

Abstract
The members of the large keratin family of cytoskeletal proteins are expressed in a carefully regulated tissue- and differentiation-specific manner. Although these proteins are thought to be involved in imparting mechanical integrity to epithelial cells, the functional significance of their complex differential expression is still unclear. Here we provide new data suggesting that the expression of particular keratins may influence cell proliferation. Specifically, we demonstrate that the ectopic expression of K10 inhibits the proliferation of human keratinocytes in culture, while K16 expression appears to promote the proliferation of these cells. Other keratins, such as K13 or K14, do not significantly alter this parameter. K10-induced inhibition is reversed by the coexpression of K16 but not that of K14. These results are coherent with the observed expression pattern of these proteins in the epidermis: basal, proliferative keratinocytes express K14; when they terminally differentiate, keratinocytes switch off K14 and start K10 expression, whereas in response to hyperproliferative stimuli, K16 replaces K10. The characteristics of this process indicate that K10 and K16 act on the retinoblastoma (Rb) pathway, as (i) K10-induced inhibition is hampered by cotransfection with viral oncoproteins which interfere with pRb but not with p53; (ii) K10-mediated cell growth arrest is rescued by the coexpression of specific cyclins, cyclin-dependent kinases (CDKs), or cyclin-CDK complexes; (iii) K10-induced inhibition does not take place in Rb-deficient cells but is restored in these cells by cotransfection with pRb or p107 but not p130; (iv) K16 efficiently rescues the cell growth arrest induced by pRb in HaCaT cells but not that induced by p107 or p130; and (v) pRb phosphorylation and cyclin D1 expression are reduced in K10-transfected cells and increased in K16-transfected cells. Finally, using K10 deletion mutants, we map this inhibitory function to the nonhelical terminal domains of K10, hypervariable regions in which keratin-specific functions are thought to reside, and demonstrate that the presence of one of these domains is sufficient to promote cell growth arrest.
Keratins are a large family of proteins which form the intermediate filament (IF) cytoskeleton of epithelial cells and their appendages, hairs and nails (reviewed in references 8 and 15). These proteins are subdivided according to biochemical criteria into two subfamilies: type I, or acidic keratins, and type II, or neutral-basic keratins. This division also has important structural and functional implications, since to build up a well-organized IF cytoskeleton, tetramers containing equimolar amounts of each keratin subtype are required. Like all IF proteins, keratins consist of a central α-helical rod domain responsible for dimerization and higher-order polymerization. The rod domain is flanked by globular head (amino) and tail (carboxyl) domains, the functions of which are still unclear. Variations in these nonhelical end domains largely account for the differences between individual keratin proteins. The presence of specific pairs of type I/type II keratins (the so-called expression pairs) in different epithelia is highly regulated in a cell type- and differentiation-specific manner. Although the primary function of keratin IF has long been thought of as structural, this hypothesis was not confirmed until the discovery that keratin mutations result in diseases characterized by epithelial fragility (reviewed in references 3, 5, 6, and 15. The variety of keratin genes differentially expressed suggests that these proteins may, however, have additional functions. From a structural point of view, we have recently reported that different keratin polypeptides, even those belonging to the same expression pair, display distinct dynamics in cell hybrids (19). To gain a deeper insight into the diversity of keratin functions, we have transfected human HaCaT keratinocytes with plasmids coding for several acidic keratins. We found that K10 expression leads to the inhibition of cell proliferation, while K16 appears to facilitate this process. Using different approaches, we also found that the molecular mechanism by which these keratins modulate cell growth seems to be linked to pRb and the molecular machinery controlling cell cycle progression during G1 and that, in the case of K10, this function resides in the nonhelical termini of the protein.
MATERIALS AND METHODS
Cells.
HaCaT and C33A cells were cultured routinely in plastic petri dishes (Nunc) in Dulbecco modified Eagle medium (Gibco) containing 10% fetal calf serum (BioWhittaker) and antibiotics at 37°C in a 5% CO2 atmosphere and 95% humidity. BMGE+H and PtK2 cells were cultured as described previously (20).
Plasmid constructs.
To construct pcDNA3K10, a 5-kb HindIII-EcoRI fragment of the whole human K10 gene from pBSKHK10 (1, 20) was inserted into the HindIII-EcoRI site of pcDNA3 (Invitrogen). pcDNA3K13 was constructed by subcloning the HindIII fragment of human K13 cDNA (11) into the HindIII site of pcDNA3. pcDNA3K16 was a generous gift from Liz Rugg (University of Dundee, Dundee, United Kingdom). pcDNA3K14 was made by subcloning the HindIII-EcoRI fragment containing the human K14 cDNA from pJK14.P (generously provided by E. Fuchs, University of Chicago) into the HindIII-EcoRI site of pcDNA3. pMTHK10 is a generous gift from M. Blessing (University of Mainz, Mainz, Germany). pHK10 Hyg was created by subcloning the Asp718-NotI fragment of pBSKHK10 containing the entire human K10 gene (1) into the Asp718-BamHI sites of pGLVP under the control of the cytomegalovirus (CMV) promoter. The pGLVP plasmid also confers hygromycin resistance. All K10 deletion constructs were generated from pBSKHK10. Δ3′UTR was obtained by subcloning a HindIII-BamHI fragment into the HindIII-BamHI sites of pcDNA3. ΔC was created by subcloning the HindIII-AflIII fragment into the HindIII-SmaI sites of pcDNA3. ΔCΔcoil was created by subcloning the 1.6-kb HindIII-EcoRI fragment of pBSKHK10 into the HindIII-EcoRI sites of pcDNA3. The in-frame deletion of the K10 amino terminus was constructed as follows: the 1.3-kb K10 SacI-EcoRI fragment was subcloned into the SphI-EcoRI site of pCMVHK10 (20), replacing the 1.6-kb fragment of K10; from this plasmid a 1.4-kb BamHI-EcoRI fragment was obtained and subcloned into the BamHI-EcoRI sites of pcDNA3, generating the plasmid p(1.4)HK10ΔN. ΔN was then generated by subcloning the 3.5-kb EcoRI fragment from pBSKHK10 into the EcoRI site of p(1.4)HK10ΔN, and ΔNΔC was obtained by subcloning the EcoRI-NotI fragment of ΔC into the EcoRI-NotI site of p(1.5)HK10ΔN. The functionality of these constructs was assayed by transient transfection experiments with PtK2 cells as recipients and analyzed by Western blotting and indirect immunofluorescence (see Fig. 3 and reference 20). The plasmids coding for pRb, p107, and p130 in pcDNA3 have been described previously (22). pCMVcdk2 and pCMVcdk4 (pRcCMV backbone) were a generous gift from M. E. Ewen. Plasmids coding for different cyclins in the pRcCMV backbone have been described previously (10).
FIG. 3.

K10 represses cell cycle progression in a dose-dependent manner. PtK2 simple epithelial cells were transfected with the different keratin-coding plasmids. (A to D) Immunofluorescence analysis showing that in all cases the transfected proteins were similarly expressed and integrated into the endogenous cytoskeleton, as determined by staining with k8.60 against K10 (A), AE8 against K13 (B), RCK107 against K14 (C), or LL025 against K16 (D). (E) Cell cycle phase distribution of transfected cells and nontransfected cells (control, immunofluorescence-negative cells sorted from the same experiments) analyzed in parallel by FACS after being stained with the above-mentioned antibodies and propidium iodide. (F) PtK2 cells were cotransfected with equimolar amounts of pMTHK10, in which the K10 gene is under the control of the methallothionein promoter, and CMVβ-Gal. Twenty-four hours after transfection, cells were split and cultured in parallel, and 24 h later ZnCl2 was added at the indicated concentrations for 18 h. Protein extracts were obtained and analyzed in Western blots with antibodies against K10, β-Gal, and the endogenous keratins K8 and K18. (G) In the above transfections, cells cultured on glass coverslips were incubated in the presence of 10 μM BrdU for 8 h after induction. At this time, the base analog incorporation in the transfected (β-Gal-positive) and nontransfected cells was analyzed by double immunofluorescence with antibodies against BrdU and β-Gal, and the relative inhibition was determined for each ZnCl2 concentration. Note that there is significant inhibition of BrdU incorporation at ZnCl2 concentrations of 25 μM or higher. Data in panels E and G are from triplicate experiments and are shown as means ± standard deviations.
Transfections.
Permanent transfections with 10 to 20 μg of total plasmid DNA per 10-cm-diameter petri dish were performed by the calcium phosphate method. In cotransfection experiments, unless otherwise stated, equimolar amounts of each plasmid were used. We used 0.5 mg of G418 per ml for single selection or 0.5 mg of G418 per ml plus 0.1 mg of hygromycin per ml for double selection for 15 to 20 days, and colonies were then fixed, stained, and scored. Controls were done with empty pcDNA3 and/or pGLVP vectors. Transiently transfected cells were fixed and processed for immunofluorescence according to conventional methods (1, 4, 19, 20). Fluorescence-activated cell sorter (FACS) analysis was performed with ethanol-fixed cells incubated with antibodies against the diverse keratins (19) and with fluorescein isothiocyanate-labelled anti-mouse secondary antibodies. DNA content was estimated with propidium iodide, and the cell cycle profile was analyzed by using multicycle software.
Immunoblotting.
Total protein extracts from cells transiently transfected were obtained by lysis in radioimmunoprecipitation assay buffer (150 mM NaCl, 1.0% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1 mM NaVO4, 1 mM NaF, 50 mM Tris HCl, pH 8.0) containing protease inhibitors (2 μg of aprotinin per ml, 2 μg of leupeptin per ml, and 100 μg of phenylmethylsulfonyl fluoride per ml) for 30 min at 4°C. Lysates were precleared by centrifugation, and supernatants were stored at −70°C. Protein content was determined colorimetrically (Bio-Rad protein assay). A 250-μg sample of total protein was electrophoresed in sodium dodecyl sulfate-polyacrylamide gels and transferred to nitrocellulose (Amersham) on semidry equipment. Membranes were blocked by incubation in Tris-buffered saline containing 0.1% Tween 20 and 5% nonfat dry milk (TBST-milk) and probed with the gal40 monoclonal antibody (MAb) against β-galactosidase (β-Gal) (1/1000; Sigma), AE1 MAb against most acidic keratins (1/10,000; ICN), IF8 anti-pRb MAb (neat supernatant; a generous gift from D. P. Lane, University of Dundee), rabbit polyclonal antibody against p16ink4a (1/1,000; a generous gift from D. Beach, Cold Spring Harbor Laboratory), and DCS-6 MAb (Novocastra Labs; 1/20) against cyclin D1. Secondary horseradish peroxidase-labelled antibodies (Jackson Immunoresearch) were used at a 1/1,000 dilution in TBST-milk. Blots were developed by using the ECL kit (Amersham) and following the manufacturer’s recommendations.
RESULTS AND DISCUSSION
Keratin K10 inhibits and K16 facilitates cell proliferation.
To study possible specific functions of keratins, we transfected human HaCaT keratinocytes with plasmids coding for K10 (specific to differentiating cells of orthokeratinized epithelia), K13 (specific for differentiating noncornified stratified epithelia), K14 (characteristic of basal cells of all stratified epithelia), and K16 (induced in epidermal cells by hyperproliferative stimuli). HaCaT cells were initially chosen as recipients for these keratins because they are not transformed, retain many epidermal differentiation characteristics, and have been extensively used in vitro as a human keratinocyte model (2, 4, 9, 21, 22). On the other hand, epidermal keratinocytes are particularly interesting for these purposes, as they present at least three differentiation stages associated with the expression of specific keratin pairs: mitotic basal cells synthesize K5-K14, terminally differentiating keratinocytes express the K1-K10 pair, and activated hyperproliferative keratinocytes induce keratins K6 and K16. All the plasmids used have the same backbone (pcDNA3), with the expression of the keratins being driven by the CMV promoter-enhancer, and also provide the neomycin resistance gene under simian virus 40 (SV40) control. We observed that the transfection of HaCaT cells with the K10-coding plasmid leads to a significant decrease in the number of G418-resistant colonies compared to that produced by the empty vector, whereas this number is increased upon transfection with the K16-coding plasmid (Fig. 1A). Transfection with the K13-coding plasmid gives rise to a mild reduction in the number of colonies, whereas the K14-coding plasmid does not alter this number compared to that of the control empty vector (Fig. 1A). Similar results were obtained with PtK2 (simple epithelial) and BMGE+H (mammary gland) cells (data not shown). The summary of different colony formation experiments is shown in Fig. 1B. These results indicate that K10 and to a lesser extent K13 appear to repress cell growth. On the contrary, K16 seems to facilitate cell proliferation, whereas K14 did not significantly alter it. The conclusion that K10 and K16 play opposite roles in cell proliferation is also reinforced by the different sizes of the colonies obtained after transfection with these keratins (Fig. 1C). Colonies that grew out after K10 transfection were smaller than those obtained in control vector transfections, while those obtained after K16 transfection were bigger. This conclusion is also supported by the observed selective inactivation of these transgenes. With Northern blot analysis, we observed that only a minor portion of the clones obtained in K10 transfections expressed the transgene (5 of 17); within these clones, immunofluorescence analysis demonstrated that K10 expression is restricted to only a few cells (Fig. 2A) and rapidly decreases with cell passage. In contrast, most of the K16 clones analyzed (12 of 13) expressed the transgene homogeneously (Fig. 2B).
FIG. 1.

Expression of keratin K10 inhibits cell proliferation. (A) Examples of HaCaT cells transfected with empty pcDNA3 (NEO) or the corresponding keratin-containing plasmids. (B) Summary of 5 to 10 independent experiments demonstrating that K10 inhibits cell proliferation and K16 produces an increase in the number of clones. Data are shown as means ± standard deviations. (C) Distribution of the clone sizes from transfections with pcDNA3 (Neo), K10, and K16 demonstrates that K16 clones are larger than vector clones and that these are larger than K10 clones.
FIG. 2.

Examples of double immunofluorescence analysis of clones isolated after K10 (A, A′, and A") and K16 (B, B′, and B") transfection. Note that K10 (A) expression is restricted to a few cells, whereas K16 (B) is expressed in most cells. Cells were stained with K8.60 antibody (A) or LL025 antibody (B). A′ and B′ are the same fields as A and B, respectively, stained with the Troma 1 anti-K8 antibody. A" and B" are the double exposures from A and A′ and B and B′, respectively, to better visualize cells positive (yellow-orange) and negative (green) for the transfected keratin.
To investigate whether the expression of these keratins operates to influence cell cycle progression, we used these constructs in transient transfections in PtK2 cells and analyzed the cell cycle profile of the transfected and nontransfected cells by FACS analysis. We chose these simple epithelial cells because they do not express keratin K10, K13, K14, or K16 and they are more efficiently transfected than HaCaT keratinocytes (typically 10 to 20% of the cells in the culture are transfected), characteristics that facilitate the experiment. As indicated in Fig. 3, in these cells the transfection efficiency and the expression of the transfected keratins were similar, and these proteins integrated into the endogenous cytoskeleton (Fig. 3A to D and results not shown). FACS analysis (Fig. 3E) of samples obtained in parallel experiments indicated that K10 expression leads to an increase in the G1 population, with a decrease in the percentage of cells in S phase. Conversely, K16 expression reduces the fraction of cells in G1 while increasing that in S phase.
To determine the levels of K10 which are required to promote the cell cycle arrest, PtK2 cells were cotransfected with a plasmid coding for β-Gal under the control of the CMV promoter and pMTHK10 plasmid, in which K10 is under the control of the metallothionein promoter. Protein extracts were biochemically analyzed for the expression of the transfected constructs. Western blots evidenced that K10 expression increases as a function of the ZnCl2 concentration, while levels of β-Gal and the endogenous keratins K8 and K18 remain constant (Fig. 3F). In parallel, cells grown on glass coverslips in the same transfection experiments were analyzed by double immunofluorescence to study the ability of the transfected (β-Gal-positive) cells to incorporate bromodeoxyuridine (BrdU). We found that, at ZnCl2 concentrations of 25 μM or higher, there is clear inhibition of BrdU incorporation in these cells (Fig. 3G). We have previously reported that K10 under the control of the CMV promoter, which provides a higher level of expression than the MT promoter used here, is not overexpressed with respect to the endogenous keratins in these cells (20). Therefore, taking into account the transfection efficiency and the experimental conditions used, these results (Fig. 3F) suggest that K10 does not seem to be overexpressed with respect to the endogenous keratins. In this context, it is important to remark that K10 and its partner K1 are among the most prevalent proteins in suprabasal cells of the normal epidermis, its natural in vivo expression site, with the other epidermal keratins, K5 and K14, present at much lower, or even undetectable, levels in these cells.
To further confirm that K16 expression facilitates cell proliferation, HaCaT cells were transfected with the empty vector or the K16-coding plasmid and colony formation experiments were performed under limiting growth conditions by decreasing the serum concentration during selection. Although serum reduction led to a decrease in the number of colonies arising from both plasmids (Fig. 4A), K16 consistently gave a much larger number of clones than the corresponding empty vector control transfections. Even in the complete absence of serum, the number of colonies obtained after K16 transfection was similar to that obtained in the control transfection with a normal serum concentration during selection (Fig. 4A).
FIG. 4.

K16 facilitates keratinocyte proliferation and specifically reverts the K10-induced cell growth arrest. (A) After transfection with pcDNA3 or K16, cells were cultured under selection with the indicated amounts of serum. Note the higher proliferative potential of K16-transfected cells in all the situations compared to the cells transfected with empty vector. (B) HaCaT cells were transfected with a fixed amount of K10 (Hyg) and increasing amounts of either K14 or K16 (in pcDNA3 [Neo], conferring neomycin resistance). After double selection colonies were fixed, stained, and scored. Note that K16, in contrast to K14, efficiently reverses the K10-induced inhibition. Data are from three independent experiments and are shown as means ± standard deviations.
Collectively, these results demonstrate that K10 and K16 have opposite functions in the modulation of cell proliferation and correlate well with the K10 and K16 expression patterns observed in vivo. When mitotically active basal epidermal keratinocytes withdrawn from the cell cycle are committed to terminal differentiation, they switch off K14 expression and induce the expression of K10 (7). In skin undergoing hyperproliferation, as during wound healing and certain disorders, including cancer, however, K10 expression is drastically reduced or even absent, and K16, which is normally absent from the interfollicular epidermis, is rapidly induced and expressed throughout the suprabasal compartment (29).
It has also been reported that K16 overexpression in transgenic mouse epidermis leads to aberrant keratinization and hyperproliferation, with the severity of the phenotype related to the K16/K10 ratio (27). These latter results led us to investigate the possibility that K10-induced arrest could be reversed by the coexpression of K16. HaCaT cells were therefore cotransfected with a fixed amount of K10 and increasing amounts of either K14 or K16. We found (Fig. 4B) that K16 coexpression efficiently reversed the inhibitory activities of K10 in a dose-dependent manner, suggesting that the changes in keratin expression observed in epidermis in vivo are relevant to the hyperproliferative response. On the other hand, K14 expression was unable to rescue the K10-induced inhibition significantly, even when K14 amounts used in transfection were twice those of K10.
K10-induced inhibition of cell proliferation requires a functional Rb protein.
The major mechanisms controlling cell cycle progression during G1 are mediated by p53 and/or the retinoblastoma (Rb) family of proteins (pRb, p107, and p130) (reviewed in references 23 and 28). We therefore investigated whether these mechanisms are responsible for the K10-induced cell cycle arrest. HaCaT cells were cotransfected with K10 and either a wild-type SV40 large T antigen (T Ag), which binds and inactivates both p53 and pRb, or a mutant form of this protein (k1T Ag), which binds to and inactivates p53 but not pRb or its relatives. We observed that the coexpression of the wild-type T Ag, but not that of the mutant k1T Ag form, reversed K10-induced arrest, suggesting that pRb (or its relatives), but not p53, is involved in the inhibition of cell growth induced by K10 (Fig. 5A). This is consistent with the fact that HaCaT cells bear mutations in both alleles of the p53 gene which render the protein transcriptionally inactive (12). We furthermore observed that from the clones derived from K10 and wild-type SV40 T Ag cotransfections, it was possible to derive permanent cell lines in which the vast majority of the cells express K10 (Fig. 6A′). This is at variance with the observation (see above) that the few clones of HaCaT cells obtained upon single K10 transfection were mostly negative for K10 expression (Fig. 2A, A′, and A").
FIG. 5.

Keratin-induced modulation of cell proliferation requires a functional Rb protein. (A) Coexpression of a wild-type SV40 large T Ag (pZIPTAg) but not a mutant form lacking the ability of pRb binding (pZIPk1TAg) results in recovery from the K10-induced arrest. (B) The specific coexpression of certain cyclins and/or cdk’s with K10 reverts the induced inhibition in HaCaT cells, as demonstrated by the increased number of colonies with respect to empty vector (Hyg) plus each cyclin construct. (C) K10 is unable to cause cell growth inhibition in pRb-deficient C33A cells. (D) The coexpression of pRb or p107, but not that of p130, along with K10 in C33A cells restores the ability of this keratin to block cell proliferation. (E) Coexpression of K16 overrides the growth inhibition promoted by pRb, but not that promoted by p107 or p130, in HaCaT cells. Data in panels A to E are from at least three independent experiments and are shown as means ± standard deviations. (F) Immunoblotting of protein extracts from C33A cells transiently cotransfected with the indicated plasmids, demonstrating the expression of transfected β-Gal, pRb, K10, K16, and the endogenous K19 keratins (with AE1 antibody), cyclin D1, and p16ink4a. Note that K10 reduces, while K16 increases, the phosphorylation of the cotransfected pRb as well as the endogenous cyclin D1 expression level.
The involvement of the pRb pathway was further confirmed by two different and independent approaches. First, since pRb is functionally inactivated by hyperphosphorylation through the activity of different complexes of cyclins and cyclin-dependent kinases (cdk’s) during normal cell cycle progression (9, 10, 28), we studied whether the coexpression of K10 with combinations of these molecules could reverse the inhibitory functions of K10. We found that compared to control transfections containing the corresponding cyclins and/or cdk’s plus empty hygromycin vector, the coexpression of cyclin E, cyclin D1, or cdk4 completely rescued K10-induced arrest, while cyclin A or cdk2 only reversed it partially and cyclin B had no significant effect (Fig. 5B). Furthermore, the expression of cyclin A, E, or D1, along with their natural respective catalytic partners, cdk2 and cdk4, totally abolished the inhibitory effects of K10.
As a second approach to study the involvement of pRb or its relatives p107 and p130, we analyzed the ability of K10 to induce cell growth arrest in Rb-deficient C33A human cervical carcinoma cells. In these cells K10, similarly to K13 and K14, did not induce a reduction in the number of G418-resistant colonies compared to empty vector, although an increase in this number was observed in K16 transfections (Fig. 5C). Similar results were obtained with human osteosarcoma Rb-deficient Saos2 cells (data not shown). Finally and most significantly, when C33A cells were transfected with K10 together with pRb, p107, or p130, we observed that pRb and p107, but not p130, restored the ability of K10 to induce cell cycle arrest (Fig. 5D). Given the above results indicating that K16 is able to rescue the K10-induced arrest (Fig. 4B) and the dependence of this process on pRb (Fig. 5A to D), we studied whether the expression of K16 may override the inhibition of the proliferation mediated by pocket proteins. HaCaT cells were cotransfected with these proteins and K16 as described above for K10, and the numbers of resulting colonies were scored. The results obtained (Fig. 5E) demonstrate that in agreement with our previous results (22), the three Rb proteins efficiently inhibit HaCaT cell growth, with p107 being the most effective. We also found that coexpression of K16 is able to overcome the growth inhibition promoted by pRb, but not that induced by p107 or p130.
Collectively, these results indicate that keratins K10 and K16 may operate by impairing or activating, respectively, the functional inactivation of pRb. In this regard, it has been shown that p107 can inhibit cell proliferation through two separate mechanisms, one similar to that displayed by pRb, requiring the pocket domain, and another that depends on its ability to bind cyclin A-cyclin E-cdk2 complexes through its C-terminal domain. It has been proposed that the first mechanism is not functional in C33A cells and that the latter mechanism is responsible for the p107- but not pRb-dependent growth arrest observed in these cells (30, 31) (Fig. 5D). In agreement with this, the increased capacity of p107 to arrest C33A cells when it is coexpressed with K10 (Fig. 5D) can be attributed to the reactivation by keratin of the pocket-dependent growth inhibitory mechanism shared by p107 and pRb. Conversely, the ability of K16 to override the pRb- but not the p107-induced growth arrest in HaCaT cells (Fig. 5E) indicates that this keratin may also act on the molecular mechanism controlling the cell cycle through the functionality of this pocket domain.
These results also show a striking parallelism to those reported from antibody-mediated knockout experiments demonstrating that cyclin D1 is dispensable for G1 progression in Rb-deficient cells and that the reintroduction of wild-type pRb restores the cyclin D1 requirement (14). This parallelism also suggested that K10-K16 expression could alter the cyclin D1. To test this hypothesis, C33A cells were cotransfected with CMVpRb and either K16 or K10 at a 1:1 molar ratio, and protein extracts were probed by Western blotting (Fig. 5F). To normalize the data, a CMVβ-Gal plasmid was included in the transfections. Immunoblotting with anti-β-Gal indicated that the transfection efficiencies were similar in all the experiments. Blots with the AE1 antibody (which recognizes an epitope present in most acidic keratins) showed that K10 and K16 were expressed at similar levels after transfection. These experiments revealed changes in the degree of phosphorylation of the transfected pRb as a function of the cotransfected keratin. pRb appeared to be less phosphorylated in the presence of K10 and more phosphorylated in the presence of K16 than in control transfections. Interestingly, the cyclin D1 levels, which are undetectable in the parental cells and increase as a consequence of reintroducing a functional Rb (see also reference 14), were also lower in K10-cotransfected cells and higher in K16-cotransfected cells than in control pRb-transfected cells. These results suggest that the keratin-induced cell cycle modulation may be mediated by altering the levels of cyclin D1 expression and, as a consequence, preventing or activating pRb hyperphosphorylation.
Finally, the observation that cdk4 can reverse the K10-induced arrest suggests the possibility that an ink4 inhibitor may be involved in this process. To study this the expression of p16ink4a was analyzed in these transfections (Fig. 5F, lower panel). We observed that p16ink4a expression is high in C33A cells transfected with β-Gal alone and that its expression decreased to very low levels with the reintroduction of pRb either alone or cotransfected with K10. These results are consistent with the reported repression of p16ink4a expression by underphosphorylated pRb (13) and indicate that the K10-induced arrest is not due to the induction of this cdk inhibitor.
The K10-induced cell cycle arrest requires nonhelical terminal domains.
IF proteins, in general, and keratins, in particular, share a common structure, with a conserved α-helical rod domain flanked by nonconserved amino (head) and carboxyl (tails) ends. To determine whether any of these domains mediates the growth inhibitory effect of K10, we generated a series of deletion constructs (Fig. 7A) affecting the divergent non-α-helical domains (ΔN, ΔC, and ΔNΔC) and also a deletion construct in which most of the rod domain was removed (ΔCΔcoil). Inspired by the recent finding that the 3′ untranslated regions (3′UTR) from certain cytoskeletal muscle differentiation genes can inhibit cell proliferation in vitro and in vivo (24, 25), we also generated a K10 deletion construct in which the last amino acid and the complete 3′UTR were removed (Δ3′UTR). All of these constructs were also made in the pcDNA3 vector, and its expression was tested by immunofluorescence of transiently transfected PtK2 cells with a K10-specific LH3 MAb. We observed that with the exception of the one affecting the rod domain (ΔCΔcoil), these truncated proteins were able to integrate into the endogenous cytoskeleton (Fig. 7B). The ΔCΔcoil mutant protein, in agreement with data from previous studies (5, 6, 8), provokes the collapse of the endogenous keratin cytoskeleton similarly to mutated keratins found in epithelial fragility diseases (3, 5, 6, 15).
FIG. 7.


Removal of both amino and carboxyl termini abolishes the inhibitory function of K10. (A) Map of the K10 gene showing some restriction sites and the deletions generated. (B) Examples of transient transfections with the mutant proteins demonstrating that all of them except ΔCΔcoil integrate into the endogenous keratin cytoskeleton in PtK2 cells, including K10 (A), Δ3′UTR (B), ΔCΔcoil (C), ΔN (D), ΔC (E), and ΔNΔC (F). (C) Summary of five independent permanent transfection experiments in HaCaT cells with the different deletion proteins, demonstrating that only the simultaneous elimination of both the amino- and carboxyl-terminal domains of K10 abolishes K10’s ability to repress cell growth.
The ability of these mutants to induce cell growth arrest was assayed in parallel with the wild-type K10 protein in HaCaT cells in colony formation experiments. Removal of either the head (ΔN) or tail (ΔC), the 3′UTR (Δ3′UTR), or the deletion involving most of the rod domain (ΔCΔcoil) did not reduce the ability of the protein to inhibit cell proliferation (Fig. 7C). The construct in which the amino and carboxyl termini were simultaneously deleted (ΔNΔC) was unable to induce cell growth arrest, demonstrating that both nonhelical termini are involved in this function and that the presence of at least one of these domains is sufficient to inhibit cell proliferation. The finding that the ΔCΔcoil mutant, which does not integrate into a well-organized cytoskeletal structure upon transfection (Fig. 7B), has a growth suppression potential similar to those of other mutants (Fig. 7C) may suggest that the K10 effect on the cell cycle does not require the proper localization of this protein. Alternatively, the growth inhibition observed could be due to the keratin cytoskeleton aggregation promoted by the mutant protein.
In conclusion, we show here that keratins K10 and K16 can modulate keratinocyte proliferation in opposite ways, depending on pRb, and probably p107, function but not that of p53. These results therefore indicate that in addition to providing mechanical integrity to the cells in the context of a tissue, keratins also participate in signaling processes fundamental for cell physiology. Although several IF proteins undergo reorganization during mitosis, this is the first reported evidence that keratins can be involved in cell cycle control during the progression from G1 to S phase. The antithetical cell cycle modulation functions of K10 and K16 correlate well with the expression pattern of these keratins in the epidermis during differentiation (characterized by K10 induction) and hyperproliferation (characterized by K10 down regulation and induction of K16 expression). These keratin-specific functions would explain the need for the differential expression of members of this protein family. We have determined that the growth inhibitory function resides at the divergent nonhelical terminal domains of K10. These regions are thought to protrude from the filament core and may be involved in the interaction with other non-IF cytoplasmic proteins (26). Given the differences in localization of keratins and the Rb pocket proteins, it can be hypothesized that K10 interacts with cytoplasmic factors involved in a pathway leading to the control of cyclin D1 expression and therefore to the functional inactivation of pRb (or p107). This interaction may lead to changes either in the cellular localization or in the proteolytic degradation of these putative factors. In this regard, it has been reported that keratins K8 and K18, characteristic of simple epithelial cells, may interact with potentially regulatory proteins such as HSP70, members of the 14-3-3 family and certain protein kinase C isozymes (discussed in reference 17), as well as with proteosomes, in a cell cycle-dependent manner (18). We are currently identifying those factors that interact with K10 and/or K16 and characterizing the mechanism by which they may control the cell cycle progression.
FIG. 6.
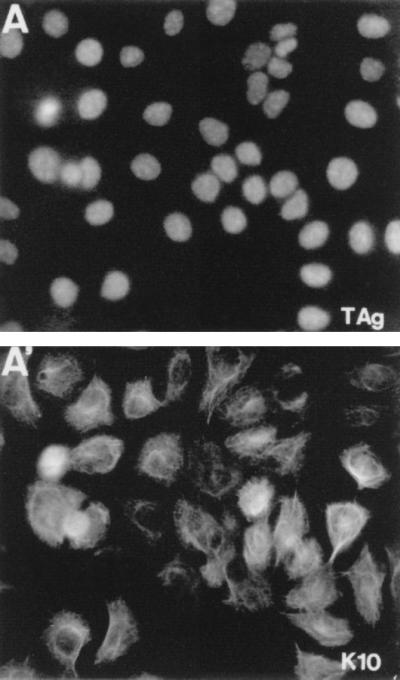
The expression of SV40 T Ag (A) and K10 (A′) in a HaCaT cell line isolated from a clone obtained in cotransfection experiments (Fig. 5A). Note that the majority of these cells express K10, in contrast with the few positive cells observed in cell lines isolated from clones arising in single K10 transfections (Fig. 2A).
ACKNOWLEDGMENTS
Our thanks go to D. Beach, M. Blessing, M. E. Ewen, E. Fuchs, E. Harlow, D. P. Lane, R. Bravo, and M. Serrano for their generous gifts of materials and helpful comments. We also thank M. Aldea and E. Cerezo for their expert technical support, J. C. Segovia for his help with FACS analysis, and C. Mark for editorial revision of the manuscript. The photographic work of S. Moreno is specially acknowledged.
This work was partially supported by grants from the DGICYT (PB 94-1230) and CRC.
REFERENCES
- 1.Blessing M, Rüther U, Franke W W. Ectopic synthesis of epidermal cytokeratins in pancreatic islet cells of transgenic mice interferes with cytoskeletal order and insulin production. J Cell Biol. 1993;120:743–755. doi: 10.1083/jcb.120.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boukamp P, Petruseva R T, Breitkreutz D, Hornung J, Markham A, Fusenig N E. Normal keratinization in a spontaneously immortalized human keratinocyte cell line. J Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Compton J G. Epidermal disease: faculty keratins take their role. Nat Genet. 1994;6:6–7. doi: 10.1038/ng0194-6. [DOI] [PubMed] [Google Scholar]
- 4.Fahraeus R, Paramio J M, Ball K L, Lain S, Lane D P. Inhibition of pRb phosphorylation and cell cycle progression by a 20-residue peptide derived from p16cdkn2/ink4a. Curr Biol. 1996;6:84–91. doi: 10.1016/s0960-9822(02)00425-6. [DOI] [PubMed] [Google Scholar]
- 5.Fuchs E. The cytoskeleton and disease: genetic disorders of intermediate filaments. Annu Rev Genet. 1996;30:197–231. doi: 10.1146/annurev.genet.30.1.197. [DOI] [PubMed] [Google Scholar]
- 6.Fuchs E. Of mice and men: genetic disorders of the cytoskeleton. Mol Biol Cell. 1997;69:899–902. doi: 10.1091/mbc.8.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fuchs E, Green H. Changes in keratin expression during terminal differentiation of the keratinocyte. Cell. 1980;19:1033–1042. doi: 10.1016/0092-8674(80)90094-x. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs E, Weber K. Intermediate filaments: structure, dynamics, function and disease. Annu Rev Biochem. 1994;63:345–382. doi: 10.1146/annurev.bi.63.070194.002021. [DOI] [PubMed] [Google Scholar]
- 9.Geng Y, Weinberg R A. Transformation growth factor b effects on expression of G1 cyclins and the cyclin-dependent kinases. Proc Natl Acad Sci USA. 1993;90:10315–10319. doi: 10.1073/pnas.90.21.10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hinds P W, Mittnacht S, Dulic S, Arnold A, Reed S I, Weinberg R A. Regulation of the retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70:993–1006. doi: 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- 11.Kuruc N, Leube R, Moll Y, Bader B L, Franke W W. Synthesis of cytokeratin 13, a component characteristic of internal stratified epithelia, is not induced in human epidermal tumors. Differentiation. 1989;42:111–123. doi: 10.1111/j.1432-0436.1989.tb00612.x. [DOI] [PubMed] [Google Scholar]
- 12.Lehman T A, Modali R, Boukamp P, Stanek J, Bennett W P, Welsh J E, Metcalf R A, Stamfer M R, Fusenig N E, Rogan E M, Harris C C. p53 mutations in human immortalized epithelial cell lines. Carcinogenesis. 1993;14:833–839. doi: 10.1093/carcin/14.5.833. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Nichols M A, Shay J W, Xiong Y. Transcriptional repression of the D-type cyclin-dependent kinase inhibitor p16 by the retinoblastoma susceptibility gene product pRb. Cancer Res. 1994;54:6078–6082. [PubMed] [Google Scholar]
- 14.Lukas J, Müller H, Bartkova J, Spitkovsky D, Kjerulff A A, Jansen-Dürr P, Strauss M, Bartek J. DNA tumor virus oncoproteins and retinoblastoma gene mutations share the ability to relieve the cell’s requirement for cyclin D1 function in G1. J Cell Biol. 1994;125:625–638. doi: 10.1083/jcb.125.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLean W H I, Lane E B. Intermediate filaments in disease. Curr Opin Cell Biol. 1995;7:118–125. doi: 10.1016/0955-0674(95)80053-0. [DOI] [PubMed] [Google Scholar]
- 16.Moll R, Franke W W, Schiller D L, Geiger B, Krepler R. The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell. 1982;31:11–24. doi: 10.1016/0092-8674(82)90400-7. [DOI] [PubMed] [Google Scholar]
- 17.Omary M B, Ku N O. Intermediate filament proteins of the liver: emerging disease association and functions. Hepatology. 1997;25:1043–1048. doi: 10.1002/hep.510250537. [DOI] [PubMed] [Google Scholar]
- 18.Palmer A, Mason G F G, Paramio J M, Knecht E W, Rivett A J. Changes in proteasome localization during cell cycle. Eur J Cell Biol. 1994;64:163–175. [PubMed] [Google Scholar]
- 19.Paramio J M, Casanova M L, Alonso A, Jorcano J L. Keratin intermediate filament dynamics in cell heterokaryons reveals diverse behavior of different keratins. J Cell Sci. 1997;110:1099–1111. doi: 10.1242/jcs.110.9.1099. [DOI] [PubMed] [Google Scholar]
- 20.Paramio J M, Jorcano J L. Assembly dynamics of epidermal keratins K1 and K10 in transfected cells. Exp Cell Res. 1994;215:319–331. doi: 10.1006/excr.1994.1348. [DOI] [PubMed] [Google Scholar]
- 21.Paramio J M, Jorcano J L. Role of protein kinases in the in vitro differentiation of human epidermal HaCaT cells. Br J Dermatol. 1997;137:44–50. [PubMed] [Google Scholar]
- 22.Paramio J M, Laín S, Segrelles C, Lane E B, Jorcano J L. Differential expression and functionally co-operative roles for the retinoblastoma family of proteins in epidermal differentiation. Oncogene. 1998;17:949–958. doi: 10.1038/sj.onc.1202031. [DOI] [PubMed] [Google Scholar]
- 23.Picksley S M, Lane D P. p53 and pRb: their cellular roles. Curr Opin Cell Biol. 1994;6:853–858. doi: 10.1016/0955-0674(94)90056-6. [DOI] [PubMed] [Google Scholar]
- 24.Rastinejad F, Blau H M. Genetic complementation reveals a novel regulatory role for 3′ untranslated regions in growth and differentiation. Cell. 1993;72:903–917. doi: 10.1016/0092-8674(93)90579-f. [DOI] [PubMed] [Google Scholar]
- 25.Rastinejad F, Conboy M J, Rando T A, Blau H M. Tumor suppression by RNA from the 3′ untranslated region of α-tropomyosin. Cell. 1993;75:1107–1117. doi: 10.1016/0092-8674(93)90320-p. [DOI] [PubMed] [Google Scholar]
- 26.Steinert P M, Rice R H, Roop D R, Trus B L, Steven A C. Complete amino acid sequence of a mouse epidermal keratin subunit and implications for the structure of intermediate filaments. Nature. 1983;302:794–800. doi: 10.1038/302794a0. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi K, Folmer J, Coulombe P. Increased expression of keratin 16 causes anomalies in cytoarchitecture and keratinization in transgenic mouse skin. J Cell Biol. 1994;127:505–520. doi: 10.1083/jcb.127.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weinberg R A. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 29.Weiss R A R, Eichner R, Sun T T. Monoclonal antibody analysis of keratin expression in epidermal diseases: a 48- and 56-kilodalton keratin as molecular markers for hyperproliferative keratinocytes. J Cell Biol. 1984;98:1397–1406. doi: 10.1083/jcb.98.4.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu L, Enders G, Lees J A, Beijersbergen R L, Bernards R, Harlow E. The pRb-related protein p107 contains two growth suppression domains: independent interactions with E2F and cyclin/cdk complexes. EMBO J. 1995;14:1904–1913. doi: 10.1002/j.1460-2075.1995.tb07182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu L, van den Heuvel S, Helin K, Fattaey A, Ewen M, Livingston D, Dyson N, Harlow E. Inhibition of cell proliferation by p107, a relative of the retinoblastoma protein. Genes Dev. 1993;7:1111–1125. doi: 10.1101/gad.7.7a.1111. [DOI] [PubMed] [Google Scholar]