

Abstract
Diabetes mellitus is etiologically classified into type 1, type 2 and other types of diabetes. Despite distinct etiologies and pathogenesis of these subtypes, many studies have suggested the presence of shared susceptibilities and underlying mechanisms in β‐cell failure among different types of diabetes. Understanding these susceptibilities and mechanisms can help in the development of therapeutic strategies regardless of the diabetes subtype. In this review, we discuss recent evidence indicating the shared genetic susceptibilities and common molecular mechanisms between type 1, type 2 and other types of diabetes, and highlight the future prospects as well.
Keywords: β‐Cell failure, Endoplasmic reticulum stress, Oxidative stress
Despite distinct etiologies and pathogenesis of type 1, type 2 and other types of diabetes, many studies have suggested the presence of shared susceptibilities and underlying mechanisms in β‐cell failure among different types of diabetes. Understanding these susceptibilities and mechanisms can help in the development of therapeutic strategies regardless of the diabetes subtype. In this review, we discuss recent evidence indicating the shared genetic susceptibilities and common molecular mechanisms between type 1, type 2 and other types of diabetes, and highlight the future prospects as well.
Introduction
Diabetes mellitus is etiologically classified into type 1, type 2 and other types of diabetes1, 2. Type 1 diabetes is caused by the immune‐mediated destruction of the pancreatic β‐cells; whereas, type 2 diabetes is caused by decreased insulin action because of impaired insulin secretion and insulin resistance. Despite the differences in the etiologies and pathogenesis of type 1 and type 2 diabetes, they both share the same pathophysiology; that is, β‐cell failure leading to the development and progression of the disease. In 2004, we proposed that type 1 and type 2 diabetes shared common susceptibilities and underlying mechanisms based on the clustering of both types of diabetes in families and animal models3. Since then, many studies have suggested that type 1 and type 2 diabetes share genetic susceptibilities and underlying mechanisms of β‐cell failure4, 5, 6. Elucidating common susceptibilities and molecular mechanisms can help in providing fundamental information regarding β‐cell failure and fragility in diabetes patients, thereby leading to the development of effective methods for the prevention and intervention of diabetes, regardless of the subtype. Thus, in this review, we discuss recent evidence indicating the shared genetic susceptibilities and molecular mechanisms between type 1, type 2 and other types of diabetes, and highlight the future prospects as well.
β‐Cell failure in diabetes: Offense versus defense
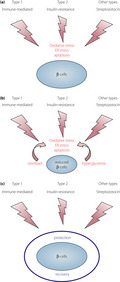
Overt diabetes develops when the β‐cells cannot satisfy the demand of insulin that is required to maintain a normal glucose metabolism. At the onset of overt diabetes, functional β‐cell mass, which is the sum of the number of β‐cells and functional state of each β‐cell, is markedly decreased to a level that is insufficient to sustain a normal glucose metabolism, and is referred to as ‘β‐cell failure.’ In general, β‐cell failure occurs when the balanced offense and defense mechanisms shift toward stronger offense and weaker defense (Figure 1). The stronger the attack and weaker the protection, more severe is the disease. An offensive attack is usually a result of external stress or insult to the system, whereas, a defensive mechanism is usually β‐cell intrinsic. Both these mechanisms contribute to β‐cell failure, ultimately leading to diabetes; however, the strength of an offensive attack differs between type 1 and type 2 diabetes. The offense mechanism in type 1 diabetes is the immune‐mediated destruction of β‐cells, and that in type 2 diabetes is an increased insulin demand due to insulin resistance. Notably, the offense mechanism is much stronger in type 1 diabetes than in type 2 diabetes. However, in both cases, the defense mechanism shares the same characteristics, and is not strong enough to protect against the offensive attack during diabetes development. Even under strong offensive attack, as in type 1 diabetes, if the defense is sufficiently strong enough to protect the β‐cells, diabetes might not manifest (Figure 1b). Thus, the relative strength or weakness of the offense and defense mechanisms determines β‐cell failure and diabetes development.
Figure 1.
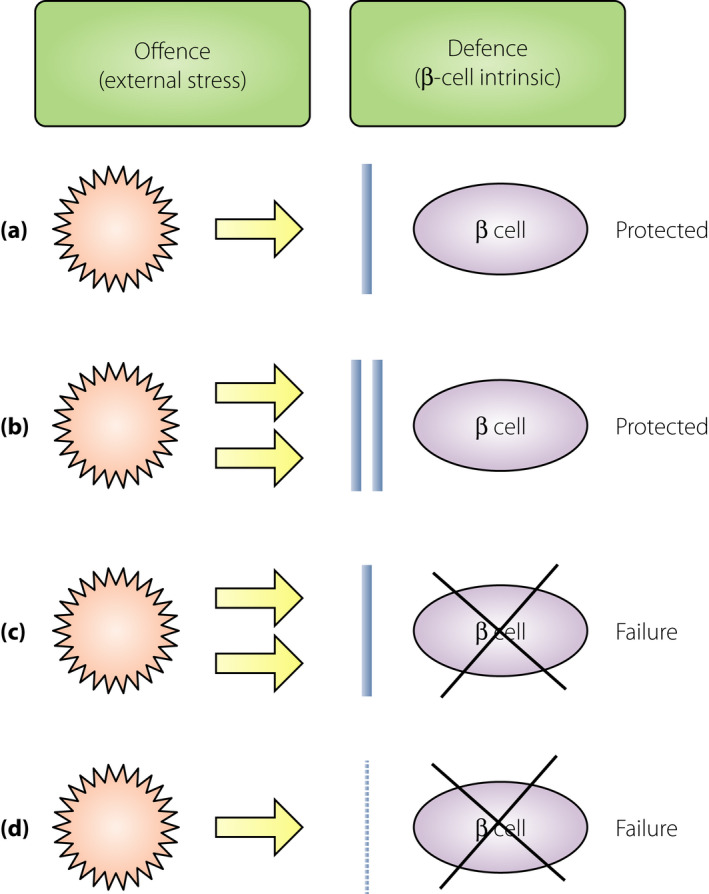
Relative balance between the offense and defense mechanisms in β‐cell failure. Offense is usually an external stress against the β‐cells, such as an immune‐mediated attack in type 1 diabetes and increased insulin demand due to obesity and insulin resistance in type 2 diabetes. Defense is usually a β‐cell intrinsic mechanism, such as protective mechanisms against oxidative stress, endoplasmic reticulum stress and apoptosis. (a) Normal balance. During a usual offensive attack (yellow arrow), normal defense (blue bar) can protect the β‐cells from failure. (b) Strong offense and defense. During a strong offensive attack, β‐cells can be protected from failure if the defense is sufficiently strong. (c) β‐Cell failure due to strong offense. Faced with a strong offensive attack, β‐cell failure manifests if the defense is not sufficiently strong. (d) β‐Cell failure due to weak defense. Even during a usual or slightly strong offensive attack, β‐cell failure can manifest if the defense is too weak.
β‐Cell failure in type 1 diabetes patients
Immune‐mediated attack against the β‐cells is the primary offensive mechanism in type 1 diabetes. An offensive attack starts before the onset of overt diabetes, and is referred to as the ‘prediabetes stage.’ During this stage, the functional β‐cell mass can maintain the normal glucose metabolism. However, progressive loss of the functional β‐cell mass is detected during this stage, as evidenced by the progressive decrease in acute insulin response to intravenous glucose in autoantibody positive twins and relatives of a type 1 diabetes proband7. Overt type 1 diabetes develops when the functional mass reduces below the critical level required to maintain the normal glucose metabolism (Figure 2a).
Figure 2.
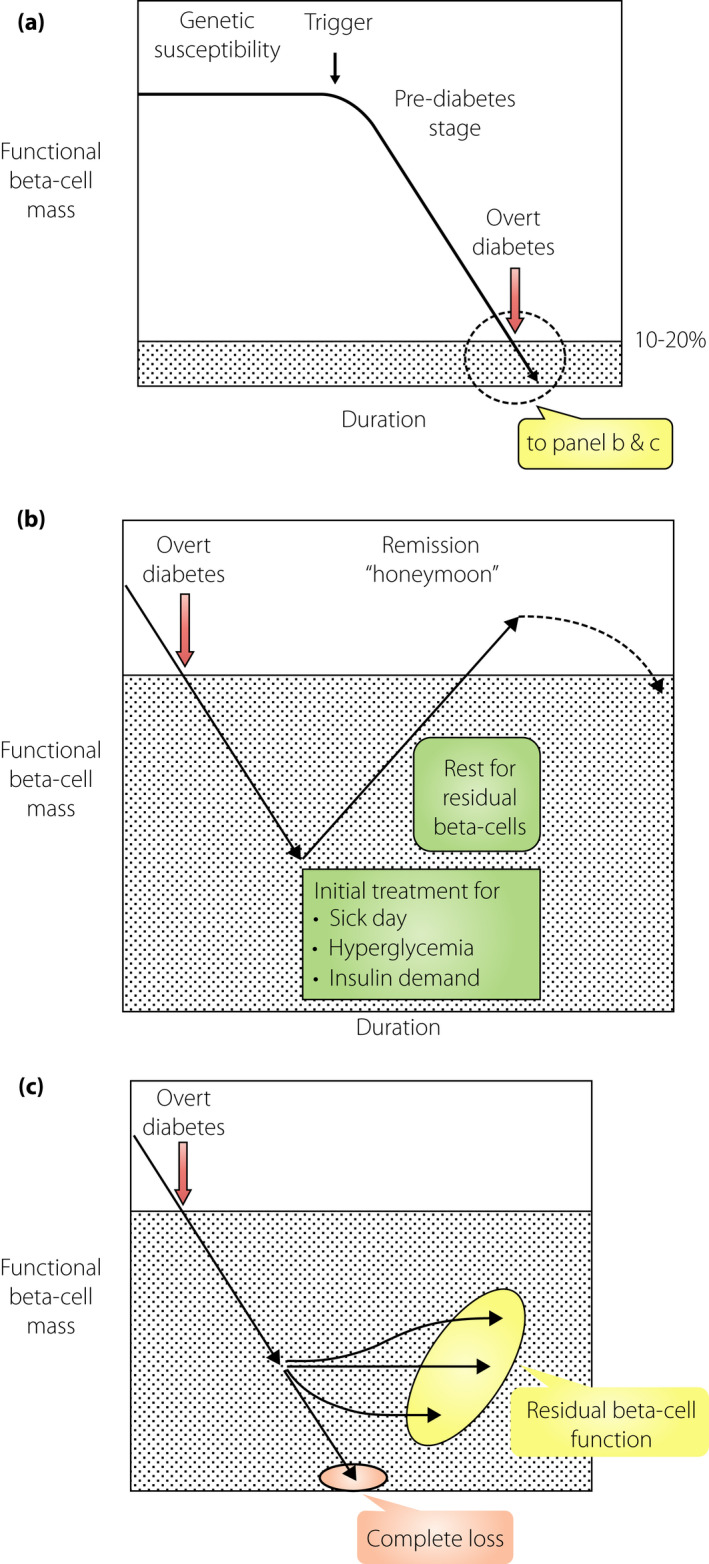
Functional β‐cell mass before and after the onset of type 1 diabetes. The vertical axis represents the functional β‐cell mass, which denotes the sum of β‐cell mass and functional status of each β‐cell. The horizontal axis is the time taken for the onset of type 1 diabetes. (a) Progressive decrease in the functional β‐cell mass toward the onset of diabetes. The functional β‐cell mass progressively decreases as the onset of type 1 diabetes approaches, even if the glucose level is within the normal range (prediabetes stage). This decrease is not necessarily linear; rather, it fluctuates from time‐to‐time, depending on the situation. In the long term, however, this decrease is progressive toward diabetic onset. When the functional β‐cell mass reaches a critically low level, overt type 1 diabetes develops with acute‐onset ketosis or ketoacidosis. (b) Partial recovery in the functional β‐cell mass soon after diabetes onset (honeymoon period). The functional β‐cell mass partially recovers after initial treatment of sick‐day conditions and near normalization of hyperglycemia can be achieved by administering sufficient quantity of insulin. (c) The functional β‐cell mass in the long term after the onset of diabetes. Changes in the functional β‐cell mass after diabetes onset vary from patient to patient; for instance, the progressive decrease in β‐cell mass results in complete depletion of endogenous insulin in some cases. In other cases, the functional β‐cell mass is preserved, albeit a very small amount, even after a long duration. However, there might be cases with sustained remission, although not clearly evidenced, that keep the functional β‐cell mass and insulin secretory function above the insulin‐dependency level.
At the onset of overt type 1 diabetes, the functional β‐cell mass is decreased to the level of insulin‐dependency, leading to acute onset of ketosis or ketoacidosis. Although the functional β‐cell mass is remarkably decreased because of β‐cell destruction, decrease in the quantity of β‐cells is not the only reason for insulin‐dependency, but the quality of the residual β‐cells is also responsible. Toward the onset of diabetes, precipitating events, such as an antecedent infection and sick‐day condition, are frequently observed, resulting in an increased insulin demand against the decreased number of β‐cells, leading to β‐cell failure and hyperglycemia. Drinking sugar‐containing soft drinks in response to thirst further accelerates the hyperglycemia. Hyperglycemia itself accelerates β‐cell failure through glucose toxicity. All these insults against the decreased number of β‐cells contribute to β‐cell failure, leading to insulin‐dependency during onset of type 1 diabetes.
Although the number of β‐cells are decreased because of immune‐mediated destruction, in most cases, they are not completely abolished at the disease onset, as evidenced by measurable C‐peptide levels and the presence of insulin‐positive cells in pancreas histological examinations8, 9, 10. Clinically, this has been recognized as the “honeymoon period” in a subset of patients, wherein after managing the sick‐day conditions, administering sufficient exogenous insulin and normalizing hyperglycemia can decrease the insulin requirement, and in some cases, normoglycemia can be maintained with no or very small exogenous insulin administration (Figure 2b). This is interpreted as the recovery of residual β‐cells whose function was impaired by external stresses at the onset of type 1 diabetes. However, the honeymoon period does not last long, because the immune‐mediated destruction of β‐cells does not subside and β‐cell failure eventually manifests as permanent insulin‐dependency (Figure 2b).
After a long duration of type 1 diabetes, the functional β‐cell mass is heterogeneous with either a complete loss in some patients or retention of minimal residual β‐cell function in others (Figure 2c)11. Evidence for the possibility of β‐cell mass recovery and long‐lasting remission is limited12. TIDE‐J (Japanese type 1 diabetes database), a prospective follow‐up study, is an ongoing collaborative effort of the National Center for Global Medicine and Health and the Committee on Type 1 Diabetes, Japan Diabetes Society, to investigate longitudinal changes in clinical parameters, such as β‐cell function from the onset of type 1 diabetes, for identifying genes and biomarkers to predict, prevent, and intervene in β‐cell failure and diabetes progression.
β‐Cell failure in type 2 diabetes patients
In type 2 diabetes patients, β‐cell failure is relative, as insulin secretion is not sufficient to compensate for the increased insulin demand mostly due to insulin resistance. The functional β‐cell mass, which reflects both the quality and quantity of the β‐cells during insulin secretion, is already decreased before the diagnosis of diabetes. We have previously reported that acute insulin response to intravenous glucose is already impaired in the prediabetes stage and further declines in type 2 diabetes13, which shares some similarities with the prediabetes stage of type 1 diabetes7. Histological examination shows a decrease in β‐cell mass at the diagnosis of type 2 diabetes14 and a progressive loss after disease onset15, 16.
Progressive β‐cell failure after diabetes onset
Progressive failure of residual β‐cells continues after the onset of the disease in both type 1 and type 2 diabetes patients. In case of type 1 diabetes, one driver of this failure is on the offensive side, which is the immune‐mediated attack against the β‐cells; however, contribution of the defensive side, which is the β‐cell intrinsic mechanism, should not be dismissed. Under normal conditions, the β‐cells can produce up to 1 million insulin molecules/minute, and this number further increases after meals or a glucose challenge17. Once the β‐cell mass is decreased by immune‐mediated destruction, each β‐cell is stressed and overworked because of an increased demand for insulin secretion; that is, production, processing, folding, packaging and excretion of the insulin molecules by each β‐cell. Such a situation increases the generation of reactive oxygen species through several pathways, including the formation of three disulfide bonds in each insulin molecule, excessive glucose metabolism and increased mitochondrial oxidative phosphorylation, resulting in oxidative stress18. Overwork in each β‐cell also increases the accumulation of unfolded or misfolded proteins in the endoplasmic reticulum (ER), leading to ER stress17. Oxidative stress and ER stress, unless properly resolved, can cause apoptotic cell death, leading to a progressive failure of the residual β‐cells17, 18. Hyperglycemia itself accelerates β‐cell failure by facilitating glucose toxicity, which is medicated by several mechanisms, including oxidative stress, the formation of advanced glycation end‐products, activation of protein kinase‐C, glyceraldehyde auto‐oxidation, increased polyol pathway activity and increased hexosamine metabolism18. Of note, these contributors of β‐cell failure are not only limited to type 1 diabetes, but are also shared by type 2 diabetes and other types of diabetes, such as partial pancreatectomy. Regardless of the etiology, once the β‐cell mass decreases and hyperglycemia manifests, each β‐cell is exposed to oxidative stress and ER stress, leading to progressive β‐cell failure. Administering a sufficient quantity of insulin and normalizing glucose metabolism can help in avoiding or reducing these stresses, as shown by better preservation of β‐cell function on using intensive insulin therapy in the Diabetes Control and Complications Trial19. However, complete normalization of glucose cannot be easily achieved, and might result in almost a complete loss of β‐cells with little or no endogenous insulin secretion in long‐standing type 1 diabetes. As residual insulin secretion, albeit a small amount, is closely associated with stable glycemic control11, 20, 21, 22, and better prognosis and outcomes of chronic complications23, preserving β‐cells during the natural history of type 1 diabetes is crucial.
Heterogeneity and ethnic differences associated with β‐cell failure
In populations of European descent, β‐cell failure in type 1 diabetes might not necessarily result in a complete loss of β‐cells; in fact, residual insulin secretion and insulin‐positive cells in the pancreas are observed in some patients even years after the onset of diabetes (Figure 3a)24, 25. In contrast, complete loss of insulin secretion is frequently observed in the Japanese population during long‐term follow up (Figure 3b)26, 27, suggesting that β‐cells are more vulnerable or fragile in the Japanese population than in the populations of European descent.
Figure 3.
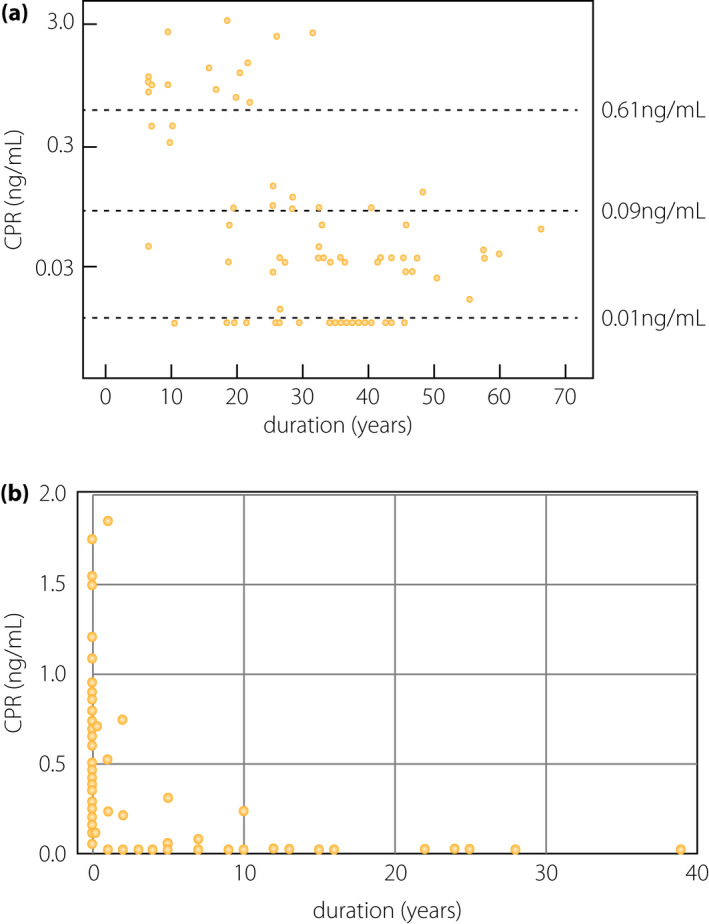
β‐Cell function relative to the duration of type 1 diabetes. (a) C‐peptide (CPR) levels at 90 min after meal tolerance test (vertical axis) relative to the duration of type 1 diabetes in European populations. Endogenous insulin secretion is preserved in a substantial number of patients with long‐standing type 1 diabetes in European populations (modified from Oram et al.25). (b) Fasting CPR levels (vertical axis) are plotted against the duration of diabetes (horizontal axis) in Japanese patients with acute‐onset type 1 diabetes (autoimmune; n = 77) enrolled at Kindai University Hospital. The endogenous insulin secretion, as assessed by CPR level, was completely lost in most patients as the duration increased. Note that the vertical axis is different from (a), in that fasting CPR levels are shown in the linear scale in this panel, whereas CPR levels at 90 min after the meal tolerance test are shown in the logarithmic scale in (a).
A similar trend has been observed for type 2 diabetes patients. Unlike the populations of European descent, Japanese and most East Asian populations develop type 2 diabetes with no or mild obesity28, 29. Furthermore, the insulin secretion capacity is much lower in the Japanese population than in the populations of European descent13, 28, 29. Thus, β‐cells in the Japanese population are easily decompensated against mild obesity, suggesting a weaker defense mechanism and more fragile β‐cells in the Japanese population than in the populations of European descent.
Weak defense mechanism or fragile β‐cells in Japanese individuals are also reflected by fulminant type 1 diabetes, which is characterized by an abrupt onset; that is, a complete loss of β‐cells at the onset of diabetes. Fulminant type 1 diabetes is mostly observed in Japan and East Asian countries, but very rarely in Western countries30. A recent genome‐wide association study in Japanese patients with fulminant type 1 diabetes identified a novel susceptibility locus, CSAD/lnc‐ITGB7‐1, on chromosome 1231. Top‐hit single‐nucleotide polymorphism is located in CSAD, which encodes for cysteine sulfinic decarboxylase, a key enzyme of taurine biosynthesis. Taurine exerts cytoprotective and anti‐inflammatory effects by membrane stabilization, osmoregulation, and anti‐oxidant and antiapoptotic activities32, suggesting its role in the defensive mechanisms of tissues and organs. CSAD is expressed in several organs, including the pancreas33. Taurine reportedly protects the pancreatic islets from destruction in autoimmune type 1 diabetes34 and attenuates streptozotocin (STZ)‐induced β‐cell failure35, 36, suggesting that CSAD contributes to fulminant type 1 diabetes by increasing the fragility of the β‐cells37.
The influence of the genetic background on β‐cell failure is indicated not only by ethnic differences in humans, but also by animal models. Single gene mutations in leptin and its receptor in mice, ob (Lepob ) and db (Leprdb), result in extreme obesity and insulin resistance38, 39, 40. However, the development of diabetes depends on the genetic background of the strain40, 41, 42, 43. For instance, despite the same degree of morbid obesity, C57BL/6J mice develop only mild and transient diabetes; whereas, C57BL/KsJ mice develop severe and life‐shortening diabetes. The C57BL/6J mice are resistant to diabetes, because insulin resistance due to morbid obesity is compensated by hypersecretion of insulin associated with hypertrophy and hyperplasia of the islets41, 42, 43. In contrast, the C57BL/KsJ mice develop severe diabetes associated with progressive failure and apoptosis of the β‐cells, resulting in disorganized islet morphology and insulin depletion. These differences likely arise from the differences in the genetically‐determined defensive strength of the β‐cells against strong external stresses caused by ob and db mutations, wherein, strong β‐cells in the C57BL/6J mice can compensate for the increased insulin demand, whereas fragile β‐cells in the C57BL/KsJ mice deteriorate, leading to subsequent β‐cell failure (Figure 1).
Molecular mechanisms in β‐cell failure
Although immune‐mediated mechanisms are the primary cause of type 1 diabetes, several mechanisms, such as oxidative stress, ER stress and apoptosis, are involved at the molecular level in the final stage of β‐cell destruction44, 45, 46. Free radicals and reactive oxygen species secreted from the immune cells and induced in β‐cells by pro‐inflammatory cytokines have been reported as effector molecules in β‐cell destruction47, 48. Intervention of oxidative stress by anti‐oxidants and scavengers reportedly preserves the endogenous insulin secretion and functional β‐cell mass48, 49, suggesting the contribution of oxidative stress in the destruction of β‐cells in type 1 diabetes. To directly investigate the protective effects of the anti‐oxidative molecules in β‐cell destruction, thioredoxin, a molecule with potent anti‐oxidative and anti‐apoptotic effects, was specifically overexpressed in the β‐cells of the NOD mouse, an animal model of autoimmune type 1 diabetes50. The development of type 1 diabetes was protected and insulin content was preserved in the NOD mice with transgenic expression of human thioredoxin gene (TRX) in the β‐cells. When the pancreatic histology was examined, insulitis was not attenuated, indicating that thioredoxin protected the β‐cells from destruction by infiltrating the immune cells, and not by attenuating the infiltration of the immune cells to the islets50. These data suggested that β‐cell failure in type 1 diabetes could be protected by increasing the defensive mechanism of the β‐cells.
Oxidative stress has been implicated in β‐cell failure in type 2 diabetes as well18, 51, 52. We studied the protective effect of β‐cell‐specific overexpression of TRX on β‐cell failure in type 2 diabetes in db/db mice53. In the db/db mouse, TRX overexpression in the β‐cells attenuated β‐cell failure, preserved insulin content and islet morphology, and resulted in better glycemic profiles53. Amelioration of β‐cell failure and diabetes has been reported by β‐cell‐specific overexpression of glutathione peroxidase, another anti‐oxidative molecule, in db/db mice54, indicating the protective effect of anti‐oxidative molecules in β‐cell failure and oxidative stress as an effector‐mediated mechanism in both type 1 and type 2 diabetes. Additionally, we studied the protective effect of β‐cell‐specific overexpression of TRX against a high dose of STZ, a well‐known β‐cell toxic reagent50. TRX overexpression attenuated the development of diabetes and β‐cell failure by STZ50, indicating that TRX overexpression protected β‐cell failure in type 1, type 2 and drug‐induced diabetes. These data suggest that a common molecular mechanism acts in the final stage of β‐cell failure in type 1, type 2 and other types of diabetes, providing a common molecular target for protection and intervention of β‐cell failure, regardless of the diabetes subtype (Figure 4).
Figure 4.
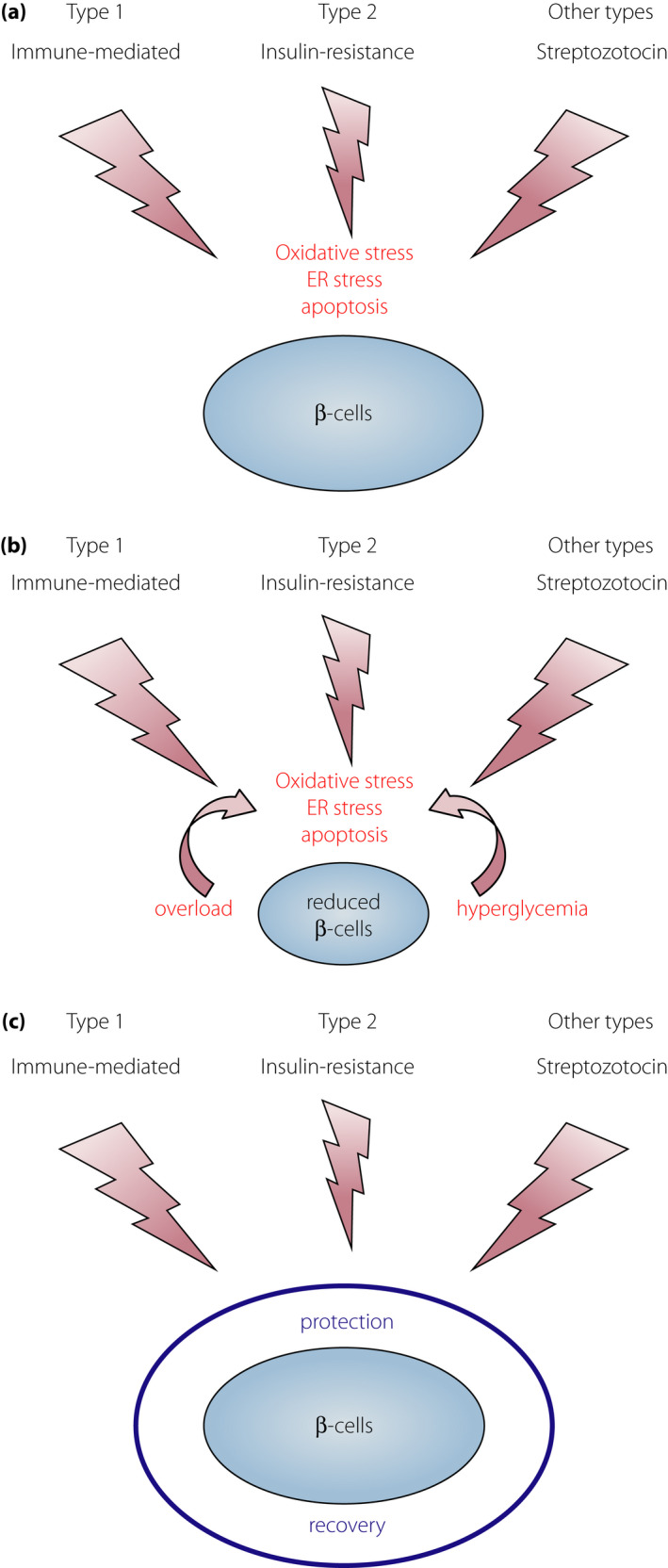
External stresses and mechanisms in β‐cell failure. (a) Different external stresses and similar final mechanisms of β‐cell failure in different types of diabetes. Different external stresses: immune‐mediated attack in type 1 diabetes, insulin resistance in type 2 diabetes and β‐cell toxic effect (streptozotocin) in other types of diabetes. Different offensive stresses share the same mechanisms, such as oxidative stress, endoplasmic reticulum stress and apoptosis in the final stage of β‐cell failure. (b) Progressive β‐cell failure after diabetes onset. Once the β‐cell mass is reduced, each β‐cell faces increased stress, such as increased insulin demand (overload) and hyperglycemia, leading to the acceleration of β‐cell failure because of oxidative stress and endoplasmic reticulum stress. (c) Sufficient protection against external stresses with a strong defensive mechanism, such as overexpression of thioredoxin, can preserve the functional β‐cell mass in type 1 (NOD mice)50, type 2 (db/db mice)53 and other types of diabetes (single high dose of streptozotocin)50.
Shared susceptibility between type 1 and type 2 diabetes
Epidemiological studies have shown an increase in the frequency of type 1 diabetes in siblings of a type 1 diabetes proband55, 56, 57. In addition, clustering of type 1 and type 2 diabetes in the same families has been reported58, 59, 60, suggesting the existence of a genetic link and shared susceptibility between type 1 and type 2 diabetes. In fact, a causative variant for type 1 diabetes, identified using genome‐wide linkage analysis in multiplex families, has been associated with type 2 diabetes as well61, 62, 63, 64.
A genetic link between type 1 and type 2 diabetes has also been suggested in animal models of diabetes. The NOD mouse, an inbred strain of mice with spontaneous development of autoimmune type 1 diabetes, is established from a closed colony of Jcl:ICR mice3, 65. From the same closed colony, the NSY mouse, an inbred animal model of type 2 diabetes, has also been established66, 67, showing clustering of type 1 and type 2 diabetes in related strains of mice derived from the same closed colony (for details please refer to Ikegami et al.3). Additionally, clustering of type 1 and type 2 diabetes in sister strains has been observed in rats. The LETL rat and its high incidence line, the KDP rat, are inbred strains of rats that show a spontaneous development of autoimmune type 1 diabetes68, 69. The LETL rat is established from a closed colony of Long‐Evans rats (Crl:LE), from which an inbred strain of rat with type 2 diabetes, the OLETF rat, has been established in the same animal facility70, showing the clustering of type 1 and type 2 diabetes in sister strains of rats and mice3. Based on these observations, in 2004, we proposed that there were common genetic susceptibilities and shared mechanisms between type 1 and type 2 diabetes (Figure 5)3. Since then, evidence supporting this has accumulated and underlying mechanisms have been identified4, 5, 6, 50, 53, 62, 63, 64.
Figure 5.
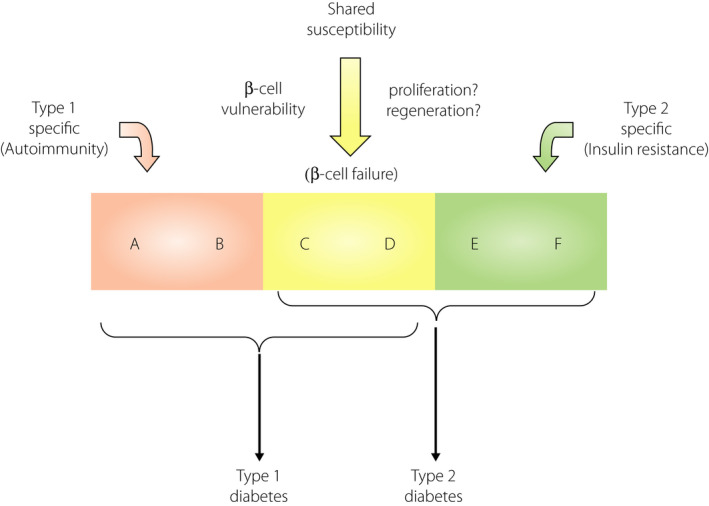
Shared genetic susceptibilities between type 1 and type 2 diabetes. Both type 1 and type 2 diabetes are multifactorial diseases caused by the interaction of genetic and environmental factors. Genetic factors consist of multiple susceptibility genes, and among them, some genes are specific to each subtype; for instance, type 1 diabetes‐specific genes (A and B), such as autoimmune‐related genes (e.g., HLA), and type 2 diabetes‐specific genes (E and F) such as obesity‐ and insulin resistance‐related genes (e.g., FTO). Additionally, there are some common genes shared between both diabetes types (C and D), such as genes related to β‐cell fragility or vulnerability (e.g., GLIS3). Modified from Ikegami et al.3 with permission.
Despite studies suggesting a genetic link between type 1 and type 2 diabetes, the actual genes that link the two subtypes are largely unknown. Genome scanning in animal models has identified many susceptibility loci for both type 1 and type 2 diabetes71, 72. Among these, chromosome 11 in mice is of particular interest (Figure 6). In our previous studies on genome scanning and congenic mapping for type 2 diabetes genes in the NSY mouse, susceptibility loci for type 2 diabetes were mapped on chromosome 1172, 73, 74, 75. To directly investigate the contribution of chromosome 11 to genetic susceptibility in type 2 diabetes, chromosome 11 of the control C3H/He mice was substituted with chromosome 11 from the NSY mice (C3H‐Chr11NSY in Figure 6a)73. This introgression converted the diabetes‐resistant C3H/He mice to diabetes‐susceptible mice (Figure 6a), indicating that chromosome 11 harbored susceptibility genes for type 2 diabetes73, 74. The NSY mouse developed type 2 diabetes along with impaired insulin secretion and mild obesity67, 76, 77. The impaired insulin secretion was accelerated under a high‐sucrose environment75, 78, suggesting the contribution of β‐cell vulnerability in the development of diabetes in this model. Susceptibility to high‐sucrose induced diabetes is also mapped to chromosome 11 (Figure 6a) and genes for impaired β‐cell function under a high‐sucrose environment was localized to the central and distal segments of chromosome 11 (Figure 6b)75. In this region, a susceptibility locus for type 1 diabetes, Idd4, was mapped by genome scanning and congenic mapping in the NOD mice71, 79, 80, 81, suggesting the presence of a common susceptibility gene for both type 1 and type 2 diabetes in this region.
Figure 6.
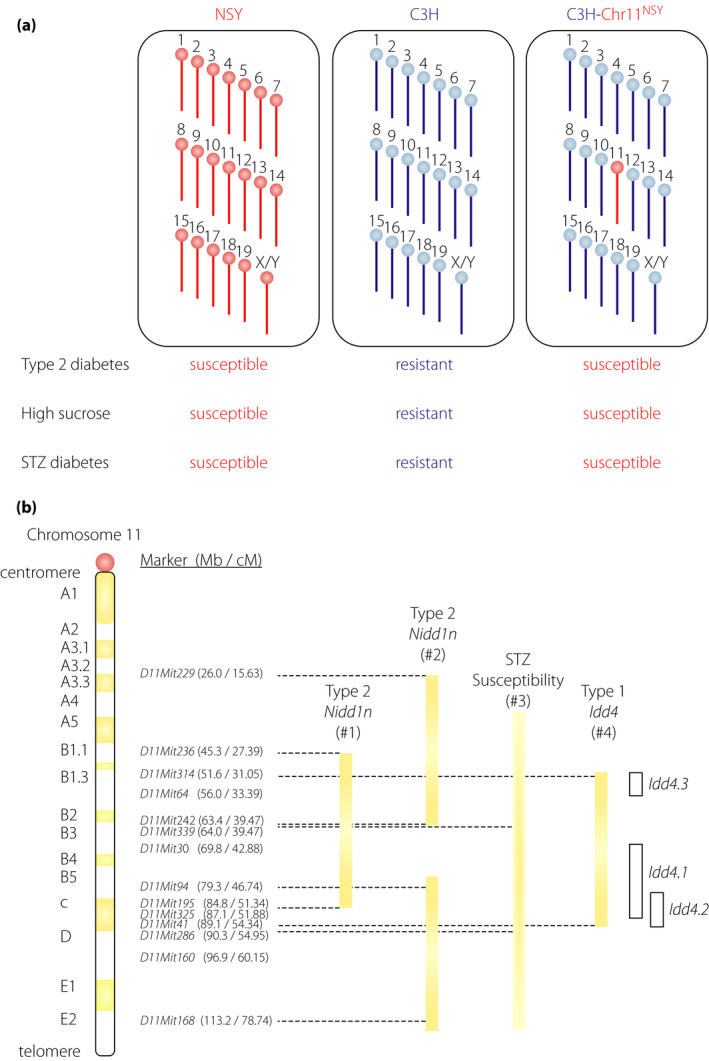
Chromosome 11 harbors genes for different types of diabetes. (a) Chromosome 11 of the NSY mouse possesses susceptibility genes for spontaneous type 2 diabetes, high‐sucrose induced diabetes and streptozotocin (STZ)‐induced diabetes. Control C3H mice are resistant to diabetes; whereas, NSY mice are susceptible to type 2 and STZ‐induced diabetes. Substitution of a single chromosome 11 of C3H mice with chromosome 11 from NSY mice (C3H‐Chr11NSY) converted the diabetes‐resistant C3H mice to diabetes‐susceptible mice, indicating that chromosome 11 harbored susceptibility genes for spontaneous type 2 diabetes73, high‐sucrose induced diabetes75 and STZ‐induced diabetes82. (b) Location of the susceptible loci for type 1 (Idd4 in NOD mice), type 2 (Nidd1n in NSY mice) and STZ‐induced diabetes on chromosome 11. #1 The support interval of quantitative trait loci for glucose intolerance72. #2 Regions of impaired insulin secretion by congenic mapping under a high‐sucrose environment75. #3 The support interval of STZ sensitivity locus in NOD mice was not clearly defined because of a limited number of markers83. The centromeric and telomeric ends of the interval are therefore shown in the graduation. #4 Idd4 is now divided into several sub‐loci (Idd4.1, Idd4.2, and Idd4.3); however, each sub‐locus was mapped by multiple research groups using different strain combinations. Therefore, the interval including all these loci is shown in the closed bar. The interval of each sub‐locus is shown in the open bar.
As aforementioned, the NOD mouse and the NSY mouse were derived from the same closed colony. In addition, both NOD and NSY mice are highly susceptible to STZ‐induced diabetes82, 83, suggesting that these strains share β‐cell vulnerability or fragility that is inherited from the original closed colony of Jcl:ICR mice. A susceptibility locus for STZ‐induced diabetes has been mapped to chromosome 11 in NOD mice83. To directly investigate the contribution of chromosome 11 to susceptibility to STZ‐induced diabetes, susceptibility to STZ was studied in C3H‐Chr11NSY mice in comparison with NSY and C3H mice (Figure 6a)82. Substitution of a single chromosome 11 of C3H mice with chromosome 11 from NSY mice (C3H‐Chr11NSY) converted the STZ‐resistant C3H mice to STZ‐susceptible mice, indicating that chromosome 11 harbored susceptibility genes responsible for STZ‐induced diabetes82. A susceptibility locus mapped to chromosome 11 in spontaneous type 2 diabetes73, high‐sucrose accelerated diabetes75 and STZ‐induced diabetes82, 83, 84 is likely to determine the intrinsic vulnerability of the β‐cells under external stress, which consequently leads to β‐cell failure and diabetes. Altogether, these data suggest that chromosome 11 harbors either a single or multiple genes for β‐cell vulnerability in type 1, type 2 and STZ‐induced diabetes (Figure 6b). Thus, identification of causative variants in this region can provide fundamental information on the genetic susceptibility and molecular mechanisms underlying β‐cell failure shared by type 1, type 2 and other types of diabetes.
Shared susceptibility according to genome‐wide association studies
Genome‐wide association studies in humans have identified many susceptibility loci for both type 1 and type 2 diabetes31, 85, 86. Susceptibility loci mapped in type 1 diabetes often harbor genes associated with immunological pathways; however, genes associated with β‐cell‐related functions and expression have also been identified87, suggesting that the latter group of genes might be candidate genes for the common susceptibility shared by type 1 and type 2 diabetes. Although many loci have been identified using genome‐wide association studies, loci identified in both types of diabetes are limited. Among the identified genes, GLIS3 has been implicated in both type 1 and type 2 diabetes85, 86, 88. GLIS3 encodes for a transcription factor, GLI‐similar family zinc‐finger protein 3. Recently, Dooley et al.5 identified a genetic variant of Glis3, a mouse homologue of human GLIS3, as a causative gene for type 1 diabetes in NOD mice by reducing Glis3 expression. Reduced Glis3 expression in NOD mice makes the β‐cells susceptible to ER stress, thereby leading to β‐cell apoptosis and failure. Reduced Glis3 expression has also been observed under a high‐fat diet5, indicating the role of the Glis3 variant in β‐cell failure in type 2 as well as type 1 diabetes89. These data showed that the same gene (Glis3) and mechanism (vulnerability or fragility of the β‐cells against ER stress) act in the development of β‐cell failure and diabetes in both type 1 and type 2 subtypes89, 90.
Lessons from other types of diabetes
Diabetes mellitus due to other specific mechanisms or disorders might provide important insight into the common genetic susceptibility and underlying mechanisms shared by different types of diabetes. These include rare monogenic forms of diabetes that are caused by mutations in critical genes, and rare genetic syndromes associated with insulin‐dependent diabetes1, 91.
Mutations with severe functional defects usually cause neonatal or early‐onset diabetes with extreme phenotypes. Rare mutations of GLIS3, a gene shared by both type 1 and type 2 diabetes, can reportedly cause neonatal diabetes with insulin‐dependency through β‐cell failure due to increased ER stress92. Several mutations in the insulin gene (INS) reportedly cause neonatal diabetes with insulin‐dependency93, and this is not autoimmune; rather, it is caused by ER stress because of accumulated unfolded or misfolded pre‐proinsulin proteins within the ER lumen, leading to β‐cell failure93. These findings were supported in studies in the Akita mouse, in which a mutation in Ins2, a mouse insulin gene, results in insulin‐deficient diabetes due to misfolded and accumulated mutant insulin molecules in the ER94, 95. Wolfram syndrome is caused by WFS1 mutations96, and insulin‐dependent diabetes is an important phenotype associated with it; it is also caused by ER stress‐mediated β‐cell apoptosis due to mutations in WFS1, encoding a negative regulator of ER stress97. Loss of ER‐resistant protein, MANF, has been recently reported to cause childhood‐onset syndromic diabetes because of increased ER stress98. Defective upregulation of Manf, a mouse homologue of human MANF, has also been reported in the genetic background of the NOD mice5.
All these are examples of β‐cell failure caused by β‐cell intrinsic defects against ER stress. Reported mutations in the abovementioned genes markedly increase the ER stress and/or impair unfolded protein response in β‐cells, leading to β‐cell failure and insulin dependency by themselves. In contrast, variants in these genes, which result in mild functional alterations, possibly increase the vulnerability and fragility of β‐cells under excess stress, thereby leading to increased susceptibility to type 1 diabetes under an autoimmune attack or type 2 diabetes under increased insulin demand due to obesity and insulin resistance. In fact, common polymorphisms in GLIS3 85, 86 and WFS1 99, 100 are associated with both type 1 and type 2 diabetes, suggesting β‐cell vulnerability or fragility as the common underlying mechanism of β‐cell failure in different types of diabetes.
Partial pancreatectomy is another example of β‐cell fragility as the underlying mechanism of β‐cell failure in diabetes. In partial pancreatectomy, approximately half of the pancreas is typically resected, leading to a marked reduction in β‐cell mass, and increase in insulin demand and stress against the remaining β‐cells. In our prospective studies on β‐cell function and glucose tolerance after pancreatectomy101, even though the same volume and portion of the pancreas were resected, we noticed considerable interindividual variation in glucose tolerance and in whether diabetes eventually developed. Similar observations have been reported in diabetes development after hemi‐pancreatectomy in living donors of pancreas transplantation102, 103. The aforementioned studies and those on pancreatectomy in rodents104 suggest the contribution of β‐cell vulnerability and failure in response to increased insulin demand due to a physical reduction in the β‐cell mass in diabetes development after pancreatectomy.
Future prospects for the protection, intervention and cure of diabetes
Given the contribution of oxidative stress and ER stress in β‐cell failure in both type 1 and type 2 diabetes, molecules and regulatory mechanisms involved in these pathways are potential therapeutic targets for the protection and intervention against β‐cell failure in diabetes. For example, pharmacological activators of Nrf2, a master regulator of cellular response to oxidative stress, are being tested for preserving β‐cell mass and treating diabetes18. The peroxiredoxin/thioredoxin anti‐oxidant system, a pathway regulated by Nrf2 18, is a primary defense mechanism of the β‐cells against oxidative stress105, which is consistent with our previous observation that overexpression of thioredoxin has a protective role in β‐cell failure in type 1, type 2 and STZ‐induced diabetes50, 53.
ER stress, oxidative stress and mitochondrial function are closely interrelated17, 18, 106. A mitochondrial DNA mutation, A3243G, causes diabetes as a part of maternally inherited diabetes and deafness, and myopathy, encephalopathy, lactic acidosis and stroke‐like episodes. Recent studies have shown that A3243G causes defects in taurine modification of mitochondrial transfer ribonucleic acid, leading to the aggregation of mitochondrial proteins in the cytosol, induction of cytotoxic unfolded protein response and cell death107, which is also a possible cause for β‐cell failure in type 1 and type 2 diabetes. Taurine supplementation can ameliorate stroke‐like episodes in myopathy, encephalopathy, lactic acidosis and stroke‐like episodes108, suggesting the role of taurine in restoring defective mitochondrial functions. These data, along with the association of the taurine biosynthesis pathway in fulminant type 1 diabetes31, suggest the possible application of taurine in β‐cell failure and diabetes due to maternally inherited diabetes and deafness, and type 1 and type 2 diabetes.
Cytotoxic unfolded protein response is another target for diabetes intervention. A taurine‐conjugated bile acid, tauroursodeoxycholic acid, suppresses these pathways and restores mitochondrial function107. Given the contribution of ER stress and cytotoxic unfolded protein responses in both mitochondrial diseases107 and β‐cell failure in type 1 and type 2 diabetes5, 89, 90, chemical chaperones for protein folding, such as tauroursodeoxycholic acid, might be potential therapeutic targets for the prevention and intervention in β‐cell failure.
While excess ER stress and oxidative stress result in β‐cell failure and death, stresses at a moderate or appropriate level can promote the functional adaptation of ER capacity and β‐cell proliferation106, 109, 110, suggesting that the degree of stress and appropriate response should be considered for the prevention and intervention of β‐cell failure.
Given that excess stress and overload promote β‐cell failure, β‐cell rest is another approach for the prevention and intervention of β‐cell failure. Currently, β‐cell rest can only be achieved by supplying a sufficient amount of exogenous insulin and reducing insulin resistance by lifestyle modifications and pharmacological treatments. Recent studies, however, suggest that β‐cell rest by optimizing glucose metabolism in β‐cells can be another target for preventing β‐cell failure. Activating mutations of glucokinase, which cause congenital hyperinsulinism, result in long‐term toxicity in β‐cells, leading to β‐cell failure through increased oxidative stress and DNA damage111, 112. In contrast, glucokinase inactivation can ameliorate β‐cell failure and diabetes by reducing the metabolic stress in β‐cells113, 114, suggesting that optimizing glucose metabolism and reducing β‐cell overload can be a target for preventing β‐cell failure. In addition to metabolic overload, β‐cell stress is also associated with alterations in messenger ribonucleic acid splicing, protein translation and protein modification, leading to the production of stress‐related modifications in β‐cell proteins. These altered proteins act as neo‐epitopes in immune‐mediated destruction of β‐cells in type 1 diabetes115. Therefore, β‐cell rest might be beneficial in preventing β‐cell failure by not only increasing the defensive power, but also decreasing the offensive attack.
Common susceptibilities and mechanisms between different types of diabetes indicate that studies on molecular mechanisms and pathways to determine β‐cell vulnerability and fragility can provide fundamental information on the prevention and intervention of β‐cell failure in all types of diabetes by increasing the defense mechanism of the β‐cells against external stresses. In addition to the similarities between type 1 and type 2 diabetes, some differences in ER signaling pathways between type 1 and type 2 diabetes have been suggested116. Further studies on the similarities and differences in β‐cell failure between different types of diabetes will clarify the whole landscape of β‐cell failure and increase our understanding of genes and molecules shared by different types of diabetes, leading to more effective methods for the prevention and intervention of β‐cell failure in diabetes.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
This research was supported by Grants‐in Aid for Scientific Research (KAKENHI) from the Japan Society for the Promotion of Science: 18K08530 and 21K08589 awarded to HI.
J Diabetes Investig 2021; 12: 1526–1539
References
- 1.The Committee of the Japan Diabetes Society on the Diagnostic Criteria of Diabetes Mellitus . Report of the committee on the classification and diagnostic criteria of diabetes mellitus. J Diabetes Investig 2010; 1: 212–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.American Diabetes Association . Diagnosis and classification of diabetes mellitus. Diabetes Care 2014; 37(suppl 1): S81–S90. [DOI] [PubMed] [Google Scholar]
- 3.Ikegami H, Fujisawa T, Ogihara T. Mouse models of type 1 and type 2 diabetes derived from the same closed colony: genetic susceptibility shared between two types of diabetes. ILAR J 2004; 45: 268–277. [DOI] [PubMed] [Google Scholar]
- 4.Boitard C, Efendic S, Ferrannini E, et al. A tale of two cousins: type 1 and type 2 diabetes. Diabetes 2005; 54(Suppl2): S1–S3. [DOI] [PubMed] [Google Scholar]
- 5.Dooley J, Tian L, Schonefeldt S, et al. Genetic predisposition for beta cell fragility underlies type 1 and type 2 diabetes. Nat Genet 2016; 48: 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Redondo MJ, Hagopian WA, Oram R, et al. The clinical consequences of heterogeneity within and between different diabetes types. Diabetologia 2020; 63: 2040–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Srikanta S, Ganda OP, Gleason RE, et al. Pre‐type I diabetes. Linear loss of beta cell response to intravenous glucose. Diabetes 1984; 33: 717–720. [DOI] [PubMed] [Google Scholar]
- 8.Imagawa A, Hanafusa T, Tamura S, et al. Pancreatic biopsy as a procedure for detecting in situ autoimmune phenomena in type 1 diabetes: close correlation between serological markers and histological evidence of cellular autoimmunity. Diabetes 2001; 50: 1269–1273. [DOI] [PubMed] [Google Scholar]
- 9.Greenbaum CJ, Anderson AM, Dolan LM, et al. Preservation of beta‐cell function in autoantibody‐positive youth with diabetes. Diabetes Care 2009; 32: 1839–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodriguez‐Calvo T, Richardson SJ, Pugliese A. Pancreas pathology during the natural history of type 1 diabetes. Curr Diab Rep 2018; 18: 124. [DOI] [PubMed] [Google Scholar]
- 11.Fukuda M, Tanaka A, Tahara Y, et al. Correlation between minimal secretory capacity of pancreatic beta‐cells and stability of diabetic control. Diabetes 1988; 37: 81–88. [DOI] [PubMed] [Google Scholar]
- 12.Oram RA, Sims EK, Evans‐Molina C. Beta cells in type 1 diabetes: mass and function; sleeping or dead? Diabetologia 2019; 62: 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoneda H, Ikegami H, Yamamoto Y, et al. Analysis of early‐phase insulin responses in nonobese subjects with mild glucose intolerance. Diabetes Care 1992; 15: 1517–1521. [DOI] [PubMed] [Google Scholar]
- 14.Butler AE, Janson J, Bonner‐Weir S, et al. Beta‐cell deficit, and increased beta‐cell apoptosis in humans with type 2 diabetes. Diabetes 2003; 52: 102–110. [DOI] [PubMed] [Google Scholar]
- 15.Matveyenko AV, Butler PC. Relationship between beta‐cell mass and diabetes onset. Diabetes Obes Metab 2008; 10(Suppl 4): 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakuraba H, Mizukami H, Yagihashi N, et al. Reduced beta‐cell mass and expression of oxidative stress‐related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia 2002; 45: 85–96. [DOI] [PubMed] [Google Scholar]
- 17.Meyerovich K, Ortis F, Allagnat F, et al. Endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. J Mol Endocrinol 2016; 57: R1–R17. [DOI] [PubMed] [Google Scholar]
- 18.Baumel‐Alterzon S, Katz LS, Brill G, et al. Nrf2: the master and captain of beta cell fate. Trends Endocrinol Metab 2021; 32: 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The Diabetes Control and Complications Trial Research Group . Effect of intensive therapy on residual beta‐cell function in patients with type 1 diabetes in the diabetes control and complications trial. A randomized, controlled trial. Ann Intern Med 1998; 128: 517–523. [DOI] [PubMed] [Google Scholar]
- 20.Shibasaki S, Imagawa A, Terasaki J, et al. Endogenous insulin secretion even at a very low level contributes to the stability of blood glucose control in fulminant type 1 diabetes. J Diabetes Investig 2010; 1: 283–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibb FW, McKnight JA, Clarke C, et al. Preserved C‐peptide secretion is associated with fewer low‐glucose events and lower glucose variability on flash glucose monitoring in adults with type 1 diabetes. Diabetologia 2020; 63: 906–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Babaya N, Noso S, Hiromine Y, et al. Relationship of continuous glucose monitoring‐related metrics with HbA1c and residual β‐cell function in Japanese patients with type 1 diabetes. Sci Rep 2021; 11: 4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lachin JM, McGee P, Palmer JP, et al. Impact of C‐peptide preservation on metabolic and clinical outcomes in the diabetes control and complications trial. Diabetes 2014; 63: 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keenan HA, Sun JK, Levine J, et al. Residual insulin production and pancreatic ß‐cell turnover after 50 years of diabetes: Joslin Medalist Study. Diabetes 2010; 59: 2846–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oram RA, Jones AG, Besser REJ, et al. The majority of patients with long‐duration type 1 diabetes are insulin microsecretors and have functioning beta cells. Diabetologia 2014; 57: 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uno S, Imagawa A, Kozawa J, et al. Complete loss of insulin secretion capacity in type 1A diabetes patients during long‐term follow up. J Diabetes Investig 2018; 9: 806–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugihara S, Kikuchi T, Urakami T, et al. Residual endogenous insulin secretion in Japanese children with type 1A diabetes. Clin Pediatr Endocrinol 2021; 30: 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kodama K, Tojjar D, Yamada S, et al. Ethnic differences in the relationship between insulin sensitivity and insulin response: a systematic review and meta‐analysis. Diabetes Care 2013; 36: 1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yabe D, Seino Y, Fukushima M, et al. β cell dysfunction versus insulin resistance in the pathogenesis of type 2 diabetes in East Asians. Curr Diab Rep 2015; 15: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanafusa T, Imagawa A. Fulminant type 1 diabetes: a novel clinical entity requiring special attention by all medical practitioners. Nat Clin Pract Endocrinol Metab 2007; 3: 36–45. [DOI] [PubMed] [Google Scholar]
- 31.Kawabata Y, Nishida N, Awata T, et al. A genome‐wide association study confirming a strong effect of HLA and identifying variants in CSAD/lnc‐ITGB7‐1 on chromosome 12q13.13 associated with susceptibility to fulminant type 1 diabetes. Diabetes 2019; 68: 665–675. [DOI] [PubMed] [Google Scholar]
- 32.Lambert IH, Kristensen DM, Holm JB, et al. Physiological role of taurine – from organism to organelle. Acta Physiol 2014; 213: 191–212. [DOI] [PubMed] [Google Scholar]
- 33.Kerr TA, Matsumoto Y, Matsumoto H, et al. Cysteine sulfinic acid decarboxylase regulation: a role for farnesoid X receptor and small heterodimer partner in murine hepatic taurine metabolism. Hepatol Res 2014; 44: E218–E228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arany E, Strutt B, Romanus P, et al. Taurine supplement in early life altered islet morphology, decreased insulitis and delayed the onset of diabetes in non‐obese diabetic mice. Diabetologia 2004; 47: 1831–1837. [DOI] [PubMed] [Google Scholar]
- 35.Lin S, Yang J, Wu G, et al. Inhibitory effects of taurine on STZ‐induced apoptosis of pancreatic islet cells. Adv Exp Med Biol 2013; 775: 287–297. [DOI] [PubMed] [Google Scholar]
- 36.Nakatsuru Y, Murase‐Mishiba Y, Bessho‐Tachibana M, et al. Taurine improves glucose tolerance in STZ‐induced insulin‐deficient diabetic mice. Diabetol Int 2018; 9: 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawabata Y, Ikegami H. Genetics of fulminant type 1 diabetes. Diabetol Int 2020; 11: 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Proenca R, Maffei M, et al. Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372: 425–432. [DOI] [PubMed] [Google Scholar]
- 39.Lee G‐H, Proenca R, Montez JM, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature 1996; 379: 632–635. [DOI] [PubMed] [Google Scholar]
- 40.Coleman DL. A historical perspective on leptin. Nat Med 2010; 16: 1097–1099. [DOI] [PubMed] [Google Scholar]
- 41.Coleman DL, Hummel KP. The influence of genetic background on the expression of the obese (Ob) gene in the mouse. Diabetologia 1973; 9: 287–293. [DOI] [PubMed] [Google Scholar]
- 42.Baetens D, Stefan Y, Ravazzola M, et al. Alteration of islet cell populations in spontaneously diabetic mice. Diabetes 1978; 27: 1–7. [DOI] [PubMed] [Google Scholar]
- 43.Joost HG, Schürmann A. The genetic basis of obesity‐associated type 2 diabetes (diabesity) in polygenic mouse models. Mamm Genome 2014; 25: 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tersey SA, Nishiki Y, Templin AT, et al. Islet β‐cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 2012; 61: 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marhfour I, Lopez XM, Lefkaditis D, et al. Expression of endoplasmic reticulum stress markers in the islets of patients with type 1 diabetes. Diabetologia 2012; 55: 2417–2420. [DOI] [PubMed] [Google Scholar]
- 46.Roep BO, Thomaidou S, van Tienhoven R, et al. Type 1 diabetes mellitus as a disease of the β‐cell (do not blame the immune system?). Nat Rev Endocrinol 2021; 17: 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mandrup‐Poulsen T, Helqvist S, Wogensen LD, et al. Cytokine and free radicals as effector molecules in the destruction of pancreatic beta cells. Curr Top Microbiol Immunol 1990; 164: 169–193. [DOI] [PubMed] [Google Scholar]
- 48.Horio F, Fukuda M, Katoh H, et al. Reactive oxygen intermediates in autoimmune islet cell destruction of the NOD mouse induced by peritoneal exudate cells (rich in macrophages) but not T cells. Diabetologia 1994; 37: 22–31. [DOI] [PubMed] [Google Scholar]
- 49.Fukuda M, Ikegami H, Kawaguchi Y, et al. Antioxidant, probucol, can inhibit the generation of hydrogen peroxide in islet cells induced by macrophages and prevent islet cell destruction in NOD mice. Biochem Biophys Res Commun 1995; 209: 953–958. [DOI] [PubMed] [Google Scholar]
- 50.Hotta M, Tashiro F, Ikegami H, et al. Pancreatic beta cell‐specific expression of thioredoxin, an antioxidative and antiapoptotic protein, prevents autoimmune and streptozotocin‐induced diabetes. J Exp Med 1998; 188: 1445–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawamori D, Kajimoto Y, Kaneto H, et al. Oxidative stress induces nucleo‐cytoplasmic translocation of pancreatic transcription factor PDX‐1 through activation of c‐Jun NH(2)‐terminal kinase. Diabetes 2003; 52: 2896–2904. [DOI] [PubMed] [Google Scholar]
- 52.Newsholme P, Cruzat VF, Keane KN, et al. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem J 2016; 473: 4527–4550. [DOI] [PubMed] [Google Scholar]
- 53.Yamamoto M, Yamato E, Toyoda S, et al. Transgenic expression of antioxidant protein thioredoxin in pancreatic beta cells prevents progression of type 2 diabetes mellitus. Antioxid Redox Signal 2008; 10: 43–49. [DOI] [PubMed] [Google Scholar]
- 54.Harmon JS, Bogdani M, Parazzoli SD, et al. Beta‐Cell‐specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 2009; 150: 4855–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rish N. Assessing the role of HLA‐linked and unlinked determinants of disease. Am J Hum Genet 1987; 40: 1–14. [PMC free article] [PubMed] [Google Scholar]
- 56.Ikegami H, Ogihara T. Genetics of insulin‐dependent diabetes mellitus. Endocr J 1996; 43: 605–613. [DOI] [PubMed] [Google Scholar]
- 57.Ikegami H, Noso S, Babaya N, et al. Genetic basis of type 1 diabetes: similarities and differences between East and West. Rev Diabet Stud 2008; 5: 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fujisawa T, Ikegami H, Kawaguchi Y, et al. Common genetic basis between type 1 and type 2 diabetes mellitus indicated by interview‐based assessment of family history. Diab Res Clin Pract 2004; 66S: S91–S95. [DOI] [PubMed] [Google Scholar]
- 59.Allen C, Palta M, D'Alessio DJ. Risk of diabetes in siblings and other relatives of IDDM subjects. Diabetes 1991; 40: 831–836. [DOI] [PubMed] [Google Scholar]
- 60.Dahlquist G, Blom L, Tuvemo T, et al. The Swedish childhood diabetes study ‐ results from a nine year case register and a one year case‐referent study indicating that type 1 (insulin‐dependent) diabetes mellitus is associated with both type 2 (non‐insulin‐dependent) diabetes mellitus and autoimmune disorders. Diabetologia 1989; 32: 2–6. [DOI] [PubMed] [Google Scholar]
- 61.Guo D, Li M, Zhang Y, et al. A functional variant of SUMO4, a new I kappa B alpha modifier, is associated with type 1 diabetes. Nat Genet 2004; 36: 837–841. [DOI] [PubMed] [Google Scholar]
- 62.Noso S, Ikegami H, Fujisawa T, et al. Genetic heterogeneity in association of the SUMO4 M55V variant with susceptibility to type 1 diabetes. Diabetes 2005; 54: 3582–3586. [DOI] [PubMed] [Google Scholar]
- 63.Noso S, Fujisawa T, Kawabata Y, et al. Association of small ubiquitin‐like modifier 4 (SUMO4) variant, located in IDDM5 locus, with type 2 diabetes in the Japanese population. J Clin Endocrinol Metab 2007; 92: 2358–2362. [DOI] [PubMed] [Google Scholar]
- 64.Tang S‐T, Peng W‐J, Wang C‐J, et al. Polymorphism M55V in gene encoding small ubiquitin‐like modifier 4 (SUMO4) protein associates with susceptibility to type 1 (and type 2) diabetes. Diabetes Metab Res Rev 2012; 28: 679–687. [DOI] [PubMed] [Google Scholar]
- 65.Ikegami H, Makino S. The NOD mouse and its related strains. In: Sima AAF, Shafrir E (eds). Animal Models of Diabetes A Primer. Harwood Academic Publishers, Amsterdam, 2000; 43–61. [Google Scholar]
- 66.Shibata M, Yasuda B. New experimental congenital diabetic mice (N.S.Y. mice). Tohoku J Exp Med 1980; 130: 139–142. [DOI] [PubMed] [Google Scholar]
- 67.Ueda H, Ikegami H, Yamato E, et al. The NSY mouse: a new animal model of spontaneous NIDDM with moderate obesity. Diabetologia 1995; 38: 503–508. [DOI] [PubMed] [Google Scholar]
- 68.Kawano K, Hirashima T, Mori S, et al. New inbred strain of Long‐Evans Tokushima lean rats with IDDM without lymphopenia. Diabetes 1991; 40: 1375–1381. [DOI] [PubMed] [Google Scholar]
- 69.Komeda K, Noda M, Terao K, et al. Establishment of two substrains, diabetes‐prone and non‐diabetic, from Long‐Evans Tokushima Lean (LETL) rats. Endocr J 1998; 45: 737–744. [DOI] [PubMed] [Google Scholar]
- 70.Kawano K, Hirashima T, Mori S, et al. Spontaneous long‐term hyperglycemic rat with diabetic complications. Otsuka Long‐Evans Tokushima Fatty (OLETF) strain. Diabetes 1992; 41: 1422–1428. [DOI] [PubMed] [Google Scholar]
- 71.Todd JA, Aitman TJ, Cornall RJ, et al. Genetic analysis of autoimmune type 1 diabetes mellitus in mice. Nature 1991; 351: 542–547. [DOI] [PubMed] [Google Scholar]
- 72.Ueda H, Ikegami H, Kawaguchi Y, et al. Genetic analysis of late‐onset type 2 diabetes in a mouse model of human complex trait. Diabetes 1999; 48: 1168–1174. [DOI] [PubMed] [Google Scholar]
- 73.Babaya N, Fujisawa T, Nojima K, et al. Direct evidence for susceptibility genes for type 2 diabetes on mouse chromosomes 11 and 14. Diabetologia 2010; 53: 1362–1371. [DOI] [PubMed] [Google Scholar]
- 74.Babaya N, Ueda H, Noso S, et al. Dose effect and mode of inheritance of diabetogenic gene on mouse chromosome 11. J Diabetes Res 2013; 2013: 608923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kobayashi M, Ueda H, Babaya N, et al. Type 2 diabetes susceptibility genes on mouse chromosome 11 under high sucrose environment. BMC Genet 2020; 21: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ueda H, Ikegami H, Kawaguchi Y, et al. Age‐dependent changes in phenotypes and candidate gene analysis in a polygenic animal model of Type II diabetes mellitus; NSY mouse. Diabetologia 2000; 43: 932–938. [DOI] [PubMed] [Google Scholar]
- 77.Hamada Y, Ikegami H, Ueda H, et al. Insulin secretion to glucose as well as non‐glucose stimuli is impaired in spontaneously diabetic Nagoya‐Shibata‐Yasuda (NSY) mice. Metabolism 2001; 50: 891–894. [DOI] [PubMed] [Google Scholar]
- 78.Nojima K, Sugimoto K, Ueda H, et al. Analysis of hepatic gene expression profile in a spontaneous mouse model of type 2 diabetes under a high sucrose diet. Endocrine J 2013; 60: 261–274. [DOI] [PubMed] [Google Scholar]
- 79.Grattan M, Mi Q‐S, Meagher C, et al. Congenic mapping of the diabetogenic locus Idd4 to a 5.2‐cM region of chromosome 11 in NOD mice: identification of two potential candidate subloci. Diabetes 2002; 51: 215–223. [DOI] [PubMed] [Google Scholar]
- 80.Steward CA, Gonzalez JM, Trevanion S, et al. The non‐obese diabetic mouse sequence, annotation and variation resource: an aid for investigating type 1 diabetes. Database (Oxford) 2013; 2013: bat032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Banday VS, Lejon K. Elevated systemic glutamic acid level in the non‐obese diabetic mouse is Idd linked and induces beta cell apoptosis. Immunology 2017; 150: 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Babaya N, Ikegami H, Fujisawa T, et al. Susceptibility to streptozotocin‐induced diabetes is mapped to mouse chromosome 11. Biochem Biophys Res Commun 2005; 328: 158–164. [DOI] [PubMed] [Google Scholar]
- 83.Gonzalez C, Cuvellier S, Hue‐Beauvais C, et al. Genetic control of non obese diabetic mice susceptibility to high‐dose streptozotocin‐induced diabetes. Diabetologia 2003; 46: 1291–1295. [DOI] [PubMed] [Google Scholar]
- 84.Maegawa T, Miyasaka Y, Kobayashi M, et al. Congenic mapping and candidate gene analysis for streptozotocin‐induced diabetes susceptibility locus on mouse chromosome 11. Mamm Genome 2018; 29: 273–280. [DOI] [PubMed] [Google Scholar]
- 85.Barrett JC, Clayton DG, Concannon P, et al. Genome‐wide association study and meta‐analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 2009; 41: 703–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cho YS, Chen C‐H, Hu C, et al. Meta‐analysis of genome‐wide association studies identifies 8 new loci for type 2 diabetes in East Asians. Nat Genet 2012; 2012: 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med 2009; 360: 1646–1654. [DOI] [PubMed] [Google Scholar]
- 88.Awata T, Yamashita H, Kurihara S, et al. A low‐frequency GLIS3 variant associated with resistance to Japanese type 1 diabetes. Biochem Biophys Res Commun 2013; 437: 521–525. [DOI] [PubMed] [Google Scholar]
- 89.Todd JA. Intolerable secretion and diabetes in tolerant transgenic mice, revisited. Nat Genet 2016; 48: 476–477. [DOI] [PubMed] [Google Scholar]
- 90.Liston A, Todd JA, Lagou V. Beta‐cell fragility as a common underlying risk factor in type 1 and type 2 diabetes. Trends Mol Med 2017; 23: 181–194. [DOI] [PubMed] [Google Scholar]
- 91.Yang Y, Chan L. Monogenic diabetes: what it teaches us on the common forms of type 1 and type 2 diabetes. Endocr Rev 2016; 37: 190–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Senée V, Chelala C, Duchatelet S, et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet 2006; 38: 682–687. [DOI] [PubMed] [Google Scholar]
- 93.Stoy J, Edghill EL, Flanagan SE, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci USA 2007; 104: 15040–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang J, Takeuchi T, Tanaka S, et al. A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta‐cell dysfunction in the Mody mouse. J Clin Invest 1999; 103: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Izumi T, Yokota‐Hashimoto H, Zhao S, et al. Dominant negative pathogenesis by mutant proinsulin in the Akita diabetic mouse. Diabetes 2003; 52: 409–416. [DOI] [PubMed] [Google Scholar]
- 96.Inoue H, Tanizawa Y, Wasson J, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet 1998; 20: 143–148. [DOI] [PubMed] [Google Scholar]
- 97.Abreu D, Urano F. Current landscape of treatments for Wolfram syndrome. Trends Pharmacol Sci 2019; 40: 711–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Montaser H, Patel KA, Balboa D, et al. Loss of MANF causes childhood‐onset syndromic diabetes due to increased endoplasmic reticulum stress. Diabetes 2021; 70: 1006–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sandhu MS, Weedon MN, Fawcett KA, et al. Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet 2007; 39: 951–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Awata T, Inoue K, Kurihara S, et al. Missense variations of the gene responsible for Wolfram syndrome (WFS1/wolframin) in Japanese: possible contribution of the Arg456His mutation to type 1 diabetes as a nonautoimmune genetic basis. Biochem Biophys Res Commun 2000; 268: 612–616. [DOI] [PubMed] [Google Scholar]
- 101.Niwano F, Babaya N, Hiromine Y, et al. Glucose metabolism after pancreatectomy: opposite extremes between pancreaticoduodenectomy and distal pancreatectomy. J Clin Endocrinol Metab 2021; 106: e2203–e2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kendall DM, Sutherland DER, Najarian JS, et al. Effects of hemipancreatectomy on insulin secretion and glucose tolerance in healthy humans. N Engl J Med 1990; 322: 898–903. [DOI] [PubMed] [Google Scholar]
- 103.Kirchner VA, Finger EB, Bellin MD, et al. Long‐term outcomes for living pancreas donors in the modern era. Transplantation 2016; 100: 1322–1328. [DOI] [PubMed] [Google Scholar]
- 104.Ebrahimi AG, Hollister‐Lock J, Sullivan BA, et al. Beta cell identity changes with mild hyperglycemia: Implications for function, growth, and vulnerability. Mol Metab 2020; 35: 100959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Stancill JS, Broniowska KA, Oleson BJ, et al. Pancreatic β‐cells detoxify H2O2 through the peroxiredoxin/thioredoxin antioxidant system. J Biol Chem 2019; 294: 4843–5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Benáková Š, Holendová B, Plecitá‐Hlavatá L. Plecitá‐Hlavatá L Redox homeostasis in pancreatic β‐cells: from development to failure. Antioxidants (Basel) 2021; 10: 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fakruddin MD, Wei F‐Y, Suzuki T, et al. Defective mitochondrial tRNA taurine modification activates global proteostress and leads to mitochondrial disease. Cell Rep 2018; 22: 482–496. [DOI] [PubMed] [Google Scholar]
- 108.Ohsawa Y, Hagiwara H, Nishimatsu S‐I, et al. Taurine supplementation for prevention of stroke‐like episodes in MELAS: a multicentre, open‐label, 52‐week phase III trial. J Neurol Neurosurg Psychiatry 2019; 90: 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sharma RB, O’Donnell AC, Stamateris RE, et al. Insulin demand regulates β cell number via the unfolded protein response. J Clin Invest 2015; 125: 3831–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Elksnis A, Martinell M, Eriksson O, et al. Heterogeneity of metabolic defects in type 2 diabetes and its relation to reactive oxygen species and alterations in beta‐cell mass. Front Physiol 2019; 10: 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tornovsky‐Babeay S, Dadon D, Ziv O, et al. Type 2 diabetes and congenital hyperinsulinism cause DNA double‐strand breaks and p53 activity in β cells. Cell Metab 2014; 19: 109–121. [DOI] [PubMed] [Google Scholar]
- 112.Tornovsky‐Babeay S, Weinberg‐Corem N, Ben‐Haroush Schyr R, et al. Biphasic dynamics of beta cell mass in a mouse model of congenital hyperinsulinism: implications for type 2 diabetes. Diabetologia 2021; 64: 1133–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Omori K, Nakamura A, Miyoshi H, et al. Glucokinase inactivation paradoxically ameliorates glucose intolerance by increasing β‐Cell mass in db/db mice. Diabetes 2021; 70: 917–931. [DOI] [PubMed] [Google Scholar]
- 114.Martin D, Bellanne‐Chantelot C, Deschamps I, et al. Long‐term follow‐up of oral glucose tolerance test‐derived glucose tolerance and insulin secretion and insulin sensitivity indexes in subjects with glucokinase mutations (MODY2). Diabetes Care 2008; 31: 1321–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Piganelli JD, Mamula MJ, James EA. The role of β cell stress and neo‐epitopes in the immunopathology of type 1 diabetes. Front Endocrinol 2021; 11: 624590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Eizirik DL, Pasquali L, Cnop M. Pancreatic β‐cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat Rev Endocrinol 2020; 16: 349–362. [DOI] [PubMed] [Google Scholar]