expression via type I interferon and thereby prevent severe immunopathology after LCMV infection. Cell Death Dis. 2016;7(11):e2446. doi: 10.1038/cddis.2016.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Veglia F, Gabrilovich DI. Dendritic cells in cancer : the role revisited. Curr Opin Immunol. 2018;45:43–51. doi: 10.1016/j.coi.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leveille S, Goulet M-L, Lichty BD, Hiscott J. Vesicular stomatitis virus oncolytic treatment interferes with tumor-associated dendritic cell functions and abrogates tumor antigen presentation. J Virol. 2011;85(23):12160–12169. doi: 10.1128/JVI.05703-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mennechet FJD, Uzé G. Interferon-λ-treated dendritic cells specifically induce proliferation of FOXP3-expressing suppressor T cells. Blood. 2006;107(11):4417–4423. doi: 10.1182/blood-2005-10-4129. [DOI] [PubMed] [Google Scholar]
- 66.Swiecki M, Wang Y, Vermi W, Gilfillan S, Schreiber RD, Colonna M. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J Exp Med. 2011;208(12):2367–2374. doi: 10.1084/jem.20110654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Webster B, Assil S, Dreux M, Pfeiffer J. Cell-Cell sensing of viral infection by plasmacytoid dendritic cells. J Virol. 2016;90(22):10050–10053. doi: 10.1128/JVI.01692-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hou W, Gibbs JS, Lu X, Brooke CB, Roy D, Modlin RL, Bennink JR, Yewdell JW. Viral infection triggers rapid differentiation of human blood monocytes into dendritic cells. Blood. 2012;119(13):3128–3131. doi: 10.1182/blood-2011-09-379479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tomczyk T, Wróbel G, Chaber R, Siemieniec I, Piasecki E, Krzystek-Korpacka M, Orzechowska BU. Immune consequences of in vitro infection of human peripheral blood leukocytes with vesicular stomatitis virus. J Innate Immun. 2018;10(2):131–144. doi: 10.1159/000485143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. 2017;17(6):349–362. doi: 10.1038/nri.2017.28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.