

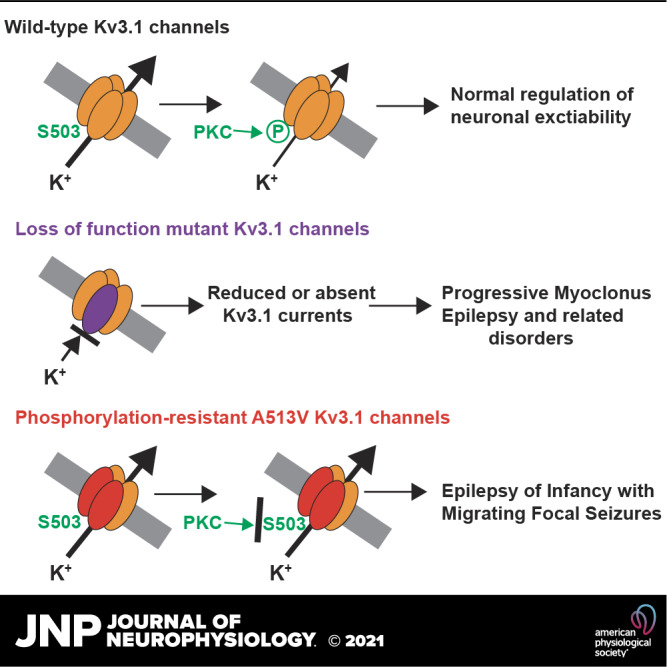
Keywords: channelopathy, epilepsy, intellectual disability, Kv3.1, protein kinase C
Abstract
Channelopathies caused by mutations in genes encoding ion channels generally produce a clear change in channel function. Accordingly, mutations in KCNC1, which encodes the voltage-dependent Kv3.1 potassium channel, result in progressive myoclonus epilepsy as well as other developmental and epileptic encephalopathies, and these have been shown to reduce or fully abolish current amplitude. One exception to this is the mutation A513V Kv3.1b, located in the cytoplasmic C-terminal domain of the channel protein. This de novo variant was detected in a patient with epilepsy of infancy with focal migrating seizures (EIFMS), but no difference could be detected between A513V Kv3.1 current and that of wild-type Kv3.1. Using both biochemical and electrophysiological approaches, we have now confirmed that this variant produces functional channels but find that the A513V mutation renders the channel completely insensitive to regulation by phosphorylation at S503, a nearby regulatory site in the C-terminus. In this respect, the mutation resembles those in another channel, KCNT1, which are the major cause of EIFMS. Because the amplitude of Kv3.1 current is constantly adjusted by phosphorylation in vivo, our findings suggest that loss of such regulation contributes to EIFMS phenotype and emphasize the role of channel modulation for normal neuronal function.
NEW & NOTEWORTHY Ion channel mutations that cause serious human diseases generally alter the biophysical properties or expression of the channel. We describe a de novo mutation in the Kv3.1 potassium channel that causes severe intellectual disability with early-onset epilepsy. The properties of this channel appear identical to those of wild-type channels, but the mutation prevents phosphorylation of the channel by protein kinase C. Our findings emphasize the role of channel modulation in normal brain function.
INTRODUCTION
The Kv3.1 potassium channel is one of four members of the Kv3 subfamily of voltage-dependent potassium channels (1). It is expressed in a subset of neurons, most of which are capable of firing at high rates (2–4). These include parvalbumin-containing interneurons within the cerebral cortex, auditory brainstem neurons, cerebellar granule cells, and neurons of the reticular nucleus of the thalamus. One feature that distinguishes Kv3 channels, and in particular Kv3.1, from most of the other 26 members of the mammalian Kv family is ultra-rapid activation and deactivation in response to depolarization. Moreover, under normal conditions, Kv3.1 activates only at membrane potentials more positive than about −15 mV (5–8). Pharmacological, genetic, and simulation studies have demonstrated that these properties promote the ability of neurons to fire at high rates of up to hundreds of hertz.
There are two major splice isoforms of the Kv3.1 channel, Kv3.1a and Kv3.1b, which differ in the length of their cytoplasmic C-termini (7). The longer Kv3.1b isoform has a serine residue, S503, that undergoes phosphorylation by protein kinase C (PKC) to suppress current amplitude. In other respects, the currents of Kv3.1a and Kv3.1b isoforms appear identical (9). In most neurons, however, Kv3.1b is the dominant isoform and the Kv3.1a is dominant only early in development before synapse formation (3, 10, 11). Sensory stimulation in vivo or high rates of synaptic stimulation of Kv3.1b-expressing neurons has been shown to produce very rapid changes in Kv3.1b current mediated by phosphorylation/dephosphorylation of the S503 residue (12, 13).
Human mutations in KCNC1, the gene that encodes Kv3.1 channels, have been found to result progressive myoclonus epilepsy, as well several other epileptic encephalopathies and ataxia and intellectual disability (14–17). All of the characterized mutations resulted in partial or complete loss-of-channel function. One exception was a variant, (c.1538C > T) A513V Kv3.1, found in a patient with epilepsy of infancy with focal migrating seizures (EIFMS, also termed malignant migrating seizures of infancy) (14). When expressed in Xenopus oocytes, this mutation gave rise to potassium currents with characteristics and amplitude identical to that of wild-type Kv3.1 channels. The A513 site is located in the cytoplasmic C-terminus that is specific to the Kv3.1b isoform. We have now found that this mutation renders the Kv3.1b channel incapable of phosphorylation at the regulatory serine S503 residue.
METHODS
Site-Directed Mutagenesis
Point mutations in Kv3.1 channel cDNA were introduced by full-length PCR amplification of plasmid DNA with sense and antisense mutated primers using Pfu polymerase (Stratagene). Parental (wild-type) DNA was digested with DpnI (New England Biolabs), and 1 μL of this reaction was used for transformation of competent Escherichia coli XL1Blue strain (Stratagene). DNA sequencing confirmed the presence of the A513V mutation and the absence of other undesired mutations.
Cell Culture
Chinese hamster ovary (CHO) cells were grown in Iscove’s modified Dulbecco’s medium (Invitrogen, Cat. No. 12440053) supplemented with 10% fetal bovine serum (heat-inactivated), 100 units/mL penicillin/streptomycin, 5% HT supplement (Invitrogen, Cat. No. 11067030) in a 5% CO2 incubator at 37°C. Cells were seeded 1 day before transient transfection. Lipofectamine 2000 (Invitrogen, Cat. No. 11668019) was used to co-transfect CHO cells with wild-type Kv3.1/pCDNA3 and N-EGFP or with mutant A513V Kv3.1 and green fluorescent protein (GFP). Currents were recorded 24 h after transfection.
Patch-Clamp Recordings
Electrodes (2–3 MΩ resistance) for whole cell patch recording were pulled from 1.5-mm OD borosilicate capillary glass (World Precision Instruments). The intracellular solution was (in mM) 97.5 potassium gluconate, 32.5 KCl, 10 HEPES, 5 EGTA, pH 7.2, with KOH. The bath solution was (in mM) 140 NaCl, 5.4 KCl, 1.3 CaCl2, 25 HEPES, 33 glucose, pH 7.4. Series resistance was 2–4 MΩ and was compensated by 80%–85%. Data were acquired at 10 kHz and filtered at 5 kHz (pClamp9 software, Molecular Devices). Reagents used were the PKC activator 12-O-tetradecanoylphorbol-13-acetate (TPA, Sigma, Cat. No. P-1585) and the PKC inhibitor GF109203X (TOCRIS, Cat. No. 0741).
Western Blot
CHO cells were harvested, washed twice in cold PBS, and resuspended in cold homogenization buffer [25 mm Tris-HCl at pH 7.4, 150 mM NaCl, 1% Nonidet P-40 (NP-40), 1 mM EDTA, 5% glycerol, with complete Mini Protease Inhibitor Tablet (Roche Applied Science, SKU No. 11836153001), and PhosSTOP EASYpack (Roche, Cat. No. 04906837001)]. After homogenization and centrifugation at 13,000 g (15 min at 4°C), supernatants were aliquoted, frozen in liquid nitrogen, and stored at −70°C until use. Protein estimation was done using Bradford’s reagent (Bio-Rad). Samples were suspended in 2× sample buffer for electrophoresis. After electrophoresis, proteins were transferred onto polyvinylidene difluoride membranes (Bio-Rad). Blots were then blocked in TBST (Tris-buffered saline, 0.1% Tween 20 detergent) containing 5% nonfat milk for 1 h at room temperature with shaking. Blots were then incubated with the respective primary antibodies phospho-Kv3.1 (Ser 503) antibody (Thermo Fisher, Cat. No. PA1-4638) or GAPDH (Santa Cruz, Cat. No. sc-32233) overnight at 4°C. After three washes with TBST buffer, blots were incubated for 1 h with goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (Invitrogen, Cat. No, 32460) or goat anti-mouse horseradish peroxidase-conjugated secondary antibody (Invitrogen, Cat. No. 32430), followed by extensive washes in TBST. Labeled proteins were detected by enhanced chemiluminescence.
Surface Biotinylation
Equal amount of wild-type and mutant Kv3.1 plasmids were transfected in CHO cells by Lipofectamine 2000. After 36 h, surface Kv3.1 protein was detected using a Pierce Cell Surface Biotinylation and Isolation Kit. Briefly, intact cells were surface biotinylated for 10 min at room temperature using a membrane impermeant Sulfo-NHS-SS-Biotin/PBS solution. After washing in ice-cold Tris-buffered saline, cells were lysed in extraction buffer (Tris-HCl 10 mM at pH 7.5, NaCl 150 mM, EDTA 10 mM, Triton X-100 1%, SDS 0.1%, mammalian protease inhibitor cocktail 1%) on ice for 30 min. After sonication and centrifugation (15,000 g for 5 min at 4°C), supernatants containing equal amounts of protein were incubated with NeutrAvidin resin at room temperature for 30 min to immunoprecipitate the surface-biotinylated proteins. After washing, proteins were eluted from NeutrAvidin resin and then resolved by SDS-PAGE and immunoblotting for Kv3.1 (Antibodies Inc., Cat. No. N16B/8).
Immunocytochemistry
CHO cells expressing wild-type Kv3.1 or mutant Kv3.1 were grown on glass coverslips to ∼80%–90% confluence. Coverslips were washed three times in PBS, and cells were fixed in PBS containing 4% paraformaldehyde for 10 min. Coverslips were washed three times with PBS, and cells were then permeabilized by incubating in PBS containing 1% BSA and 0.2% Triton X-100 for 10 min. This buffer was used for all subsequent incubations. Primary antibodies were added to the coverslips for 1 h at room temperature. Coverslips were washed and incubated with Alexa Fluor 488 goat anti-rabbit or Alexa Fluor 594 goat anti-mouse antibodies for 30 min and then washed and mounted on glass slides with proLong Gold antifade Mountant with DAPI (Cat. No. p36931) for viewing by confocal imaging. Levels of phospho-immunostaining were quantified both by measuring total fluorescence signals in cells and by comparing the distribution of pixel intensities for several images of fields for cells in each condition.
RESULTS
The amino acid sequence of human KCNC1 gene that encodes Kv3.1b channels is 99.83% identical to that of the rat and mouse genes, which have been used for characterization of the properties and modulation of Kv3.1 channels (6, 9, 13), and the sequence of the C-terminal cytoplasmic region containing the A513V mutation is identical in the rat and human channels. We, therefore, generated a rat Kv3.1b construct bearing this mutation, expressed it transiently in CHO cells, and compared properties of the A513V Kv3.1b currents and protein with those in cells expressing the wild-type channel.
Whole cell patch-clamp recordings of cells expressing A513V Kv3.1b revealed current that appeared identical to those of wild-type Kv3.1 (Fig. 1, A and B). In response to voltage steps from a holding potential of −70 mV to test voltages between −80 and +70 mV, both constructs gave rise to rapidly activating and deactivating currents that did not inactivate during pulses lasting 600 ms. There was no statistically significant difference in overall current amplitudes between cells expressing A513V Kv3.1b and wild-type Kv3.1b (wild-type Kv3.1b current density, 0.48 ± 0.02 nA/nF at +70 mV, n = 7; A513V Kv3.1b current density, 0.49 ± 0.01 nA/nF at + 70 mV, n = 7). The voltage dependence of the potassium current was identical in the two sets of cells, as revealed by plotting normalized I-V relations (Fig. 1C).
Figure 1.
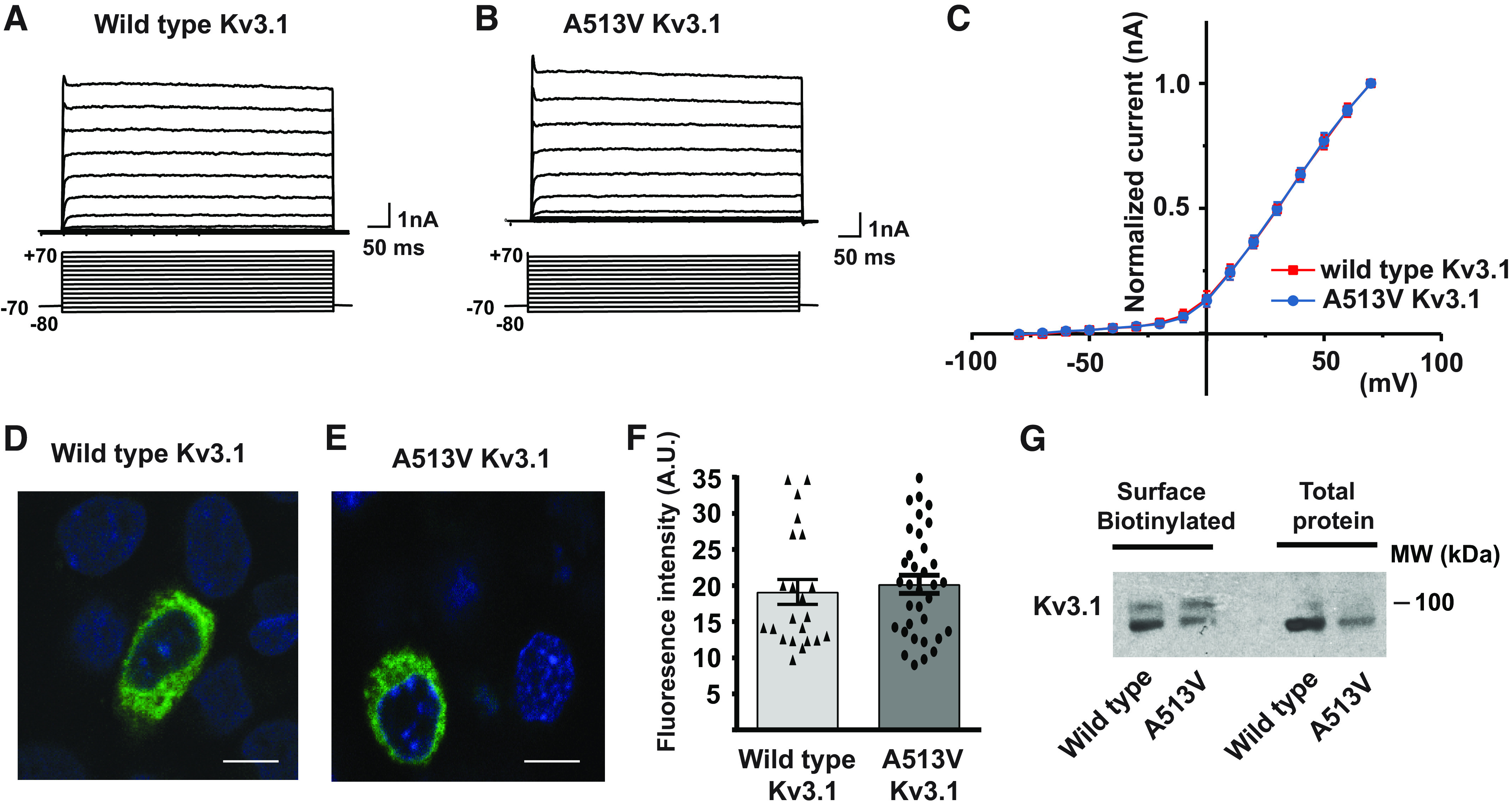
The A513V Kv3.1 mutation does not affect Kv3.1 current amplitude or current-voltage dependence. A and B: representative voltage-clamp traces for the wild-type and mutant A513V Kv3.1 currents. Currents were evoked from a holding potential of−70 mV to test potentials between −80 and +70 mV. C: current-voltage relationship for CHO cells transfected with wild-type Kv3.1 and mutant A513V Kv3.1. Values are means ± SE; n = 7, 7. D and E: immunostaining for Kv3.1 in CHO cells transfected with wild-type and A513V Kv3.1 plasmids, scale bar 10 µm. F: quantification of the fluorescence intensity of Kv3.1 immunostaining in CHO cells transfected with wild-type and mutant A513V Kv3.1 genes using NIH image J software. Values are means ± SE; n = 22 cells for wild-type; n = 34 cells for mutant A513V Kv3.1. G: blots showing levels of Kv3.1 channels detected in the plasma membrane after surface biotinylation of membrane proteins in wild-type and mutant A513V Kv3.1 expressing CHO cells. A.U., arbitrary units; CHO, Chinese hamster ovary.
As a further test for expression levels of the wild-type and mutant channels, transfected cells were immunostained for total Kv3.1b protein (Fig. 1, D and E). The overall pattern of staining was similar in A513V Kv3.1 and wild-type Kv3.1b expressing cells, and the quantification of overall fluorescence revealed no difference in expression (Fig. 1F). Finally, we carried out one additional experiment to determine whether the mutation of the A513 residue resulted in a change in the amount of Kv3.1b protein incorporated into the plasma membrane. Intact, nonpermeabilized A513V Kv3.1b- and wild-type Kv3.1-expressing cells were subjected to a surface biotinylation protocol before lysis. The ratio of the intensity of the band corresponding to biotinylated (surface) Kv3.1 to that for total Kv3.1 was similar in both types of cells (1.03 vs. 1.18 for wild-type vs. A513V Kv3.1 channels, respectively, Fig. 1G).
There are multiple phosphorylation sites in Kv3.1 that are common to both the Kv3.1a and Kv3.1b isoforms (9). There is an additional phosphorylation site, serine 503, that is specific to the Kv3.1b isoform, and this site is required for adjusting the amplitude of potassium current by activation of PKC. Consistent with previous studies (6, 9), we found that exposure of cells expressing wild-type Kv3.1b to the PKC activator TPA (12-O-tetradecanoylphorbol-13-acetate, 100 nM) resulted in a significant reduction in current amplitude measured using whole cell patch-clamp recordings (Fig. 2, A and B).
Figure 2.
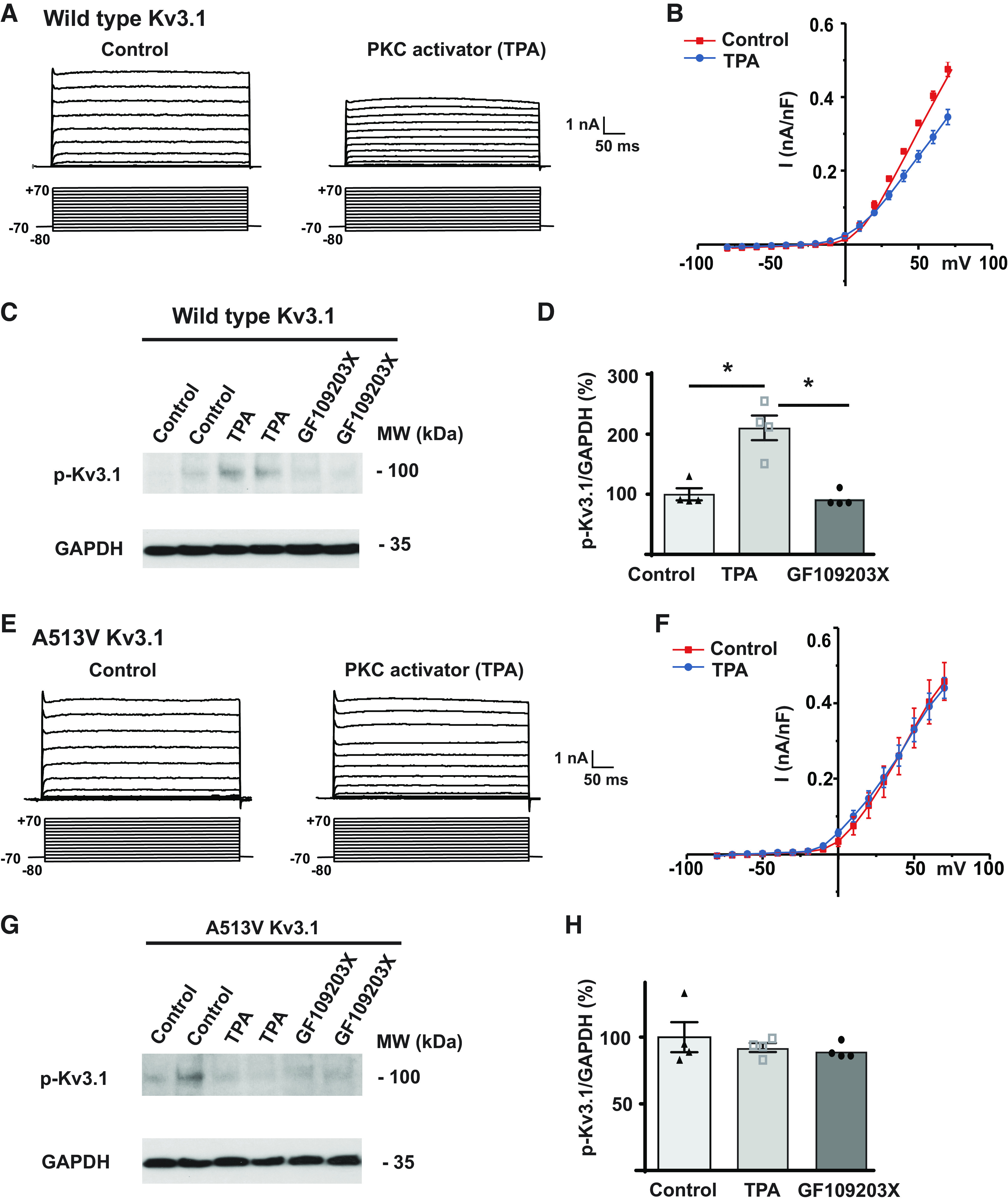
A PKC activator suppresses wild-type Kv3.1 currents, but not those of the A513V Kv3.1 mutant. A: representative voltage-clamp traces for wild-type Kv3.1 expressed in CHO cells before and after treatment with 100 nM TPA. Currents were evoked from a holding potential of −70 mV to test potentials between −80 and +70 mV. B: current-voltage relationship for CHO cells expressing wild-type Kv3.1 gene treated with 100 nM TPA. Currents were normalized by cell capacitance. C: immunoblots for phospho-S503 Kv3.1 in CHO cells expressing wild-type Kv3.1 and treated with either carrier solution DMSO alone, 100 nM TPA for 30 min, or pretreated with PKC inhibitor, 1 µM GF109203X for 30 min before exposure to 100 nM TPA with inhibitor for 30 min. D: quantification of phospho-S503 Kv3.1 levels in the three conditions. One-way ANOVA Turkey’s multiple-comparison test was performed. *P < 0.05. Values are means ± SE; n = 4, 4, 4 independent experiments. E–G: as for A–C but for CHO cells expressing 513V Kv3.1. H: quantification of phospho-S503 Kv3.1 levels in 513V Kv3.1 cells for the three conditions. One-way ANOVA Turkey’s multiple-comparison test was performed. Values are means ± SE; n = 4, 4, 4 independent experiments. CHO, Chinese hamster ovary; PKC, protein kinase C; TPA, 12-O-tetradecanoylphorbol-13-acetate.
To examine the phosphorylation of Kv3.1b protein directly, we carried out Western blotting on extracts of cells using a previously described phospho-specific antibody that recognizes Kv3.1b channels only when phosphorylated at serine residue 503 (12, 13). In extracts of cells expressing wild-type Kv3.1b, a very clear increase in channel immunoreactivity was detected in cells treated with 100 nM TPA for 30 min (Fig. 2, C and D). Consistent with this increase in phosphorylation being mediated by PKC, the effect of TPA was abolished by the pretreatment with the PKC inhibitor, 1 µM GF109203X for 30 min before application of TPA in the continued presence of the inhibitor (Fig. 2, C and D).
We next repeated the same experiments using cells expressing A513V Kv3.1b. In whole cell voltage-clamp experiments, TPA had no effect on either the amplitude or voltage dependence of the potassium current (Fig. 2, E and F). Moreover, Western blotting of extracts of A513V Kv3.1b cells with the S503-phosphospecific antibody revealed no significant change in immunoreactivity between untreated cells, those exposed to TPA for 30 min or those treated with TPA in the presence of GF109203x (Fig. 2, G and H).
We further examined the differences in the response of A513V Kv3.1b and wild-type Kv3.1b cells to phosphorylation by immunofluorescent staining using the S503-phosphospecific antibody (Fig. 3). Little or no immunostaining could be detected in wild-type Kv3.1b cells treated only with the carrier medium DMSO alone. Exposure to TPA for 30 min, however, produced very clear labeling at the plasma membrane in these cells (Fig. 3A). This was quantified two ways, both using the same exposure times and laser intensity for all images. We first measured the difference in amount of immunofluorescence over background between DMSO- and TPA-treated cells in regions of interest (ROIs) corresponding to individual cells in each field [relative immunofluorescence 27.4 ± 4.04 vs. 61.08 ± 5.09 arbitrary units (AU), n = 8, 9 fields of cells, respectively, P < 0.0001, Student’s unpaired t test]. For the second approach, we compared the distribution of total fluorescence in multicell images of fields of the two sets of cells (Fig. 3B), and this also yielded a significant difference (P < 0.0001, χ2-test).
Figure 3.

Immunostaining of phospho-S503 Kv3.1 protein in CHO cells transfected with wild-type and mutant A513V Kv3.1. A: phospho-S503 Kv3.1 staining in CHO cells expressing wild-type Kv3.1 treated with DMSO for 30 mins (top) or treated with 100 nM TPA for 30 mins (bottom), scale bar 50 µm. B: distribution of fluorescence intensity of pixels in images of phospho-S503 immunostaining in CHO cells transfected with wild-type Kv3.1. C and D: as for A and B but for cells expressing A513V Kv3.1. Distributions shown in B and D are combined from images of 200–250 cells from 3 independent fields each. A.U., arbitrary units; CHO, Chinese hamster ovary; TPA, 12-O-tetradecanoylphorbol-13-acetate.
When cells expressing the A513V Kv3.1b mutant were subject to immunofluorescent staining using the S503-phosphospecific antibody, no clear signal could be detected in images of the cells either in cells treated with DMSO or TPA (Fig. 3C). Quantification of immunofluorescence between individual DMSO- and TPA-treated cells confirmed that, in contrast to wild-type Kv3.1b cells, activation of PKC produced no change in the phospho-S503 signal (relative immunofluorescence, 17.00 ± 4.6 vs. 24.75 ± 4.4 AU, n = 4 fields of cells). We also found no significant difference in the distribution of total fluorescence in multicell images of fields of the mutant cells with or without TPA (Fig. 3D, N.S. χ2-test).
DISCUSSION
Previous studies have provided compelling data that human mutations in the KCNC1 gene, which encodes Kv3.1 voltage-dependent potassium channels, result in several epileptic encephalopathies, and these can be coupled to severe intellectual disability (14–17). One de novo mutation, found in a patient with EIFMS, had previously been found to encode functional channels that did not appear to differ from wild-type Kv3.1 channels. This mutation, A513V Kv3.1, was therefore considered to represent a variant of uncertain significance (14). Using mammalian cell lines, we have confirmed that A513V Kv3.1 channels are functional with basal properties similar to those of the wild-type channel. Our data clearly show, however, that a physiological mechanism that regulates the amplitude of Kv3.1 currents both in vivo and in vitro, that is, phosphorylation by PKC, is completely disrupted by this mutation. Our findings, therefore, support that this mutation is closely linked to the pathophysiology of EIMFS.
The inability of the A513V Kv3.1 to be regulated by PKC is shared by other channel mutations that result in EIMFS. The major known genetic cause of EIMFS, an intractable infantile epilepsy associated with severe intellectual disability (18), is de novo mutations in KCNT1, a gene that encodes another potassium channel, Slack, also termed KNa1.1 (19–26). Mutations in the Slack channel also result in other epilepsies, mainly focal and including autosomal dominant nocturnal frontal lobe epilepsy (ANDFLE) (24, 27–29) and in sudden unexpected death in epilepsy (SUDEP) (24). In common with A513V Kv3.1, nearly all of these KCNT1 mutations produce functional channels, although current amplitude is increased over that of wild-type channels (19, 20, 26). Like Kv3.1b channels, the amplitude of wild-type Slack currents is regulated by phosphorylation of a C-terminal serine residue by PKC (19, 30). A key feature of disease-causing KCNT1 mutations in which responses to PKC have been examined is that the mutant channels fail to respond normally to the activation of this kinase (19). Thus, inability to modulate channel activity by signaling pathways may be a key feature of certain channelopathies, and particularly for pathogenic variants of genes causing EIMFS.
There are several potential mechanisms by which the A513V Kv3.1 mutation, as well as those in Slack channels, could prevent phosphorylation. The simplest hypothesis is that they induce a conformational change in the cytoplasmic C-terminal that obstructs the binding of PKC isozymes required for basal phosphorylation (12). There are, however, other possibilities. For example, the mutation could result in the constitutive binding of a protein phosphatase or alter the spatial relationship between the kinase and target serine residue.
Kv3.1 is primarily expressed in the central nervous system and particularly high levels are found in fast-spiking neurons such as GABAergic parvalbumin-positive cortical interneurons rates (2–4). Most of the disease-causing channel mutations reduce Kv3.1 current, and dominant-negative mutations are expected to reduce current in channels that are comprised of tetramers containing wild-type Kv3.1 with other Kv3 family subunits (14–17). A central hypothesis for epileptogenesis is therefore that the mutations reduce the ability of cortical inhibitory neurons to fire at high rates, leading to an overexcitation of excitatory pyramidal cells. This hypothesis is based, however, on cortical localization data obtained using rodents (31, 32). In the neocortex of macaque monkeys, Kv3.1b expression is detected in a wider variety of cortical neurons including subsets of pyramidal cells (33, 34). The implications of the mutations for cortical circuit dynamics in primate brain may, therefore, be more complex than that expected from rodent models.
In summary, we have identified the functional consequence of a Kv3.1 mutation associated with EIMFS. The A513V Kv3.1 mutation is located adjacent to a key phosphorylation site that normally adjusts the amplitude of Kv3.1 current in response to different patterns of synaptic stimulation. Experiments in which this mutation is expressed in mice, or in neurons derived from human stem cells, will help to determine whether the overall effect of this mutation on neuronal excitability resembles that of other models of EIMFS (35–37).
GRANTS
This work was supported by NIH Grants DC01919 and NS102239 to L.K.K.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.Z. and L.K.K. conceived and designed research; Y.Z. and S.R.A. performed experiments; Y.Z. and L.K.K. analyzed data; G.B., R.N. and L.K.K. interpreted results of experiments; Y.Z. and L.K.K. prepared figures; Y.Z. and L.K.K. drafted manuscript; R.N. and L.K.K. edited and revised manuscript; Y.Z., S.R.A., R.N., G.B., and L.K.K. approved final version of manuscript.
REFERENCES
- 1.Kaczmarek LK, Zhang Y. Kv3 channels: enablers of rapid firing, neurotransmitter release, and neuronal endurance. Physiol Rev 97: 1431–1468, 2017. doi: 10.1152/physrev.00002.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weiser M, Bueno E, Sekirnjak C, Martone ME, Baker H, Hillman D, Chen S, Thornhill W, Ellisman M, Rudy B. The potassium channel subunit KV3.1b is localized to somatic and axonal membranes of specific populations of CNS neurons. J Neurosci 15: 4298–4314, 1995. doi: 10.1523/JNEUROSCI.15-06-04298.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perney TM, Marshall J, Martin KA, Hockfield S, Kaczmarek LK. Expression of the mRNAs for the Kv3.1 potassium channel gene in the adult and developing rat brain. J Neurophysiol 68: 756–766, 1992. doi: 10.1152/jn.1992.68.3.756. [DOI] [PubMed] [Google Scholar]
- 4.Sekirnjak C, Martone ME, Weiser M, Deerinck T, Bueno E, Rudy B, Ellisman M. Subcellular localization of the K+ channel subunit Kv3.1b in selected rat CNS neurons. Brain Res 766: 173–187, 1997. doi: 10.1016/S0006-8993(97)00527-1. [DOI] [PubMed] [Google Scholar]
- 5.Grissmer S, Nguyen AN, Aiyar J, Hanson DC, Mather RJ, Gutman GA, Karmilowicz MJ, Auperin DD, Chandy KG. Pharmacological characterization of five cloned voltage-gated K+ channels, types Kv1.1, 1.2, 1.3, 1.5, and 3.1, stably expressed in mammalian cell lines. Mol Pharmacol 45: 1227–1234, 1994. [PubMed] [Google Scholar]
- 6.Kanemasa T, Gan L, Perney TM, Wang LY, Kaczmarek LK. Electrophysiological and pharmacological characterization of a mammalian Shaw channel expressed in NIH 3T3 fibroblasts. J Neurophysiol 74: 207–217, 1995. doi: 10.1152/jn.1995.74.1.207. [DOI] [PubMed] [Google Scholar]
- 7.Luneau CJ, Williams JB, Marshall J, Levitan ES, Oliva C, Smith JS, Antanavage J, Folander K, Stein RB, Swanson R. Alternative splicing contributes to K+ channel diversity in the mammalian central nervous system. Proc Natl Acad Sci USA 88: 3932–3936, 1991. doi: 10.1073/pnas.88.9.3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Macica CM, Kaczmarek LK. Casein kinase 2 determines the voltage dependence of the Kv3.1 channel in auditory neurons and transfected cells. J Neurosci 21: 1160–1168, 2001. doi: 10.1523/JNEUROSCI.21-04-01160.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macica CM, von Hehn CAA, Wang L-Y, Ho C-S, Yokoyama S, Joho RH, Kaczmarek LK. Modulation of the Kv3.1b potassium channel isoform adjusts the fidelity of the firing pattern of auditory neurons. J Neurosci 23: 1133–1141, 2003. doi: 10.1523/JNEUROSCI.23-04-01133.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu SJ, Kaczmarek LK. The expression of two splice variants of the Kv3.1 potassium channel gene is regulated by different signaling pathways. J Neurosci 18: 2881–2890, 1998. doi: 10.1523/JNEUROSCI.18-08-02881.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu SJ, Kaczmarek LK. Depolarization selectively increases the expression of the Kv3.1 potassium channel in developing inferior colliculus neurons. J Neurosci 18: 8758–8769, 1998. doi: 10.1523/JNEUROSCI.18-21-08758.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song P, Kaczmarek LK. Modulation of Kv3.1b potassium channel phosphorylation in auditory neurons by conventional and novel protein kinase C isozymes. J Biol Chem 281: 15582–15591, 2006. doi: 10.1074/jbc.M512866200. [DOI] [PubMed] [Google Scholar]
- 13.Song P, Yang Y, Barnes-Davies M, Bhattacharjee A, Hamann M, Forsythe ID, Oliver DL, Kaczmarek LK. Acoustic environment determines phosphorylation state of the Kv3.1 potassium channel in auditory neurons. Nat Neurosci 8: 1335–1342, 2005. doi: 10.1038/nn1533. [DOI] [PubMed] [Google Scholar]
- 14.Cameron JM, Maljevic S, Nair U, Aung YH, Cogné B, Bézieau S, Blair E, Isidor B, Zweier C, Reis A, Koenig MK, Maarup T, Sarco D, Afenjar A, Huq AHMM, Kukolich M, Billette de Villemeur T, Nava C, Héron B, Petrou S, Berkovic SF. Encephalopathies with KCNC1 variants: genotype-phenotype-functional correlations. Ann Clin Transl Neurol 6: 1263–1272, 2019. doi: 10.1002/acn3.50822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim H, Lee S, Choi M, Kim H, Hwang H, Choi J, Chae JH, Kim KJ, Lim BC. Familial cases of progressive myoclonic epilepsy caused by maternal somatic mosaicism of a recurrent KCNC1 p.Arg320His mutation. Brain Dev 40: 429–432, 2018. doi: 10.1016/j.braindev.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 16.Muona M, Berkovic SF, Dibbens LM, Oliver KL, Maljevic S, Bayly MA, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet 47: 39–46, 2015. doi: 10.1038/ng.3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park J, Koko M, Hedrich UBS, Hermann A, Cremer K, Haberlandt E, Grimmel M, Alhaddad B, Beck-Woedl S, Harrer M, Karall D, Kingelhoefer L, Tzschach A, Matthies LC, Strom TM, Ringelstein EB, Sturm M, Engels H, Wolff M, Lerche H, Haack TB. KCNC1-related disorders: new de novo variants expand the phenotypic spectrum. Ann Clin Transl Neurol 6: 1319–1326, 2019. doi: 10.1002/acn3.50799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coppola G, Plouin P, Chiron C, Robain O, Dulac O. Migrating partial seizures in infancy: a malignant disorder with developmental arrest. Epilepsia 36: 1017–1024, 1995. doi: 10.1111/j.1528-1157.1995.tb00961.x. [DOI] [PubMed] [Google Scholar]
- 19.Barcia G, Fleming MR, Deligniere A, Gazula VR, Brown MR, Langouet M, Chen H, Kronengold J, Abhyankar A, Cilio R, Nitschke P, Kaminska A, Boddaert N, Casanova JL, Desguerre I, Munnich A, Dulac O, Kaczmarek LK, Colleaux L, Nabbout R. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet 44: 1255–1259, 2012. doi: 10.1038/ng.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim GE, Kronengold J, Barcia G, Quraishi IH, Martin HC, Blair E, Taylor JC, Dulac O, Colleaux L, Nabbout R, Kaczmarek LK. Human slack potassium channel mutations increase positive cooperativity between individual channels. Cell Rep 9: 1661–1672, 2014. doi: 10.1016/j.celrep.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shimada S, Hirano Y, Ito S, Oguni H, Nagata S, Shimojima K, Yamamoto T. A novel KCNT1 mutation in a Japanese patient with epilepsy of infancy with migrating focal seizures. Hum Genome Var 1: 14027, 2014. doi: 10.1038/hgv.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanderver A, Simons C, Schmidt JL, Pearl PL, Bloom M, Lavenstein B, Miller D, Grimmond SM, Taft RJ. Identification of a novel de novo p.Phe932Ile KCNT1 mutation in a patient with leukoencephalopathy and severe epilepsy. Pediatr Neurol 50: 112–114, 2014. doi: 10.1016/j.pediatrneurol.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mikati MA, Jiang YH, Carboni M, Shashi V, Petrovski S, Spillmann R, Milligan CJ, Li M, Grefe A, McConkie A, Berkovic S, Scheffer I, Mullen S, Bonner M, Petrou S, Goldstein D. Quinidine in the treatment of KCNT1-positive epilepsies. Ann Neurol 78: 995–999, 2015. doi: 10.1002/ana.24520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Møller RS, Heron SE, Larsen LH, Lim CX, Ricos MG, Bayly MA, van Kempen MJ, Klinkenberg S, Andrews I, Kelley K, Ronen GM, Callen D, McMahon JM, Yendle SC, Carvill GL, Mefford HC, Nabbout R, Poduri A, Striano P, Baglietto MG, Zara F, Smith NJ, Pridmore C, Gardella E, Nikanorova M, Dahl HA, Gellert P, Scheffer IE, Gunning B, Kragh-Olsen B, Dibbens LM. Mutations in KCNT1 cause a spectrum of focal epilepsies. Epilepsia 56: e114–e120, 2015. doi: 10.1111/epi.13071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rizzo F, Ambrosino P, Guacci A, Chetta M, Marchese G, Rocco T, Soldovieri MV, Manocchio L, Mosca I, Casara G, Vecchi M, Taglialatela M, Coppola G, Weisz A. Characterization of two de novo KCNT1 mutations in children with malignant migrating partial seizures in infancy. Mol Cell Neurosci 72: 54–63, 2016. doi: 10.1016/j.mcn.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 26.Tang QY, Zhang FF, Xu J, Wang R, Chen J, Logothetis DE, Zhang Z. Epilepsy-related slack channel mutants lead to channel over-activity by two different mechanisms. Cell Rep 14: 129–139, 2016. doi: 10.1016/j.celrep.2015.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heron SE, Smith KR, Bahlo M, Nobili L, Kahana E, Licchetta L, Oliver KL, Mazarib A, Afawi Z, Korczyn A, Plazzi G, Petrou S, Berkovic SF, Scheffer IE, Dibbens LM. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 44: 1188–1190, 2012. doi: 10.1038/ng.2440. [DOI] [PubMed] [Google Scholar]
- 28.Martin HC, Kim GE, Pagnamenta AT, Murakami Y, Carvill GL, Meyer E, Copley RR, Rimmer A, Barcia G, Fleming MR, Kronengold J, Brown MR, Hudspith KA, Broxholme J, Kanapin A, Cazier JB, Kinoshita T, Nabbout R; WGS500 Consortium, Bentley D, McVean G, Heavin S, Zaiwalla Z, McShane T, Mefford HC, Shears D, Stewart H, Kurian MA, Scheffer IE, Blair E, Donnelly P, Kaczmarek LK, Taylor JC. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet 23: 3200–3211, 2014. doi: 10.1093/hmg/ddu030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohba C, Kato M, Takahashi N, Osaka H, Shiihara T, Tohyama J, Nabatame S, Azuma J, Fujii Y, Hara M, Tsurusawa R, Inoue T, Ogata R, Watanabe Y, Togashi N, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Tanaka F, Saitsu H, Matsumoto N. De novo KCNT1 mutations in early-onset epileptic encephalopathy. Epilepsia 56: e121–e128, 2015. doi: 10.1111/epi.13072. [DOI] [PubMed] [Google Scholar]
- 30.Santi CM, Ferreira G, Yang B, Gazula VR, Butler A, Wei A, Kaczmarek LK, Salkoff L. Opposite regulation of Slick and Slack K+ channels by neuromodulators. J Neurosci 26: 5059–5068, 2006. doi: 10.1523/JNEUROSCI.3372-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chow A, Erisir A, Farb C, Nadal MS, Ozaita A, Lau D, Welker E, Rudy B. K+ channel expression distinguishes subpopulations of parvalbumin- and somatostatin-containing neocortical interneurons. J Neurosci 19: 9332–9345, 1999. doi: 10.1523/JNEUROSCI.19-21-09332.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martina M, Schultz JH, Ehmke H, Monyer H, Jonas P. Functional and molecular differences between voltage-gated K+ channels of fast-spiking interneurons and pyramidal neurons of rat hippocampus. J Neurosci 18: 8111–8125, 1998. doi: 10.1523/JNEUROSCI.18-20-08111.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Constantinople CM, Disney AA, Maffie J, Rudy B, Hawken MJ. Quantitative analysis of neurons with Kv3 potassium channel subunits, Kv3.1b and Kv3.2, in macaque primary visual cortex. J Comp Neurol 516: 291–311, 2009. doi: 10.1002/cne.22111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ichinohe N, Watakabe A, Miyashita T, Yamamori T, Hashikawa T, Rockland KS. A voltage-gated potassium channel, Kv3.1b, is expressed by a subpopulation of large pyramidal neurons in layer 5 of the macaque monkey cortex. Neuroscience 129: 179–185, 2004. doi: 10.1016/j.neuroscience.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 35.Quraishi IH, Mercier MR, McClure H, Couture RL, Schwartz ML, Lukowski R, Ruth P, Kaczmarek LK. Impaired motor skill learning and altered seizure susceptibility in mice with loss or gain of function of the Kcnt1 gene encoding Slack (KNa1.1) Na+-activated K+. Sci Rep 10: 3213, 2020. doi: 10.1038/s41598-020-60028-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quraishi IH, Stern S, Mangan KP, Zhang Y, Ali SR, Mercier MR, Marchetto MC, McLachlan MJ, Jones EM, Gage FH, Kaczmarek LK. An epilepsy-associated KCNT1 mutation enhances excitability of human iPSC-derived neurons by increasing Slack KNa currents. J Neurosci 39: 7438–7449, 2019. doi: 10.1523/JNEUROSCI.1628-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shore AN, Colombo S, Tobin WF, Petri S, Cullen ER, Dominguez S, Bostick CD, Beaumont MA, Williams D, Khodagholy D, Yang M, Lutz CM, Peng Y, Gelinas JN, Goldstein DB, Boland MJ, Frankel WN, Weston MC. Reduced GABAergic neuron excitability, altered synaptic connectivity, and seizures in a KCNT1 gain-of-function mouse model of childhood epilepsy. Cell Rep 33: 108303, 2020. doi: 10.1016/j.celrep.2020.108303. [DOI] [PMC free article] [PubMed] [Google Scholar]