

Abstract
The transition from normal forage to a highly fermentable diet to achieve rapid weight gain in the cattle industry can induce ruminal acidosis. The molecular host mechanisms that occur in acidosis are largely unknown. Therefore, the histology and transcriptome profiling of rumen epithelium was investigated in normal and acidosis animals to understand the molecular mechanisms involved in the disease. The rumen epithelial transcriptome from acidosis (n=3) and control (n=3) Holstein steers was obtained using RNA-sequencing. The mean values of clean reads were 70,975,460±984,046 and 71,142,189±834,526 in normal and acidosis samples, respectively. In total, 1,074 differentially expressed genes were identified in the two groups (P<0.05), of which 624 and 450 genes were up- and down-regulated in the acidosis samples, respectively. Functional analysis indicated that the majority of the up-regulated genes had a function in filament organization, positive regulation of epithelial and muscle fiber concentration, biomineral tissue development, negative regulation of fat cell differential, regulation of ion transmembrane transport, regulation of cell adhesion and butyrate, as well as short-chain fatty acid absorption that was metabolized as an energy source. Functional analysis of the down-regulated genes revealed effects in immune response, positive regulation of T-cell migration, regulation of metabolic processes, and localization. Furthermore, the results showed a differential expression of genes involved in the Map Kinase and Toll-like receptor signaling pathways. The IL1B, CXCL5, IL36A, and IL36B were significantly down-regulated in acidosis rumen tissue samples. The results suggest that rapid shifts to rich fermentable carbohydrates diets cause an increase in the concentration of ruminal volatile fatty acids, tissue damage, and significant changes in transcriptome profiles of rumen epithelial.
Keywords: Acidosis, Cattle, Ruminal epithelial tissue, Transcriptome
Introduction
In ruminant animals, such as cattle, healthy rumen plays an important role in production and economic efficiency. Economics of feedlot production dictates that cattle must gain weight at their maximum potential rate. This involves getting them quickly onto a full feed of a diet containing a high concentration of grain ( Alston and Pardey, 2014 ; Kahn and Cottle, 2014 ). Economics also favors the processing of grain to increase the digestibility of starch ( Durunna et al., 2011 ). All of these factors set the stage for grain overload in feedlot cattle and may induce ruminal acidosis disorder. Ruminal acidosis is defined as a reduction in ruminal PH below the normal levels and is increasingly recognized as a significant disorder of ruminants ( Enemark, 2008 , Blanch et al., 2010 ). Acidosis is rich due to ingesting large amounts of unaccustomed diets in a rapidly fermentable feed (high grain) to maximize energy intake and weight gain ( Hernandez et al., 2014 ). The resulting production of large quantities of volatile fatty acids (VFA) needs to be offset by VFA absorption through the ruminal wall ( Ash and Baird, 1973 ). One of the most important reasons for the appearance of acidosis is a decreased capacity of the rumen to absorb VFA ( Sehested et al., 1999 ). Consequently, the ruminal PH drops and subsequently causes damage to the rumen tissue. This condition increases the morbidity and mortality of stock, markedly reduces weight gains in the feedlot, complicates drought feeding strategies for cattle, and is increasingly recognized in livestock breeding ( Kleen et al., 2003 ; Hernandez et al., 2014 ). Several in vivo studies have demonstrated changes in gene expression in rumen tissue in response to acidosis ( Penner et al., 2011 ; Gressley, 2014 ); however, consequences and functional mechanisms are still largely unknown constituting an important knowledge gap in our understanding of the underlying biology. Therefore, this study aimed to investigate the differences between normal and acidosis rumen epithelial tissues of cattle in terms of gene expression using ribonucleic acid (RNA)-sequencing technology combined with system biological analysis through advanced bioinformatics tools. Previous studies investigated gene expression changes in rumen epithelium under high grain concentration diets using microarray data or polymerase chain reaction (PCR) assay in dairy cattle. However, to our knowledge, this is the first study to assay global transcriptional changes using RNA-seq based analysis. It is hypothesized that the rumen tissue in acidosis cases has greater expression of genes involved in rapid structural changes to maintain hemostasis, nutrient absorption, and energetic metabolism.
Material and Methods
Experimental Design and Animal Management
In total, six Holstein steers (230 days old) were purchased and kept in the farm of Agriculture resources of the University of Tehran, Tehran, Iran. Animals were randomly divided into two gropes of control and acidosis. In order to induce acidosis, a diet was used with a high proportion of grain for 130 days composed of 10.5% alfalfa hay, 14.5% corn silage, 34% barley grain, 23.3% corn grain ground, 9% soybean meal, 3% wheat bran, 2.5% rice bran, and 3.2% supplement. The final mean body weight values before the slaughter were 529±20 and 570±10kg in the control and acidosis groups, respectively. Clinical signs of acidosis were observed in all three animals. The ruminal pH was measured in the 30th and 129th days (Table 1).
Table 1.
pH of normal and acidosis groups
| Group | 30th day | 129th day |
|---|---|---|
| Control | 5.57- 5.97- 5.87 | 6.03- 6.17- 6.0 |
| Acidosis | 5.97- 5.17- 5.57 | 5.19- 5.10- 5.24 |
Sample Collection for Next Generation Sequencing Data
After slaughter, for each sample, a 4 cm2 piece of rumen tissue was obtained from the central region of the ventral sac and rinsed with sterilized PBS buffer (pH= 6.8) before being placed in a 50 ml tube containing RNA later solution (Invitrogen, Carlsbad, CA). The samples were then stored at -80 °C until further processing. The ventral sac of the rumen was chosen as a sampling site since it had the highest capillary blood flow per unit weight mucosa of any location within the rumen ( O'Shea et al., 2016 ).
Sample Collection for Histopathological Analysis
In order to assay any histopathological change, tissue segments with 2×2 cm size and 0.5 cm thickness were removed from the ventral sac ( Steele et al., 2011 ). After washing, the samples were fixed with 10% formaldehyde for 48 h. Subsequently, it was rinsed in flowing water overnight and dehydrated using ethyl alcohol. Following that, the samples were cleared with xylenol for 4 h and then impregnated with liquid paraffin. After impregnation, paraffin sample blocks were cut in 5 µm slices using a rotary microtome and stained with hematoxylin and eosin. Morphometric measurements of the tissue were carried out by computer-aided light microscopy image analysis. Seven rumen tissues per slide were randomly selected for analysis.
Ribonucleic Acid Extraction and Sequencing
Total RNA was extracted from the tissue samples using Trizol Reagent kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Moreover, the RNA concentration and purity were determined by measuring the absorbance at 260 nm and calculating the A260/A280 ratio using a Nanodrop ND-2000 spectrophotometer. In the next stage, the RNA samples were stored at -80 °C until RNA-seq library preparation. For each sample, a cDNA library was generated from 100 ng of total RNA. The mRNA was enriched by removing rRNA from the total RNA with Ribo-Zero TM Magnetic Kit and fragmented into short fragments (about 200~700 bp). Additionally, positive-strand cDNA was synthesized by random hexamer primers using the fragments as templates. Buffer, dNTPs, RNase H, and DNA polymerase I was added to synthesize the negative-strand cDNA. The double-stranded cDNA was purified using the QiaQuick PCR extraction kit and then used for end-polishing. Sequencing adapters were ligated to fragments, and the second strand was degraded using Uracil-N-Glycosylase. The fragments were purified by Agarose gel electrophoresis and enriched using PCR amplification. The Illumina HiSeqTM 2000 sequencing was performed to obtain high quality 100 bp paired-end reads.
Differential Gene Expression Analysis
The quality of sequencing was assessed using FastQC, and the data were preprocessed using Trimmomatic to filter out low-quality reads, remove adaptors, and trim low-quality regions of the reads. Transcriptome mapping and alignment of reads to the bovine reference genome (UMD3.1) was performed using TopHat2 (v 2.0.9) and Bowite2 (v 2.0.1) of the TopHat package ( Langmead and Salzberg, 2012 ; Kim et al., 2013 ). Differential gene expression analysis was performed using the Tuxedo protocol in the statistical environment R (version 3.4.4). Transcript assembly was done using Cufflinks, and all assemblies were merged into a single transcriptome annotation using Cuffmerge. The cuffdiff was used to identify differentially expressed genes using default parameters in every step. Results were visualized using the cummeRbund R library by estimating the Jensen-Shannon distance ( Trapnell et al., 2012 ).
Functional Gene Annotation
Additional analysis was performed to understand the potential functional implications of significant genes in rumen tissue. To this purpose, Database for Annotation Visualization, and Integrated Discovery (DAVID), as well as gene ontology (GO), were utilized to functionally annotate DE genes ( Huang da et al., 2009 ). In the same line, enrichment analysis was conducted in functional categories, such as KEGG-Pathway, SP-PIR-Keywords, and GO using DAVID (molecular function, biological process, and cellular component) ( Kanehisa and Goto, 2000 ; Kanehisa et al., 2014 ). Biological terms were considered significant if the adjusted P-value was less than 0.05. The list of significant genes was further screened using PANTHER for further functional annotation ( Mi et al., 2013 ).
Results
Identification of Rumen Epithelial Transcriptome
The mean clean reads (after removing adaptor sequence and low-quality reads) were 71 million reads with mean values of 70,975,460±984,046 and 71,142,189±834, 526 reads in normal and acidosis samples, respectively. Moreover, the mean values of Q20 and GC percentages were 97.96% and 50.87%, respectively (Additional file 1). The total number of expressed genes (genes with at least 3 reads in one sample) included 24,494 for rumen tissue, 27,633 for total isoforms, 25,434 for transcriptional start site, 21,965 for coding sequence, 24,494 for promoters, and 25,434 for splicing sites. The overall read alignment rate to the bovine reference genome (UMD 3.1.1) was 87%±3.43%.
Differential Gene Expression between Normal and Acidosis Group
Differential expression (DE) analysis was performed using the Tuxedo pipeline to determine which genes were significantly associated with acidosis. In total, 1,077 differentially expressed genes were identified in rumen tissues of normal and acidosis samples using the FDR-corrected p-value with a significance level of a=0.05 (Additional file 2). Out of the DE genes, 627 and 450 genes were up- and down-regulated in acidosis group, respectively (Table 2).
Table 2.
Ribonucleic acid-sequencing mapping result
| Feature | Total assigned tag | Total significant | Up-regulated | Down-regulated |
|---|---|---|---|---|
| Gene | 24494 | 1077 | 627 | 450 |
| Isoform | 27633 | 978 | 584 | 394 |
| Transcription start site | 25434 | 1049 | 635 | 414 |
| Coding DNA sequence | 21965 | 998 | 607 | 391 |
| Promoter | 24494 | 2 | 1 | 1 |
Table 3 tabulates the most significantly up- and down-regulated DE genes. Functional analysis by PANTHER using only the down-regulated genes pointed to biological processes, such as “biological regulation” (CXCL20, SLCO4A1, IL1B), “positive regulation of T cell migration” (CCL20, IL1B), “regulation of metabolic process, such as gluconeogenesis, pyruvate biosynthesis” (SDS, LPO, SLC5A8, CCL20), “cellular process” (SDS, LAP, CCL20, SAA3, LPO, SLC7A11), and “localization” (SLC7A11, SDS, LAP, CCL20) (Figure 1.a). Conversely, PANTHER analysis using only up-regulated genes resulted in “filament organization” (DES, TPM1, SYNM, LMOD1), “positive regulation of epithelial and muscle fiber concentration” (TGFB1I1, LMOD1), “biomineral tissue development” (SPP1), “negative regulation of fat cell differential” (TGF1I1, LMO3), “regulation of ion transmembrane transport” (KCNF1, KCNMB1, CHRNA3, AKAP6, SLCO4A1), “regulation in cell adhesion” (MACAM), and “negative regulation of inflammatory response” (PTGIS) (Figure 1.A, B). Among the 1077 DE genes, only 984 gene IDs could be assigned DAVID IDs (52 genes unknown). Enrichment analysis was performed using SP-PIR-Keywords, GO terms, and KEGG-Pathway terms resulting in 20 KEGG pathways, 15 biological function terms, and 8 GO terms that were significantly enriched for DE genes (Table 4). The results obtained from KEGG-Pathway showed that 28.1% of the DE genes are involved in the calcium signaling pathway, mitogen-activated protein kinase (MAPK) signaling pathway, and focal adhesion. A total of 24 genes were involved in the calcium signaling pathway, 3 and 21 of which were down- (i.e., CHP1, GNA15, P2RX5) and up-regulated, respectively.
Table 3.
List of most significantly up- and down-regulated of DE genes
| Symbol | Description | Log2 FC | P-value | Gene-id |
|---|---|---|---|---|
| Up-regulated | ||||
| DES | desmin | 2.42768 | 5.00E-05 | XLOC_010237 |
| TPM1 | tropomyosin 1 | 2.31574 | 5.00E-05 | XLOC_001837 |
| SYNM | synemin | 2.29625 | 5.00E-05 | XLOC_011765 |
| PRUNE2 | prune homolog 2 | 2.2446 | 5.00E-05 | XLOC_022447 |
| FBXO32 | F-box protein 32 | 2.25528 | 5.00E-05 | XLOC_004459 |
| MCAM | melanoma cell adhesion molecule | 2.31794 | 5.00E-05 | XLOC_005572 |
| LMOD1 | leiomodin 1 | 2.53754 | 5.00E-05 | XLOC_006508 |
| SLCO4A1 | solute carrier organic anion transporter family member 4A1 | -2.0286 | 5.00E-05 | XLOC_004166 |
| MSRB3 | methionine sulfoxide reductase B3 | 2.3615 | 5.00E-05 | XLOC_019383 |
| ZMYM4 | zinc finger MYM-type containing 4 | 1.01485 | 5.00E-05 | XLOC_017616 |
| PTGIS | prostaglandin I2 synthase | 2.58484 | 5.00E-05 | XLOC_004336 |
| CALML4 | calmodulin like 4 | 2.0944 | 0.0001 | XLOC_001570 |
| LMO3 | LIM domain only 3 | 2.42625 | 5.00E-05 | XLOC_018938 |
| TGFB1I1 | transforming growth factor beta 1 induced transcript 1 | 2.05071 | 5.00E-05 | XLOC_014022 |
| KCNMB1 | potassium calcium-activated channel subfamily M regulatory beta subunit 1 | 2.39365 | 5.00E-05 | XLOC_011133 |
| BPIFA2A | BPI fold containing family A | 6.41295 | 5.00E-05 | XLOC_003810 |
| AKAP6 | A-kinase anchoring protein 6 | 1.92865 | 5.00E-05 | XLOC_011502 |
| ACTN2 | actinin alpha 2 | 2.00658 | 5.00E-05 | XLOC_015278 |
| SPP1 | secreted phosphoprotein 1 | 2.13256 | 0.00025 | XLOC_020268 |
| GAS1 | growth arrest specific 1 | 2.34304 | 5.00E-05 | XLOC_022601 |
| CHRNA3 | cholinergic receptor nicotinic alpha 3 subunit | 2.56782 | 5.00E-05 | XLOC_011859 |
| HAND2 | heart and neural crest derivatives expressed 2 | 2.87881 | 0.0002 | XLOC_022317 |
| KCNF1 | potassium voltage-gated channel modifier subfamily F member 1 | 3.26246 | 5.00E-05 | XLOC_002913 |
| DUSP26 | dual specificity phosphatase 26 | 3.23648 | 5.00E-05 | XLOC_015190 |
| Down-regulated | ||||
| TAP | tracheal antimicrobial peptide | -3.22231 | 5.00E-05 | XLOC_015120 |
| LAP | lingual antimicrobial peptide | -2.78825 | 5.00E-05 | XLOC_014982 |
| CCL20 | C-C motif chemokine ligand 20 | -5.22138 | 5.00E-05 | XLOC_010261 |
| SAA3 | serum amyloid A 3 | -3.2735 | 5.00E-05 | XLOC_016010 |
| KRT36 | keratin 36 | -4.18681 | 5.00E-05 | XLOC_009675 |
| CHI3L2 | chitinase 3 like 2 | -3.84164 | 5.00E-05 | XLOC_017282 |
| PRSS53 | serine protease 53 | -2.41518 | 5.00E-05 | XLOC_014392 |
| PI15 | peptidase inhibitor 15 | -4.13219 | 5.00E-05 | XLOC_004529 |
| SERPINB7 | serpin family B member 7 | -2.35488 | 5.00E-05 | XLOC_013597 |
| SLC5A8 | solute carrier family 5 member 8 | -4.09189 | 5.00E-05 | XLOC_019503 |
| PTX3 | pentraxin 3 | -3.49976 | 5.00E-05 | XLOC_000811 |
| IL1B | interleukin 1 beta | -2.94266 | 5.00E-05 | XLOC_002760 |
| SLC7A11 | solute carrier family 7 member 11 | -2.47759 | 5.00E-05 | XLOC_006731 |
| LPO | lacto peroxidase | -2.90729 | 5.00E-05 | XLOC_008599 |
| SDS | serine dehydratase | -2.88622 | 5.00E-05 | XLOC_006861 |
| CXCL5 | chemokine (C-X-C motif) ligand 5 | -2.1685 | 5.00E-05 | XLOC_020068 |
| SLC6A14 | solute carrier family 6 member 14 | -2.28041 | 5.00E-05 | XLOC_023913 |
| PRSS22 | serine protease 22 | -1.73141 | 0.0002 | XLOC_013847 |
| 20ALPHA-HSD | placental and ovary 20alpha hydroxysteroid dehydrogenase protein | -2.28909 | 0.00015 | XLOC_003667 |
| CXCL3 | chemokine (C-X-C motif) ligand 3 | -1.91816 | 0.00015 | XLOC_020072 |
Figure 1.
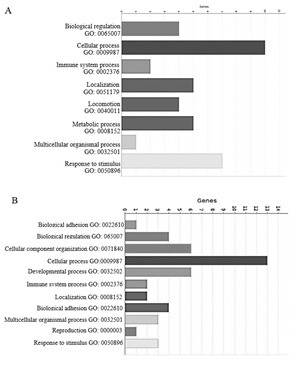
Biological process of the most significantly DE genes as identified by PANTHER A. Down-regulated genes B. Up-regulated genes
Table 4.
Enrichment analysis using KEGG, SP-PIR, and GO. Reported p-values were corrected for multiple testing using the Benjamini-Hochberg procedure. Enrichment analysis was performed using DAVID with default parameters
| DAVID functional category | Biological term | Number of genes | P-value | |
|---|---|---|---|---|
| KEGG-Pathway | Calcium signaling pathway | 24 | 4.80E-05 | 2.5 |
| Focal adhesion | 24 | 2.40E-04 | 2.3 | |
| Vascular smooth muscle contraction | 17 | 3.00E-04 | 2.8 | |
| Dilated cardiomyopathy | 12 | 4.10E-03 | 2.7 | |
| Regulation of actin cytoskeleton | 21 | 5.70E-03 | 1.9 | |
| Hypertrophic cardiomyopathy (HCM) | 11 | 7.40E-03 | 2.7 | |
| ECM-receptor interaction | 11 | 9.80E-03 | 2.6 | |
| Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 10 | 1.10E-02 | 2.7 | |
| Arachidonic acid metabolism | 9 | 1.20E-02 | 2.8 | |
| Hematopoietic cell lineage | 11 | 1.30E-02 | 2.5 | |
| Steroid biosynthesis | 5 | 1.40E-02 | 5.1 | |
| Toll-like receptor signaling pathway | 12 | 1.80E-02 | 2.2 | |
| Aminoacyl-tRNA biosynthesis | 7 | 2.50E-02 | 3 | |
| PPAR signaling pathway | 9 | 3.60E-02 | 2.3 | |
| Axon guidance | 12 | 6.10E-02 | 1.8 | |
| MAPK signaling pathway | 22 | 6.10E-02 | 1.5 | |
| Adipocytokine signaling pathway | 8 | 6.70E-02 | 2.2 | |
| Arginine and proline metabolism | 7 | 7.50E-02 | 2.3 | |
| Cardiac muscle contraction | 8 | 9.20E-02 | 2 | |
| Apoptosis | 9 | 9.60E-02 | 1.9 | |
| Gastric acid secretion | 6 | 4.50E-02 | 3.1 | |
| SP-PIR-Keyword | calcium | 36 | 5.40E-04 | 1.8 |
| glycoprotein | 92 | 8.70E-04 | 1.4 | |
| phosphoprotein | 160 | 1.00E-03 | 1.2 | |
| Ionic channel | 17 | 2.20E-03 | 2.3 | |
| Signal | 82 | 4.00E-03 | 1.3 | |
| Transmembrane | 129 | 4.50E-03 | 1.2 | |
| Ion transport | 25 | 6.50E-03 | 1.8 | |
| membrane | 149 | 8.30E-03 | 1.2 | |
| disulfide bond | 68 | 1.10E-02 | 1.3 | |
| cell membrane | 44 | 1.10E-02 | 1.5 | |
| cell junction | 17 | 1.40E-02 | 1.9 | |
| magnesium | 15 | 7.10E-02 | 1.6 | |
| receptor | 40 | 7.90E-02 | 1.3 | |
| cytoskeleton | 19 | 8.30E-02 | 1.5 | |
| oxidoreductase | 31 | 9.90E-02 | 1.3 | |
| GO terms | ATP binding | 74 | 6.60E-03 | 1.3 |
| nucleotide binding | 108 | 1.20E-02 | 1.2 | |
| glucose transmembrane transporter activity | 30 | 5.10E-02 | 3.2 | |
| growth factor binding | 70 | 5.60E-02 | 2.5 | |
| carbohydrate binding | 140 | 5.70E-02 | 1.7 | |
| protein kinase inhibitor activity | 40 | 6.10E-02 | 4.3 | |
| cAMP metabolic process | 30 | 8.10E-02 | 6.2 | |
| inflammatory response | 13 | 9.70E-03 | 2.3 |
Of the 22 genes involved in the MAPK signaling pathway, 7 genes were down-regulated (i.e., CD14, CHP1, PLA2G2D1, DUSP14, NFKB1, RELB), and 15 genes were up-regulated in diseased rumen tissues (Figure 2). Penner (2014) found that out of the 5,200 differentially expressed genes from normal and acidosis groups, 96 genes were multi-functional. Gene ontogeny analysis elucidated that the MAPK signaling pathway, regulation of the actin cytoskeleton, and focal adhesion were dominantly affected pathways in ruminal acidosis induced by high fermentable diets Penner, 2014) . These pathways are all involved in epithelial barrier function. The MAPK family members are one of the most crucial signal transduction mediators that respond to acidosis ( Ki et al., 2013 ). Zhao et al. (2018) demonstrated the ruminal mRNA, protein levels of NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells), and MAPK signaling pathway were markedly higher in acidosis cows than in control cows. Moreover, the overactivation of these pathways could promote the transcription expression of genes that coded inflammatory cytokines. The results of SP-PIR-Keywords showed that 47% of these genes associated significantly with enriched terms (FDR<0.05), such as phosphoprotein, membrane, transmembrane, glycoprotein, or disulfide bund. Li et al. (2012) indicated that an increase in carbohydrate binding typically occurred concurrently with acidosis leading to increased concentrations of short-chain fatty acid and ruminal lipopolysaccharide, as well as a reduction in pH, and damage to the rumen epithelial tissue and inflammatory responses.
Figure 2.

KEGG pathway for MAPK signaling pathway. The output of DAVID analysis showing the MAPK signaling pathway significantly enriched for DE genes. Genes within the significant differential expression in acidosis rumen tissue are shown in red.
Comparison of Transcriptome Profiles
A CummeRbund package was used in R to display differentially expressed genes between control and acidosis groups. The boxplots show the distribution of gene expression levels (log10 FPKM) for each sample (Figure 3A) confirming that our RNA-sequencing data in the control (C_0, C_1, C_2) and acidosis groups (A_0, A_1, A_2) were reproducible and of high quality. The density plot shows the number of genes of each sample at mean FPKM value indicating the different distribution of FPKM scores among each sample of the two groups (Figure 3B). The hierarchical clustering of samples (Figure 3C) showed that all control samples clustered together and were clearly separated from acidosis samples. The MA-plot was used for quality control (Figure 3D). Finally, Figure 3E shows a volcano plot of our data depicting -log10 p-values over log2 fold changes. The genes that significantly and differentially expressed were shown in red dots.
Figure 3.
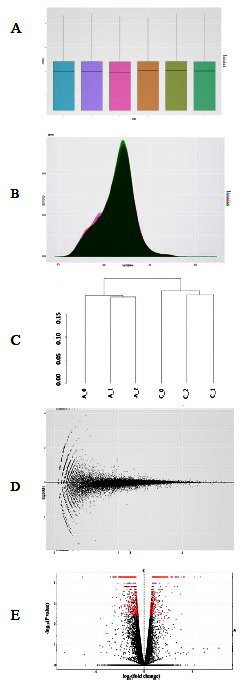
A. Boxplot showing range and distribution of the gene expression level (log10 FPKM) in each sample, B. Density plot displaying distributions of FPKM scores across samples, C. Dendrogram of hierarchical clustering analysis of gene expression levels (FPKM) for each sample, D. M-A-plot, E. Volcano plot representing the relationship between fold-change and significance. Red points identifying differentially expressed genes between control and acidosis samples.
Histopathological Changes of Rumen Tissue between two Groups
The data of tissue histology were analyzed using GLM procedures of SAS and the following Equation: Yij= µ + Ti + eij where Yij is the measured (observed) value, µ signifies the overall mean, Ti identifies the disease effect, and eij indicates the residual error assumed to be normally distributed with mean zero. The result of the histology of ventral sac rumen tissue in control and acidosis samples is presented in table 5 and figure 4. Ruminal papillae count, length, and width in the control group under the usual diet were significantly more than papillae of acidosis animal samples (P<0.05) (Additional file 2). The ruminal epithelium is responsible for physiological function, such as nutrient absorption and transport, as well as the metabolism of VFA, and protection. The destruction of epithelial rumen tissue and papillae is one of the most important responses to acidosis disease. The result of the histology analysis markedly shows that animals receiving a high grain diet significantly decrease the number and size of the ruminal papilla (Figure 5). The size of ruminal papillae has effects on the absorption of VFA and regulation of ruminal pH. Since VFAs are weak acids with a pKa ~4.8, at the physiological pH of rumen fluid between 90% and 99%, the acids that are in the dissociated forms are absorbed through the rumen wall as base conjugate (VFA-) and H+. The capacity for absorption is depended on the surface area of the papillae.
Table 5.
Histology effects of over grain diet on ruminal ventral sac tissue in cattle
| Control | Acidosis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Response | Sample1 | Sample2 | Sample3 | Average | STD | Sample4 | Sample5 | Sample6 | Average | STD |
| Papillae count, No./mm | 16.14 | 15.87 | 13.6 | 15.2 | 1.14 | 12 | 11.91 | 11.83 | 11.91 | 0.08 |
| Papillae height, µm | 5547.49 | 4308.11 | 4569.55 | 4808.38 | 533.41 | 3555.94 | 2707.5 | 2447.24 | 3555.94 | 277.01 |
| Papillae width, µm | 980.5 | 966.2713 | 929.386 | 958.72 | 21.54 | 905.18 | 809.39 | 713.59 | 809.39 | 95.79 |
Figure 4.

Comparison of the control and acidosis groups regarding the ruminal papillae count, length, and width (P<0.05).
Figure 5.

A light micrograph of papillae to compare the histology results between the control group (C_0, C_1, C_2) and acidosis group (A_0, A_1, A_2) (scale bar = 200 µm).
Discussion
Regarding the cow breeding, it is of utmost importance to understand transcriptional profiles in rumen tissue and their shift during acidosis. Ruminal acidosis is a digestive disorder that is known to be associated with highly fermentable diets.
The effects of acidosis go beyond decreased ruminal pH and include rumen epithelial damage, laminitis, inflammation, depressing animal performance, production efficiency, and liver abscesses ( Kleen et al., 2003 ; Blanch et al., 2010 ; Gressley, 2014 ; Hernandez et al., 2014 ). Ruminal tissue has many functions, such as nutrient absorption, metabolism, pH regulation, immune system regulation, and barrier functions. Recent management practices strive to minimize ruminal acidosis occurrence; however, a better understanding of the disease’s etiology, prevention, and treatment is still required ( Enemark, 2008 ). Genome sequencing has the potential to provide information on system-wide transcriptional and regulatory alterations; however, there are challenges to identify the underlying mechanisms from such data. This is the first study to describe how gene expression changes during ruminal acidosis using RNA sequencing. For this propose, the transcriptome was assembled, and gene expression patterns were compared in rumen epithelial tissue of normal and acidosis animals. The result of this analysis shows that 4.38% of the total genes in rumen epithelial tissue are significantly and differently expressed between control and acidosis cows. Li et al. (2019) conducted a study on a total of 672 genes of rumen epithelial transcriptome of eleven Holstein bull calves. They showed significant differential expression in the rumen acidosis group, compared to the control group. According to hierarchical clustering results, expression profiles were clustered together in acidosis and control samples indicating global alterations in these groups. This signifies the similarities in the gene expression profiles of animals in each group that may be associated with the type of diet or the occurrence of disease. Volcano and density plots of gene expression profiles reflected morphological and functional differences of the rumen epithelial tissue from control and acidosis groups. Notably, 58.37% of the significant genes were upregulated, and the top genes (i.e., DES, TPM1, KCNMB1, KCNF1, S100A7, SLCO4A1, SLC6A4, TGFB1I1. DES [Desmin], and TPM1 [tropomyosin]) were essential for proper muscular structure and function; moreover, they were involved in filament organization. Desmin plays a crucial role in maintaining the structure of sarcomeres and providing strength for muscle fiber during activity. Up-regulated expression of these genes may indicate involvement in rumen epithelial tissue morphological changes during acidosis and increase the activity to maintain the structure of rumen papilla. Calcium-activated potassium channel subunit beta-1 is a protein in bovine that is encoded by the KCNMB1 gene and modulates the calcium sensitivity and gating kinetics of KCNMA1, thereby contributing to KCNMA1 channel diversity. In addition, it leads to increases in the apparent Ca2+/voltage sensitivity of the KCNMA1 channel. KCNF1 (potassium voltage-gated channel modifier subfamily F member 1), S100A7 (S100 calcium-binding protein A7), and SLCO4A1 (solute carrier organic anion transporter family member 4A1) are involved in regulating ion transmembrane transport. Acidosis stimulates physicochemical mineral dissolution and subsequently cell-mediated bone resorption. In response to acidosis, a decrease is observed in mineral elements, such as sodium, potassium, carbonate, and phosphate, which induces calcium efflux from bones causing laminitis. TGFB1I1 (transforming growth factor beta-1-induced transcript 1 protein) functions as a molecular adapter at the focal adhesion complex and regulates the activity of SLC6A4. When cattle are fed higher levels of butyrate and energy, the plasma concentration of these genes increases correlated with rumen papillae growth. The rumen epithelium tissue is the greatest consumer of the total viscera energy, and the results of this study show that a higher expression of genes (i.e., CDS2, DGKB, INPPL1, PLCB4, and PRKCB) are involved in butyrate and short-chain fatty acids absorption that are metabolized as an energy source. According to a study by Baldwin et al. (2018) , the effects of butyrate infusion were evaluated on rumen epithelial transcriptome of dairy cattle in the dry periods. In addition to the maximal effect of butyrate on day 7, they revealed that 117 genes were responsive consistently from day 1 to day 14, and 42 genes were lasting through day 7. Moreover, temporal effects induced by butyrate infusion indicated that the transcriptomic alterations were very dynamic. The CDS2 (phosphatidate cytidylyltransferase 2) provides CDP-diacylglycerol and plays an important role in the metabolism and biosynthesis of lipids and phospholipids. It was also found that an up-regulation in genes (i.e., ARHGEF6, AB12, ACTN2, CHRM3, and CLF2 MYL9) was involved in the regulation of the actin cytoskeleton pathway that was vital to many processes, such as absorption of nutrients, cell migration, tissue remodeling, and maintenance of tissue integrity. Dionissopoulos et al. (2014) investigated how the capacity for fat metabolism changed in rumen epithelia immediately before and after the onset of lactation in dairy cows. The microarray analysis results of total RNA from rumen epithelial biopsies revealed 1476 differentially expressed genes at a false discovery rate of 10%. These results were filtered for genes that were directly related to the immune system and fat metabolism/homeostasis. The GO enrichment analysis in biological processes identified that significant GO terms were changed in acidosis epithelial rumen tissue that related to the various metabolism, such as ATP binding, glucose transmembrane transporter activity, carbohydrate binding, protein kinase inhibitor activity, growth factor binding, cAMP metabolic process, and inflammatory response. One of the most important biological processes that involved DE genes is related to immune response. The obtained results from this study suggest that immune function is differently regulated between normal and acidosis animals. Previous studies indicated that lowered immune function might negatively affect rumen tissue function.
It was found in this study that the most abundant GO pathways enriched by down-regulated significant genes in acidosis samples were related to MAPK signaling and Toll-like receptor (TLR) signaling. Furthermore, the results showed that the expression of genes, including interleukin 1 beta (IL1B), C-X-C chemokine ligand 5(CXCL5), interleukin 36 alpha (IL36A), interleukin 36 beta (IL36B) are significantly down-regulated in acidosis rumen tissue samples. In general, the findings suggested that rapid shifts and use of diets rich in fermentable carbohydrates (high grain) increased the concentration of ruminal VFA and toxins causing cell damage and significant changes in transcriptome profiles of rumen epithelial tissue.
This study aimed to investigate changes in gene expression in rumen epithelium under acidosis. To this purpose, it was imperative to evaluate the rumen epithelium histology and transcriptome data in the normal and acidosis samples. Initially, acidosis was naturally induced by changes in the diet. The analysis of RNA-sequencing data of acidosis epithelium showed significantly increased expression of genes involved in focal adhesion, gastric acid secretion, calcium, and cAMP signaling pathways. Furthermore, the results showed a significant decrease in the expression of genes involved in phosphate metabolic process, TLR, and B-cell receptor signaling pathway. It is suggested that further studies be undertaken to evaluate liver and lung tissues to provide more detailed information regarding the effects of acidosis on cows since acidosis causes liver abscessation and lung infections, which are not recognizable until death. This is the first study to assay rumen epithelium cattle under acidosis using transcriptome sequencing. The obtained results indicate that the use of a high grain diet could elicit a strong change in histology, gene expression, and transcriptome response in ruminal tissue. Moreover, the findings provide a fundamental understanding to reveal which genes are associated with acidosis leading to the identification of biomarkers to select the best cows for animal breeding.
Ethics
We hereby declare all ethical standards have been respected in preparation of the submitted article.
Grant Support
This work was supported by Agricultural Sciences and Natural Resources University of Khuzestan (student grant No. 9324203).
Authors’ Contribution
Conceptualization, Fayazi, J. and Gholizadeh, M.
Methodology, Gholizadeh, M.; Fayazi, J.; Zali, H. and Asgari, Y.
Investigation, Gholizadeh, M. and Zali, H.
Resources, Zali, H.; Gholizadeh, M., Fayazi, J.
Writing-Original Draft Preparation, Gholizadeh, M.
Writing-Review & Editing, All authors
Supervision, Fayazi, J.
Conflict of Interest:
The authors declare that they have no conflict of interest.
Acknowledgement
We thank Mohsen Sari, Mehdi Dehghan and Javad Khalifeh for their help and advice in animal raring period and sample preparation.
References
- 1.Alston JM, Pardey PG. Agriculture in the global economy. J Econ Perspec. 2014; 28: 121–146. [Google Scholar]
- 2.Ash R, Baird GD. Activation of volatile fatty acids in bovine liver and rumen epithelium. Evidence for control by autoregulation. Biochem J. 1973;136(2):311–9. doi: 10.1042/bj1360311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baldwin RL 6th, Li RW, Jia Y, Li CJ. Transcriptomic Impacts of Rumen Epithelium Induced by Butyrate Infusion in Dairy Cattle in Dry Period. Gene Regul Syst Bio. 2018;12:1177625018774798. doi: 10.1177/1177625018774798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blanch M, Calsamiglia S, Devant M, Bach A. Effects of acarbose on ruminal fermentation, blood metabolites and microbial profile involved in ruminal acidosis in lactating cows fed a high-carbohydrate ration. J Dairy Res. 2010;77(1):123–8. doi: 10.1017/S0022029909990562. [DOI] [PubMed] [Google Scholar]
- 5.Dionissopoulos L, AlZahal O, Steele MA, Matthews JC, McBride BW. Transcriptomic changes in ruminal tissue induced by the periparturient transition in dairy cows. Am J Anim Vet Sci. 2014;9(1):36–45. [Google Scholar]
- 6.Durunna ON, Mujibi FD, Goonewardene L, Okine EK, Basarab JA, Wang Z, Moore SS. Feed efficiency differences and reranking in beef steers fed grower and finisher diets. J Anim Sci. 2011;89(1):158–67. doi: 10.2527/jas.2009-2514. [DOI] [PubMed] [Google Scholar]
- 7.Enemark JM. The monitoring, prevention and treatment of sub-acute ruminal acidosis (SARA): a review. Vet J. 2008;176(1):32–43. doi: 10.1016/j.tvjl.2007.12.021. [DOI] [PubMed] [Google Scholar]
- 8.Gressley TF. Inflammatory responses to sub-acute ruminal acidosis. 25th Annual Florida Ruminant Nutrition Symposium, Florida, USA; 2014 [Google Scholar]
- 9.Hernández J, Benedito JL, Abuelo A, Castillo C. Ruminal acidosis in feedlot: from aetiology to prevention. Sci World J. 2014;2014: 702572. doi: 10.1155/2014/702572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 11.Kahn L, Cottle D. Beef cattle production and trade, Clayton, Australia: Csiro Publishing; 2014 p. 221 [Google Scholar]
- 12.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28: 27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42(1):199–205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ki YW, Park JH, Lee JE, Shin IC, Koh HC. JNK and p38 MAPK regulate oxidative stress and the inflammatory response in chlorpyrifos-induced apoptosis. Toxicol Lett. 2013;218(3):235–45. doi: 10.1016/j.toxlet.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 15.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleen JL, Hooijer GA, Rehage J, Noordhuizen JP. Subacute ruminal acidosis (SARA): a review. J Vet Med A Physiol Pathol Clin Med. 2003 Oct;50(8):406–14. doi: 10.1046/j.1439-0442.2003.00569.x. [DOI] [PubMed] [Google Scholar]
- 17.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li S, Khafipour E, Krause DO, Kroeker A, Rodriguez-Lecompte JC, Gozho GN, Plaizier JC. Effects of subacute ruminal acidosis challenges on fermentation and endotoxins in the rumen and hindgut of dairy cows. J Dairy Sci. 2012; 95(1):294–303. doi: 10.3168/jds.2011-4447. [DOI] [PubMed] [Google Scholar]
- 19.Li W, Gelsinger S, Edwards A, Riehle C, Koch D. Transcriptome analysis of rumen epithelium and meta-transcriptome analysis of rumen epimural microbial community in young calves with feed induced acidosis. Sci Rep. 2019;9(1):4744. doi: 10.1038/s41598-019-40375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2012;41(1):377–86. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Shea E, Waters SM, Keogh K, Kelly AK, Kenny DA. Examination of the molecular control of ruminal epithelial function in response to dietary restriction and subsequent compensatory growth in cattle. J Anim Sci Biotechnol. 2016;7:53. doi: 10.1186/s40104-016-0114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Penner GB. Mechanisms of volatile fatty acid absorption and metabolism and maintenance of a stable rumen environment. 25th Florida Ruminant Nutrition Symposium; 2014 pp. 92-104 [Google Scholar]
- 23.Penner GB, Steele MA, Aschenbach JR, McBride BW. Ruminant Nutrition Symposium: Molecular adaptation of ruminal epithelia to highly fermentable diets. J Anim Sci. 2011;89(4):1108–19. doi: 10.2527/jas.2010-3378. [DOI] [PubMed] [Google Scholar]
- 24.Sehested J, Diernaes L, Møller PD, Skadhauge E. Ruminal transport and metabolism of short-chain fatty acids (SCFA) in vitro: effect of SCFA chain length and pH. Comp Biochem Physiol A Mol Integr Physiol. 1999;123(4):359–68. doi: 10.1016/s1095-6433(99)00074-4. [DOI] [PubMed] [Google Scholar]
- 25.Steele MA, Vandervoort G, AlZahal O, Hook SE, Matthews JC, McBride BW. Rumen epithelial adaptation to high-grain diets involves the coordinated regulation of genes involved in cholesterol homeostasis. Physiol Genomics. 2011;43(6):308–16. doi: 10.1152/physiolgenomics.00117.2010. [DOI] [PubMed] [Google Scholar]
- 26.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao C, Liu G, Li X, Guan Y, Wang Y, Yuan X, et al. Inflammatory mechanism of Rumenitis in dairy cows with subacute ruminal acidosis. BMC Vet Res. 2018;14(1):1–8. doi: 10.1186/s12917-018-1463-7. [DOI] [PMC free article] [PubMed] [Google Scholar]