

Abstract
Despite impressive and durable responses, nonsmall cell lung cancer (NSCLC) patients treated with anaplastic lymphoma kinase (ALK) inhibitors (ALK‐Is) ultimately progress due to development of resistance. Here, we have evaluated the clinical utility of circulating tumor DNA (ctDNA) profiling by next‐generation sequencing (NGS) upon disease progression. We collected 26 plasma and two cerebrospinal fluid samples from 24 advanced ALK‐positive NSCLC patients at disease progression to an ALK‐I. These samples were analyzed by NGS and digital PCR. A tool to retrieve variants at the ALK locus was developed (VALK tool). We identified at least one resistance mutation in the ALK locus in ten (38.5%) plasma samples; the G1269A and G1202R mutations were the most prevalent among patients progressing to first‐ and second‐generation ALK‐Is, respectively. Overall, 61 somatic mutations were detected in 14 genes: TP53, ALK, PIK3CA, SMAD4, MAP2K1 (MEK1), FGFR2, FGFR3, BRAF, EGFR, IDH2, MYC, MET, CCND3, and CCND1. Specifically, a deletion in exon 19 in EGFR, a non‐V600 BRAF mutation (G466V), and the F129L mutation in MAP2K1 were identified in four patients who showed no objective survival benefit from ALK‐Is. Potential ALK‐I‐resistance mutations were also found in PIK3CA and IDH2. Finally, a c‐MYC gain, along with a loss of CCND1 and FGFR3, was detected in a patient progressing on a first‐line treatment with crizotinib. We conclude that NGS analysis of liquid biopsies upon disease progression identified different putative ALK‐I‐resistance mutations in most cases and could be a valuable approach for therapy decision making.
Keywords: ALK‐TKI, EML4‐ALK, liquid biopsy, NGS, NSCLC
Despite impressive and durable responses, nonsmall cell lung cancer patients treated with ALK inhibitors (ALK‐Is) ultimately progress due to the development of resistance mechanisms. In this study, we show that next‐generation sequencing (NGS) analysis of liquid biopsies upon disease progression is feasible and informative as it can identify different mechanisms underlying resistance to ALK‐Is by NGS profiling of ctDNA.
Abbreviations
- AF
allele frequency
- ALK
anaplastic lymphoma kinase
- ALK‐Is
Anaplastic lymphoma kinase inhibitors
- cfDNA
cell‐free DNA
- CNS
central nervous system
- CNVs
copy‐number variations
- CSF
cerebrospinal fluid
- CTC
circulating tumor cell
- dPCR
digital PCR
- ECOG
cooperative oncology group
- LOD
limit of detection
- MAF
mutant allele frequency
- MNP
multiple‐nucleotide polymorphism
- NGS
next‐generation sequencing
- NPA
negative percentage agreement
- NSCLC
nonsmall cell lung cancer
- ORA
overall rates of agreement
- OS
overall survival
- PFS
progression‐free survival
- PPA
positive percentage agreement
- SNVs
single nucleotide variants
- UMIs
unique molecular identifiers
- wt
wild‐type
1. Introduction
Anaplastic lymphoma kinase (ALK) inhibitors (ALK‐Is) have dramatically improved outcomes of nonsmall cell lung cancer (NSCLC) patients whose tumors harbor an ALK translocation [1, 2]. A broad therapeutic arsenal is currently available to treat ALK‐positive NSCLC tumors, and sequential treatment with different ALK‐Is is the best therapeutic option for NSCLC patients with an ALK translocation [3, 4]. However, it remains unclear how ALK‐Is should be sequenced. It has been proposed that treatment sequencing can be established according to clinical characteristics of the patients or toxicity profile. In this way, second‐generation ALK‐Is have shown impressive central nervous system (CNS) efficacy in ALK‐positive NSCLC patients [5, 6]. On the other hand, crizotinib is associated with adverse events dominated by gastrointestinal and visual effects, increased transaminases and edema, whereas the most common adverse events of alectinib are anemia, myalgia, and increased blood bilirubin [7]. The tumor molecular profile can also determine the treatment response. In this way, among patients treated with the ALK‐I lorlatinib, the ALK fusion variant 3 was associated with significantly longer progression‐free survival (PFS) than variant 1 [8]. Finally, several resistance mutations have been identified upon progression to an ALK‐I [9, 10]. While some of them confer resistance to specific ALK‐Is, others do not [11, 12, 13]. Paradoxically, therapy is seldom decided based on the tumor molecular profile upon disease progression, and ALK‐Is are usually prescribed empirically. Conversely, biomarker testing after treatment failure is routinely performed in EGFR‐positive NSCLC patients, as recommended by clinical guidelines [14].
Repeat tumor biopsy upon disease progression is not always feasible. Nevertheless, there is considerable evidence showing that genotyping cell‐free DNA (cfDNA) is a valid and significantly faster approach than genotyping solid biopsies. Next‐generation sequencing (NGS) enables the interrogation of a large number of mutations and can be used with liquid biopsies [15].
In this study, we have analyzed 26 plasma and two cerebrospinal fluid (CSF) samples, from 24 patients, collected upon disease progression while being treated with an ALK‐I, in order to determine the clinical utility of liquid biopsies for ALK‐Is sequencing. In addition, we provide a pipeline specifically designed to detect somatic mutations in the ALK domain. Finally, we evaluate the clinical significance of somatic alterations in genes other than ALK.
2. Methods
2.1. Study population
Between June 2015 and July 2019, 24 stage IV, ALK‐positive NSCLC patients progressing on an ALK‐I (the ALK cohort), were prospectively recruited from six hospitals across Spain. The study protocol was approved by the Hospital Puerta de Hierro Ethics Committee (internal code 79‐18) and was conducted in accordance with the precepts of the Code of Ethics of The World Medical Association (Declaration of Helsinki). All patients provided their appropriate written informed consent to participate in the study prior to enrollment. Briefly, patients who were 18+ years of age and with a pathologically confirmed diagnosis of stage IV NSCLC with an EML4‐ALK translocation were eligible for inclusion. The identification of EML4‐ALK rearrangement was carried out by different methodologies, according to each participant center (Table S1). Specifically the ALK testing was performed by immunohistochemistry (IHC) using the Ventana ALK (D5F3) CDx assay on a Ventana BenchMark XT automated slide‐processing system (Ventana Medical Systems, Tucson, AZ, USA) or the mAb ALK (5A4) (Novocastra™; Leica Biosystems, Newcastle Upon Tyne, UK); Fluorescence In Situ Hybridization (FISH) using Vysis LSI ALK Dual Color Break Apart FISH probe kit (Vysis, Downers Grove, IL, USA) or ALK (2P23) Break Apart FISH probe kit (cYTOtEST, Rockville, MD, USA). The nCounter analysis system (NanoString Technologies, Seattle, WA, USA) was also used to detect EML4‐ALK translocation. All plasma samples were collected upon disease progression, which was assessed according to RECIST criteria v.1.1. In total, 26 plasma and two CSF specimens were collected and analyzed.
2.2. Laboratory procedures
Peripheral whole‐blood samples were collected in a 10‐mL Streck cfDNA BCT® (Streck, Omaha, NE, USA) tube for cfDNA. CSF samples were collected in a 10‐mL sterile tube with no additives or anticoagulants. CSF samples were taken from patients progressing at the brain level. Samples were centrifuged at room temperature in two consecutive centrifugations of 1500 g for 10 min and 5000 g for 20 min in order to separate plasma or CSF from the cellular fraction. cfDNA was isolated using QIAamp Circulating Nucleic Acid Kit (QIAgen, Valencia, CA, USA) according to the manufacturer's instructions. Libraries were prepared from at least 15ng of input cfDNA using the Oncomine™ Pan‐Cancer Cell‐Free Assay kit (Thermo Fisher, Palo Alto, CA, USA) according to the manufacturer's instructions. According to manufacturer, this amplicon‐based targeted sequencing assay allows the detection of multiple variants in ctDNA isolated from liquid biopsy samples with a limit of detection (LOD) down to 0.1% mutant allele frequency (MAF). Specifically, the panel interrogates variants in 52 genes (Table S2). AMPureXP magnetic beads (Beckman Coulter, Inc., Brea, CA, USA) were used to purify all libraries. Subsequently, the individual libraries were quantified using the Ion Library TaqMan® Quantitation Kit (Thermo Fisher, Palo Alto, CA, USA) in a StepOnePlus™ qPCR machine (Thermo Fisher) and adjusted to a final concentration of 50 pm. Eight samples were pooled. Templating and Ion 550™ Chip loading were carried out with an Ion Chef™ System (Thermo Fisher). Finally, an Ion GeneStudio™ S5 Sequencer (Thermo Fisher) was used to sequence loaded Ion 550™ chips. torrent suite Software (v5.12) was used to analyze the raw sequencing data. The coverage analysis (v. 5.12.0.0) plugin was used for sequencing coverage analysis (Thermo Fisher). Raw reads were aligned to the human reference genome hg19. Variant calling, annotation, and filtering were performed on the Ion Reporter (v5.10, Thermo Fisher) platform using the Oncomine TagSeq Pan‐Cancer Liquid Biopsy workflow (v2.1, Thermo Fisher). All candidate mutations were manually reviewed using the Integrative Genomics Viewer v.2.3.40, (Broad Institute, Cambridge, MA, USA). The clinical significance of somatic variants was determined according to the Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer [16].
The mutations identified by NGS were confirmed by digital PCR (dPCR) using a QuantStudio® 3D dPCR System (Applied Biosystems, South San Francisco, CA, USA). In accordance with the manufacturer's specifications, dPCR reactions were performed in an 18‐μL volume comprising 9 μL of 20X QuantStudio 3D Master Mix, 0.45 μL of 40× commercially available predesigned or custom TaqMan® assays and 8.55 μL of cfDNA (minimum amount 2 ng). Subsequently, 14.5 μL of the PCR reaction was loaded onto a QuantStudio 3D dPCR 20K chip using QuantStudio™ 3D dPCR Chip Loader. Each dPCR run included a negative control DNA, as a wild‐type (wt) control, a blank (with no cfDNA) and a positive control. PCR reactions were performed in a thermal cycler (Applied Biosystems) at 96 °C for 10 min, then 40 cycles at 56 °C for 2 min and 98 °C for 30 s, and a final elongation step at 72 °C for 10 min. Finally, samples were maintained at 22 °C for at least 30 min. Chips fluoresce was read twice two independent QuantStudio™ 3D dPCR Instruments. Results were visualized and analyzed using QuantStudio® 3D Analysis Suite™ Cloud Software (Thermo Fisher). The automatic call assignments for each data cluster were manually adjusted when needed. The MAF was calculated as the ratio of mutant DNA molecules to the sum of mutant and wt DNA molecules.
The LOD and limit of quantitation (LOQ) of the dPCR assays for ALK variants were evaluated for four custom TaqMan® assays by mixing DNA from two different plasmids an one patient sample harboring the specific ALK mutations in a background of wt DNA (from healthy donors) at different mutant allele concentrations (i.e., 1%, 0.5%, 0.1%, 0.05%). LOD and LOQ were estimated for an input of cfDNA of 7 ng. Specifically, LOD and LOQ for ALK G1202R assay were estimated using a patient sample. The mutation was previously confirmed by NGS. LOD and LOQ for ALK S1206Y were estimated using ALK_1 plasmid. Finally, LOD and LOQ for ALK L1196M and G1269A assays were estimated using ALK_3 plasmid. Plasmids were designed by Classic GeneArt Gene Synthesis Portal (Thermo Fisher). Schematic plasmid maps are available upon request. LOD and LOQ were calculated based on the standard deviation of the response and the slope according to ICH Q2 (R1) guidelines (Validation of analytical procedures: text and methodology). The standard deviation of the response was calculated based on standard error of the y‐intercept. Results of the detection sensitivity of TaqMan assays are displayed in Data S1. Overall, mutant allele frequencies correlated with the expected mutant allele frequencies in all cases (Pearson's correlation coefficient, 0.9999, 0.9991, 0.9994, and 0.9994 for G1202R, S1206Y, L1196M, and G1269A, respectively). LOD were 0.05%, 0.23%, 0.19%, and 0.18% for G1202R, S1206Y, L1196M, and G1269A, respectively. Additionally, 10 wt cfDNA from healthy donors were used to evaluate the false‐positive signals. ALK mutations were not detected in any of the wt samples.
Samples were considered to be positive when the MAF was greater than or equal to 0.1% and when there were at least 300 copies·mL−1 of wt DNA. A negative control DNA (wt) and a blank sample (containing no DNA) were included in every run.
In order to discard that mutations detected by NGS were not clonal hematopoiesis‐derived mutations (specifically those in TP53), DNA from peripheral blood mononuclear cells (PBMCs) was analyzed in 10 samples by dPCR or Sanger sequencing (Table S3). To this aim, DNA was isolated from PBMCs using Maxwell® RSC Whole Blood DNA Kit (Promega Corporation, Madison, WI, USA) according to the manufacturer's instructions. Custom primer sequences to target specific TP53 regions (exon 2, exon 4, exon 7, and exon 8 splice site) were designed using the Primer3plus software. PCR reactions were performed in a 15‐μL volume comprising 7.5 μL of 20X QuantStudio 3D Master Mix, 5.5 μL of H2O, 0.5 μL of forward primer, 0.5 μL of reverse primer, and 1 μL of isolated DNA. PCR amplifications were carry out in the VeritiPro Thermal Cycler (Applied Biosystems). The thermal program included an initial step of 94 °C for 10 min, follow by 40 cycles of denaturation at 94 °C for 30 s, annealing at 60 °C for 1 min and extension at 72 °C for 30 s, and finally, at 72 °C for 7 min. Amplified DNA samples were subjected to electrophoresis with 2% agarose gel, stained with GelRed ™ Nucleic Acid Gel Stain, 10 000X in Water (Biotium, Hayward, CA, USA), and examined under an UV transilluminator. Products revealing clear PCR bands were purified by adding 2 µL of ExoSAP‐IT reagent (GE Healthcare®, Madison, WI, USA) for each 5 µL of preamplification volume and incubated for 15 min at 37 °C followed by 15 min at 80 °C. One microliter of PCR product was subjected to Sanger sequencing using PCR primers and the BigDye™ Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems). The thermal program included an initial step of 94 °C for 10 min, follow by 25 cycles of denaturation at 94 °C for 10 s, annealing at 50 °C for 10 s and extension at 60 °C for 2 min. The PCR product was purified with the protocol of the BigDye® XTerminator™ Purification Kit (Applied Biosystems) and sequenced with the forward and reverse PCR primers on ABI PRISM 310 Genetic Analyzer (Applied Biosystems). Sequence data were analyzed on sequencing analysis Software 7 v7.0 (Applied Biosystems).
To increase the detection rate for variants at the ALK locus, we have developed a bioinformatic pipeline, called the VALK tool (available at GitHub: https://github.com/AtochaHUPH/VALK‐tool‐), which is capable of fully automating the filtering generating a.csv file containing the output variants and a list of their properties. Specific conditions for single nucleotide variants (SNVs), indels, multiple‐nucleotide polymorphisms (MNP), fusions and copy‐number variation (CNV) calls were defined. The nonfiltered‐oncomine.tsv file with variants in ‘Variant Call Format' was obtained for the 28 samples. All variants that have passed the Oncomine Variants (v.5.12, Thermo Fisher) filter were included in the final analysis. In addition, we performed a second filtering process in order to rescue other somatic variants ruled out by the Oncomine Variants (v.5.12) filter. Specifically, parameters such as the overall error of the NGS assay, the LOD, the coverage depth, the percentage of targeted bases sequenced at that coverage depth, the total number of target reads covering a variant region, the number of reads supporting a specific variant, and the clinical significance, among others, were taken into account for the selection of the different thresholds. Figure S1 shows the selection criteria based on certain variables as presented in the nonfiltered‐oncomine.tsv file.
All computations were performed in r v.3.6.3 (R Foundation for Statistical Computing, Vienna, Austria) using additional packages. Specifically, the Tcl/Tk package that was used to provide the end‐user an intuitive graphical interface to carry out the entire filtering process. In addition, the Scales (v1.1.1; https://scales.r‐lib.org/) package, which contains functions that convert data values to perceptual properties, was used to transform the raw data obtained from the nonfiltered‐oncomine.tsv files into interpretable values for the end‐user. Positive and negative percentage agreement (PPA and NPA) and overall rates of agreement (ORA) of the VALK tool for detecting the ALK mutations specified in Table S4 were calculated considering the imperfect reference standard the dPCR result and using the two independent data sets, the ALK cohort and the Valencia cohort, which consists of 54 cfDNA samples from NSCLC patients.
2.3. Statistical analysis
Median follow‐up was estimated by the reverse Kaplan–Meier method. Overall survival (OS) was defined as the time from the start of treatment with an ALK inhibitor to death or last follow‐up. PFS was defined as the time between the start of an ALK inhibitor and disease progression (as ascertained by RECIST criteria), death, or the censored date of the last assessment, whichever occurred first. The log‐rank test was used to assess statistical differences between Kaplan–Meier survival curves. Hazard ratios were estimated from the Cox model using a multivariable approach adjusted for sex, cooperative oncology group (ECOG) performance status, and lines of ALK‐TKI. Association between clinicopathological variables and genomic features was assessed by Mann–Whitney and Fisher's exact test as needed. Values of P < 0.05 were considered statistically significant. Statistical analyses were performed using Stata 15.1 (Stata Corporation, College Station, TX, USA).
3. Results
3.1. Study cohort
We collected and analyzed 26 plasma and two CSF specimens from 24 metastatic patients diagnosed with an ALK‐positive NSCLC who were progressing on an ALK‐I. Baseline clinicopathological characteristics of the study population (N = 24) are presented in Table 1. The median age at diagnosis was 53 years (range, 36–72 years) and 58.3% were females. The majority of the patients were never smokers (62.5%) and the most frequent histology was adenocarcinoma (95.8%). ECOG Performance Status at study entry varied from 0 to 2. As shown in Fig. 1, two samples from two patients were collected upon disease progression while on two consecutive lines of treatment with an ALK‐I; three samples were obtained from one patient upon failure to three consecutive lines of ALK‐I; all 21 other members of the cohort each provided a single sample.
Table 1.
Baseline characteristics of the study cohort.
| Feature | N | % | |
|---|---|---|---|
| Age of diagnosis (years) | Median (range) | 53 (36–72) | |
| Sex | Male | 10 | 41.7 |
| Female | 14 | 58.3 | |
| Smoking status | Current smoker | 3 | 12.5 |
| Ex‐smoker | 6 | 25 | |
| Never‐smoker | 15 | 62.5 | |
| ECOG performance status | 0 | 12 | 50 |
| 1 | 11 | 45.8 | |
| 2 | 1 | 4.2 | |
| Histology | Adenocarcinoma | 23 | 95.8 |
| Neuroendocrine carcinoma | 1 | 4.2 | |
| Clinical stage at diagnosis | III | 6 | 25 |
| IV | 18 | 75 |
Fig. 1.
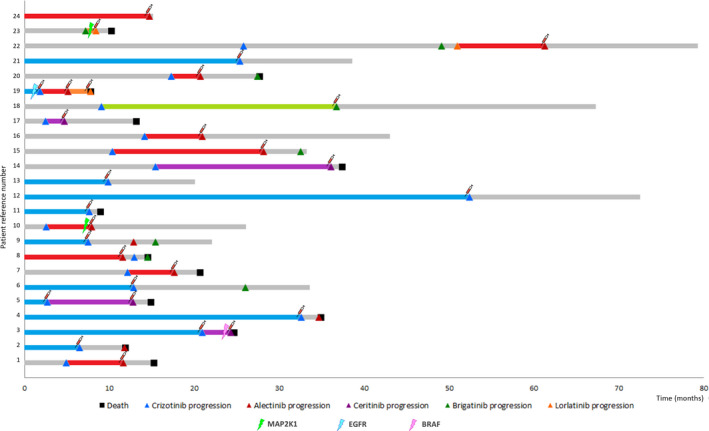
Swimmer chart showing the individual treatment responses of the study cohort. Blue, red, purple, green, and orange bars correspond to response duration of patients who were treated with crizotinib, alectinib, ceritinib, brigatinib, and lorlatinib, respectively, and from whom a blood sample was taken at the time of disease progression (syringe icon). Gray bars denote treatments responses to therapy for which samples at disease progression could not be analyzed (out of study). Tumor progression is denoted by triangle, and patient's death is denoted by a squared. Mutations in BRAF, EGFR, and MAP2K1 are indicated by a lightning.
As presented in Fig. 1, 13 samples corresponded to ALK‐I‐naïve patients who progressed on a first‐line crizotinib (N = 11) or alectinib (N = 2) treatment. For these patients, the median PFS and OS were 11.6 months (95% CI: 6.5–20.9 months) and 24.6 months (95% CI: 11.8–NR months), respectively. In addition, 12 samples corresponded to patients who had received previously crizotinib and were treated with a second‐generation ALK‐I. Finally, the cohort included two samples from patients progressing on lorlatinib after failure of a prior second‐generation ALK‐I and one patient progressing on alectinib who had previously received crizotinib plus two second‐generation ALK‐I. Detailed information about treatment lines is presented in Table S5. The median PFS and OS for patients progressing on a second or subsequent line with an ALK‐I were 5.4 months (95% CI: 2–9.1) and 11.2 months (95% CI: 3–NR) months, respectively.
3.2. Next‐generation sequencing analysis upon disease progression
Overall plasma samples yielded higher cfDNA concentrations than CSF. Despite the small sample size of the study cohort, the median concentration of cfDNA isolated from CSF was significantly lower (0.20 ng·μL−1) than median concentration of cfDNA obtained from plasma samples (1.94 ng·μL−1; P = 0.039; calculated by Mann–Whitney test).
The average mapped reads per sample was 9 775 623, resulting in a median overall sequencing depth of 25 322. The median read coverage per sample was 21 261 and the median molecular coverage per sample was 1670.8. Regarding the two CSF samples, taken from patients with CNS disease, the average mapped reads per sample was 8 268 194, resulting in a median overall sequencing depth of 9318.
In total, 61 somatic variants in ctDNA from 24 samples were detected. In three patients, no mutations were found. One of the patients with undetectable plasma ctDNA had progressed exclusively at the brain level. The average number of mutations per patient was 2.18 and the median MAF was 0.39%, with a minimum MAF of 0.02% and a maximum of 5.09%. As expected, SNPs were the most frequent mutation type (N = 48). In addition, we identified 10 indels and three CNVs (Fig. 2). Specifically, a c‐MYC gain in conjunction with a CCND1 and an FGFR3 loss was detected in a patient progressing on a first line with crizotinib. This patient also harbored a mutation in TP53 (Fig. 2).
Fig. 2.
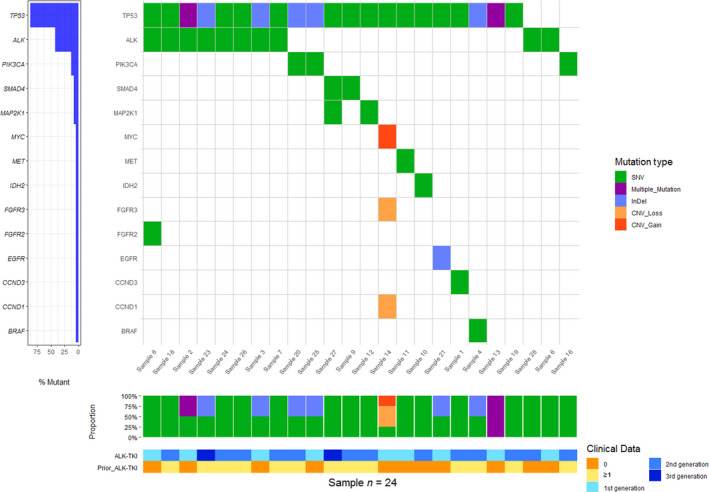
Co‐mutation plot according to ctDNA profiling by NGS. Each column represents data from a single patient. Each row represent data from a specific gene. Indels are represented in blue whereas missense mutations are represented in green. CNVs are colored in orange. Heat map at the bottom illustrates type of treatment received.
As illustrated in Fig. 2, somatic mutations were detected in 14 genes: TP53, ALK, PIK3CA, SMAD4, MAP2K1 (MEK) FGFR2, FGFR3, BRAF, EGFR, IDH2, MYC, MET, CCND3, and CCND1. All the variants detected are listed in Table S6. Thirteen variants (12 of which were in the ALK locus and one was in the EGFR gene) were categorized as being of strong clinical significance. Mutations in TP53 gene were further tested in PBMCs in order to discard a clonal hematopoietic origin (Table S3).
3.3. Identification of acquired resistance mutations in the ALK locus upon disease progression
To increase the sensitivity for detecting somatic mutations in the ALK locus, we developed an algorithm named VALK tool. The tool has been specifically designed for the analysis of the NGS data obtained from liquid biopsies. Among other parameters, the algorithm takes into account the molecular depth and molecular counts as well as specific regions that are more likely for false‐positive calls. In order to test the analytical performance of the tool, all SNPs in the ALK locus that were present in the nonfiltered‐oncomine.tsv file were analyzed by dPCR. In total, 19 ALK variants from 22 samples were evaluated (Table S4). Considering the nonreference standard the dPCR result PPA, NPA, and overall percent agreement of ALK mutation detection for the VALK tool were 67% (95% CI: 35–90%), 93% (95% CI: 75–99%), and 85% (95% CI: 69–94%), respectively (Table S7). To validate the final algorithm (locked prior to analyses), a second independent batch of 54 samples provided by a different laboratory (Valencia cohort) was used. In this case, the PPA, NPA, and ORA were 100% (95% CI: 29–100%), 98% (95% CI: 90–100%), and 98% (95% CI: 90–100%), respectively. Cross‐tables describing PPA, NPA, and ORA for the VALK tool as well as for the Oncomine variants v5.10 using ALK and Valencia cohorts are presented in Table S7–S10.
Overall, in the ALK cohort we detected at least one ALK mutation in 10 (38.5%) plasma samples collected upon disease progression (Table 2, and Fig. S2). Notably, the Oncomine variants v5.10 filter only detected ALK mutations in three patients (Table 2).
Table 2.
Somatic mutations detected at the ALK locus upon treatment failure.
| No. of patient | Nucleotide change | Amino acid change | Progression to | Line of treatment | Filter | MAF NGS (%) |
MAF dPCR (%) |
Total dPCR input (ng) |
|---|---|---|---|---|---|---|---|---|
| Patient 2 | c.3599C>T | p.A1200V | Crizotinib | First line | nonfiltered‐oncomine.tsv | 0.02 | 0.04 | 42.07 |
| Patient 3 | c.3806G>C | p.G1269A | Crizotinib | First line | Oncomine 5.10/VALK tool | 3.36 | 2.82 | 4.64 |
| Patient 5 | c.3806G>C | p.G1269A | Crizotinib | First line | Oncomine 5.10/VALK tool | 0.88 | 0.42 | 7.20 |
| Patient 5 | c.3604G>A | p.G1202R | Ceritinib | Second line | Oncomine 5.10/VALK tool | 1.28 | 2.12 | 54.04 |
| Patient 6 | c.3586C>A | p.L1196M | Crizotinib | First line | nonfiltered‐oncomine.tsv | 0.02 | 0.06 | 27.10 |
| Patient 16 | c.3617C>A | p.S1206Y | Alectinib | Second line | VALK tool | 0.06 | 0.01 | 26.50 |
| Patient 16 | c.3604G>A | p.G1202R | Alectinib | Second line | VALK tool | 0.04 | 0.04 | 26.50 |
| Patient 19 | c.3586C>A | p.L1196M | Lorlatinib | Third line | nonfiltered‐oncomine.tsv | 0.05 | 0.05 | 29.24 |
| Patient 19 | c.3824G>A | p.R1275Q | Lorlatinib | Third line | nonfiltered‐oncomine.tsv | 0.03 | 0.04 | 21.80 |
| Patient 20 | c.3604G>A | p.G1202R | Alectinib | Second line | VALK tool | 0.05 | 0.32 | 14.11 |
| Patient 22 | c.3604G>A | p.G1202R | Alectinib | Fourth line | VALK tool | 0.03 | 0.01 | 48.05 |
| Patient 24 | c.3538G>C | p.V1180L | Alectinib | First line | nonfiltered‐oncomine.tsv | 0.37 | 0.35 | 16.67 |
The G1202R mutation was identified in four patients who had progressed on alectinib (N = 3) and ceritinib (N = 1; patient number 5). Specifically, in case of the patient progression on ceritinib (patient 5), this mutation was not detected at treatment initiation confirming its role as a resistance mutation (Table S11). In addition, the S1206Y mutation was detected along with the G1202R mutation in one of the aforementioned alectinib‐progressing patients. This patient had been treated with crizotinib before initiating alectinib treatment. The low MAF of the S1206Y mutation suggests that it could be responsible for the previous crizotinib failure and it could have been decreased since alectinib therapy. In addition, the G1269A mutation was detected upon crizotinib failure in two cases and the L1196M mutation was identified after progression to crizotinib and lorlatinib. The latter was detected together with the R1275Q mutation in a patient diagnosed with an ALK‐positive neuroendocrine carcinoma (patient 19). Of note, neither the L1196M mutation nor the R1275Q mutation was detected in the pre‐treatment plasma sample from patient 19 (Table S11). Finally, the A1200V mutation (N = 1) was detected in a sample collected after crizotinib failure, and the V1180L mutation (N = 1) was detected in a patient progressing on a first‐line treatment with alectinib (Table 2).
3.4. Other molecular mechanisms underlying resistance to ALK‐I
A deletion in exon 19 of the EGFR gene, a non‐V600 BRAF mutation, and the F129L mutation in MAP2K1 (MEK1) were identified in four patients who showed no objective survival benefit from ALK‐Is. None of these patients had a secondary mutation in ALK locus.
Notably, the patient harboring the E746_A750del mutation in the EGFR gene had a PFS time of 1.8 months under first‐line crizotinib treatment. The patient was subsequently treated with alectinib but tumor progression was assessed 3.1 months later prompting a switch of treatment to lorlatinib, but that also failed after 1.8 months, suggesting that the tumor had primary resistance to ALK‐Is (Fig. 1, Table 3). Similarly, a non‐V600 BRAF mutation, namely G466V, was identified in the CFS collected upon disease progression to ceritinib. While PFS with first‐line crizotinib was 21 months, disease progression was assessed within 3 months of starting second‐line ceritinib treatment (Fig. 1). Remarkably, the non‐V600 BRAF mutation was absent in the plasma sample collected before ceritinib initiation (Table S11). Likewise, two patients harboring the F129L mutation in MAP2K1 (MEK1) obtained little benefit from second‐line ALK‐I (Fig. 1, Table 3). This mutation was detected upon disease progression to alectinib (patient 10) and lorlatinib (patient 23). In case of patient 10, we could confirm that this mutation was not present in the pre‐treatment plasma sample and tumor biopsy (Table S11). Noteworthy, the median PFS and OS for second‐line treatment for these patients were less than one month (0.97) and 3 months, respectively, whereas median the PFS and OS for patients progressing on a second or subsequent lines with an ALK‐I but without mutations in MAP2K1 were 5.9 and 11.2 months (P log‐rank < 0.05 in both cases; Fig. S3).
Table 3.
Resistance mutations detected in loci other than ALK upon tumor progression.
| Patient | Treatment | Treatment line | Sample | HUGO symbol | Amino acid change | Nucleotide change/CNV | Type | Variant class (Tier) | Transcript | rs | COSM |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 8 | Alectinib | 1st | Sample 10 | IDH2 | p.R140Q | c.419G>A | SNV | Potential clinical significance | NM_002168.3 | – | COSM41590 |
| Patient 14 | Ceritinib | 2nd | Sample 16 | PIK3CA | p.E545K | c.1633G>A | SNV | Potential clinical significance | NM_006218 | rs104886003 | COSM763 |
| Patient 18 | Brigatinib | 2nd | Sample 20 | PIK3CA | p.E545A | c.1634A>C | SNV | Potential clinical significance | NM_006218 | rs121913274 | COSM12458 |
| Patient 3 | Ceritinib | 2nd | Sample 4 | BRAF | p.G466V | c.1397G>T | SNV | Potential clinical significance | NM_004333.4 | rs121913351 | COSM451 |
| Patient 10 | Alectinib | 2nd | Sample 12 | MAP2K1 | p.F129L | c.385T>C | SNV | Potential clinical significance | NM_002755.3 | rs1057519805 | COSM1570285 |
| Patient 12 | Crizotinib | 1st | Sample 14 | MYC | Gain (3.08) | CNV | Potential clinical significance | NM_005359.5 | – | ||
| Patient 19 | Crizotinib | 1st | Sample 21 | EGFR | p.E746_A750del | c.2235_2249delGGAATTAAGAGAAGC | InDel | Strong clinical significance | NM_005228.4 | – | COSM6223 |
| Patient 23 | Lorlatinib | 2nd | Sample 27 | MAP2K1 | p.F129L | c.385T>C | SNV | Potential clinical significance | NM_002755.3 | rs1057519805 | COSM1570285 |
Potential ALK‐I resistance mutations were also found in IDH2, PIK3CA, and MYC (Table 3). Specifically, the oncogenic mutations E545K and E545A in the PIK3CA gene were detected in the plasma sample of two patients progressing on ceritinib and brigatinib (Table 3). These mutations were not detected in the pre‐treatment sample (Table S11). Likewise, the gain‐of‐function mutation in IDH2, R140Q, was detected upon disease progression to first‐line alectinib treatment. Finally, as previously mentioned, a c‐MYC amplification was detected jointly with a loss of CCND1 and of FGFR3.
4. Discussion
ALK‐Is have dramatically improved outcomes in NSCLC patients [17]. However, despite the impressive responses they elicit, patients invariably relapse due to acquired resistance mutations. Solid biopsies remain the gold standard for biomarker testing. However, logistics for obtaining repeat tumor biopsies are complicated and seldom feasible leading to an empirical prescription of sequential ALK‐Is. Nevertheless, blinding treatment sequential strategies might have a deleterious effect on patient's survival due to the incompletely overlapping ALK mutation coverage of different ALK‐Is. In this exploratory analysis, we show that plasma NGS is feasible and we propose several different mechanisms which may underlie resistance to ALK Inhibitors (ALK‐Is). Unfortunately, we did not have available data or plasma/tissue samples collected at baseline in 18 of the 24 patients included in the study, which may constitute an important limitation. Importantly, we also provide an algorithm capable of retrieving somatic mutations in the ALK locus that would otherwise be discarded by the commercial bioinformatic pipeline. As presented in Table 2, the commercial pipeline only detected three out of 12 mutations. MAFs of variants detected by the commercial pipeline were 2.8%, 2.1%, and 0.4%. According to the manufacturer's specifications, the LOD, in terms of MAF, for mutations is 0.1%. However, in our hands, mutations with a MAF below 0.5% are seldom detected by the commercial pipeline. By using the VALK pipeline, some mutations that would otherwise have been missed can be rescued. Yet, confirmation using an alternative technique such as dPCR would be required to rule out false‐positive calls.
Regarding acquired mutations in the ALK locus, our results are consistent with those of previous studies. Specifically, secondary mutations were detected in the plasma samples of 4 of the 11 (36%) patients treated with first‐line crizotinib, with the G1269A mutation being detected in two cases. In this regard, mutation detection rate after crizotinib failure might vary from 60% [18] to 24% [19], G1269A being the most prevalent mutation. We also detected the L1196M and S1206Y mutations, which have been reported to occur in 7% and 2% of cases, respectively, of ALK‐positive NSCLC patients treated with crizotinib [11]. Finally, we detected the A1200V mutation after crizotinib failure in one patient. This mutation is also known to appear upon crizotinib progression [18]. In addition, we found that the G1202R mutation was identified in three of the 10 patients (30%) progressing on alectinib. This mutation is known to arise after treatment with second‐generation ALK‐Is [11]. Recently, Noé et al. [20] reported a 53% ALK mutation detection rate in samples obtained post‐progression on alectinib in which G1202R was the most frequent mutation. In our cohort, more than one mutation in ALK locus was detected in two samples collected during second‐ and third‐generation ALK‐I treatment. Likewise, it has been described that ALK resistance mutations become more frequent with each successive generation of ALK‐I as sequential treatment may promote the appearance of resistance mutation at the ALK locus [21].
A reduced number of studies analyzing samples collected upon progression to an ALK‐I by NGS have so far been conducted [11, 18]. To our knowledge, there are only two studies describing NGS analysis of EML4‐ALK NSCLC liquid biopsy samples using Oncomine™ Pan‐Cancer Cell‐Free Assay and both are case reports [22, 23]. Overall all, our results are in agreement with other studies [11]. However, some genes such as IGF‐1R and SRC, which have been described to be relevant for ALK resistance, were not included in the used panel. Genomic alterations responsible of treatment failure can be detected analyzing different components of bloodstream, most notably circulating tumor cells (CTCs) and cfDNA [19, 24, 25]. However, it has been proposed that cfDNA is the best strategy for sequencing analyses [26]. Nonetheless, assays for the detection of EML4‐ALK fusion protein in CTCs have been developed [27]. Consistent with our results, mutations in TP53, FGFR2, PIK3CA, and MET have been identified in the tumor biopsy of patients progressing on ceritinib [11]. The E545K and E545A mutations in PIK3CA have not only been detected upon progression in advanced ALK‐positive NSCLC patients, but also in EGFR‐positive NSCLC patients [28, 29]. The IDH2 R140Q detected in our cohort is known to transform cells in vitro and induces myeloid and lymphoid neoplasms in mice [30, 31]. This mutation is also frequent in angioimmunoblastic T‐cell lymphoma [32]. In NSCLC, IDH1/2 mutations are rarely detected in primary tumors but it has been suggested that they could be branching drivers leading to subclonal evolution, based on the MAFs at which these mutations are detected [33]. In this way, Zhao et al. [34] described a case of an ALK‐positive tumor in which an IDH1 variant was detected upon disease progression.
Also, we found the E746_A750del mutation in one patient who did not benefit from treatment with ALK‐Is. Unfortunately, we could not orthogonally validate the presence of the ALK translocation nor the EGFR variant at baseline due to lack of tissue, which may constitute a limitation of the present study. However, it has been previously reported that mutations in EGFR in some NSCLC tumors coexist alongside ALK rearrangements, although this is, at best, a rare event [35, 36]. Concomitant ALK and EGFR alterations may lead to primary resistance to ALK‐I [37]. Likewise, a non‐V600 BRAF mutation was detected after 3 months of treatment with second‐line ceritinib treatment, suggesting that resistance of the tumor to the ALK‐I could be due to the acquisition of the BRAF mutation. It has been reported that ceritinib enhances the efficacy of trametinib, a MEK inhibitor, in BRAF/NRAS‐wt melanoma cell lines [38], which makes it plausible that ceritinib would not have any effect in BRAF‐mutated cells. Finally, two patients in whose plasma sample the F129L‐activating mutation in MAP2K1 (MEK1) was detected, exhibited marked resistance to second‐ and third‐generation ALK‐Is. Of note, we did not detect this mutation at baseline in one of these patients, supporting its role as a resistance mutation. In this way, this mutation has been identified as the molecular mechanism underlying MEK/ERK pathway activation in resistant clones of human HT‐29 colon cancer cells [39]. Moreover, the activation of this downstream pathway is critical to the survival of ALK‐positive NSCLC cells [40, 41]. Indeed, the combination of ALK and MEK inhibition was highly effective at suppressing tumor growth in a preclinical model of EML4‐ALK NSCLC [42]. Taken together, it is plausible that the F129L‐activating mutation in MAP2K1 is an acquired mutation that leads to tumor resistance to ALK‐Is.
Mutations in the FGFR2 and FGFR3 genes were detected in two patients progressing on ALK‐Is, suggesting sensitivity to fibroblast growth factor receptor inhibitors. It has been reported that alectinib, despite being a potent ALK‐I, has limited inhibitory activity against other protein kinases such as FGFR2 [43]. In this way, clinical trials evaluating the efficacy of combinations of ALK‐Is with FGFR inhibitors would be of particular interest.
Three CNVs in c‐MYC, CCND1, and FGFR3 were detected upon disease progression in one patient, who was being treated with crizotinib. Remarkably, c‐MYC amplification determines many oncogenic effects [44] and it has been identified as a potential mechanism of primary resistance to crizotinib in ALK‐rearranged NSCLC patients [45]. It has been previously suggested by Alidousty et al. that co‐occurrence of early TP53 mutations in ALK + NSCLC can lead to chromosomal instability. Specifically, authors reported that, in a subset of 53 ALK + tumors, up to a quarter of TP53‐mutated tumors showed amplifications of known cancer genes such as MYC or CCND1 [46]. Consistent with this, we detected the P92A and V157F mutations in the TP53 gene in the same plasma sample of this patient.
5. Conclusions
In conclusion, our data show that molecular mechanisms underlying treatment failure seem to involve different pathways. NGS analysis of liquid biopsies collected upon disease progression is feasible and a valuable approach toward personalized that will lead to better care for ALK‐positive NSCLC patients.
Conflict of interest
MP reports personal fees from Roche, BMS, MSD Pfizer, Lilly, Novartis, and Takeda grants and personal fees from AstraZeneca, and Boehringer during the conduct of the study. VC reports personal fees from Roche BMS, MSD, Pfizer, Lilly, AstraZeneca, Boehringer, Novartis, Takeda, during the conduct of the study. MD reports personal fees from Astra‐Zeneca, BMS, Boehringer Ingelheim, MSD, Pfizer and Roche. The rest of the authors have declared no conflict of interest.
Author contributions
AR and MP conceived and /or designed the work. ES and RS have carried out statistical analyses. VI has developed VALK tool. All authors have made substantial contributions to the acquisition and interpretation of data. AR and ES have drafted and revised the manuscript. All authors have approved the final version. Each author agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/1878‐0261.13033.
Supporting information
Fig. S1. Flowchart of the bioinformatic pipeline optimized for the processing and assessment of variants at the ALK gene locus.
Fig. S2. Frequency of ALK missense mutations identified in the study population.
Fig. S3. PFS and OS curves according to F129L (MAP2K1) mutation status.
Table S1. Identification of EML4‐ALK translocation.
Table S2. Genes included in the NGS panel used.
Table S3. DNA genotyping of PBMCs.
Table S4. List of mutations in ALK locus tested by dPCR.
Table S5. ALK‐Is treatments of the study cohort.
Table S6. List of all somatic mutations detected in the study cohort.
Table S7. Cross‐table describing PPA, NPA and ORA for ALK cohort using VALK tool.
Table S8. Cross‐tables describing PPA, NPA and ORA for Valencia cohort using VALK tool.
Table S9. Cross‐tables describing PPA, NPA and ORA for ALK cohort using the Oncomine Filter.
Table S10. Cross‐tables describing PPA, NPA and ORA for Valencia cohort using the Oncomine Filter.
Table S11. List of the mutations detected at disease progression and status at baseline or in previous sample.
Acknowledgements
The authors wish to thank the donors, and the BIOBANK HOSPITAL UNIVERSITARIO PUERTA DE HIERRO MAJADAHONDA (HUPHM)/INSTITUTO DE INVESTIGACIÓN SANITARIA PUERTA DE HIERRO‐SEGOVIA DE ARANA (IDIPHISA) (PT17/0015/0020 in the Spanish National Biobanks Network) for the human specimens used in this study. This study has been funded by Instituto de Salud Carlos III through the project ‘PI17/01977’ (Co‐funded by European Regional Development Fund/European Social Fund ‘A way to make Europe’/‘Investing in your future’). The work presented in this paper also received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No 875160. ES was funded by the Consejería de Ciencia, Universidades e Innovación of the Comunidad de Madrid (Doctorados Industriales of the Comunidad de Madrid IND2019/BMD‐17258). RS was funded by the Consejería de Educación, Juventud y Deporte of the Comunidad de Madrid and by the Fondo Social Europeo (Programa Operativo de Empleo Juvenil, and Iniciativa de Empleo Juvenil, PEJD‐2018‐PRE/BMD‐8640).
Data accessibility
This paper is available as a preprint in Research Square server, https://doi.org/10.21203/rs.3.rs‐86055/v1 (https://www.researchsquare.com/article/rs‐86055/v1).
References
- 1.Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA, Shapiro GI, Costa DB, Ou S‐HI, Butaney Met al. (2011) Effect of crizotinib on overall survival in patients with advanced non‐small‐cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol 12, 1004–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall Fet al. (2013) Crizotinib versus chemotherapy in advanced ALK‐positive lung cancer. N Engl J Med 368, 2385–2394. [DOI] [PubMed] [Google Scholar]
- 3.Gadgeel SM, Gandhi L, Riely GJ, Chiappori AA, West HL, Azada MC, Morcos PN, Lee R‐M, Garcia L, Yu Let al. (2014) Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib‐resistant ALK‐rearranged non‐small‐cell lung cancer (AF‐002JG): results from the dose‐finding portion of a phase 1/2 study. Lancet Oncol 15, 1119–1128. [DOI] [PubMed] [Google Scholar]
- 4.Duruisseaux M, Besse B, Cadranel J, Pérol M, Mennecier B, Bigay‐Game L, Descourt R, Dansin E, Audigier‐Valette C, Moreau Let al. (2017) Overall survival with crizotinib and next‐generation ALK inhibitors in ALK‐positive non‐small‐cell lung cancer (IFCT‐1302 CLINALK): a French nationwide cohort retrospective study. Oncotarget 8, 21903–21917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huber RM, Hansen KH, Paz‐Ares Rodríguez L, West HL, Reckamp KL, Leighl NB, Tiseo M, Smit EF, Kim D‐W, Gettinger SNet al. (2020) Brigatinib in crizotinib‐refractory ALK+ NSCLC: 2‐year follow‐up on systemic and intracranial outcomes in the phase 2 ALTA trial. J Thorac Oncol 15, 404–415. [DOI] [PubMed] [Google Scholar]
- 6.Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, Ou SI, Pérol M, Dziadziuszko R, Rosell Ret al. (2017) Alectinib versus crizotinib in untreated ALK‐positive non–small‐cell lung cancer. N Engl J Med 377, 829–838. [DOI] [PubMed] [Google Scholar]
- 7.Camidge DR, Dziadziuszko R, Peters S, Mok T, Noe J, Nowicka M, Gadgeel SM, Cheema P, Pavlakis N, de Marinis Fet al. (2019) Updated efficacy and safety data and impact of the EML4‐ALK fusion variant on the efficacy of alectinib in untreated ALK‐positive advanced non‐small cell lung cancer in the global phase III ALEX study. J Thorac Oncol 14, 1233–1243. [DOI] [PubMed] [Google Scholar]
- 8.Lin JJ, Zhu VW, Yoda S, Yeap BY, Schrock AB, Dagogo‐Jack I, Jessop NA, Jiang GY, Le LP, Gowen Ket al. (2018) Impact of EML4‐ALK variant on resistance mechanisms and clinical outcomes in ALK‐positive lung cancer. J Thorac Oncol 14, 1233–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WAet al. (2012) Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non‐small cell lung cancer. Clin Cancer Res 18, 1472–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Katayama R, Shaw AT, Khan TM, Mino‐Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes Cet al. (2016) Cancer: mechanisms of acquired crizotinib resistance in ALK‐rearranged lung cancers. Sci Transl Med 4, 120ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, Dagogo‐Jack I, Gadgeel S, Schultz K, Singh Met al. (2016) Molecular mechanisms of resistance to first‐ and second‐generation ALK inhibitors in ALK ‐rearranged lung cancer. Cancer Discov 6, 1118–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okada K, Araki M, Sakashita T, Ma B, Kanada R, Yanagitani N, Horiike A, Koike S, Oh‐hara T, Watanabe Ket al. (2019) Prediction of ALK mutations mediating ALK‐TKIs resistance and drug re‐purposing to overcome the resistance. EBioMedicine 41, 105–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li J, Huang Y, Wu M, Wu C, Li X & Bao J (2018) Structure and energy based quantitative missense variant effect analysis provides insights into drug resistance mechanisms of anaplastic lymphoma kinase mutations. Sci Rep 8, 10664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ettinger DS, Wood DE, Aisner DL, Akerley W, Bauman J, Chirieac LR, D'Amico TA, DeCamp MM, Dilling TJ, Dobelbower Met al. (2017) Non‐small cell lung cancer, version 5.2017: clinical practice guidelines in oncology. J Natl Compr Canc Netw 15, 504–535. [DOI] [PubMed] [Google Scholar]
- 15.Provencio M, Pérez‐Barrios C, Barquin M, Calvo V, Franco F, Sánchez E, Sánchez R, Marsden D, Cristóbal Sánchez J, Martin Acosta Pet al. (2020) Next‐generation sequencing for tumor mutation quantification using liquid biopsies. Clin Chem Lab Med 58, 306–313. [DOI] [PubMed] [Google Scholar]
- 16.Li MM, Datto M, Duncavage EJ, Kulkarni S, Lindeman NI, Roy S, Tsimberidou AM, Vnencak‐Jones CL, Wolff DJ, Younes Aet al. (2017) Standards and guidelines for the interpretation and reporting of sequence variants in cancer. J Mol Diagnostics 19, 4–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gambacorti Passerini C, Farina F, Stasia A, Redaelli S, Ceccon M, Mologni L, Messa C, Guerra L, Giudici G, Sala Eet al. (2014) Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase‐positive lymphoma patients. J Natl Cancer Inst 106, djt378. [DOI] [PubMed] [Google Scholar]
- 18.Yanagitani N, Uchibori K, Koike S, Tsukahara M, Kitazono S, Yoshizawa T, Horiike A, Ohyanagi F, Tambo Y, Nishikawa Set al. (2020) Drug resistance mechanisms in Japanese anaplastic lymphoma kinase‐positive non–small cell lung cancer and the clinical responses based on the resistant mechanisms. Cancer Sci 111, 932–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horn L, Whisenant JG, Wakelee H, Reckamp KL, Qiao H, Leal TA, Du L, Hernandez J, Huang V, Blumenschein GRet al. (2019) Monitoring therapeutic response and resistance: analysis of circulating tumor DNA in patients with ALK+ lung cancer. J Thorac Oncol 14, 1901–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noé J, Lovejoy A, Ou SHI, Yaung SJ, Bordogna W, Klass DM, Cummings CA & Shaw AT (2020) ALK mutation status before and after alectinib treatment in locally advanced or metastatic ALK‐positive NSCLC: Pooled analysis of two prospective trials. J Thorac Oncol 15, 601–608. [DOI] [PubMed] [Google Scholar]
- 21.Dagogo‐Jack I, Rooney M, Lin JJ, Nagy RJ, Yeap BY, Hubbeling H, Chin E, Ackil J, Farago AF, Hata ANet al. (2019) Treatment with next‐generation ALK inhibitors fuels plasma ALK mutation diversity. Clin Cancer Res 25, 6662–6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sánchez‐Herrero E, Clemente MB, Calvo V, Provencio M & Romero A (2020) Next‐generation sequencing to dynamically detect mechanisms of resistance to ALK inhibitors in ALK‐positive NSCLC patients: a case report. Transl Lung Cancer Res 9, 366–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berger LA, Janning M, Velthaus JL, Ben‐Batalla I, Schatz S, Falk M, Iglauer P, Simon R, Cao R, Forcato Cet al. (2018) Identification of a high‐level MET amplification in CTCs and cfTNA of an ALK‐positive NSCLC patient developing evasive resistance to crizotinib. J Thorac Oncol 13, e243–e246. [DOI] [PubMed] [Google Scholar]
- 24.Pailler E, Faugeroux V, Oulhen M, Mezquita L, Laporte M, Honore A, Lecluse Y, Queffelec P, NgoCamus M, Nicotra Cet al. (2019) Acquired resistance mutations to ALK inhibitors identified by single circulating tumor cell sequencing in ALK‐rearranged non–small‐cell lung cancer. Clin Cancer Res 25, 6671–6682. [DOI] [PubMed] [Google Scholar]
- 25.Alix‐Panabières C, Pantel K & Authors C (2021) Liquid biopsy: from discovery to clinical application. Cancer Discov 11, 858–873. [DOI] [PubMed] [Google Scholar]
- 26.Hofman P (2021) Detecting resistance to therapeutic ALK inhibitors in tumor tissue and liquid biopsy markers: an update to a clinical routine practice. Cells 10, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossi E, Aieta M, Tartarone A, Pezzuto A, Facchinetti A, Santini D, Ulivi P, Ludovini V, Possidente L, Fiduccia Pet al. (2021) A fully automated assay to detect the expression of pan‐cytokeratins and of EML4‐ALK fusion protein in circulating tumour cells (CTCs) predicts outcome of non‐small cell lung cancer (NSCLC) patients. Transl Lung Cancer Res 10, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Romero A, Serna‐Blasco R, Alfaro C, Sánchez‐Herrero E, Barquín M, Turpin MC, Chico S, Sanz‐Moreno S, Rodrigez‐Festa A, Laza‐Briviesca Ret al. (2020) ctDNA analysis reveals different molecular patterns upon disease progression in patients treated with osimertinib. Transl Lung Cancer Res 9, 532–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaft JE, Arcila ME, Paik PK, Lau C, Riely GJ, Catherine Pietanza M, Zakowski MF, Rusch V, Sima CS, Ladanyi Met al. (2012) Coexistence of PIK3CA and other oncogene mutations in lung adenocarcinoma‐rationale for comprehensive mutation profiling. Mol Cancer Ther 11, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang F, Travins J, DeLaBarre B, Penard‐Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser Cet al. (2013) Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 340, 622–626. [DOI] [PubMed] [Google Scholar]
- 31.Mylonas E, Janin M, Bawa O, Opolon P, David M, Quivoron C, Bernard OA, Ottolenghi C, DeBotton S & Penard‐Lacronique V (2014) Isocitrate dehydrogenase (IDH)2 R140Q mutation induces myeloid and lymphoid neoplasms in mice. Leukemia 28, 1343–1346. [DOI] [PubMed] [Google Scholar]
- 32.Cairns RA, Iqbal J, Lemonnier F, Kucuk C, De Leval L, Jais JP, Parrens M, Martin A, Xerri L, Brousset Pet al. (2012) IDH2 mutations are frequent in angioimmunoblastic T‐cell lymphoma. Blood 119, 1901–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez EF, De Marchi F, Lokhandwala PM, Belchis D, Xian R, Gocke CD, Eshleman JR, Illei P & Li MT (2020) IDH1 and IDH2 mutations in lung adenocarcinomas: evidences of subclonal evolution. Cancer Med 9, 4386–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao P, Peng L, Wu W, Zheng Y, Jiang W, Zhang H, Tong Z, Liu L, Ma R, Wang Let al. (2019) Carcinoma of unknown primary with EML4‐ALK fusion response to ALK inhibitors. Oncologist 24, 449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang JJ, Zhang XC, Su J, Xu CR, Zhou Q, Tian HX, Xie Z, Chen HJ, Huang YS, Jiang BYet al. (2014) Lung cancers with concomitant egfr mutations and ALK rearrangements: Diverse responses to EGFR‐TKI and crizotinib in relation to diverse receptors phosphorylation. Clin Cancer Res 20, 1383–1392. [DOI] [PubMed] [Google Scholar]
- 36.Ulivi P, Chiadini E, Dazzi C, Dubini A, Costantini M, Medri L, Puccetti M, Capelli L, Calistri D, Verlicchi Aet al. (2016) Nonsquamous, non‐small‐cell lung cancer patients who carry a double mutation of EGFR, EML4‐ALK or KRAS: frequency, clinical‐pathological characteristics, and response to therapy. Clin Lung Cancer 17, 384–390. [DOI] [PubMed] [Google Scholar]
- 37.Tiseo M, Gelsomino F, Boggiani D, Bortesi B, Bartolotti M, Bozzetti C, Sammarelli G, Thai E & Ardizzoni A (2011) EGFR and EML4‐ALK gene mutations in NSCLC: a case report of erlotinib‐resistant patient with both concomitant mutations. Lung Cancer 71, 241–243. [DOI] [PubMed] [Google Scholar]
- 38.Verduzco D, Kuenzi BM, Kinose F, Sondak VK, Eroglu Z, Rix U & Smalley KSM (2018) Ceritinib enhances the efficacy of trametinib in BRAF/NRAS‐wild‐type melanoma cell lines. Mol Cancer Ther 17, 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H, Daouti S, Li WH, Wen Y, Rizzo C, Higgins B, Packman K, Rosen N, Boylan JF, Heimbrook Det al. (2011) Identification of the MEK1(F129L) activating mutation as a potential mechanism of acquired resistance to MEK inhibition in human cancers carrying the B‐Raf V600E mutation. Cancer Res 71, 5535–5545. [DOI] [PubMed] [Google Scholar]
- 40.Hrustanovic G & Bivona TG (2015) RAS‐MAPK in ALK targeted therapy resistance. Cell Cycle 14, 3661–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S, Blakely CM, Okimoto RA, Lin L, Neel DS, Sabnis Aet al. (2015) RAS‐MAPK dependence underlies a rational polytherapy strategy in EML4‐ALK‐positive lung cancer. Nat Med 21, 1038–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger Pet al. (2014) Patient‐derived models of acquired resistance can identify effective drug combinations for cancer. Science 346, 1480–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sakamoto H, Tsukaguchi T, Hiroshima S, Kodama T, Kobayashi T, Fukami TA, Oikawa N, Tsukuda T, Ishii N & Aoki Y (2011) CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell 19, 679–690. [DOI] [PubMed] [Google Scholar]
- 44.Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI & Young RA (2012) Transcriptional amplification in tumor cells with elevated c‐Myc. Cell 151, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rihawi K, Alfieri R, Fiorentino M, Fontana F, Capizzi E, Cavazzoni A, Terracciano M, La Monica S, Ferrarini A, Buson Get al. (2019) MYC amplification as a potential mechanism of primary resistance to crizotinib in ALK‐rearranged non‐small cell lung cancer: a brief report. Transl Oncol 12, 116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alidousty C, Baar T, Martelotto LG, Heydt C, Wagener S, Fassunke J, Duerbaum N, Scheel AH, Frank S, Holz Bet al. (2018) Genetic instability and recurrent MYC amplification in ALK‐translocated NSCLC: a central role of TP53 mutations. J Pathol 246, 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Flowchart of the bioinformatic pipeline optimized for the processing and assessment of variants at the ALK gene locus.
Fig. S2. Frequency of ALK missense mutations identified in the study population.
Fig. S3. PFS and OS curves according to F129L (MAP2K1) mutation status.
Table S1. Identification of EML4‐ALK translocation.
Table S2. Genes included in the NGS panel used.
Table S3. DNA genotyping of PBMCs.
Table S4. List of mutations in ALK locus tested by dPCR.
Table S5. ALK‐Is treatments of the study cohort.
Table S6. List of all somatic mutations detected in the study cohort.
Table S7. Cross‐table describing PPA, NPA and ORA for ALK cohort using VALK tool.
Table S8. Cross‐tables describing PPA, NPA and ORA for Valencia cohort using VALK tool.
Table S9. Cross‐tables describing PPA, NPA and ORA for ALK cohort using the Oncomine Filter.
Table S10. Cross‐tables describing PPA, NPA and ORA for Valencia cohort using the Oncomine Filter.
Table S11. List of the mutations detected at disease progression and status at baseline or in previous sample.
Data Availability Statement
This paper is available as a preprint in Research Square server, https://doi.org/10.21203/rs.3.rs‐86055/v1 (https://www.researchsquare.com/article/rs‐86055/v1).