

Abstract
Pulmonary hypertension (PH) is a devastating disease characterized by progressive elevation of pulmonary vascular resistance, right ventricular failure, and ultimately death. We have shown previously that insulin receptor substrate 2 (IRS2), a molecule highly critical to insulin resistance and metabolism, has an anti-inflammatory role in Th2-skewed lung inflammation and pulmonary vascular remodeling. Here, we investigated the hypothesis that IRS2 has an immunomodulatory role in human and experimental PH. Expression analysis showed that IRS2 was significantly decreased in the pulmonary vasculature of patients with pulmonary arterial hypertension and in rat models of PH. In mice, genetic ablation of IRS2 enhanced the hypoxia-induced signaling pathway of Akt and Forkhead box O1 (FOXO1) in the lung tissue and increased pulmonary vascular muscularization, proliferation, and perivascular macrophage recruitment. Furthermore, mice with homozygous IRS2 gene deletion showed a significant gene dosage-dependent increase in pulmonary vascular remodeling and right ventricular hypertrophy in response to hypoxia. Functional studies with bone marrow-derived macrophages isolated from homozygous IRS2 gene-deleted mice showed that hypoxia exposure led to enhancement of the Akt and ERK signaling pathway followed by increases in the pro-PH macrophage activation markers, vascular endothelial growth factor-A and arginase 1. Our data suggest that IRS2 contributes to anti-inflammatory effects by regulating macrophage activation and recruitment, which may limit the vascular inflammation, remodeling, and right ventricular hypertrophy that are seen in PH pathology. Restoring the IRS2 pathway may be an effective therapeutic approach for the treatment of PH and right heart failure.
Keywords: hypoxia, macrophage, pulmonary hypertension, pulmonary vascular remodeling
INTRODUCTION
Despite major advances in the diagnosis and treatment of pulmonary hypertension (PH) over the past several decades, its underlying mechanisms remain enigmatic. Disease progression is inevitable, and mortality remains unacceptably high (1, 2). An expanding body of knowledge has related PH pathogenesis to a variety of factors, including genetic susceptibility, inflammation, and metabolic/glycolytic shifts (3). Because of this complexity, the progress in PH drug development suffers from poor translation to clinical application, and current therapy is limited mainly to symptomatic relief.
In our present study, we targeted one of the more proximal signaling networks in the pathogenesis of PH: insulin receptor substrate 2 (IRS2). IRS2 is a member of the insulin receptor substrate family of adaptor proteins that are critical to insulin resistance and cellular energy homeostasis. IRS2 is the main regulator of insulin and insulin growth factor (IGF) signaling, and loss of its expression promotes insulin resistance and type II diabetes (4). IRS2 also mediates T helper 2 (Th2) signaling and macrophage activation via type I interleukin (IL)-4 receptor (IL-4Rα) (5, 6). IRS2 serves as an intermediate docking platform to transit signaling of multiple receptors such as 1) insulin receptors for insulin signaling, 2) IGF-1 receptor (IGF-1R) for IGF signaling, 3) erythropoietin (EPO) receptor for EPO signaling, and 4) IL-4Rα for signaling of Th2 cytokines, IL-4 and IL-13 (4–8). Overall, the loss of IRS2 appears to be deleterious in multiple cell types and disease conditions. There is also evidence that the loss of insulin signaling and IRS protein exacerbates diabetic complications (9). Recently, an important association between insulin resistance and PH has been identified (3, 10). Both conditions occur in the presence of chronic inflammation and vascular dysfunction. Although IRS2 has been well studied in diabetes responses, its contribution to the pathophysiology of PH is unknown.
Our previous gene expression profiling of peripheral blood mononuclear cells (PBMCs) from patients with pulmonary arterial hypertension (PAH) revealed that impaired erythroid differentiation was strongly correlated with hemodynamic measures of increasing disease severity (11). Similar observations have been reported from multiple institutions (11), suggesting that impaired hematopoietic cell differentiation may be prevalent in this disease. Indeed, gene expression profiling has shown that IRS2 expression is significantly decreased in PBMCs of patients with idiopathic PAH (IPAH) and scleroderma-PAH (Ssc-PAH) as compared with that in control subjects (11). These data indicate that impaired IRS2 in hematopoietic cells is detrimental and that functional IRS2 may protect against disease progression. We also showed that the loss of IRS2 in macrophages significantly enhances Th2-skewed pulmonary vascular remodeling in mice, suggesting a regulatory role in macrophage activation and function (12). Thus, IRS2 in macrophages plays a role in limiting inflammation and pathological vascular remodeling, which can be protective for PH.
Our group has shown that hypoxia-induced mitogenic factor [HIMF, also known as FIZZ1/resistin-like molecule α (RELMα)] plays a critical role in pulmonary vascular remodeling and PH development (13–17). HIMF/FIZZ1 is a marker for alternatively activated (M2) macrophages (18) and is considered a mediator for both hypoxia-induced and Th2-mediated pulmonary vascular remodeling (12, 19–22). Inhibition of M2 (HIMF/FIZZ1-expressing) macrophage recruitment to the lung also significantly ameliorates hypoxia-induced PH, highlighting the importance of this phenotype in PH (19). However, the role of IRS2 in macrophage activation and vascular remodeling under hypoxic conditions is unknown.
In this study, we examined whether IRS2 is dysregulated in human and experimental PH. We also tested our hypotheses that 1) IRS2 has an immunomodulatory role in experimental PH and 2) IRS2 in macrophages preserves anti-inflammatory properties by limiting macrophage activation in vivo and in vitro under hypoxic conditions.
METHODS
The authors declare that all supporting data are available within the article (and in the supplementary file). All animal housing and animal-related experimental procedures complied with the guidelines issued by the Institutional Animal Care and Use Committee at the Johns Hopkins University.
Human Tissue Samples
Paraffin-embedded sections of human lung specimens from control subjects (n = 5), patients with IPAH (n = 5), and patients with Ssc-PAH (n = 5) were obtained from the Johns Hopkins University Scleroderma Center as described previously (23). The lung tissue obtained from patients with IPAH and Ssc-PAH was collected after lung transplantation. Lung tissue from control subjects was obtained from organ donors or resections of lung tumors. Approval from the Johns Hopkins Medicine Institutional Review Board was obtained before the start of these studies.
Rat Models of PH
We used two different rat models of PH to analyze lung perivascular IRS2 expression. One was created by combining vascular injury [by Sugen 5416 (SU)] and hypoxia (the SU/Hx model) and the second was created by treating athymic rats (which have dysregulated immunity) with Sugen 5416 (the SU/athymic model). For the SU/Hx study, adult male Sprague–Dawley rats (200–250 g, 6–8 wk old, Hilltop Lab Animals, Scottsdale, PA) were injected subcutaneously with a single dose of Sugen 5416 (20 mg/kg, Sigma, St. Louis, MO) dissolved in carboxymethylcellulose sodium (CMC), or with CMC (vehicle) alone, and exposed to hypoxia (10% O2) or normal room air (20.8% O2) for 3 wk (14, 24, 25). To achieve the desired internal Po2, oxygen concentration was regulated by solenoid-controlled nitrogen flow linked to a Clark electrode (Pro-Ox controller; BioSpherix, Redfield, NY). The chambers were continuously scavenged for CO2 and ammonia. PH establishment was confirmed by hemodynamic measurements. For the SU/athymic study, the experimental protocol was approved by the Veterans Affairs Palo Alto Animal Care and Use Committee at Stanford University, and the study was performed as described previously (25). Athymic nude rats (male, 180–220 g, Biomedical Research Models, Inc., Worcester, MA) were injected subcutaneously with a single dose of either Sugen 5416 (10 mg/kg dissolved in DMSO) or DMSO (vehicle) alone. All animals were maintained under normoxic conditions for 3 wk, and PH establishment was confirmed by hemodynamic measurements.
Mice and In Vivo Hypoxia
B6;129-IRS2tm1Mfw/J mice were purchased from The Jackson Laboratory (Bar Harbor, ME), and IRS2+/− mice were backcrossed to the C57BL/6 NTac background for 10 generations before being used to establish breeding colonies as previously described (12). The IRS2−/− mice show mild insulin resistance, develop increasing blood glucose levels over time (10–12 wk of age), and eventually develop diabetes (26). Therefore, in all of our in vivo experiments, we used male mice that were less than 8 wk of age. In vivo hypoxia stimulation was performed as described previously (27). Briefly, animals were placed in a Plexiglas chamber maintained at 10% O2 or in a chamber open to room air for 4 days with a 12:12-h, light-dark cycle. We chose this period because both perivascular proliferative activity and recruitment of inflammatory cells and FIZZ1/HIMF/RELMα-expressing macrophages to the lung become maximal at 4 days of hypoxia. Hypoxia was maintained as same as chronic hypoxia stimulation for the rat SU/Hx model. Mice were euthanized by exsanguination, and the lungs were removed en bloc. For histology, the left lung lobe was inflated under constant pressure with 1% low melting point agarose in phosphate-buffered saline and placed on ice. The inflated lung was then placed in 4% paraformaldehyde and subsequently processed for histology as described previously (22, 28). The right lung lobe was stored at −80°C for use in immunoblot analysis or in RNAlater (Qiagen, Hilden, Germany) for quantitative RT-PCR (QRT-PCR). All in vivo experiments were repeated at least three times.
Hemodynamic Analysis and Fulton Index Measurement
Right ventricular function of mice was assessed in vivo by a pressure-volume catheter as described previously (28). Briefly, mice were anesthetized with 75 mg/kg intraperitoneal (ip) urethane, 5 mg/kg intraperitoneal etomidate, and 1 mg/kg intraperitoneal morphine. Then they underwent tracheostomy and were ventilated with 6–7 μL/g tidal volume at 130 breaths/min. Right ventricular systolic pressure (RVSP) was measured and data were collected and analyzed with the AD Instruments Powerlab 8/35 (ADInstruments, Colorado Springs, CO) and Millar MPVS Ultra (Millar Instruments, Houston, TX). Right heart hypertrophy induced by hypoxia was also assessed. The heart was dissected free of all major vessels, separated into right ventricle (RV) and left ventricle (LV) plus septum (S), and weighed. The Fulton index (RV/LV + S) was determined as described previously (28).
Pulmonary Vascular Remodeling Analysis
We determined pulmonary vascular remodeling of the mice as previously published (28). Lung sections were dual labeled with antibodies to von Willebrand factor (vWF; A008202, Dako, Santa Clara, CA) and α-smooth muscle actin (α-SMA; M085129, Dako), which stain endothelium and vascular smooth muscle with horseradish peroxidase-diaminobenzidine (HRP-DAB) (brown, Vector Laboratories, Burlingame, CA, SK-41001) and alkaline phosphatase (red, SK-51001, Vector Labs) systems, respectively. An investigator blinded to treatment group assessed remodeling of the lung arteries and arterioles. Approximately, 100 randomly selected arteries with an internal diameter of <80 μm were examined. These vessels were classified as nonmuscular, partially muscular, or fully muscular, according to α-SMA staining. The examination procedure and criteria have been described previously (28).
Immunofluorescence and Confocal Microscopy
Immunofluorescence staining was carried out as described previously (22). Briefly, the paraffin sections were blocked with appropriate blocking serum (Vector Laboratories) and then treated with anti-IRS2 (Abcam, Cambridge, UK), anti-α-SMA, anti-vWF, anti-rat RELMα (Enzo Biochem Inc., New York, NY), anti-mouse RELMα (R&D Systems, Minneapolis, MN), anti-F4/80, anti-CD163, or anti-rat CD68 (Bio-Rad, Hercules, CA) antibodies. Then, the sections were incubated with the appropriate fluorochrome-coupled secondary antibody (Jackson ImmunoResearch, West Grove, PA). Negative control sections for the immunohistochemical experiments received identical treatments but were not exposed to the primary antibody. Staining was imaged with a Zeiss LSM 700 confocal microscope (Carl Zeiss Microscopy, Thornwood, NY) at the Johns Hopkins School of Medicine Microscope Core Facility. Immunofluorescence intensity in the pulmonary vessels was quantified by Fiji-Image J software. We also quantified colocalization of Ki67+ cells in the pulmonary vasculature on lower-magnification photomicrographs (×20 objective) from each group.
Murine Bone Marrow-Derived Macrophage Culture and Cell Marker Analysis
Murine bone marrow-derived (BMD) macrophages were generated from IRS2+/+ and IRS2−/− female littermates with a well-established method (22). Briefly, bone marrow was isolated from femurs and tibias of 4- to 6-wk-old mice. To deplete adherent stromal cells, we cultured the harvested BMD cells in MEMα supplemented with 10% heat-inactivated fetal bovine serum and 100 U/mL penicillin and 100 μg/mL streptomycin overnight. Nonadherent cells were harvested, depleted of red blood cells, and then cultured in 6-well plates for 10 days with recombinant murine macrophage-colony stimulating factor (20 ng/mL, R&D Systems) to differentiate BMD macrophages. Cells were then exposed to hypoxia (2% O2) and collected for later immunoblot or QRT-PCR analysis. For QRT-PCR analysis, recombinant IL-4 (50 ng/mL) was used as a positive control for M2 activation.
QRT-PCR Analysis
QRT-PCR analysis was carried out as previously described (28). Total RNA was isolated from mouse lung tissue or BMD macrophages with the RNeasy method (Qiagen). Total RNA was reverse transcribed with the iScript cDNA Synthesis Kit (Bio-Rad) for RT-PCR. QRT-PCR was carried out with the TaqMan assay system (Applied Biosystems, Foster City, CA) using a CFX Connect Real-Time PCR Detection System (Bio-Rad). Probes and primers were designed and synthesized by Applied Biosystems. Average fold change 2-averageΔΔCt was used to show differences in gene expression of treated samples relative to that of controls.
Immunoblotting
Mouse lung tissue or cell lysate was prepared and immunoblotting was carried out as described previously (22). Briefly, lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and protein was transferred onto nitrocellulose membrane. Membranes were incubated with primary antibodies against Akt (No. 9272), phospho (p)-Akt (Ser473, No. 9271), p44/43 MAPK (ERK, No. 4695), p-ERK (No. 4370), ribosomal protein S6 (S6, No. 2217), p-S6 (No. 4858), FOXO1 (No. 2880), p-FOXO1 (Thr24 and Ser256, No. 9464 and No. 9461) (all from Cell Signaling Technology, Danvers, MA), mouse HIF-1α (No. NB100-105, Novus Biologicals, Littleton, CO), and β-actin (Santa Cruz Biotechnology, Dallas, TX). After membranes were incubated with horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology), the signal was visualized with ECL Western Blotting Detection substrate kit (GE Healthcare, Buckinghamshire, England). Each band was quantified by Image J or ImageQuant (Cytiva, Marlborough, MA) software.
Statistical Analysis
All data are presented as means ± SD. Differences between multiple groups were compared by analysis of variance (ANOVA) followed by Bonferroni’s multiple comparison test. Two-group analysis was performed by Student’s t test. A value of P < 0.05 was considered statistically significant.
RESULTS
IRS2 Expression Is Dysregulated in the Pulmonary Vasculature of Patients with IPAH and Ssc-PAH
To determine the expression and localization of IRS2 in human PAH, we performed immunohistochemical analysis on lung tissue isolated from healthy donors (control) and patients with IPAH or Ssc-PAH. Immunofluorescence staining of the lung tissue revealed that IRS2 expression was significantly decreased in obliterated lumen of pulmonary arteries from patients with IPAH and Ssc-PAH as compared with that in control subjects (Fig. 1, A and B). In the pulmonary arteries of control subjects, IRS2 expression was evident in the α-smooth muscle actin (SMA)-positive cells and immune cells surrounding the vascular media. Most of the IRS2+ immune cells were macrophages, as evidenced by colabeling with anti-CD163 antibody in the pulmonary vasculature of control subjects (Fig. 1C). Our results suggest that most IRS2 is expressed in SMA+ and CD163+ cells in pulmonary arteries and that it decreases or is absent in patients with PAH.
Figure 1.
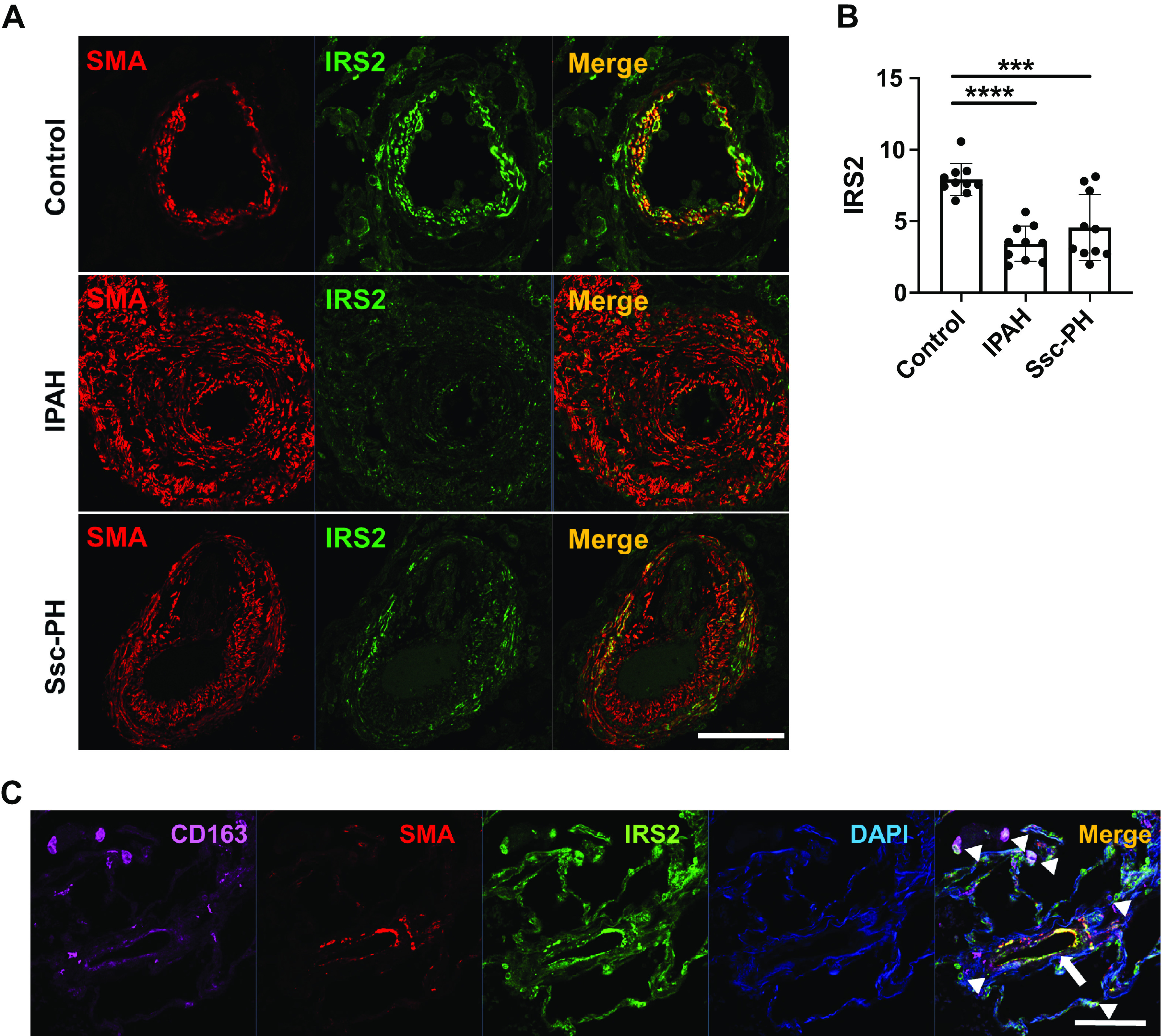
IRS2 expression is decreased in the pulmonary vasculature (PV) of patients with pulmonary arterial hypertension (PAH). A: expression of IRS2 and smooth muscle actin (SMA) in pulmonary arteries of lung tissue from a normal control subject and patients with idiopathic PAH (IPAH) and scleroderma-associated PAH (Ssc-PAH). IRS2 expression is seen in the PV media and surrounding cells of control lung but is substantially reduced in PAH lung. B: quantification of IRS2 expression in the pulmonary vessels. Fluorescence intensity data are expressed as means ± SD. ***P < 0.001, ****P < 0.0001 (n = 10 vessels/group). Immunofluorescence intensity was significantly diminished in pulmonary vessels of patients with IPAH and Ssc-PAH. C: IRS2-expressing cells colocalize with CD163-positive cells (arrowheads) and SMA-positive cells (arrow). Representative data are shown from normal control subjects (n = 5), subjects with IPAH (n = 5), and subjects with Ssc-PH (n = 5). Scale bars = 100 µm. IRS2, insulin receptor substrate 2.
IRS2 Expression Is Dysregulated in the Pulmonary Vasculature of Rats with Experimental PH
The vascular wall of pulmonary arteries during PH is characterized by endothelial cell dysfunction, accumulation of inflammatory cells, and hyperproliferative vascular smooth muscle cells. Accordingly, growth factor upregulation and metabolic and mitochondrial abnormalities are present in PH (29). In rats, exposure to SU/Hx leads to human PAH-like occlusive vascular lesions and severe right ventricular pressure overload (30). We observed IRS2 expression in the vascular media and surrounding immune cells of the control animals; however, the IRS2 signal was significantly decreased in SU/Hx rats (Fig. 2, A and B). In T cell-deficient (athymic) rats, PH was induced by Sugen 5416 alone (SU/athymic). The mechanism of this model reflects that of human PH from autoimmune diseases, in which pulmonary arterioles become occluded and surrounded by collections of macrophages, B cells, and mast cells (31). In this SU/athymic rat model, FIZZ1/RELMα expression was elevated in pulmonary vessels at 3 wk of PH development phase (Fig. 2C). Furthermore, IRS2 expression was significantly lower in the remodeling pulmonary vasculature than it was in control animals. These results suggest that IRS2 expression in the pulmonary vasculature is decreased in rat models of PH, and that it negatively correlates with increased FIZZ1/RELMα expression in the pulmonary vasculature of SU/athymic-treated rats. We did not measure FIZZ1/RELMα in rats with SU/Hx-induced PH because expression is transient and no longer evident in their lungs after 3 wk of hypoxia (31).
Figure 2.

IRS2 expression is diminished in the pulmonary vasculature (PV) of rats with experimental PH. A: IRS2 expression in the PV of SU5416/hypoxia (SU/Hx)-treated rats. Representative images show IRS2 and smooth muscle actin (SMA) in pulmonary arteries of lung tissues from controls and SU/Hx-treated rats (at 4 wk). B: quantification of IRS2 expression in the pulmonary vessels. Fluorescence intensity data are expressed as means ± SD. **P < 0.01 (n = 5 rats/group). C: FIZZ1/RELMα and IRS2 expression in the PV of SU/athymic rats at 3 wk. Representative images show increased perivascular RELMα in the PV of SU-treated athymic rats. Endothelial cells were labeled by anti-von Willebrand factor (vWF) antibody to detect PV. D: quantification of FIZZ1/RELMα and IRS2 expression in the pulmonary vessels. Increased perivascular RELMα and decreased IRS2 expression are features of SU/athymic rats. Fluorescence intensity data are expressed as means ± SD. ***P < 0.001, ****P < 0.0001 (n = 5 rats/group). Scale bars = 100 µm. IRS2, insulin receptor substrate 2; RELMα, resistin-like molecule α; SU/Hx; Sugen 5416 and hypoxia.
IRS2 Knockdown Exacerbates Hypoxia-Induced Macrophage Accumulation and Muscularization in the Pulmonary Vasculature
We have published that IRS2 is a key upstream regulator of M2 markers in macrophages, and that loss of IRS2 macrophages significantly increases FIZZ1/RELMα expression and pulmonary vascular remodeling in response to Th2 stimulation (12). Here, we examined whether a similar mechanism is present in wild-type (IRS2+/+) and IRS2+/− mice exposed to hypoxia stimulation. After 4 days of hypoxia, we observed increased perivascular FIZZ1/RELMα signal and partially muscularized vessels, as expected, in IRS2+/+ mice (Fig. 3, A and B). To our surprise, IRS2+/− mice showed significant increases in fully muscularized pulmonary vessels at 4 days of hypoxia stimulation, accompanied by increased FIZZ1/RELMα-expressing macrophages, whereas full muscularization usually requires at least 3 wk in mice.
Figure 3.

Perivascular FIZZ1/RELMα expression, cell proliferation, and muscularization are exacerbated in the lungs of IRS2+/− mice by 4 days of hypoxia. A: macrophage recruitment and muscularization in IRS2+/+ and IRS2+/− mice in response to 4 days of hypoxia (10% O2). Colocalization of perivascular muscularized cells (SMA+, red), macrophages (F4/80+, green), and RELMα (marker for M2 macrophages, far red) is indicated by arrows. B. quantification of FIZZ1/RELMα and SMA expression in the pulmonary vessels. Fluorescence intensity data are expressed as means ± SD. ##P < 0.01, ####P < 0.0001, ****P < 0.0001 versus indicated group (n = 5–6 animals). Scale bars = 100 µm. C: representative images show perivascular muscularization (SMA+, far red), cell proliferation (Ki67+, red), and macrophages (F4/80+, green) in the pulmonary vasculature of hypoxia-stimulated IRS2+/+ (top) and IRS2+/- (bottom) mice. D: hypoxia induced significantly more vascular cell proliferation in IRS2+/− mice than in IRS2+/+ mice. Data are expressed as means ± SD. ####P < 0.001 (n = 15 vessels). E: hypoxia induction of Retnla gene expression in whole lung tissue from IRS2+/+ and IRS2+/− mice. Data are expressed as means ± SD. **P < 0.01, ****P < 0.0001, ††P < 0.001 versus indicated group (n = 5–6 animals). IRS2, insulin receptor substrate 2; HYP, hypoxia; NOR, normoxia; RELMα, resistin-like molecule α.
Hypoxia-induced cell proliferation activity within pulmonary arteries was significantly higher in IRS2+/− mice than in IRS2+/+ mice, and the proliferating (Ki67+) cells were located in muscularized vessels (Fig. 3, C and D).
We also analyzed FIZZ1/RELMα and resistin expression in whole lung tissue (Fig. 3E) because lung epithelial cells are known to produce high levels of FIZZ1/RELMα in the lung in response to 4 days of hypoxia. However, whole lung tissue from the two mouse genotypes exhibited no significant difference in hypoxia-induced FIZZ1/RELMα expression. We also analyzed resistin expression because resistin is a family member of RELM proteins and is a prodiabetic and proinsulin resistance cytokine in mice (32). We thought that resistin expression might be significantly higher in IRS2-deficient mice because these mice are known to develop insulin resistance (4). As expected, resistin expression was significantly higher in IRS2+/− mice than in IRS2+/+ mice at baseline. These results suggest that decreased IRS2 increases perivascular FIZZ1//RELMα expression and proliferative activities in the lung.
IRS2 Deficiency Significantly Enhances Hypoxia-Induced Pulmonary Vascular Remodeling and Right Heart Hypertrophy
We further analyzed and compared the hypoxia-induced pulmonary vascular remodeling and PH development in IRS2+/+, IRS2+/−, and IRS2−/− mice. RVSP was measured at 3 wk of hypoxia exposure, but we observed no significant difference between IRS2+/+ and IRS2+/− mice (Fig. 4A). To our surprise, the IRS2−/− mice were unable to survive hemodynamic analysis and died as soon as the catheter was inserted into the heart. Accordingly, the hypoxia-induced right heart hypertrophy was significantly greater in IRS2−/− mice than in IRS2+/+ mice (Fig. 4B). Moreover, histological data showed that IRS2+/− and IRS2−/− mice had greater pulmonary vessel thickening during the late PH development stage than did hypoxic IRS2+/+ mice (Fig. 4C). In addition, IRS2+/− and IRS2−/− mice showed a gene dosage effect in pulmonary arterial muscularization, in that IRS2+/− mice developed intermediate levels of pulmonary vascular remodeling (Fig. 4D). These data suggest that loss of IRS2 exacerbates hypoxia-induced pulmonary vascular remodeling and right heart hypertrophy.
Figure 4.

Hypoxia-induced PH development and pulmonary vascular remodeling in IRS2+/+, IRS2+/−, and IRS2−/− mice. A: hemodynamic analysis. IRS2+/− mice subjected to hypoxia exhibited increased right ventricular systolic pressure (RVSP) similar to that of IRS2+/+ mice. RVSP results for hypoxia-treated IRS2−/− mice are not available because the measurement procedure caused sudden death of these mice. B: Fulton index. Right ventricular hypertrophy was determined as RV/LV+S ratio in each genotype. IRS2−/− mice showed a significantly greater increase in hypoxia-induced right ventricular hypertrophy than did IRS2+/+ mice. Data are expressed as means ± SD. ***P < 0.001, ****P < 0.0001, #P < 0.05 versus indicated group (n = 5) C: representative hematoxylin and eosin-stained images of small pulmonary vessels of each genotype. Scale bars = 100 µm. D: percent muscularization of small pulmonary vessels in each genotype. IRS2 gene deletion caused a significant increase in vascular remodeling. Comparison of hypoxia-induced pulmonary vascular remodeling in IRS2+/+, IRS2+/−, and IRS2−/− mice. Bar graphs show the percentage of small pulmonary arteries in each genotype that were nonmuscularized (NM), partially muscularized (PM), or fully muscularized (FM). Data are expressed as means ± SD. ***P < 0.001, ****P < 0.0001. At least 100 vessels were counted in each lung section (n = 5 animals/group). IRS2, insulin receptor substrate 2; LV, left ventricle; PH, pulmonary hypertension; RV, right ventricle.
IRS2 Negatively Regulates the Akt-FOXO1 Pathway but Not the ERK Pathway in Lung under Hypoxic Conditions
Although IRS2 mediates many signaling pathways, its predominant effect is on the phosphoinositide 3-kinase/Akt and p44/43 MAPK (ERK) cascades, which regulate many downstream effectors (9). In this context, we examined how loss of IRS2 function affects downstream signaling in lung tissue under hypoxic conditions. Our results showed that the hypoxia-induced phosphorylation of Akt, but not ERK, was significantly greater in IRS2+/− mice than in IRS2+/+ mice (Fig. 5). Ribosomal protein S6 and hypoxia-inducible factor (HIF)-1α are downstream of Akt and ERK signaling, respectively, and are activated in response to hypoxia, but we observed no significant differences in these proteins between genotypes. Furthermore, we analyzed the phosphorylation of FOXO1 because it lies downstream of the IRS2-Akt pathway and its phosphorylation and degradation have been implicated in human and experimental PH (33). Interestingly, hypoxia-induced phosphorylation of FOXO1 at both Thr24 and Ser256 was significantly increased in IRS2+/− mice as compared with that in IRS2+/+ mice; baseline Thr24 phosphorylation was also significantly higher in IRS2+/− mice. These data suggest that IRS2 negatively regulates the Akt-FOXO1 pathway under hypoxic conditions in the lung.
Figure 5.

Expression levels of Akt-FOXO1 pathway proteins are elevated in the lungs of IRS2+/− mice. Immunoblotting was used to analyze downstream signaling of IRS2 in the lungs of IRS2+/+ and IRS2+/- mice after 4 days of hypoxia stimulation. Expression levels of total (T) and phosphorylated (P) Akt, ERK, S6, FOXO1 (Thr24 and Ser256), and HIF-1α were assessed. Data are expressed as means ± SD. *P < 0.05, **P < 0.01, #P < 0.05, ##P < 0.01 versus indicated group (n = 3–4). Immunoblots shown are from one of three independent experiments conducted. IRS2, insulin receptor substrate 2; FOXO1, Forkhead box O1.
IL-4 and Hypoxia Stimulation Decrease IRS2 Gene Transcription, and IRS2 Deficiency Enhances Tumor-Associated Macrophage-Like Markers in Response to Hypoxia
Our previous and current results suggest that decreased macrophage IRS2 might exacerbate vascular inflammation and remodeling in response to hypoxia and Th2 stimulation. In this context, we tested whether Th2 cytokine IL-4 or hypoxia can regulate IRS2 expression in BMD macrophages isolated from IRS2−/− mice. Both IL-4 and hypoxia caused a significant decrease in Irs2 gene expression in BMD macrophages (Fig. 6A), suggesting that those stimuli may impair IRS2 function. Furthermore, we tested whether complete deletion of IRS2 (IRS2−/−) in macrophages would regulate arginase 1 (Arg1) and vascular endothelial growth factor (VEGF), which are markers of pro-PH macrophages (34), wound healing (18), and tumor-associated macrophages (TAM) (35). We have previously shown that IL4/IL-13 stimulation dramatically enhances Arg1 expression in IRS2−/− macrophages (12). Here, both IL-4 and hypoxia significantly increased Arg1 and Vegfa expression in IRS2−/− macrophages as compared with levels in IRS2+/+ macrophages (Fig. 6B). Thus, IRS2 in macrophages may play a critical anti-inflammatory role by modulating the expression of these markers and macrophage activation under hypoxic conditions.
Figure 6.

In response to hypoxia, pro-PH macrophage markers arginase 1 (Arg1) and vascular endothelial growth factor-A (Vegfa) are significantly increased in bone marrow-derived (BMD) macrophages from IRS2−/− mice. A: isolated BMD macrophages from IRS2+/+ mice were stimulated with IL-4 (50 ng/mL) or hypoxia (2% O2) for 24 h. Irs2 gene expression was analyzed by quantitative RT-PCR. B: quantitative RT-PCR was used to analyze gene expression of Arg1 and Vegfa in BMD macrophages from IRS2+/+ and IRS2−/− mice. Data are expressed as means ± SD (n = 6). **P < 0.01; ****P < 0.0001; ###P < 0.001; ####P < 0.0001 versus indicated group. HYP, hypoxia; IRS2, insulin receptor substrate 2; NOR, normoxia; PH, pulmonary hypertension.
IRS2 Negatively Regulates the Akt and ERK Pathway in BMD Macrophages in Response to Hypoxia
To further our understanding of IRS2-induced cell signaling in macrophages under hypoxic conditions, we analyzed the phosphorylation of Akt and ERK in BMD macrophages isolated from IRS2+/+ and IRS2−/− mice. We found that the hypoxia-induced phosphorylation of Akt, ERK, and S6 proteins was significantly greater in IRS2−/− cells than in IRS2+/+ cells (Fig. 7). These data suggest that IRS2 negatively regulates the Akt-ERK pathway in BMD macrophages under hypoxic conditions.
Figure 7.
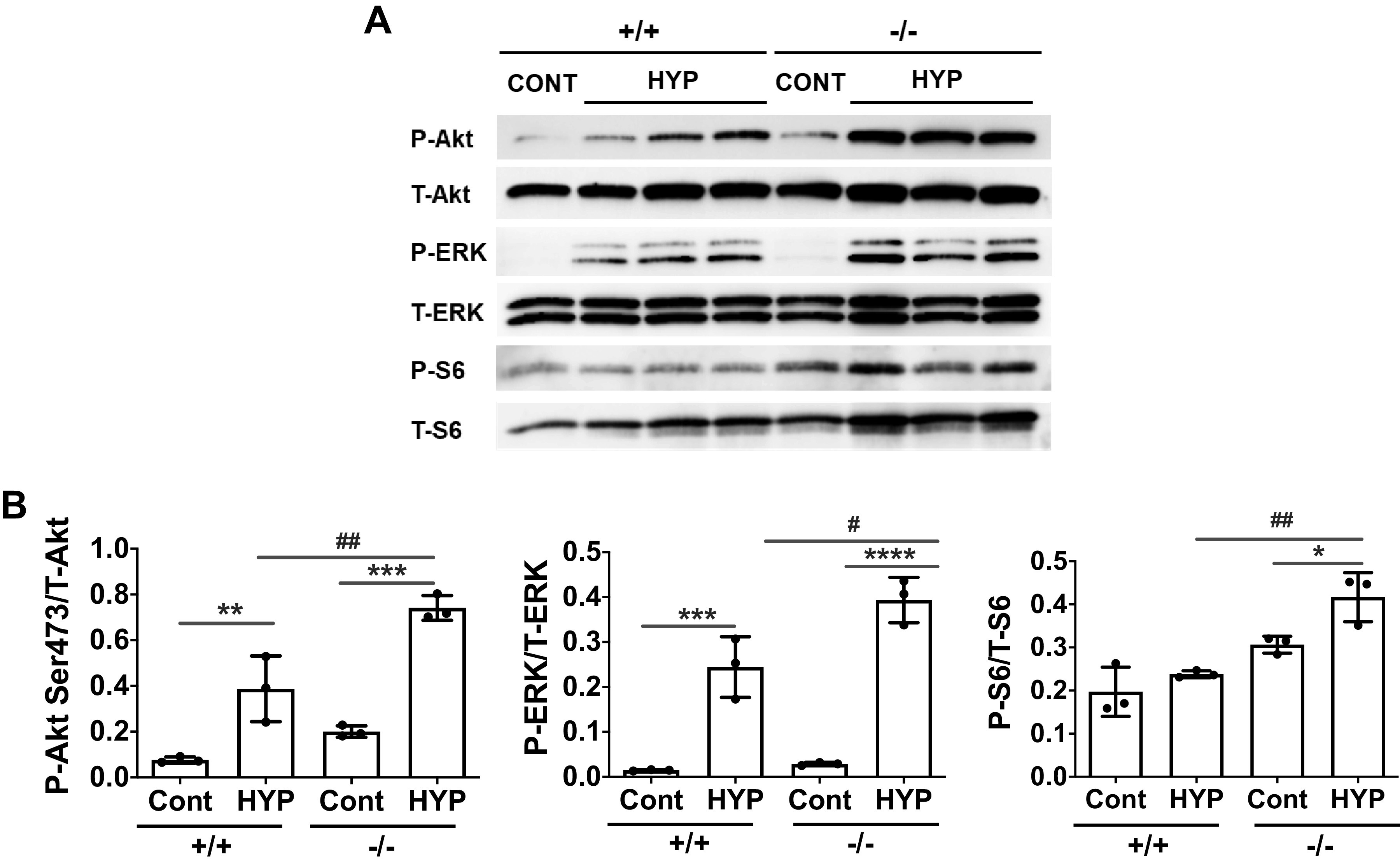
Hypoxia-induced phosphorylation of Akt and ERK pathway proteins is significantly increased in IRS2−/− bone marrow-derived (BMD) macrophages. Isolated BMD macrophages from IRS2+/+ and IRS2−/− mice were stimulated with hypoxia (2% O2) for 2 h. A: immunoblotting was used to analyze downstream signaling of IRS2 in the BMD macrophages. The representative immunoblot shows expression of total (T) and phosphorylated (P) Akt, ERK, and S6. B: relative expression of pAkt, pERK, and pS6 was quantified. Data are expressed as means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P< 0.0001, #P < 0.05, ##P < 0.01 versus indicated group (n = 3). Data are representative from one of three independent experiments. IRS2, insulin receptor substrate 2.
DISCUSSION
This study provides strong evidence that IRS2 is downregulated in the small pulmonary arteries during clinical PH, and that loss of IRS2 function exacerbates pulmonary vascular remodeling and right heart hypertrophy in response to hypoxia in mice. Indeed, IRS2+/− and IRS2−/− mice showed a gene dosage effect in pulmonary arterial muscularization and right heart hypertrophy in response to hypoxia. Hypoxia-induced phosphorylation of Akt and FOXO1, but not ERK, was significantly increased in the lung of IRS+/− mice as compared with that in IRS2+/+ mice, indicating that the Akt-FOXO1-dependent signaling pathway is augmented in the lung in response to hypoxia. These data imply that under hypoxic conditions, IRS2 levels decrease, leading to augmented activation of the Akt-FOXO1 signaling pathway, increased muscularization of small pulmonary arteries, and ultimately to development of PH. Furthermore, hypoxia stimulation significantly reduced IRS2 gene transcription in BMD macrophages, and expression levels of Arg1 and VEGF-A in IRS2−/− BMD macrophages were significantly increased compared with those in IRS2+/+ BMD macrophages. Our BMD macrophage study also showed that loss of IRS2 significantly enhanced Akt and ERK pathway signaling in response to hypoxia. The graphical summary of our findings is shown in Fig. 8.
Figure 8.
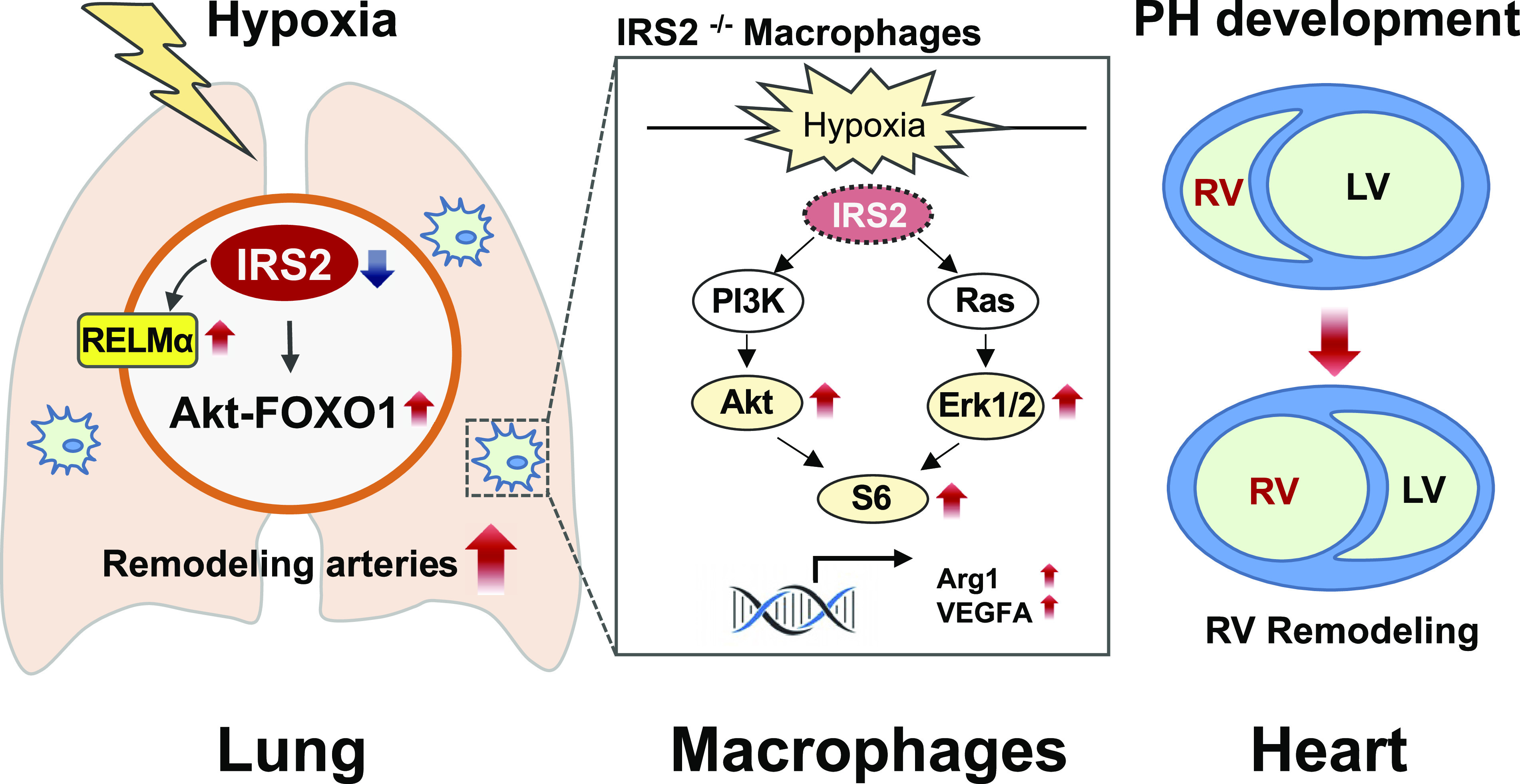
Graphical summary of the role of IRS2 in hypoxia-induced pulmonary hypertension (PH). Our data suggest that IRS2 dysfunction enhances the Akt and ERK pathway and then leads to macrophage activation, which may intensify the vascular remodeling and right ventricular hypertrophy that are seen in PH pathology. Arg1, arginase 1; IRS2, insulin receptor substrate 2; LV, left ventricle; PH, pulmonary hypertension; RELMα, resistin-like molecule α; RV, right ventricle; VEGFA, vascular endothelial growth factor A.
We have shown previously that IRS2−/− mice develop greater pulmonary inflammation and undergo more airway and vascular remodeling than do wild-type mice upon allergen stimulation. These responses depend in part on M2 macrophage-intrinsic IRS2 signaling (12). Similarly, in the current study, mice with a partial loss of IRS2 exhibited exaggerated pulmonary vascular proliferation along with increased recruitment of perivascular M2 macrophages in response to hypoxia, suggesting that IRS2 reduces hypoxia-induced vascular remodeling.
We chose to conduct our study at 4 days of hypoxia because the transient macrophage infiltration and RELMα expression are maximal in the lung at that time point (19). Additionally, HIF-1α expression and phosphorylation of Akt and ERK are significantly increased in mouse lung tissue after 4 days (27). Akt and ERK are important because 1) they are part of the downstream signaling pathway of IRS2 and 2) activation of this pathway contributes to the development of PH (36). After 4 days of hypoxia, a period known as the proliferative phase in the pulmonary vasculature, both Akt and ERK pathway proteins, including S6 protein, HIF-1α, and FOXO1, were phosphorylated in the lung. In particular, hypoxia led to significant phosphorylation of FOXO1 at both Thr24 and Ser256 in mice with a partial loss of IRS2. Importantly, phosphorylation and nuclear exclusion of FOXO1 are the initial steps of FOXO1 degradation, and FOXO1 downregulation is a hallmark of human and experimental PH. Dysregulated FOXO1 in the pulmonary vasculature increases inflammatory and pro-proliferative properties in vascular remodeling during PH development (33). Thus, our data suggest that IRS2 contributes to anti-inflammatory and antiproliferative effects during hypoxia by suppressing the Akt-FOXO1 activation pathway in the lung. It is reasonable to speculate that under hypoxic conditions, the inflammatory and antiapoptotic properties of FOXO1 are mediated, at least in part, by upstream molecules including IRS2.
In our in vitro studies, we showed that, in response to hypoxia, BMD macrophages isolated from IRS2−/− mice exhibited significantly higher levels of pro-PH macrophage and TAM activation markers, such as Arg1 and VEGF-A, than did IRS2+/+ macrophages (34, 35). The role of M2 macrophages in PH pathophysiology is controversial, and M2 macrophages are known to be protective in certain experimental PH models (37–39). M1 and M2 macrophages display great heterogeneity and change their phenotypes dramatically in response to their circumstances. Thus, a more detailed classification of macrophages is warranted to facilitate target identification for therapeutic interventions in various pathological conditions (40).
Notably, the pro-PH macrophage markers have been established to include both Arg1 and VEGF-A (41). These pro-PH macrophages are distinct from M2 macrophages and are rather similar to those recently described for hypoxic tumor-driven TAM polarization and wound-healing macrophages (42). Thus, pro-PH macrophage activation not only promotes wound healing but also provides an environment for tumor growth and pathological tissue remodeling. Most importantly, our data show that IRS2 negatively regulates both Akt and ERK signaling pathways before upregulation of Arg1 and VEGF-A expression in response to hypoxia. These observations suggest that IRS2 has an anti-inflammatory role in macrophages during pulmonary vascular remodeling in hypoxic and hypoxic-like vascular microenvironments.
Although the IRS family includes multiple proteins (IRS1-4), we chose to focus on the function of IRS2 based on our previous microarray analysis, which showed that the expression of IRS2, but not that of IRS1 or IRS4, was significantly lower in PBMCs of patients with PAH than in those of control subjects (IRS3 was undetectable) (11). IRS1 and IRS4 did not show any correlation in these groups. Additionally, IRS2 deficiency, but not IRS1 deficiency, enhanced Akt and its downstream signaling in BMD macrophages (12). These data suggest that the detrimental effect is specific to IRS2 deficiency in human and animal PH.
In the present study, one important limitation was that we were unable to measure RVSP in IRS2−/− mice because they all died as soon as the catheter was inserted. We also used IRS2+/− mice for the in vivo PH study because some IRS2−/− mice developed mild insulin resistance and eventual diabetes (26), which is known to contribute to PH development (10, 43). However, IRS2+/− mice maintain normal circulating glucose levels throughout their lives. Notably, IRS2+/− mice exhibited fully muscularized pulmonary vessels at day 4 of hypoxia. This result was surprising because it usually takes at least 3 wk of hypoxia before full muscularization is observed in wild-type mice. These data highlight the importance of IRS2 to pulmonary vascular remodeling in response to hypoxia. Additionally, hypoxia-induced right ventricular hypertrophy was significantly greater in IRS2−/− mice than in IRS2+/+ mice. This finding strongly suggests that IRS2 negatively regulates right ventricular remodeling during hypoxia-induced PH development. Our study also suggests that the dysregulation of IRS2 increased activation of shared mediators, such as FIZZ1/RELMα, as well as Akt and ERK signaling in macrophages. This activation might produce similar morphological alterations in the pulmonary arteries in response to Th2-mediated pulmonary arterial remodeling as seen in schistosomiasis.
In conclusion, we showed that IRS2 expression is dysregulated in the lung pulmonary vasculature in both human and experimental PH. In our mouse hypoxia study, knockdown of IRS2 exacerbated vascular remodeling and proliferating activity dependent on a macrophage-intrinsic mechanism. Complete lack of IRS2 in BMD macrophages enhanced the expression of Arg1 and VEGF-A, which are also associated with TAM and wound-healing macrophages, suggesting that IRS2 has an anti-inflammatory role in macrophages under hypoxic conditions. A more cell-specific analysis of IRS2 during PH development that encompasses cell types other than macrophages is warranted in the future. A new therapeutic approach for restoring IRS2 and its function may be helpful in developing new biomarkers and PH treatments that target multiple downstream neoplastic and metabolic mediators of this pathway.
GRANTS
This work was supported by a PHA Proof of Concept Research Grant, a Stimulating and Advancing ACCM Research (StAAR) Grant from the Johns Hopkins University Department of Anesthesiology and Critical Care Medicine, and National Institutes of Health (NIH) Grant 1R01HL135022 (to K.Y.-K.); by NIH Centers for Advanced Diagnostics and Experimental Therapeutics in Lung Diseases (CADET I) Grant P50HL107182 (to R.A.J.); by NIH Grant R56AI122631 and US Veterans Administration Merit Award I01 BX001850 (to A.D.K.); by NIH Grants HL122887, HL014985, HL125739, HL120001, and HL138473 (to M.R.N.); and by NIH Shared Instrumentation Grant S10OD016374 (to the Johns Hopkins University Microscope Facility).
DISCLAIMERS
This article was prepared while K. Kegan was employed at University of Maryland Baltimore. The opinions expressed in this article are the authors’ own and do not reflect the views of the National Institutes of Health, the Department of Health and Human Services, or the US Government.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.I. and K.Y.-K. conceived and designed research; M.N., H.I., J.T.S., R.T., and K.Y.-K. performed experiments; M.N., H.I., J.T.S., R.T., and K.Y.-K. analyzed data; M.N., H.I., R.T., M.R.N., and K.Y.-K. interpreted results of experiments; M.N., Q.L., and K.Y.-K. prepared figures; M.N., H.I., and K.Y.-K. drafted manuscript; Q.L., M.R.N., A.D.K., R.A.J., and K.Y.-K. edited and revised manuscript; M.N., H.I., J.T.S., Q.L., R.T., M.R.N., A.D.K., R.A.J., and K.Y.-K. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Claire F. Levine for editing the manuscript. We also thank Lyric Ramsue for technical assistance.
Present addresses: M. Nakahara, Kagoshima University School of Medicine, 8-35-1, Sakuragaoka, Kagoshima, Japan; H. Ito, Jichi Medical University, 3311-1 Yakushiji, Shimotsuke, Tochigi, Japan; K. Yamaji-Kegan, National Heart, Lung, and Blood Institute, National Institutes of Health, 6705 Rockledge Dr., Bethesda, MD 20817.
REFERENCES
- 1.Dorfmuller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Respir J 22: 358–363, 2003. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- 2.Tuder RM, Voelkel NF. Pulmonary hypertension and inflammation. J Lab Clin Med 132: 16–24, 1998. doi: 10.1016/S0022-2143(98)90020-8. [DOI] [PubMed] [Google Scholar]
- 3.Rabinovitch M, Guignabert C, Humbert M, Nicolls MR. Inflammation and immunity in the pathogenesis of pulmonary arterial hypertension. Circ Res 115: 165–175, 2014. doi: 10.1161/CIRCRESAHA.113.301141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White MF. IRS2 integrates insulin/IGF1 signalling with metabolism, neurodegeneration and longevity. Diabetes Obes Metab 16,Suppl 1: 4–15, 2014. doi: 10.1111/dom.12347. [DOI] [PubMed] [Google Scholar]
- 5.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol 17: 701–738, 1999. doi: 10.1146/annurev.immunol.17.1.701. [DOI] [PubMed] [Google Scholar]
- 6.Heller NM, Qi X, Junttila IS, Shirey KA, Vogel SN, Paul WE, Keegan AD. Type I IL-4Rs selectively activate IRS-2 to induce target gene expression in macrophages. Sci Signal 1: ra17, 2008. doi: 10.1126/scisignal.1164795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaw LM. The insulin receptor substrate (IRS) proteins: at the intersection of metabolism and cancer. Cell Cycle 10: 1750–1756, 2011. doi: 10.4161/cc.10.11.15824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verdier F, Chretien S, Billat C, Gisselbrecht S, Lacombe C, Mayeux P. Erythropoietin induces the tyrosine phosphorylation of insulin receptor substrate-2. An alternate pathway for erythropoietin-induced phosphatidylinositol 3-kinase activation. J Biol Chem 272: 26173–26178, 1997. doi: 10.1074/jbc.272.42.26173. [DOI] [PubMed] [Google Scholar]
- 9.Lavin DP, White MF, Brazil DP. IRS proteins and diabetic complications. Diabetologia 59: 2280–2291, 2016. doi: 10.1007/s00125-016-4072-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zamanian RT, Hansmann G, Snook S, Lilienfeld D, Rappaport KM, Reaven GM, Rabinovitch M, Doyle RL. Insulin resistance in pulmonary arterial hypertension. Eur Respir J 33: 318–324, 2009. doi: 10.1183/09031936.00000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheadle C, Berger AE, Mathai SC, Grigoryev DN, Watkins TN, Sugawara Y, Barkataki S, Fan J, Boorgula M, Hummers L, Zaiman AL, Girgis R, McDevitt MA, Johns RA, Wigley F, Barnes KC, Hassoun PM. Erythroid-specific transcriptional changes in PBMCs from pulmonary hypertension patients. PloS One 7: e34951, 2012. doi: 10.1371/journal.pone.0034951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dasgupta P, Dorsey NJ, Li J, Qi X, Smith EP, Yamaji-Kegan K, Keegan AD. The adaptor protein insulin receptor substrate 2 inhibits alternative macrophage activation and allergic lung inflammation. Sci Signal 9: ra63, 2016. doi: 10.1126/scisignal.aad6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teng X, Li D, Champion HC, Johns RA. FIZZ1/RELMalpha, a novel hypoxia-induced mitogenic factor in lung with vasoconstrictive and angiogenic properties. Circ Res 92: 1065–1067, 2003. doi: 10.1161/01.RES.0000073999.07698.33. [DOI] [PubMed] [Google Scholar]
- 14.Angelini DJ, Su Q, Yamaji-Kegan K, Fan C, Skinner JT, Champion HC, Crow MT, Johns RA. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) induces the vascular and hemodynamic changes of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 296: L582–L593, 2009. doi: 10.1152/ajplung.90526.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamaji-Kegan K, Su Q, Angelini DJ, Champion HC, Johns RA. Hypoxia-induced mitogenic factor has proangiogenic and proinflammatory effects in the lung via VEGF and VEGF receptor-2. Am J Physiol Lung Cell Mol Physiol 291: L1159–L1168, 2006. doi: 10.1152/ajplung.00168.2006. [DOI] [PubMed] [Google Scholar]
- 16.Yamaji-Kegan K, Su Q, Angelini DJ, Myers AC, Cheadle C, Johns RA. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) increases lung inflammation and activates pulmonary microvascular endothelial cells via an IL-4-dependent mechanism. J Immunol 185: 5539–5548, 2010. doi: 10.4049/jimmunol.0904021. [DOI] [PubMed] [Google Scholar]
- 17.Angelini DJ, Su Q, Kolosova IA, Fan C, Skinner JT, Yamaji-Kegan K, Collector M, Sharkis SJ, Johns RA. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELM alpha) recruits bone marrow-derived cells to the murine pulmonary vasculature. PLoS One 5: e11251, 2010. doi: 10.1371/journal.pone.0011251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8: 958–969, 2008[Erratum inNat Rev Immunol10: 460,2010]. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vergadi E, Chang MS, Lee C, Liang OD, Liu X, Fernandez-Gonzalez A, Mitsialis SA, Kourembanas S. Early macrophage recruitment and alternative activation are critical for the later development of hypoxia-induced pulmonary hypertension. Circulation 123: 1986–1995, 2011. doi: 10.1161/CIRCULATIONAHA.110.978627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Angelini DJ, Su Q, Yamaji-Kegan K, Fan C, Skinner JT, Poloczek A, El-Haddad H, Cheadle C, Johns RA. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMα) in chronic hypoxia- and antigen-mediated pulmonary vascular remodeling. Respir Res 14: 1, 2013. doi: 10.1186/1465-9921-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daley E, Emson C, Guignabert C, de Waal Malefyt R, Louten J, Kurup VP, Hogaboam C, Taraseviciene-Stewart L, Voelkel NF, Rabinovitch M, Grunig E, Grunig G. Pulmonary arterial remodeling induced by a Th2 immune response. J Exp Med 205: 361–372, 2008. doi: 10.1084/jem.20071008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johns RA, Takimoto E, Meuchel LW, Elsaigh E, Zhang A, Heller NM, Semenza GL, Yamaji-Kegan K. Hypoxia-inducible factor 1α is a critical downstream mediator for hypoxia-induced mitogenic factor (FIZZ1/RELMα)-induced pulmonary hypertension. Arterioscler Thromb Vasc Biol 36: 134–144, 2016. doi: 10.1161/ATVBAHA.115.306710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Angelini DJ, Su Q, Yamaji-Kegan K, Fan C, Teng X, Hassoun PM, Yang SC, Champion HC, Tuder RM, Johns RA. Resistin-like molecule-beta in scleroderma-associated pulmonary hypertension. Am J Respir Cell Mol Biol 41: 553–561, 2009. doi: 10.1165/rcmb.2008-0271OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, Patterson AJ, Agrawal R, Rabinovitch M, Ambler K, Long CS, Voelkel NF, Nicolls MR. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res 109: 867–879, 2011. doi: 10.1161/CIRCRESAHA.110.236927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tian W, Jiang X, Tamosiuniene R, Sung YK, Qian J, Dhillon G, Gera L, Farkas L, Rabinovitch M, Zamanian RT, Inayathullah M, Fridlib M, Rajadas J, Peters-Golden M, Voelkel NF, Nicolls MR. Blocking macrophage leukotriene b4 prevents endothelial injury and reverses pulmonary hypertension. Sci Transl Med 5: 200ra117, 2013. doi: 10.1126/scitranslmed.3006674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, Bonner-Weir S, White MF. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391: 900–904, 1998. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- 27.Yamaji-Kegan K, Su Q, Angelini DJ, Johns RA. IL-4 is proangiogenic in the lung under hypoxic conditions. J Immunol 182: 5469–5476, 2009. doi: 10.4049/jimmunol.0713347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamaji-Kegan K, Takimoto E, Zhang A, Weiner NC, Meuchel LW, Berger AE, Cheadle C, Johns RA. Hypoxia-induced mitogenic factor (FIZZ1/RELMa) induces endothelial cell apoptosis and subsequent interleukin-4-dependent pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 306: L1090–L1103, 2014. doi: 10.1152/ajplung.00279.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tuder RM, Davis LA, Graham BB. Targeting energetic metabolism: a new frontier in the pathogenesis and treatment of pulmonary hypertension. Am J Respir Crit Care Med 185: 260–266, 2012. doi: 10.1164/rccm.201108-1536PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicolls MR, Mizuno S, Taraseviciene-Stewart L, Farkas L, Drake JI, Al Husseini A, Gomez-Arroyo JG, Voelkel NF, Bogaard HJ. New models of pulmonary hypertension based on VEGF receptor blockade-induced endothelial cell apoptosis. Pulm Circ 2: 434–442, 2012. doi: 10.4103/2045-8932.105031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taraseviciene-Stewart L, Nicolls MR, Kraskauskas D, Scerbavicius R, Burns N, Cool C, Wood K, Parr LE, Boackle SA, Voelkel NF. Absence of T cells confers increased pulmonary arterial hypertension and vascular remodeling. Am J Respir Crit Care Med 175: 1280–1289, 2007. doi: 10.1164/rccm.200608-1189OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature 409: 307–312, 2001. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 33.Savai R, Al-Tamari HM, Sedding D, Kojonazarov B, Muecke C, Teske R, Capecchi MR, Weissmann N, Grimminger F, Seeger W, Schermuly RT, Pullamsetti SS. Pro-proliferative and inflammatory signaling converge on FoxO1 transcription factor in pulmonary hypertension. Nat Med 20: 1289–1300, 2014. doi: 10.1038/nm.3695. [DOI] [PubMed] [Google Scholar]
- 34.El Kasmi KC, Pugliese SC, Riddle SR, Poth JM, Anderson AL, Frid MG, Li M, Pullamsetti SS, Savai R, Nagel MA, Fini MA, Graham BB, Tuder RM, Friedman JE, Eltzschig HK, Sokol RJ, Stenmark KR. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. J Immunol 193: 597–609, 2014. doi: 10.4049/jimmunol.1303048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, Cline GW, Phillips AJ, Medzhitov R. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513: 559–563, 2014. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang H, Chen J, Fraidenburg DR, Song S, Sysol JR, Drennan AR, Offermanns S, Ye RD, Bonini MG, Minshall RD, Garcia JGN, Machado RF, Makino A, Yuan JX-J. Deficiency of Akt1, but not Akt2, attenuates the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L208–L220, 2015. doi: 10.1152/ajplung.00242.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tamosiuniene R, Nicolls MR. Regulatory T cells and pulmonary hypertension. Trends Cardiovasc Med 21: 166–171, 2011. doi: 10.1016/j.tcm.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graham BB, Bandeira AP, Morrell NW, Butrous G, Tuder RM. Schistosomiasis-associated pulmonary hypertension: pulmonary vascular disease: the global perspective. Chest 137: 20S–29S, 2010. doi: 10.1378/chest.10-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graham BB, Mentink-Kane MM, El-Haddad H, Purnell S, Zhang L, Zaiman A, Redente EF, Riches DWH, Hassoun PM, Bandeira A, Champion HC, Butrous G, Wynn TA, Tuder RM. Schistosomiasis-induced experimental pulmonary hypertension: role of interleukin-13 signaling. Am J Pathol 177: 1549–1561, 2010. doi: 10.2353/ajpath.2010.100063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity 32: 593–604, 2010. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 41.Stenmark KR, Tuder RM, El Kasmi KC. Metabolic reprogramming and inflammation act in concert to control vascular remodeling in hypoxic pulmonary hypertension. J Appl Physiol (1985) 119: 1164–1172, 2015. doi: 10.1152/japplphysiol.00283.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11: 723–737, 2011. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation 115: 1275–1284, 2007. doi: 10.1161/CIRCULATIONAHA.106.663120. [DOI] [PubMed] [Google Scholar]