

Abstract
Moyamoya disease (MD) is a rare vaso-occlusive disorder that primarily affects intracranial cerebral arteries. The involvement of extracranial vessels is unusual. However, there are previous reports suggesting MD to be a systemic disorder, causing disease manifestations in vessels of other parts of the body. We report the case of a female patient with MD and multiple episodes of ischemic strokes followed by bypass surgery of cerebral arteries during infancy. Due to corresponding ischemic lesions the girl showed global retardation of psychomotor development and central right sided movement disorder. At the age of 10 years the girl was admitted to our hospital with recurrent syncope. While cranial MRI excluded any newly added ischemic lesions, electrocardiography revealed evidence of right ventricular hypertrophy, and subsequent echocardiography then indicated pulmonary hypertension, which was confirmed by cardiac catheterization. Despite an upfront combination pulmonary vasodilating therapy, the pulmonary vascular disease appeared to be progressive. Genetic analysis showed heterozygous c.12341C>T mutation in the RNF213 gene. This case presentation demonstrates that pulmonary arterial hypertension is a rare comorbidity in patients with MD, especially in patients with genetic predictors such as the RNF213 mutation. Thus, regular echocardiographic screening for early signs of pulmonary arterial hypertension in patients with MD should be part of regular clinical work-up. Early detection and treatment of pulmonary arterial hypertension in MD might help to improve the long-term outcome in the individual patient.
Keywords: Moyamoya disease (MD), pulmonary arterial hypertension (PAH), arteriopathy, angiography, case report
Introduction
Moyamoya disease (MD) is a rare non-atherosclerotic and non-inflammatory arterial disorder that primarily involves medium-sized and small arteries. Arterial hyperplasia involves the three layers—intima, media or adventitia—causing progressive dysplastic stenosis of main cerebrovascular vessels and leading to an exaggerated development of a collateral network, which gives the angiographical aspect of a “puff of smoke”. Usually this results in only minor ischemic strokes or transient ischemic attacks (1). Besides this, few patients experience serious complications of these ischemic strokes. The prognosis is usually closely connected to the burden of cerebrovascular impairment. In most cases, the origin is idiopathic, and involvement of extracranial vessels is unusual. However, there are reports suggesting MD to be a systemic disorder, causing disease manifestations in vessels of other parts of the body. We present the following case in accordance with the CARE reporting checklist (available at http://dx.doi.org/10.21037/cdt-20-249).
Case report
Following three episodes of ischemic strokes in a girl at the age of 12 months (frontal right), 14 months (parietal right) and 15 months (parietal left), MD was diagnosed by the characteristic presentation at magnetic resonance angiography. Bilateral bypass surgery [right side: superficial temporal artery to middle cerebral artery (STA-MCA); left side: superficial temporal artery to anterior cerebral artery (STA-ACA)] was performed at the age of 16 and 20 months (Figure 1, preoperative, age 12 months; Figures 2 and 3, postoperative, age 16 months). Due to the ischemic lesions the girl presented with global retardation of psychomotor development and central movement disorder, accentuated to the right side. She was able to walk alone at the age of 24 months and started to talk at the age of 18 months with a delayed but progressing psychomotor development. The girl received regular logopedic therapy, physiotherapeutic and occupational therapy. A structured cognitive testing at the age of 6 years showed mild cognitive impairment (Snijders-Oomen non-verbal test (SON-R 2½-7) 65 points, Hannover-Wechsler-Test (HAWIVA III) 61 points. Regular visits in our clinic did not show additional signs or symptoms of MD regarding on additional ischemic attacks. She received ASS 100 mg per day and Clopidogrel 25 mg/day for inhibition of platelet aggregation. Repeated echocardiograms showed normal structured heart without pathologic findings and with normal biventricular function.
Figure 1.
Cerebral MRI angiogram at age of 12 months showing an exaggerated development of a collateral network as classical finding in Moyamoya disease.
Figure 2.
Cerebral MRI angiogram at age of 16 months after first bypass surgery (right side: superficial temporal artery to middle cerebral artery (STA-MCA).
Figure 3.
Transversal image showing cortical hyperperfusion on the left hemisphere and the right sided STA-MCA shunt.
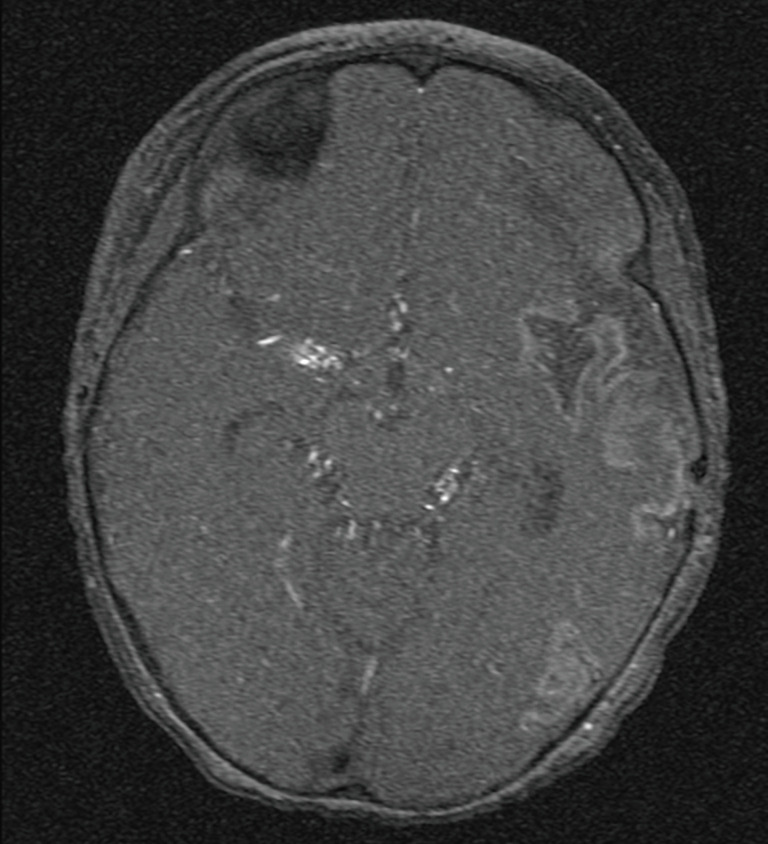
At the age of 10 years, the girl was admitted to our pediatric emergency department because of recurrent syncope and loss of consciousness for about one minute during the day, this time after getting up in the morning. On admission, general appearance was stable, heart rate was 104/min, respiratory rate 17/min, body temperature 36.9 °C, blood pressure 120/77 mmHg, oxygen saturation 99% at room air. Physical examination showed normal findings. In the neurological examination the known neurological impairment was evident, but no additional symptoms. Laboratory findings were normal (AST, ALT, CK, D-dimer, blood count, clotting time, Quick), except an elevation of NT-proBNP [144 pg/mL (Ref. <112 pg/mL)] and Creatinine [66 µmol/L (29–56 µmol/L)]. Cranial MRI did not show any newly added ischemic lesion. Remarkably, resting electrocardiography revealed criteria of right ventricular hypertrophy. In the subsequent echocardiographic examination there were indirect signs of elevated pulmonary pressure, in detail endsystolic flattened interventricular septum, mild RV hypertrophy, and shortened acceleration time of pulmonary flow curve. The estimated systolic pulmonary arterial (PA) pressure assessed by Doppler of the tricuspid valve regurgitation jet was approximately 50 mmHg (plus systemic venous pressure), RV function assessed by TAPSE was appropriate (16 mm). The chest X-ray revealed enlargement of the pulmonary trunk and its branches and mild infiltrates at both sides. To confirm pulmonary hypertension, the patient underwent cardiac catheterization for hemodynamic study and acute vasoreactivity test. The corresponding mean PA pressure was 44 mmHg and pulmonary vascular resistance index (PVRI) 9.1 WU*m2. The ratio of PVRI to systemic vascular resistance index was 0.64 (see also Table 1). The acute vasoreactivity testing showed distinct reduction of pulmonary vascular resistance and PA pressure under nitric oxide (NO) and oxygen (PVR/SVR ratio 0.47, mPAP 35 mmHg), therefore the patient was considered as responder and a pulmonary vasodilating combination therapy including Amlodipin (starting with 2.5 mg/day, then uptitrated during the following months) and Sildenafil 60 mg/day was started in addition to the ongoing platelet inhibition (ASS, Clopidogrel). For other etiologies of pulmonary hypertension, evaluations including thorax high-resolution computed tomography, pulmonary function test, serologic tests for connective tissue disorders and coagulation studies, thyroid functions, and Doppler ultrasound for liver were performed, which were all normal.
Table 1. Hemodynamic data during the first cardiac catheterization at the age of 10 years (at baseline and during acute vasoreactivity testing).
| Parameter | Baseline | O2 100% + NO 20 ppm |
|---|---|---|
| mPAP (mmHg) | 44 | 35 |
| mSAP (mmHg) | 58 | 61 |
| mPAP/mSAP | 0.75 | 0.57 |
| PAWP (mmHg) | 7 | 6 |
| CI (L/min/m2) | 3.2 | 4.5 |
| PVRI (WU*m2) | 9.1 | 6.5 |
| PVRI/SVRI | 0.64 | 0.47 |
mPAP, mean pulmonary arterial pressure; mSAP, mean systemic arterial pressure; PAWP, pulmonary arterial wedge pressure; CI, cardiac index; PVRI, pulmonary vascular resistance index; SVRI, systemic vascular resistance index; NO, nitric oxide.
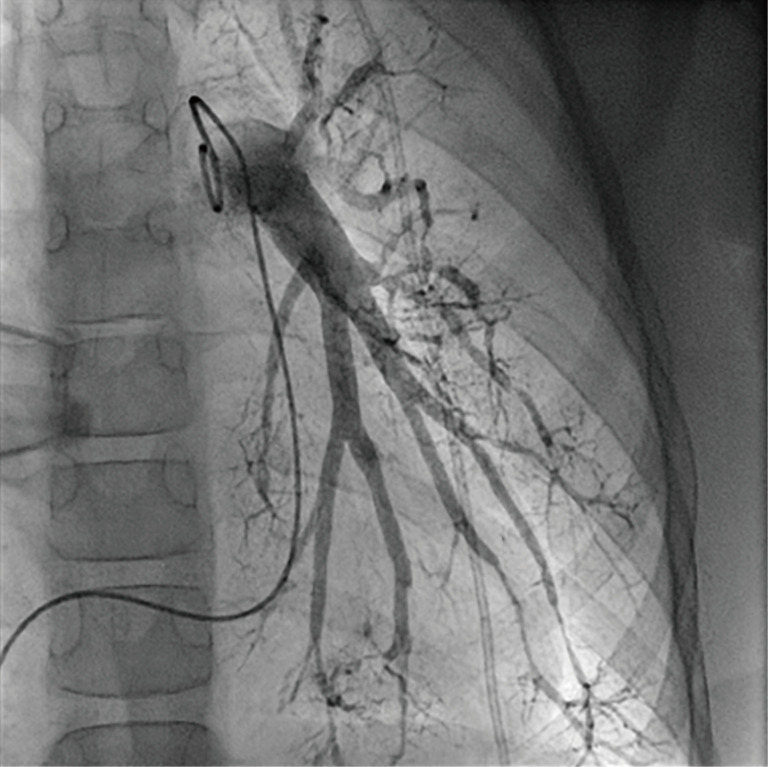
Regular echocardiographic examinations after initiation of therapy showed signs of disease progression, therefore repeat catheterization was performed 2 years after the pulmonary hypertension diagnosis, confirming an increase of PA pressure to a near systemic pressure level. Angiography revealed significant rarefication of pulmonary arteries with irregular gauge of peripheral vessels and vessel break-off (Figures 4,5). Due to the progredient character of the invasive measurements we decided for treatment escalation with Bosentan (125 mg/day). Complementary genetic analysis revealed heterozygous c.12341C>T mutation in the RNF213 gene (p.Thr4114Ile, NM_001256071.3).
Figure 4.
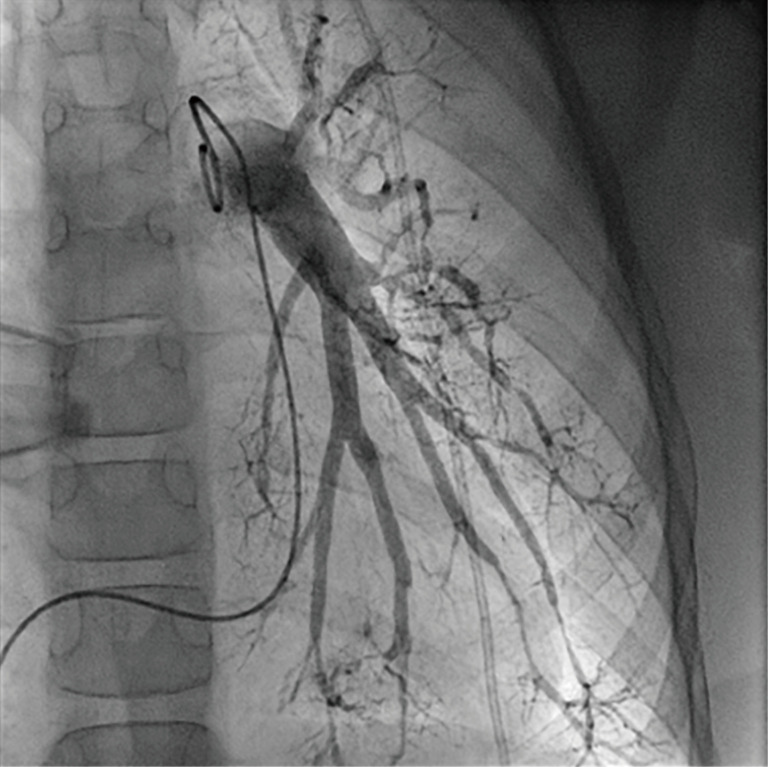
Invasive pulmonary angiography with significant rarefication of pulmonary arteries, irregular gauge of peripheral vessels and vessel break-off.
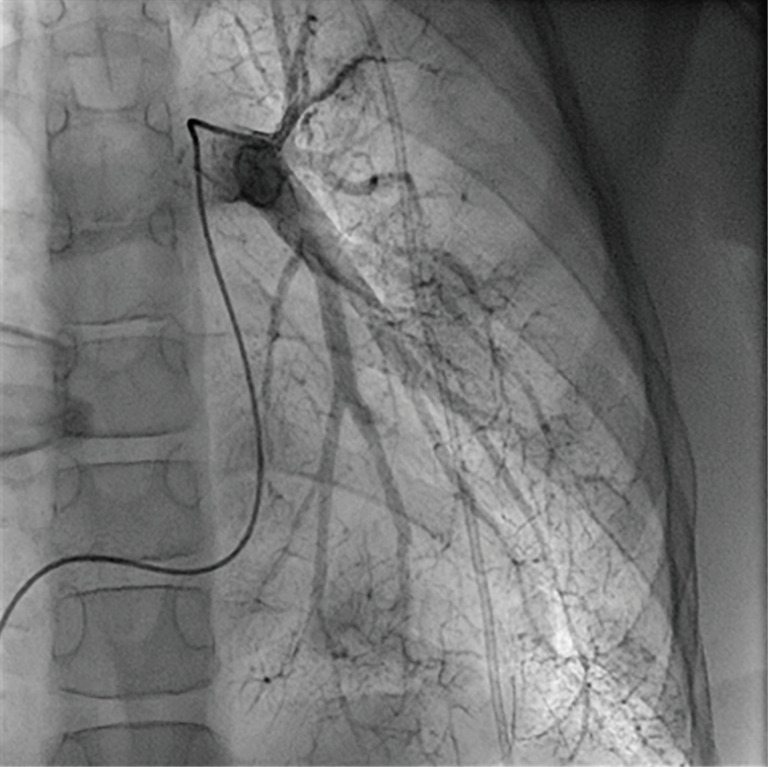
Figure 5.
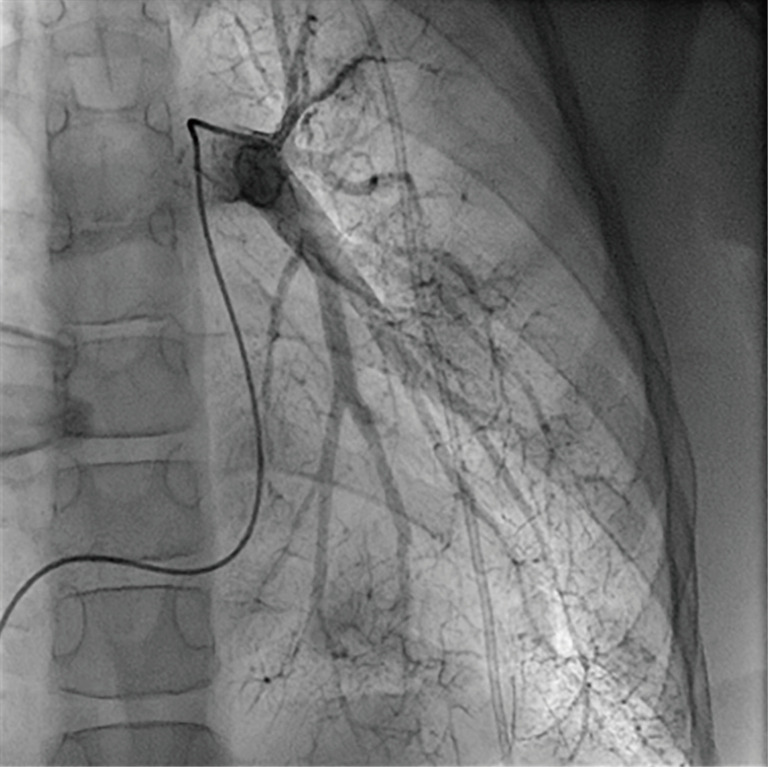
Similar to the Moyamoya-typical collateral network in cerebral vessels, pulmonary angiography shows corresponding findings in peripheral pulmonary arterial vessels (“puff of smoke”).
Discussion
Only a few cases have been published describing the association of MD with pulmonary arterial hypertension (PAH) (2-5) or renovascular hypertension (6). In the reported case, the late onset of PAH was very remarkable. Preceding echocardiographic examinations did not show any abnormalities and no sign of PAH. First evidence of PAH was found after onset of associated clinical symptoms (syncope) at the age of 10 years. In this specific case, the very first signs of reduced exercise capacity might have been blurred by the neurological impairment and the delay of psychomotor development. Therefore, it seems appropriate to screen patients with MD regularly for early signs of PAH even in absence of clinical symptoms, and consequently perform invasive hemodynamic diagnostics for confirmation if echocardiography suggests elevated PA-pressure. Non-invasive MRI-based methods can provide additional diagnostic information (7).
In the presented case, it was also remarkable, that despite an upfront combination pulmonary vasodilating therapy, the PAH was progressive within a relatively short period of time. This supports the theory of a rather progressive character of the pulmonary vascular disease in MD, with considerable poor prognosis particularly in patients carrying the RNF213 mutation (8). Therefore, advanced treatment (i.e., dual/triple combination therapy, or even parenteral treatment with prostacyclin) might be reasonable options.
Heterozygous RNF213 mutations have been described in the literature as showing a strong association to MD and being a risk allele for vascular diseases, such as pulmonary arterial hypertension (9-12). The RNF213 is a 591 kDa protein with two functional domains (AAAb ATPase and RING domain) (13) and plays important roles in cellular function (14). A recent case presentation suggests that the NF-κB- and apoptosis-inducing functions of RNF213 may be negatively regulated and result in MD onset (15). The variant which was found in our patient has not yet been described, but results in change of protein structure (p.Thr4114Ile) which is close to other described MD-related mutations (16) suggesting a change of functional relevance.
The understanding of MD as a multisystemic arteriopathy should result in more awareness of PAH as a rare, but incisive complication of the disease (17). Regular echocardiographic or MRI angiographic screening for early signs of PAH in patients with MD should be part of regular clinical work-up, even in absence of clinical symptoms. This should be considered in particular previous to planned procedures (i.e., operations, interventions, imaging), as there is usually a higher complication rate due to general anesthesia in patients with PAH (18).
Acknowledgments
Funding: None.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee(s) and with the Helsinki Declaration (as revised in 2013). Written informed consent was obtained from the patient for publication of this case report and accompanying images. A copy of the written consent is available for review by the editorial office of this journal.
Open Access Statement: This is an Open Access article distributed in accordance with the Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License (CC BY-NC-ND 4.0), which permits the non-commercial replication and distribution of the article with the strict proviso that no changes or edits are made and the original work is properly cited (including links to both the formal publication through the relevant DOI and the license). See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Footnotes
Provenance and Peer Review: This article was commissioned by the editorial office, Cardiovascular Diagnosis and Therapy for the series “Pediatric Pulmonary Hypertension”. The article has undergone external peer review.
Reporting Checklist: The authors have completed the CARE reporting checklist (available at http://dx.doi.org/10.21037/cdt-20-249).
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/cdt-20-249). The series “Pediatric Pulmonary Hypertension” was commissioned by the editorial office without any funding or sponsorship. CA served as the unpaid Guest Editor of the series. CA reports personal fees from Actelion, outside the submitted work. JK reports Speaker honoraria from Sanofi, Biomarin and Shire-Takeda. The authors have no other conflicts of interest to declare.
References
- 1.Natori Y, Ikezaki K, Matsushima T, et al. ‚Angiographic moyamoya’ its definition, classification and therapy. Clin Neurol Neurosurg 1997;99:S168-72. 10.1016/S0303-8467(97)00052-8 [DOI] [PubMed] [Google Scholar]
- 2.Kızılkaya MH, Uysal F, Gürbüz E, et al. Atypical presentation of moyamoya disease with pulmonary hypertension: A case report. Anatol J Cardiol 2018;19:350-1. 10.14744/AnatolJCardiol.2018.65642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kapusta L, Daniels O, Renier WO. Moya-Moya syndrome and primary pulmonary hypertension in childhood. Neuropediatrics 1990;21:162-3. 10.1055/s-2008-1071486 [DOI] [PubMed] [Google Scholar]
- 4.Tokunaga K, Hishikawa T, Sugiu K, et al. Fatal outcomes of pediatric patients with moyamoya disease associated with pulmonary arterial hypertension. Report of two cases. Clin Neurol Neurosurg 2013;115:335-8. 10.1016/j.clineuro.2012.05.002 [DOI] [PubMed] [Google Scholar]
- 5.Schranz D, Kerst G, Menges T, et al. Transcatheter creation of a reverse Potts shunt in a patient with severe pulmonary arterial hypertension associated with Moyamoya syndrome. EuroIntervention 2015;11:121-5. 10.4244/EIJV11I1A21 [DOI] [PubMed] [Google Scholar]
- 6.Choi Y, Kang BC, Kim KJ, et al. Renovascular hypertension in children with moyamoya disease. J Pediatr 1997;131:258-63. 10.1016/S0022-3476(97)70163-X [DOI] [PubMed] [Google Scholar]
- 7.Reiter G, Reiter U, Kovacs G, et al. Blood flow vortices along the main pulmonary artery measured with MR imaging for diagnosis of pulmonary hypertension. Radiology 2015;275:71-9. 10.1148/radiol.14140849 [DOI] [PubMed] [Google Scholar]
- 8.Hiraide T, Kataoka M, Suzuki H, et al. Poor outcomes in carriers of the RNF213 variant (p.Arg4810Lys) with pulmonary arterial hypertension. J Heart Lung Transplant 2020;39:103-12. 10.1016/j.healun.2019.08.022 [DOI] [PubMed] [Google Scholar]
- 9.Bang OY, Chung JW, Kim DH, et al. Moyamoya Disease and Spectrums of RNF213 Vasculopathy. Transl Stroke Res 2020;11:580-9. 10.1007/s12975-019-00743-6 [DOI] [PubMed] [Google Scholar]
- 10.Koizumi A, Kobayashi H, Hitomi T, et al. A new horizon of moyamoya disease and asso-ciated health risks explored through RNF213. Environ Health Prev Med 2016;21:55-70. 10.1007/s12199-015-0498-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guey S, Kraemer M, Herve D, et al. Rare RNF213 variants in the C-terminal region encompassing the RING-finger domain are associated with moyamoya angiopathy in Caucasians. Eur J Hum Genet 2017;25:995-1003. 10.1038/ejhg.2017.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi H, Kabata R, Kinoshita H, et al. Rare variants in RNF213, a susceptibility gene for moyamoya disease, are found in patients with pulmonary hypertension and aggravate hypoxia-induced pulmonary hypertension in mice. Pulm Circ 2018;8:2045894018778155. 10.1177/2045894018778155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu W, Morito D, Takashima S, et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS One 2011;6:e22542. 10.1371/journal.pone.0022542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hitomi T, Habu T, Kobayashi H, et al. The moyamoya disease susceptibility variant RNF213 R4810K (rs112735431) induces genomic instability by mitotic abnormality. Biochem Biophys Res Commun 2013;439:419-26. 10.1016/j.bbrc.2013.08.067 [DOI] [PubMed] [Google Scholar]
- 15.Takahashi K, Nakamura J, Sakiyama S.A histopathological report of a 16-year-old male with peripheral pulmonary artery stenosis and Moyamoya disease with a homozygous RNF213 mutation. Respir Med Case Rep 2019;29:100977. 10.1016/j.rmcr.2019.100977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cecchi AC, Guo D, Ren Z, et al. RNF213 Rare Variants in an Ethnically Diverse Population With Moyamoya Disease. Stroke 2014;45:3200-7. 10.1161/STROKEAHA.114.006244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Vries RR, Nikkels PG, van der Laag J, et al. Moyamoya and extracranial vascular involvement: fibromuscular dysplasia? A report of two children. Neuropediatrics 2003;34:318-21. 10.1055/s-2003-44664 [DOI] [PubMed] [Google Scholar]
- 18.Bernier ML, Jacob AI, Collaco JM, et al. Perioperative events in children with pulmonary hypertension undergoing non-cardiac procedures. Pulm Circ 2018;8:2045893217738143. 10.1177/2045893217738143 [DOI] [PMC free article] [PubMed] [Google Scholar]