

Abstract
An earlier described three-component variant of the Castagnoli-Cushman reaction employing homophthalic anhydrides, carbonyl compound and ammonium acetate was applied towards the preparation of 1-oxo-3,4-dihydroisoquinoline-4-carboxamides with variable substituent in position 3. These compounds displayed inhibitory activity towards poly(ADP-ribose) polymerase (PARP), a clinically validated cancer target. The most potent compound (PARP1/2 IC50 = 22/4.0 nM) displayed the highest selectivity towards PARP2 in the series (selectivity index = 5.5), more advantageous ADME prameters compared to the clinically used PARP inhibitor Olaparib.
Keywords: Castagnoli-Cushman reaction; 1-oxo-3,4-dihydroisoquinoline-4-carboxamides; poly(ADP-ribose) polymerase; PARP1/2 selectivity; NAD+ mimetics; druglikeness
Graphical Abstract
Introduction
Poly(ADP-ribose) polymerase (PARP) enzymes are regarded as important targets for the development of anticancer drugs owing to the clinical success of PARP inhibitors Olaparib, Talazoparib, Niraparib and Rucaparib approved for cancer treatment1. PARP1 and PARP2 are two key enzymes that are critical for repairing single-strand breaks (“nicks”) in the DNA – a mechanism which is critical for the survival of both normal and cancer cells2. It is generally accepted that, despite the non-selectivity of the approved PARP inhibitors, it is the inhibition of PARP1 that is responsible for the manifestation of their clinical efficacy3. Normal cells do not divide as frequently as cancer cells, which allows them to survive PARP1 inhibition. However, tumour cells with certain mutations that are synthetically lethal with PARP1 inhibition4 are efficiently killed by the drugs of this class. This notion motivated the development of selective PARP1 inhibitors such as NMS-P118 reported by Nerviano Medical Sciences5.
The majority of PARP1 inhibitors, including the above-mentioned “paribs”, were designed to mimic the nicotinamide moiety of NAD+ (from which the adenine ribose unit of poly(ADP-ribose) originates) with which the inhibitors compete for the NAD+-binding site of PARP1. This mimicry is achieved via the use of either a rotationally constrained primary benzamide (as in Niraparib and NMS-P118) or a benzamide motif embedded in a ring (as in Olaparib, Talazoparib and Rucaparib). Another characteristic feature noticeable in some of the advanced PARP1 inhibitors is the presence of a fluorine atom (the “magic fluorine” highlighted in blue) in the meta-position of the NAD+-mimicking benzamide moiety (Figure 1). This “magic fluorine” has been shown to enhance binding to the target5.
Figure 1.

Clinically used PARP1 inhibitors, advanced clinical candidate NMS-P118 (NAD+-mimicking motif is highlighted in red, NAD+ structure shown).
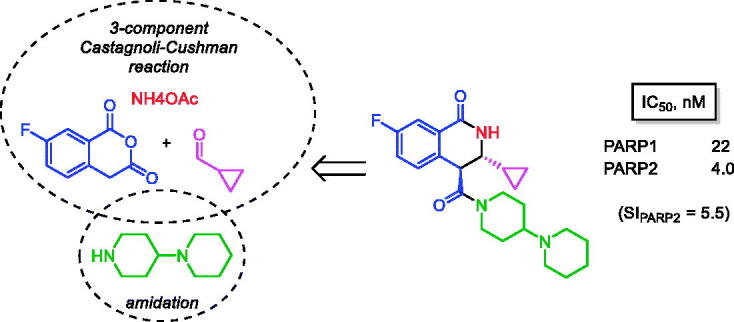
Recently, we discovered 1-oxo-3,4-dihydroisoquinoline-4-carboxamides 1 as a novel chemotype capable of delivering potent PARP inhibitors. After screening the amide residues around the basic core, we identified compound 1a as a lead structure for further development. It displayed a potent inhibition profile towards PARP1 (IC50 = 156 nM) and PARP2 (IC50 = 70.1 nM). Moreover, compound 1a displayed a better microsomal and plasma stability in vitro compared to clinically used PARP1 inhibitor Olaparib6. Encouraged by this finding, we continued looking for ways to improve the potency profile of the 1-oxo-3,4-dihydroisoquinoline-4-carboxamide series. Carboxamide 1a bore no substituents in position 3 of the 1-oxo-3,4-dihydroisoquinoline core. Therefore, we considered exploring the structure-activity relationships within the series 2 where the 3-mono- and 3,3-disubstituted scaffolds could be screened while the 1,4′-bipiperidine carboxamide would remain unchanged. Access to derivatives 2 was envisioned via the amidation of carboxylic acids 3 which, in turn, could be prepared via the recently reported three-component Castagnoli-Cushman reaction (3 C-CCR) of homophthalic anhydride (HPA), carbonyl compounds and ammonium acetate7 (Figure 2). Herein, we describe the results obtained in the course of realising this strategy.
Figure 2.

Previously discovered series of PARP1/2 inhibitors 1 with the lead compound 1a, the newly designed series of inhibitors 2 bearing substitutions at position 3 and its envisioned synthesis via the 3 C-CCR.
Results and discussion
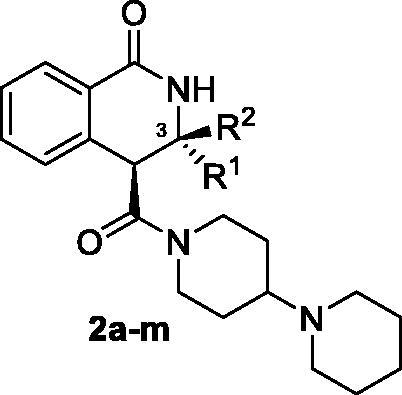
Twelve carboxylic acids 3a-l were synthesised in diastereomerically pure (trans-configured, based on their vicinal coupling constants of methine protons) form as described previously7 and subjected to amidation with 1,4′-bipiperidine using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate (HATU) to activate the carboxylic group6. Resulting amides 2a-l were obtained in moderate to excellent yields (Scheme 1).
Scheme 1.

Synthesis of 3-substituted 1,4'-bipiperidine 1-oxo-3,4-dihydroisoquinoline-4-carboxamides 2a-l.
In order to probe for the minimal substitution at position 3, we aimed to synthesise 3-methyl-substituted 1-oxo-3,4-dihydroisoquinoline-4-carboxylic acid 3 m. Unfortunately, the 3 C-CCR protocol did not furnish the desired product, likely due to the high volatility of acetaldehyde. Thus, for the preparation of 3 m, we resorted to the alternative approach recently reported by Shaw and co-workers8. It involves in situ generation of N-(4-nitrobenzene)sulfonyl (4-nosyl) imine of acetaldehyde from α-aminosulfone 49 on treatment with equimolar amount of base. N-Sulfonyl imine 4 thus generated can be reacted with HPA in the Castagnoli-Cushman fashion. Indeed, treatment of 4 with 1 equiv. of DBU followed by the addition of HPA furnished 4-nosyl CCR adduct which was esterified to give ester 5 in modest yield. Removal of the 4-nosyl protecting group followed by ester hydrolysis furnished the target acid (3 m) which was immediately introduced in the amidation reaction with 1,4′-bipiperidine to give compound 2 m in modest 26% yield over 3 steps (Scheme 2).
Scheme 2.

Synthesis of 3-methyl-substituted 1-oxo-3,4-dihydroisoquinoline-4-carboxamide 2 m.
Compounds 2a-m (confirmed to retain the trans-configuration based on the values of 3 J(H3-H4) coupling constants observed in their 1H NMR spectra) were tested for inhibitory activity towards PARP1 and PARP2 using the commercially available colorimetric activity assay kit from BPS Bioscience (San Diego, CA) in full accordance of the supplier’s method description10–11. The initial screening was performed against PARP1 at 1 µM concentration of each compound, in triplicate (n = 3) measurements. Compounds which displayed over 80% inhibition of the enzyme activity were in dose-response mode (n = 3) against PARP1 and PARP2 using Olaparib as the reference inhibitor in order to determine the compounds’ half-maximal inhibitory concentration (IC50) and assess the isoform selectivity (Table 1).
Table 1.
Inhibitory activity of compounds 2a-m vs.
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | IC50, nM (or % inhibition at 1 µM)a |
SIb | |
| PARP1 | PARP2 | ||||
| 2a | Ph | H | 87.9 ± 10.2 | 69.5 ± 12.8 | 1.26 |
| 2b | 4-FC6H4 | H | 55.4 ± 8.8 | 39.9 ± 7.3 | 1.39 |
| 2c | Et | H | 44.6 ± 5.6 | 11.0 ± 2.2 | 4.05 |
| 2d | i-Pr | H | 159.9 ± 18.6 | NDc | – |
| 2e | Cyclopropyl | H | 26.4 ± 4.1 | 18.3 ± 4.5 | 1.44 |
| 2f | t-Bu | H | 24.1 ± 6.0d | – | – |
| 2g | i-Bu | H | 87.0 ± 8.3 | 104.9 ± 20.8 | 0.83 |
| 2h | Me | Me | 4.0 ± 7.4d | – | – |
| 2i | -(CH2)5- | 12.1 ± 7.2d | – | – | |
| 2j | -(CH2)2O(CH2)2- | 5.1 ± 6.6d | – | – | |
| 2k | -(CH2)4- | 10.6 ± 9.7d | – | – | |
| 2l | Cyclopentyl | H | >500 | >500 | – |
| 2m | Me | H | 95.7 ± 17.7 | 29.2 ± 3.4 | 3.28 |
| 1a e | H | H | 156 ± 5.8 | 70.1 ± 7.6 | 2.23 |
| Olaparib | 2.8 ± 0.3 | 0.7 ± 0.2 | 3.98 | ||
aThe data are presented as the mean value ± SD obtained in triplicate (n = 3) measurement of the enzyme activity.
bSelectivity index defined as IC50(PARP1)/IC50(PARP2), i.e. values greater than 1.0 indicate inhibitor’s selectivity towards PARP2.
cNot determined.
dThe value = % inhibition at 1 µM concentration of the inhibitor.
eData from [6].
PARP1 and PARP2 enzymes.
It is apparent from the data presented in Table 1 that some small alkyl groups introduced in position 3 of the 1-oxo-3,4-dihydroisoquinoline-4–1,4′-bipiperidine carboxamide scaffold increase compounds’ potency towards PARP1 and PARP2 (cf. 2c, 2e (the most potent inhibitor in the series) and 2 m vs. 1a) while bulkier (cyclo)alkyl groups do not change (2d) or ablate (2f and 2 l) the inhibitor’s potency. Similarly, disubstituted (2 h) and spirocyclic (2i-k) analogs do not inhibit PARP1 in the concentration range relevant for drug development12. In light of the bulky substituents at position 3 being detrimental to the inhibitor’s activity, it came as a surprise that 3-aryl-substituted analogs 2a-b displayed better potency compared to their unsubstituted counterpart (1a).
We attempted to rationalise some of the observed structure-activity relationships by performing docking simulation of the inhibitors’ binding to the PARP1 active site. Figure 3 displays the binding poses of the most potent compound 2e as well as its active (2 b) and inactive (2 h and 2 l) analogs. Compounds 2e and 2 b displayed a favourable network of lipophilic contacts in the PARP-1 active cavity (Figure 2(A,B)). The 3,3-dimethyl substitution in 2 h causes a transition to another metastable conformation with the loss of hydrophobic interactions with Val762 and destabilisation of electrostatic contacts with Glu763/Asp766 (Figure 2(C)). In the case of 2 l, the cyclopentyl moiety appears to induce a conformational rearrangement of molecule. The resulting reorientation of the 1,4′-bipiperidine moiety leads to the loss of the hydrophobic contacts with Val762 and π-stacking interaction with Tyr907 (Figure 2(D)).
Figure 3.

Binding poses of compounds 2e (A), 2 b (B), 2 h (C) and 2 l (D) in the active site of PARP1; crystal structure of PARP1 with inhibitor NMS-P118 (PBD ID 4ZZZ) was used for docking.
Thus, we have identified a new lead compound based on unsubstituted 1-oxo-3,4-dihydroisoquinoline core, 2e, which inhibited PARP1 with an IC50 of 26.4 ± 4.1 nM and PARP2 with an IC50 of 18.3 ± 4.5 nM, which corresponded to the selectivity index (SI) of 1.44. Mindful of the advantageous influence of the fluorine atom positioned in the meta-position relative to the NAD+-mimicking lactam (or carboxamide) moiety, we set off to synthesise a 7-fluoro analog of compound 2e, 4-([1,4′-bipiperidine]-1′-carbonyl)-3-cyclopropyl-7-fluoro-3,4-dihydroisoquinolin-1(2H)-one (6). This required that 7-homophthalic anhydride (7) be synthesised. Adaptation of its synthesis from the literature13 involved 1) the preparation of 3–(4-fluorophenyl)propionic acid (8) via the Knoevenagel condensation and hydrogenation, 2) indanone ring closure followed by methoxalylation to give compound 9, 3) oxidative fragmentation of the latter on treatment with hydrogen peroxide and cyclodehydration of 2-(carboxymethyl)-5-fluoro benzoic acid (10). The 3 C-CCR involving 7-F-HPA (7) followed by amidation (as described above for the preparation of compounds 2a-l) furnished 7-fluoro analog 6 in modest yield over 2 steps (Scheme 3).
Scheme 3.

Synthesis of 3-cyclopropyl-substituted 1-oxo-7-fluoro-3,4-dihydroisoquinoline-4-carboxamide 6. Reagents and conditions: a. malonic acid, morpholine (10 mol%), pyridine, 110 °C, 4 h; b. H2 (1 atm), 10% Pd/C, MeOH/THF, r. t., 12 h; c. (COCl)2, DMF (cat.), DCM, r. t., 2 h; d. AlCl3, DCM, 0 °C then reflux, 2 h; e. (COCl)2, NaOMe/MeOH, TolH, r. t., 16 h; f. 30% aq. H2O2, KOH/H2O, r. t. then 50 °C, 3 h; g. TFAA, EtOAc, r. t., 3 h; h. cyclopropane carboxaldehyde, NH4OAc, MeCN, 80 °C; i. 1,4'-bipiperidine, HATU, Et3N, DMF, r. t., 16 h.
To our delight, when tested for inhibition of PARP1 and PARP2, compound 6 indeed displayed an improved potency profile towards both enzymes (PARP1 IC50 = 22.0 ± 3.2 nM, PARP2 IC50 = 4.3 ± 0.5 nM, SI = 5.1). Moreover, this compound can be regarded as a relatively rare PARP2-selective inhibitor which can enrich the toolbox of compounds needed for the investigation of the physiological role of PARP2 inhibition14–16.
We were curious to compare the physicochemical and ADME properties of the newly discovered lead compound (6) to those of more potent, clinically used PARP1/2 inhibitor Olaparib. Indeed, as was noted previously, the sheer potency of pharmacological agents is not a sole determinant of the pharmacodynamic efficacy and should be considered in combination with the overall candidate’s profile17.
While, of course, the approved PARP1 inhibitor Olaparib is well within the limits of druglikeness as defined by Lipinsky18 (and so is compound 6), the two compounds are quite similar in terms of molecular weight and lipophilicity. Quite reassuringly, compound 6 displayed similar stability in plasma to that of Olaparib. However, the metabolic stability of the former is significantly higher (Table 2).
Table 2.
Experimentally determined ADME parameters and calculated molecular characteristics for the lead compound 6 as well as clinically used drug Olaparib.
| Compound | Solubilitya | Stability |
PPBe | Calculated parameters19 |
|||||
|---|---|---|---|---|---|---|---|---|---|
| S9b | HLMc | Plasmad | MW | cLogP | HBD | HBA | |||
| Olaparib | >100 | 1.1 | 15 | 100 | 90.2 | 434.5 | 2.52 | 1 | 7 |
| 6 | >100 | 0.1 | 3.8 | 100 | 71.0 | 399.4 | 2.59 | 1 | 5 |
aSolubility (µM) in pH 7.4 0.01 M phosphate buffer solution; bStability in the presence of hepatic S9 fraction (µL/min/mg); cStability in the presence of human liver microsomes (µL/min/mg); d% compound remaining after incubation (4 h) with human plasma; eplasma protein binding (% bound).
In summary, we have further optimised the inhibitory potency of an earlier discovered lead 1a based on a 1-oxo-3,4-dihydroisoquinoline-4-carboxamide scaffold by exploring various substitutions at position 3. This exploration was reliant on the earlier described three-component variant of the Castagnoli-Cushman reaction of ammonium acetate. The profiling of various 3-mono- and 3,3-disubstituted analogs of compound 1a for PARP1 and PARP2 inhibition allowed establishing structure-activity relationships and led to the identification of 3-cyclopropyl substituent as the preferred periphery pattern. Introduction of a fluorine atom in the position 7 of the 1-oxo-3,4-dihydroisoquinoline core (inspired by the presence of this “magic fluorine” in several clinical candidates and approved drugs) further improved the potency profile and led to 1-oxo-7-fluoro-3,4-dihydroisoquinoline-4-carboxamide 6 as the new lead compound. It displayed 5.5-fold selectivity towards PARP2, good metabolic and plasma stability as well as better plasma protein binding, compared to Olaparib.
Acknowledgements
We thank the Research Center for Magnetic Resonance, the Center for Chemical Analysis and Materials Research, and Research Resource Center for Molecular and Cell Technologies of Saint Petersburg State University Research Park for obtaining the analytical data.
Funding Statement
This research was supported by the Russian Foundation for Basic Research [grant 21–53-12001]. Maxim Gureev acknowledges the funding from the Ministry of Science and Higher Education of the Russian Federation [grant 075–15-2020–926].
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- 1.Park M-T, Lee S-J.. Cell Cycle and Cancer. J Biochem Mol Biol 2003;36:60–5. [DOI] [PubMed] [Google Scholar]
- 2.Gibson BA, Kraus WL.. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 2012;13:411–24. [DOI] [PubMed] [Google Scholar]
- 3.Mateo J, Lord CJ, Serra V, et al. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol 2019;30:1437–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lord CJ, Ashworth A.. PARP inhibitors: synthetic lethality in the clinic. Science 2017;355:1152–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papeo G, Posteri H, Borghi D, et al. Discovery of 2-[1-(4,4-Difluorocyclohexyl)piperidin-4-yl]-6-fluoro-3-oxo-2,3-dihydro-1H-isoindole-4-carboxamide (NMS-P118): a potent, orally available, and highly selective PARP-1 inhibitor for cancer therapy. J Med Chem 2015;58:6875–98. [DOI] [PubMed] [Google Scholar]
- 6.Safrygin A, Zhmurov P, Dar’in D, et al. 1-Oxo-3,4-dihydroisoquinoline-4-carboxamides as novel druglike inhibitors of poly(ADP-ribose) polymerase (PARP) with favorable ADME characteristics. J Enzyme Inhib Med Chem. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Safrygin A, Bakulina O, Dar’in D, Krasavin M.. Three-component reaction of homophthalic anhydride with carbonyl compounds and ammonium acetate: new developments. Synthesis 2020;52:2190–5. [Google Scholar]
- 8.Laws SW, Moore LC, Di Maso MJ, et al. Diastereoselective base-catalyzed formal [4 + 2] cycloadditions of N-sulfonyl imines and cyclic anhydrides. Org Lett 2017;19:2466–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hernando E, Arrayas RG, Carretero JC.. Catalytic asymmetric Mannich reaction of glycine Schiff bases with α-amido sulfones as precursors of aliphatic imines. Chem Commun 2012;48:9622–4. [DOI] [PubMed] [Google Scholar]
- 10.https://bpsbioscience.com/pub/media/wysiwyg/PARPs/80580_1.pdf(PARP1).
- 11.https://bpsbioscience.com/pub/media/wysiwyg/PARPs/80581_1.pdf(PARP2).
- 12.Hughes JP, Rees S, Kalindjian SB, Philpott KL.. Principles of early drug discovery. Br J Pharmacol 2011;162:1239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang B-R, Wang J, Li H, et al. Synthesis and antitumor activity evaluation of 2-arylisoquinoline-1,3(2H,4H)-diones in vitro and in vivo. Med Chem Res 2014;23:1340–9. [Google Scholar]
- 14.Pellicciari R, Camaioni E, Costantino G, et al. On the way to selective PARP-2 inhibitors. Design, synthesis, and preliminary evaluation of a series of isoquinolinone derivatives. ChemMedChem 2008;3:914–23. [DOI] [PubMed] [Google Scholar]
- 15.Sunderland PT, Woon ECY, Dhami A, et al. 5-Benzamidoisoquinolin-1-ones and 5-(ω-carboxyalkyl)isoquinolin-1-ones as isoform-selective inhibitors of poly(ADP-ribose) polymerase 2 (PARP-2). J Med Chem 2011;54:2049–59. [DOI] [PubMed] [Google Scholar]
- 16.Gui B, Gui F, Takai T, et al. Selective targeting of PARP-2 inhibits androgen receptor signaling and prostate cancer growth through disruption of FOXA1 function. Proc Natl Acad Sci U S A 2019;116:14573–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gleeson MP, Hersey A, Montanari D, Overington J.. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat Rev Drug Discov 2011;10:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ.. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 2001;46:3–26. [DOI] [PubMed] [Google Scholar]
- 19.Calculated using Molinspiration Chemoinformatics tool ( https://www.molinspiration.com/).