

Keywords: diaphragm, neuropathology, Pompe disease, respiratory, spinal cord
Abstract
Pompe disease (PD) is a neuromuscular disorder caused by a mutation in the acid alpha-glucosidase (GAA) gene. Patients with late-onset PD retain some GAA activity and present symptoms later in life, with fatality mainly associated with respiratory failure. This case study presents diaphragm electrophysiology and a histological analysis of the brainstem, spinal cord, and diaphragm, from a male PD patient diagnosed with late-onset PD at age 35. The patient was wheelchair dependent by age 38, required nocturnal ventilation at age 40, 24-h noninvasive ventilation by age 43, and passed away from respiratory failure at age 54. Diaphragm electromyography recorded using indwelling “pacing” wires showed asynchronous bursting between the left and right diaphragm during brief periods of independent breathing. The synchrony declined over a 4-yr period preceding respiratory failure. Histological assessment indicated motoneuron atrophy in the medulla and rostral spinal cord. Hypoglossal (soma size: 421 ± 159 µm2) and cervical motoneurons (soma size: 487 ± 189 µm2) had an atrophied, elongated appearance. In contrast, lumbar (soma size: 1,363 ± 677 µm2) and sacral motoneurons (soma size: 1,411 ± 633 µm2) had the ballooned morphology typical of early-onset PD. Diaphragm histology indicated loss of myofibers. These results are consistent with neuromuscular degeneration and the concept that effective PD therapy will need to target the central nervous system, in addition to skeletal and cardiac muscle.
NEW & NOTEWORTHY This case study offered a unique opportunity to investigate longitudinal changes in phrenic neurophysiology in an individual with severe, ventilator-dependent, late-onset Pompe disease. Additional diaphragm and neural tissue histology upon autopsy confirmed significant neuromuscular degeneration, and it provided novel insights regarding rostral to caudal variability in the neuropathology. These findings suggest that a successful treatment approach for ventilator-dependent Pompe disease should target the central nervous system, in addition to skeletal muscle.
INTRODUCTION
Glycogen Storage Disease Type II (GSD2, Pompe disease) is an autosomal recessive disorder characterized by a mutation in the gene encoding acid α-glucosidase (GAA), an enzyme responsible for lysosomal glycogen degradation. Diminished or absent levels of GAA lead to impaired autophagy and accumulation of glycogen in skeletal muscle, visceral organs, and the central nervous system (1, 2). Mutational severity corresponds to residual enzyme activity, which often corresponds to the extent and rate of clinical progression (1, 3). In the most severe infantile-onset form, patients with no residual enzyme activity present with life-threatening symptoms within the first year. In contrast, less-severe mutations that enable modest enzyme activity present as a slower-progressing, late-onset Pompe disease (LOPD) which presents in adulthood.
Although infantile and adult onset Pompe differ in the onset and rate of progression, respiratory muscle weakness presents in both situations. Dysfunction of the phrenic motor system has also been well documented. For example, transdiaphragmatic pressure evoked by magnetic stimulation, an indication of diaphragm function, is significantly reduced in adults and appears to correspond to the severity of ventilatory involvement (4–6). Neuropathology in the phrenic motor system is well documented in Pompe animal models (2, 7) and is present in the mid-cervical spinal cord (including putative phrenic motoneurons) of early onset Pompe patients (2). Neuropathologic changes in the later-onset phenotype are less clear, and studies to date have focused on fibrosis and oxidative stress of neural structures and glycogenosis of CNS arterial supply (8–11).
This case study presents novel clinical, electrophysiological, and histological evidence of phrenic motor system pathology from an adult patient with a decade of chronic ventilatory failure secondary to Pompe disease before passing.
CASE HISTORY
The patient was a male diagnosed with Pompe disease at age 35 (Fig. 1). Despite alguocosidase alfa administration, his clinical progression of Pompe disease was notable for wheelchair dependence by age 38 and progressive respiratory insufficiency with requirement for nocturnal ventilation at age 40 and 24-h noninvasive ventilation by age 43. Medical comorbidities included hypertension, type 2 diabetes, hyperlipidemia, stroke (age 40, no additional permanent paralysis), and renal calculi. The patient presented to our team for respiratory and neurological evaluation at age 48.4 yr, which revealed profound pulmonary dysfunction [maximal inspiratory pressure, PIMAX: 13.5 cmH2O; maximal expiratory pressure, PEMAX: 6.0 cmH2O; unassisted tidal volume (VT) 2.0 mL/kg for <1 min; end-tidal CO2: 44 mmHg]. Notably, he rapidly desaturated within 30–40 s of unsupported breathing, disrupting the ability to eat or complete oral hygiene. Three months of inspiratory strength training did not improve his respiratory function [PIMAX: 12.8, PEMAX: 7.0 cmH2O, vital capacity (VC): 310 mL (10% predicted), unassisted VT: 2.1 mL/kg for 1 min]. Supramaximal bilateral anterolateral phrenic nerve magnetic stimulation produced a negligible response [maximal transdiaphragmatic pressure (PDIMAX): <2 cmH2O, evoked diaphragm compound muscle action potential: 0.26 mV). Additionally, the patient had diffuse quadriparesis (Medical Research Council scale: 1/5) and retained bladder and bowel control. The patient underwent placement of a diaphragm pacemaker (NeuRx, Synapse Biomedical) and tracheostomy placement at age 48.8 yr. The patient provided informed consent to participate in an institutional review board-approved, rare disease natural history study that enabled our team to periodically evaluate changes in function.
Figure 1.

Timeline of relevant case events. DC, discontinued; dx, disease; EMG, electromyogram; MV, mechanical ventilation; NIV, noninvasive ventilation.
Postoperative diaphragm conditioning consisted of progressive increases in the use of the stimulator daily, stimulation amplitude, and periods of breathing with reduced ventilator support. The patient tolerated 24 h of pacing by 6 wk postimplant, with minimal change of function (PIMAX: 12.5 cmH2O, unassisted VT: 1.1 mL/kg for <1 min). By day 180 postimplant, the patient tolerated paced breathing for up to 60 min daily, without mechanical ventilation. However, high grade tracheal and left mainstem bronchial malacia were detected, necessitating the use of long-term positive pressure support to prevent airway collapse with inspiratory efforts. Nevertheless, the patient was able to maintain minute ventilation over the following two years with ventilator inspiratory pressure reduced from 29 cmH2O to 15 cmH2O during daytime hours. Respiratory function peaked two years after diaphragm pacer placement (age 50.7 yr, PIMAX: 17.2 cmH2O, PEMAX: 18.7 cmH2O, unassisted VT: 2.4 mL/kg for 10 min with pacer, 2.0 mL/kg for 5 min no support).
Focal seizures were witnessed at age 50.8 yr, during intravenous administration of alguocosidase alfa (20 mg/kg, every other week). At 51.4 yr, the patient was hospitalized for nearly 2 mo, for a lower respiratory tract infection that triggered status epilepticus. Seizure activity was initially controlled with levetiracetam and valproic acid. Antiepileptics were subsequently transitioned to lamotrigine (200 mg BID) over the following year, but the patient reported new memory deficits that did not abate. By 6 mo postdischarge (age 51.9 yr), the patient resumed pacing and achieved intermittently lower ventilator settings [peak inspiratory pressure: (18 cmH2O) for 2–4-h periods during the daytime]. Four years postdiaphragm pacer implant (age 52.8 yr), we observed a modest decline in respiratory function: PIMAX: 13.7 cmH2O, PEMAX: 16.5 cmH2O, vital capacity, VC: 406 mL (12% of predicted), unassisted VT: 2.7 mL/kg for 2 min with pacer, 2.5 mL/kg for 2 min no support.
At 52.9 yr, a second episode of status epilepticus was triggered by an acute outbreak of shingles, and the patient suffered a proximal femur fracture during or after the seizure activity. During a 6-wk hospitalization for seizure management and surgical repair of the fracture, the patient was also treated for pseudomonas pneumonia. Upon hospital discharge, respiratory function was returned to baseline and seizures were controlled with lamotrigine (200 mg BID) and valproic acid (500 mg BID). However, the patient and family noted residual memory impairment and intermittent confusion that persisted into the following year. The patient elected to discontinue use of the pacer and to remain on full-time ventilatory support at age 53.8 yr.
At age 54.2 yr, the patient exhibited progressive neurological deterioration, including disturbed sleep, agitation, and encephalopathy. The patient experienced multiple hospitalizations over the following two months as progressively greater periods of delirium and altered consciousness impacted his ability to protect his airway. The patient was transitioned into palliative care and did not regain consciousness; he passed away from respiratory failure at age 54.4 yr. In advance of the patient’s death, informed consent was obtained for the patient to participate as a tissue donor in the University of Florida Neuromedicine Tissue Bank in accordance with IRB.
Neurophysiological Data Analysis
At 6 wk, 2 yr, and 4 yr postpacemaker implant, diaphragm activity was recorded from the right and left indwelling diaphragm pacemaker wires during spontaneous breathing on full ventilator support, off-ventilator breathing, and maximal inspiratory pressure efforts. EMG was sampled at 20 samples/s and band-pass filtered at 1–5,000 Hz, with respiratory effort bands (Respitrace, 6-wk and 2-yr tests) or respiratory flow (4-yr test) sampled concurrently at 400 samples/s (PowerLab S30-16, ADInstruments). EMG signals were rectified and integrated, and peak amplitude averaged. Data were analyzed off-line using Lab Chart Pro v8.1 (ADInstruments, Colorado Springs, CO) and MATLAB 2019a. Cross-correlation was conducted to evaluate the timing of the bursts on the right and left hemidiaphragm during off-ventilator breathing. Motor unit discrimination was conducted to evaluate timing of motor unit activity across the respiratory cycle.
Histopathology and Immunochemistry
Autopsy was performed ∼8 h after death; the brain and spinal cord were preserved, along with muscle specimens including the costal right diaphragm. The brain was sectioned at midline through the corpus callosum, cerebellar vermis, and brainstem. One-centimeter-thick serial coronal sections of the right hemisphere were frozen at −70°C. The left hemisphere, brainstem, and spinal cord were fixed in 20% formalin solution, buffered to neutral. Spinal cord tissue sections (4 µm thickness) were stained using periodic acid-Schiff (PAS) as described (12) and counterstained with cresyl violet. Diastase incubation was used to verify if the PAS staining indicated glycogen or other intracellular material (e.g., lipofuscin). Additional tissue sections stained with hematoxylin and eosin were used to evaluate putative motor neurons. These cells were identified based on morphology and location (e.g., hypoglossal motor nucleus in the medulla or the anterior horn of the spinal cord, lamina IX). To ensure a diverse sampling across each region images were captured from three separate blocks of spinal tissue across each region. Images were captured with a ×10 objective on a Ziess AxioImager.A2 microscope and AxioCam HRc camera, and motoneuron soma were manually traced and area calculated using MATLAB software. Microglia were assessed using a rabbit anti-IBA-1 antibody (019-19741, FUJIFILM Wako Chemicals; 1:1,000) and visualized using a HRP/DAB (ABC) Detection IHC kit - Micro-polymer (ab236469, Abcam). For GFAP immunohistochemistry, paraffin sections were dewaxed, treated with Roche Protease 2 solution for epitope retrieval and incubated with anti-GFAP antibody (1:1,600, Rabbit polyclonal, Agilent). Antigen was visualized by using the Ultra View DAB detection kit (Roche Diagnostic). Slides were counterstained with hematoxylin. Diaphragm paraffin sections were cut at 4 µm, dewaxed, and epitope retrieval was performed with Ventana CC1 retrieval solution. Anti-Myosin 2 (MY-32 clone from Biogenex) was then applied to sections at a 1:10 dilution. Antigen was visualized with the Ventana Ultra View DAB detection kit. A second round of immunohistochemistry was performed with anti-Myosin 1 (1:200 dilution, clone NOQ7.54D from Millipore Sigma). Antigen was visualized with the Ultra View red detection kit (Roche Diagnostics Corporation). Slides were counterstained with hematoxylin.
RESULTS
Diaphragm EMG Recordings
Figure 2 shows diaphragm EMG activity recorded from the indwelling wire electrodes which had been used for direct diaphragmatic pacing. The indwelling electrodes enabled collection of excellent signal-to-noise EMG recordings at 2-yr intervals. Figure 2A illustrates EMG recordings from the right and left hemidiaphragm during spontaneous breathing without ventilator support. The respiratory effort traces were obtained using respiratory effort bands or airflow, and they showed a peak during the inspiratory phase. At the initial recording 6 wk postimplant, asynchronous burst patterns were detected, characterized by prominent burst activity during the expiratory period in the left but not right diaphragm EMG recording. However, peak output of the left and right EMG signal were correlated (inset panel, R = 0.68). The recordings obtained two years later had a reduced respiratory rate during the period of spontaneous breathing, with continued evidence of asynchronous left versus right diaphragm EMG bursting. The correlation of peak left versus right EMG output (R = 0.70) was similar to the value 6 wk postimplant. By 4 yr postimplant, the peak EMG activity of the two hemidiaphragms became less correlated (R = 0.53), and there was an emergence of tonic motor unit firing in the left hemidiaphragm that persisted across the entire respiratory cycle.
Figure 2.
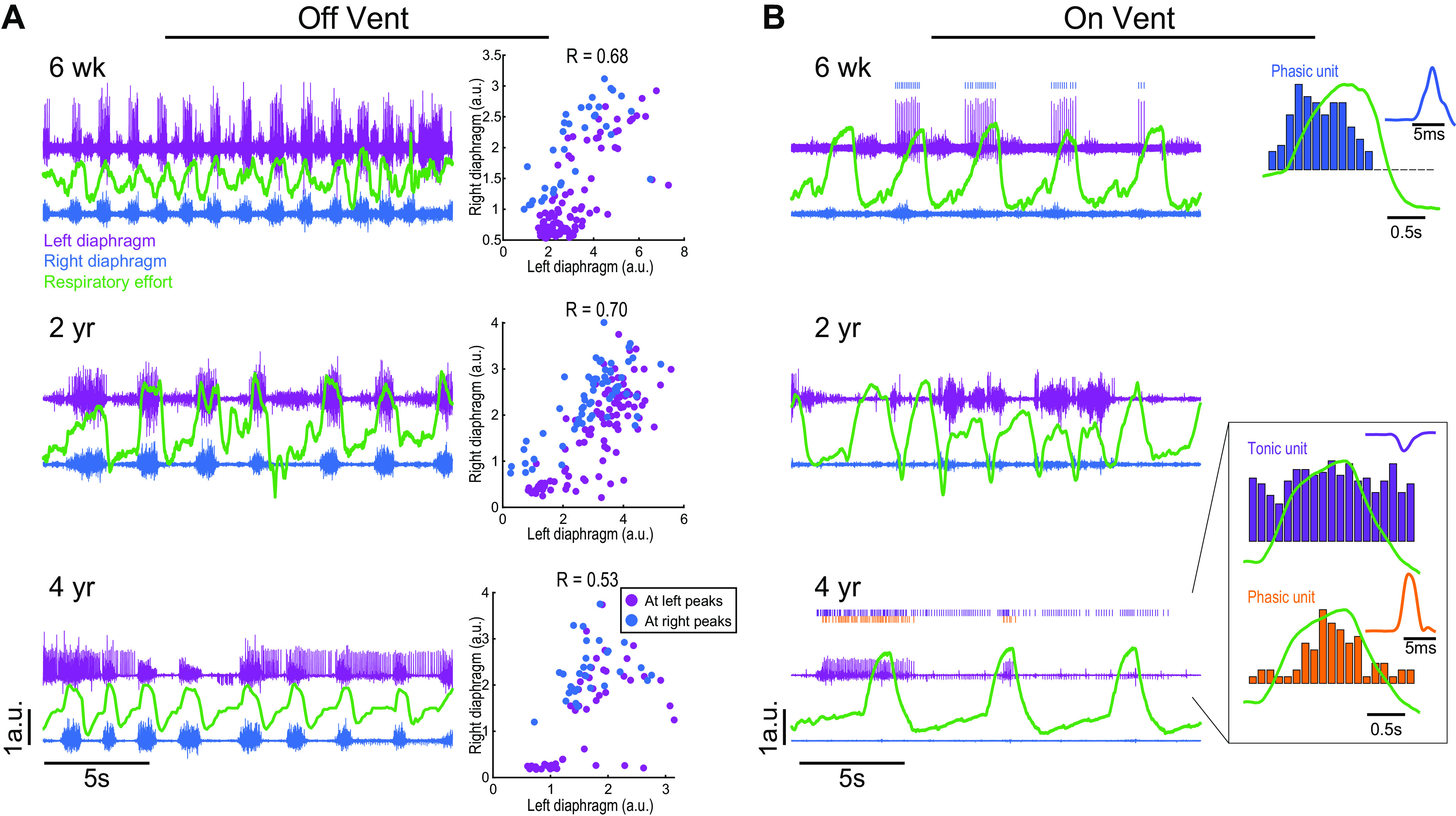
Representative diaphragm EMG recordings over a 3-yr period. EMG was recorded while the patient was spontaneously breathing without ventilator support (left) and during ventilator assistance (right). The traces show bilateral diaphragm EMG activity (purple is left side, blue is right) and respiratory efforts (green; recorded via chest band or pneumotachography) 6-wk, 2-yr, and 4-yr postimplant. In all panels, the scaling (arbitrary units, y-axis) is the same between traces. A shows EMG recordings obtained while the patient was breathing independently, without mechanical ventilator support. Clear inspiratory bursting can be seen, as well as additional asynchronous patterns between the left vs. right diaphragm. Six weeks postimplant, the left diaphragm showed small bursts of activity during expiratory period; two years later, a similar observation was noted in the left diaphragm. Four years postimplant, tonic discharge occurred in the left diaphragm throughout the respiratory cycle. The plots at the right of A show the correlation between left and right peak diaphragm inspiratory burst amplitude. Note that the left and right hemidiaphragm burst amplitude became less correlated over the next four years. The recordings shown in B were obtained while the patient was receiving mechanical ventilator support and confirm that the diaphragm was less active during these conditions. The low activity enabled discrimination of individual motor unit activity. The unit traces on the right of the figure show a histogram of burst rate overlaid with the respiratory cycle, and an average of the unit waveform. Both phasic and tonic motor units were detected in the left hemidiaphragm but were otherwise unremarkable in appearance. a.u., arbitrary units; EMG, electromyogram.
Diaphragm EMG activity was also recorded while the patient received ventilator support (Fig. 2B). The overall diaphragm EMG output was markedly reduced while mechanical ventilation was provided. During these periods, single diaphragm motor unit discharge could be identified. As the recordings progressed over four years, we noted the appearance of tonic motor units in the left diaphragm, similar to observations during spontaneous breathing.
Gross Inspection of CNS
Gross examination revealed extensive atherosclerosis extending to the distal arterial branches of the cerebrum and cerebellum. The striatum, lentiform nucleus, and subthalamic nucleus showed evidence of moderate to serve arteriolosclerosis. Significant widening of the sulci and narrowing of the gyri in a frontal and rostral parietal lobe distribution was observed. Areas of cortex appeared atrophic with marked thinning of the corpus callosum and enlargement of the ventricles. Sections of brainstem and cerebellum revealed adequately pigmented substantia nigra and locus coeruleus. Serial sections of the cerebral hemisphere revealed adequate demarcation of gray and white matter. The hippocampus was markedly atrophic with what appeared to be organizing necrosis of CA1 and the subiculum. The amygdala was markedly atrophic. No other discrete lesions were identified with gross inspection of the brain or spinal cord.
Histopathology
A striking difference in cellular morphology was observed proceeding rostral-caudal along the neuraxis. This can be appreciated by the cresyl violet staining shown in Fig. 3. Hypoglossal motoneurons in the medulla (soma size: 421 ± 159 µm2) and motoneurons in the cervical (soma size: 487 ± 189 µm2) and thoracic spinal cord (soma size: 468 ± 299 µm2) had an elongated, atrophied appearance (Fig. 3, A–C). In contrast, motoneurons in the lumbar (soma size: 1,363 ± 677 µm2) and sacral spinal cord (soma size: 1,411 ± 633 µm2) had the prototypical swollen appearance seen in lysosomal storage diseases (Fig. 3D). Figure 3 also shows immunohistochemical staining to recognize IBA-1 as a marker for microglia. Staining for IBA-1 was prominent in the lateral white matter of the medulla, cervical, and thoracic spinal cord (Fig. 3, A–C), but not in more caudal regions (Fig. 3D).
Figure 3.

Histological sections stained with cresyl violet and IBA-1 antibodies. Immunohistochemistry to recognize IBA-1 was performed to provide a marker for microglia; tissues were counterstained using cresyl violet to enable visualization of neuronal morphology. The boxed areas in the left (indicated by i and ii) are shown at a higher magnification in the middle and right. Within those panels, additional higher magnification views are provided in the insets. The circle in Ai indicates the approximate location of the hypoglossal motor nucleus. Neuronal soma were larger in the lumber spinal cord as compared to the more rostral locations. IBA-1 staining was prominent in white matter in the medulla, cervical, and thoracic cord (e.g., arrows in Aii and Bii; inset panels in Bii and Cii). Calibration bars: A–D left: 1 mm, middle (i): 500 µm, inset: 100 µm; right (ii): 500 µm, inset: 100 µm.
Figure 4 shows the distribution of motoneuron soma size across the neuraxis. Soma size was largest in the lumbar and sacral spinal cord. Another common feature, seen across the rostral-caudal extent of spinal cord and medulla, was the presence of PAS-positive coarse granules in neurons (Fig. 5). This is indicative of lipofuscin accumulation (13). Incubation with diastase did not impact the lipofuscin PAS staining, providing further confirmation that this did not reflect glycogen accumulation (data not shown). Additional histological assessment of the mid-cervical spinal cord suggested atrophy of the cervical ventral roots, and accumulation of GFAP-positive cells in the lateral white matter and near putative motoneurons (Fig. 6). Lastly, in the medulla, the inferior olive was noted to have an atrophic appearance.
Figure 4.

Motoneuron soma size was smaller in the medulla, cervical, and thoracic spinal cord compared to the lumbar and sacral spinal cord. Sample sizes: medulla, n = 49; cervical, n = 50; thoracic, n = 23; lumbar, n = 63; sacral, n = 31. One-way ANOVA on ranks indicated a statistical difference across the neuraxis: lumbar and sacral soma size were different than medulla, cervical, and thoracic (P < 0.001). Cerv., cervical; Thor., thoracic.
Figure 5.

PAS staining indicates neuronal lipofuscin accumulation. A shows putative motoneurons from the anterior horn of the thoracic spinal cord. B shows putative motoneurons from the lumbar spinal cord. Neuronal lipofuscin accumulation was prevalent (arrows). Note also the striking difference in the size of motoneurons between the thoracic and lumbar spinal cord. Scale bars = 100 µm. PAS, periodic acid-Schiff.
Figure 6.

Additional histological examples from the mid cervical spinal cord. Tissues shown in A were stained with H&E and luxol fast blue. The dorsal root can be clearly observed (i), as well as ventral rootlets (ii). The relative small size of the ventral rootlets may indicate atrophy and/or loss of motoneuron axons. In B, tissues were incubated with anti-GFAP antibodies to provide a marker for astrocytes. There is a greater density of GFAP staining in dorsal and ventral lateral white matter. Higher magnification views of gray matter (right) show GFAP-positive cells in the immediate vicinity of putative motoneurons. Calibration bars: A, left: 1 mm, right i and ii: 250 µm; B, left: 1 mm, right: 50 µm. GFAP, glial fibrillary acidic protein; H&E, hematoxylin-eosin.
Diaphragm tissue sections are shown in Fig. 7. Diaphragm histology showed widespread, complete vacuolization with “lacework” appearance of membranes and little positive stain in the space normally containing myofibers. The few remaining fibers were atrophic and stained primarily for type 1 MHC (Fig. 7A). Fiber remnants and connective tissue stained heavily for glycogen (Fig. 7B).
Figure 7.

Diaphragm histology indicates profound pathology with apparent loss of myofiber protein. The tissues in A were stained for Myosin-1 (red) and Myosin-2 (brown). The most striking observation is the complete lack of stain in many putative myofibers. These “ghost fibers” are seen to have a cell membrane but do not stain for Myosin. B shows the results of PAS staining to recognize glycogen. Calibration bars: A, left: 100 µm; right: 50 µm; B, left: 200 µm; right: 50 µm. PAS, periodic acid-Schiff.
DISCUSSION
This case report details neurophysiological and histopathological evidence that widespread, severe neural and muscular deterioration accompanied chronic ventilatory failure in LOPD. The placement of a clinical diaphragm pacemaker enabled us to longitudinally assimilate diaphragm activation with function. Although neurophysiological involvement of the limbs is well-studied in Pompe disease, the potential role of diaphragm neuromuscular function is less-known. In particular, the progressive loss of synchronized, phasic diaphragm bursting was accompanied by end stage changes in diaphragm muscle fibers. These findings are consistent with EMG evidence of diaphragm functional denervation reported in ventilator-dependent infantile disease (14). The diaphragm electrophysiology data corresponded to autopsy samples from the medulla and mid-cervical spinal cord suggesting a reduced number of motoneurons, and surviving cells having an atrophied appearance. In addition, lipofuscin accumulation was prominent in motoneurons, a finding that is consistent with a prior case report in LOPD (8). This accumulation of lipopigments, also known as neuronal lipfuscinosis, is a hallmark feature in the family of progressive neurodegenerative lysosomal storage diseases known as neuronal ceroid-lipofuscinosis (15). Raben and colleagues have also shown that lipofuscin accumulation is a hallmark of skeletal myofibers in Pompe disease (16). Our results may indicate that motoneuron lipofuscin accumulation is common in late onset Pompe disease, possibility indicative of neurodegenerative processes, as occurs in neuronal ceroid-lipofuscinosis (15).
Cervical white matter contained a high density of IBA-1 staining, indicating increase presence of microglia as has been reported in the spinal cord of advanced stage amyotrophic lateral sclerosis (ALS) (17). These observations, along with the suggestion of mid-cervical ventral root atrophy and severe diaphragm pathology may indicate motoneuron death in the region of the phrenic motor nucleus. In turn, concurrent neural and muscular pathology are likely to have contributed to the ventilatory failure of this patient with LOPD.
Prior autopsy studies of patients with LOPD have reported minimal evidence of neuronal glycogen accumulation (8–11). We also did not find evidence of neuronal glycogen, and the positive neuronal PAS staining was indicative of lipofuscin accumulation (e.g., Fig. 5). It is possible, however, that the tissue harvesting and storage procedures were not ideal for preserving neuronal glycogen. Kretzschmar (8) reported clinical and autopsy findings of a patient who presented with an acute neurological event and severe respiratory insufficiency. A clinical EMG evaluation was notable for spontaneous fibrillation potentials in the tested muscles, an indicator of muscle denervation-regeneration processes (18), along with coexistent neurogenic and myopathic changes during voluntary movements. Autopsy reported myonecrosis in the diaphragm. Similar to the current report, lipofuscinosis has been detected in neurons and glia of the spinal cord and cerebellum (8). The presence of fibrillary gliosis of the anterior horns without concurrently enhanced glycogen accumulation was also reported in the case of a young adult with Pompe disease who died from recurrent respiratory problems (9).
The rostral to caudal variability in the histological appearance of motoneurons merits particular comment. Cells in the medulla as well as cervical and thoracic spinal cord had the appearance of atrophy and neurodegeneration (e.g., Figs. 3 and 4). In contrast, the lumbar and sacral spinal cord of this LOPD patient contained anterior horn cells displaying the prototypical histopathologic appearance that is well described for lyosomal storage diseases, including Pompe (19). A rostral to caudal gradient in IBA-1 staining, recognizing microglia, was also noted. The regions that had an atrophied motoneuron phenotype showed extensive IBA-1 staining in ventral and dorsal white matter. A similar pattern of IBA-1 immunoreactivity in spinal white matter has been described in autopsy material from advanced stage ALS patients (17) and animal models of spinal degeneration including trauma (20) and ALS (21). Staining for GFAP (astrocytes) was only completed for the mid-cervical cord, but it also displayed a predominance for lateral white matter.
Traditionally, ventilatory defects in LOPD have been attributed exclusively to myofiber dysfunction secondary to extensive glycogen accumulation leading to lysosomal rupture and protein degradation. In support of this, Speisshofer et al. (6) noted variability and a lack of statistically significant difference between adult Pompe patients and controls, in response to cortical and cervical stimulation of the phrenic nerves. These findings were interpreted as an absence of neurological involvement in ventilatory disturbances occurring in LOPD. However, other studies suggest neurological factors may contribute to ventilatory deficits in LOPD. We evaluated the neuromuscular responses to cervical magnetic stimulation of the phrenic nerves in a small sample of adults and noted variable responses that reflected clinical variability of the sample (5). Diaphragm pressure and evoked motor responses largely decreased in subjects who required external support. In contrast, normal to increased compound motor action potentials were observed in patients who retained normal pulmonary function, despite reduced evoked transdiaphragmatic pressure. Although limited by a small sample, the results suggested a preserved neural ventilatory drive in asymptomatic patients with progressive decline in more severe respiratory involvement (5). Indeed, central ventilatory drive impairments have also been reported in some adult patients, in conjunction with chronic hypercapnia that cannot be explained by underlying lung disease, mechanics, or weakness (22, 23).
Although these reports may initially seem incongruent with Speisshofer et al. (6), a proposed unified model may account for neuromuscular involvement in ventilatory failure from Pompe disease (24). The model theorizes that respiratory muscle fiber degeneration predominates early in Pompe disease. Loss of fiber force initially triggers compensatory plasticity, marked by altered respiratory neural activity and extradiaphragmatic recruitment to preserve ventilatory neuromuscular function and independent breathing (5, 7, 25). However, eventually neurological function also declines, as suggested by Pompe animal models (2, 26) and patient studies (5, 27). Over time, losses of both muscular force and neurological activity reach a critical point where patients require external ventilatory support to avoid ventilatory failure and death. Muscle and neural function are tightly coupled, and neural pathology is becoming increasingly recognized as a contributor to ventilatory failure in other conditions traditionally attributed to muscle degeneration (28).
We acknowledge several possible limitations. The diagnosis of Pompe disease occurred at another institution through GAA activity measurements, and these original records were unavailable. Therefore, the mutational composition of this patient is unknown. Since the patient resided a considerable distance from our institution, we were only able to evaluate the patient periodically and received limited updates from his local neurological and respiratory providers. The last diaphragm recordings and respiratory measures were obtained more than one year before his death. Also, these findings occurred in the midst of other neurological conditions, including a remote stroke and intermittent episodes of significant seizure activity as well as histopathological evidence of cerebral infarct, widespread atherosclerosis, and cerebellar dysfunction. Therefore, we cannot rule out a multifactorial etiology for the observed respiratory pathophysiology and histopathology. Lastly, putative motoneurons were histologically identified based on location (e.g., XII motor nucleus, spinal anterior horn) and appearance, but in these autopsy samples it is not possible to unequivocally verify the identity of every neuron.
In summary, this LOPD case report describes EMG activity and histopathology of the diaphragm, and apparent neurodegeneration in hypoglossal and mid-cervical motoneurons. We suggest that these findings are consistent with the concept that effective Pompe disease therapy for respiratory insufficiency will need to target the central nervous system in addition to skeletal muscle.
GRANTS
This work was supported by NIH Grants 5R21HD090752 to B. K. Smith and R01HD052682 to D. Fuller, B. J. Byrne.
DISCLOSURES
B.J.B. is a member of the Pfizer Rare Disease Therapeutic Advisory Board and Sanofi Pompe Registry Board as well as co-founder of AavantiBio; however, the manuscript does not evaluate therapeutic interventions in Pompe disease. The author is an inventor of intellectual property related to Pompe disease, which is owned by the University of Florida. B.K.S. is an external consultant for Amicus Therapeutics.
AUTHOR CONTRIBUTIONS
D.D.F. and B.K.S. conceived and designed research; D.D.F., J.A.T.-L., A.T.Y., V.E.B., B.J.B., and B.K.S. performed experiments; J.A.T.-L., A.T.Y., M.D.S., S.R., V.E.B., and B.K.S. analyzed data; D.D.F., J.A.T.-L., A.T.Y., M.D.S., B.J.B., and B.K.S. interpreted results of experiments; D.D.F., M.D.S., S.R., and B.K.S. prepared figures; D.D.F. and B.K.S. drafted manuscript; D.D.F., J.A.T.-L., A.T.Y., M.D.S., S.R., V.E.B., B.J.B., and B.K.S. edited and revised manuscript; D.D.F., J.A.T.-L., A.T.Y., M.D.S., S.R., V.E.B., B.J.B., and B.K.S. approved final version of manuscript.
REFERENCES
- 1.Raben N, Plotz P, Byrne BJ. Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease). Curr Mol Med 2: 145–166, 2002. doi: 10.2174/1566524024605789. [DOI] [PubMed] [Google Scholar]
- 2.DeRuisseau LR, Fuller DD, Qiu K, DeRuisseau KC, Donnelly WH Jr, Mah C, Reier PJ, Byrne BJ. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc Natl Acad Sci USA 106: 9419–9424, 2009. doi: 10.1073/pnas.0902534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kroos M, Hoogeveen-Westerveld M, van der Ploeg A, Reuser AJ. The genotype-phenotype correlation in Pompe disease. Am J Med Genet C Semin Med Genet 160C: 59–68, 2012. doi: 10.1002/ajmg.c.31318. [DOI] [PubMed] [Google Scholar]
- 4.Prigent H, Orlikowski D, Laforet P, Letilly N, Falaize L, Pellegrini N, Annane D, Raphael J-C, Lofaso F. Supine volume drop and diaphragmatic function in adults with Pompe disease. Eur Respir J 39: 1545–1546, 2012. doi: 10.1183/09031936.00169011. [DOI] [PubMed] [Google Scholar]
- 5.Smith BK, Corti M, Martin AD, Fuller DD, Byrne BJ. Altered activation of the diaphragm in late-onset Pompe disease. Respir Physiol Neurobiol 222: 11–15, 2016. doi: 10.1016/j.resp.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Speisshofer J, Henke C, Kabitz HJ, Brix T, Gorlich D, Herkenrath S, Randerath W, Young P, Boentert M. The nature of respiratory muscle weakness in patients with late-onset Pompe disease. Neuromuscul Disord 29: 618–627, 2019. doi: 10.1016/j.nmd.2019.06.011. [DOI] [PubMed] [Google Scholar]
- 7.Turner SMF, Hoyt AK, ElMallah MK, Falk DJ, Byrne BJ, Fuller DD. Neuropathology in respiratory-related motoneurons in young Pompe (Gaa−/−) mice. Respir Physiol Neurobiol 227: 48–55, 2016. doi: 10.1016/j.resp.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kretzschmar HA, Wagner H, Hubner G, Danek A, Witt TN, Mehraein P. Aneurysms and vacuolar degeneration of cerebral arteries in late-onset acid maltase deficiency. J Neurol Sci 98: 169–183, 1990. doi: 10.1016/0022-510x(90)90258-o. [DOI] [PubMed] [Google Scholar]
- 9.Martin JJ, de Barsy T, den Tandt WR. Acid maltase deficiency in non-identical adult twins. A morphological and biochemical study. J Neurol 213: 105–118, 1976. doi: 10.1007/BF00313272. [DOI] [PubMed] [Google Scholar]
- 10.Matsuoka Y, Senda Y, Hirayama M, Matsui T, Takahashi A. Late-onset acid maltase deficiency associated with intracranial aneurysm. J Neurol 235: 371–373, 1988. doi: 10.1007/BF00314237. [DOI] [PubMed] [Google Scholar]
- 11.Hobson-Webb LD, Proia AD, Thurberg BL, Banugaria S, Prater SN, Kishnani PS. Autopsy findings in late-onset Pompe disease: a case report and systematic review of the literature. Mol Genet Metab 106: 462–469, 2012. doi: 10.1016/j.ymgme.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 12.Fu DA, Campbell-Thompson M. Periodic acid-Schiff staining with diastase. In: Alpha-1 Antitrypsin Deficiency: Methods in Molecular Biology, edited by Borel F, Mueller C.. New York: Humana Press, 2017, p. 145–149. doi: 10.1007/978-1-4939-7163-3_14. [DOI] [PubMed] [Google Scholar]
- 13.De Biase D, Paciello O. Essential and current methods for a practical approach to comparative neuropathology. Folia Morphol (Warsz) 74: 137–149, 2015. doi: 10.5603/FM.2015.0024. [DOI] [PubMed] [Google Scholar]
- 14.Smith BK, Collins SW, Conlon TJ, Mah CS, Lawson LA, Martin AD, Fuller DD, Cleaver BD, Clement N, Phillips D, Islam S, Dobjia N, Byrne BJ. Phase I/II trial of adeno-associated virus-mediated alpha-glucosidase gene therapy to the diaphragm for chronic respiratory failure in Pompe disease: initial safety and ventilatory outcomes. Hum Gene Ther 24: 630–640, 2013. doi: 10.1089/hum.2012.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jalanko A, Braulke T. Neuronal ceroid lipofuscinoses. Biochim Biophys Acta 1793: 697–709, 2009. doi: 10.1016/j.bbamcr.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 16.Feeney EJ, Austin S, Chien Y-H, Mandel H, Schoser B, Prater S, Hwu W-L, Ralston E, Kishnani PS, Raben N. The value of muscle biopsies in Pompe disease: identifying lipofuscin inclusions in juvenile- and adult-onset patients. Acta Neuropathol Commun 2: 2, 2014. doi: 10.1186/2051-5960-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brettschneider J, Toledo JB, Van Deerlin VM, Elman L, McCluskey L, Lee VM-Y, Trojanowski JQ. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PloS One 7: e39216, 2012. doi: 10.1371/journal.pone.0039216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pond A, Marcante A, Zanato R, Martino L, Stramare R, Vindigni V, Zampieri S, Hofer C, Kern H, Masiero S, Piccione F. History, mechanisms and clinical value of fibrillation analyses in muscle denervation and reinnervation by single fiber electromyography and dynamic echomyography. Eur J Transl Myol 24: 3297, 2014. doi: 10.4081/ejtm.2014.3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Byrne BJ, Fuller DD, Smith BK, Clement N, Coleman K, Cleaver B, Vaught L, Falk DJ, McCall A, Corti M. Pompe disease gene therapy: neural manifestations require consideration of CNS directed therapy. Ann Transl Med 7: 290, 2019. doi: 10.21037/atm.2019.05.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu D, Miyamoto O, Shibuya S, Okada M, Igawa H, Janjua NA, Norimatsu H, Itano T. Different expression of macrophages and microglia in rat spinal cord contusion injury model at morphological and regional levels. Acta Med Okayama 59: 121–127, 2005. doi: 10.18926/AMO/31950. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez-Trapero M, Espejo-Porras F, Rodriguez-Cueto C, Coates JR, Perez-Diaz C, de Lago E, Fernandez-Ruiz J. Upregulation of CB2 receptors in reactive astrocytes in canine degenerative myelopathy, a disease model of amyotrophic lateral sclerosis. Dis Model Mech 10: 551–558, 2017. doi: 10.1242/dmm.028373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Vito EL, Monteiro SG, Aruj PK. Blunted hypercapnic respiratory drive response in subjects with late-onset Pompe disease. Respir Care 61: 930–935, 2016. doi: 10.4187/respcare.03940. [DOI] [PubMed] [Google Scholar]
- 23.Berger KI, Chan Y, Rom WN, Oppenheimer BW, Goldring RM. Progression from respiratory dysfunction to failure in late-onset Pompe disease. Neuromuscul Disord 26: 481–489, 2016. doi: 10.1016/j.nmd.2016.05.018. [DOI] [PubMed] [Google Scholar]
- 24.Fuller DD, ElMallah MK, Smith BK, Corti M, Lawson LA, Falk DJ, Byrne BJ. The respiratory neuromuscular system in Pompe disease. Respir Physiol Neurobiol 189: 241–249, 2013. doi: 10.1016/j.resp.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee K-Z, Qiu K, Sandhu MS, Elmallah MK, Falk DJ, Lane MA, Reier PJ, Byrne BJ, Fuller DD. Hypoglossal neuropathology and respiratory activity in Pompe mice. Front Physiol 2: 31, 2011. doi: 10.3389/fphys.2011.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sidman RL, Taksir T, Fidler J, Zhao M, Dodge JC, Passini MA, Raben N, Thurberg BL, Cheng SH, Shihabuddin LS. Temporal neuropathologic and behavioral phenotype of 6neo/6neo Pompe disease mice. J Neuropathol Exp Neurol 67: 803–818, 2008. doi: 10.1097/NEN.0b013e3181815994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corti M, Smith BK, Falk DJ, Lawson LA, Fuller DD, Subramony SH, Byrne BJ, Christou EA. Altered activation of the tibialis anterior in individuals with Pompe disease: Implications for motor unit dysfunction. Muscle Nerve 51: 877–883, 2015. doi: 10.1002/mus.24444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burns DP, Roy A, Lucking EF, McDonald FB, Gray S, Wilson RJ, Edge D, O'Halloran KD. Sensorimotor control of breathing in the mdx mouse model of Duchenne muscular dystrophy. J Physiol 595: 6653–6672, 2017[Erratum inJ Physiol596: 343-344, 2018]. doi: 10.1113/JP274792. [DOI] [PMC free article] [PubMed] [Google Scholar]