

Addendum to: Nature 10.1038/s41586-020-2308-7 Published online 27 May 2020
This analysis explores the extent of loss-of-function (LoF) tolerance in human disease genes.
Databases of human population genetic variation, such as the Genome Aggregation Database (gnomAD), are generally expected to be depleted for variation with severe effects on health. As such, it is expected that genes that carry highly disruptive changes, predicted (p)LoF variants, in these databases are less likely to be responsible for severe human disease. However, the precise relationship between pLoF tolerance and human disease causation is not well-characterized.
In our Article, we reported a total of 2,636 variants in 1,815 genes that were homozygous in at least one individual and annotated as pLoF after applying both automated filtering and manual curation of both sequencing quality and functional annotation. We labelled these genes as ‘LoF-tolerant’, indicating that total functional loss of these genes appears to be compatible with life. This does not exclude the involvement of these genes in diseases compatible with presence in individuals in gnomAD1. Neither the ‘LoF Transcript Effect Estimator’ (LOFTEE) nor manual curation took previous gene–phenotype associations into account, as this would create a bias that affects downstream analyses and also may result in the spurious exclusion of true LoF-tolerant genes owing to previous false-positive reported associations with disease. This unbiased approach is appropriate for permitting downstream analyses, but it means that the enrichment of pLoF artefacts will remain higher in genes for which genetic disruption is genuinely associated with severe disease.
Prompted by comments on our original Article, we explored the degree to which our LoF-tolerant list includes genes associated with disease by manually curating the 158 genes (with 217 pLoF variants) on the LoF-tolerant list associated with autosomal recessive and X-linked traits in ‘Online Mendelian Inheritance in Man’ (OMIM) by an additional biocurator1.
Of these genes, 71% (n = 112) are associated with phenotypes that are likely to be found in gnomAD, on the basis of gnomAD inclusion criteria. These are phenotypes such as infertility, hearing or visual impairment, benign or mild metabolic or haematological phenotypes, expected at similar frequency as the general population (95 phenotypes) and, to a lesser extent, traits that are likely to be depleted from gnomAD, but for which someone with the condition may participate in a common disease study (17 phenotypes). We observed an overrepresentation of traits that are likely to be found (60% versus 33%) and an underrepresentation of traits that are not expected to be found (29% versus 53%) in gnomAD (early-onset severe or lethal rare disease that generally would restrict participation in genetic studies) versus a control set of 100 random selected autosomal recessive and X-linked OMIM traits (P = 3.0 × 10−5, Fisher’s exact test) (Fig. 1a). We performed a thorough literature review of the 46 phenotypes that were initially not expected to be found in gnomAD, which revealed that 35% (16 out of 46) can be explained by evidence of mechanism of disease not being LoF (n = 2), variable expressivity (n = 5) or penetrance (n = 3), phenotype being responsive to treatment (n = 4) and onset after age of the individual in gnomAD (n = 2) (Fig. 1b, blue).
Fig. 1. Assessment of pLoF variants in LoF-tolerant genes associated with autosomal recessive and X-linked phenotypes in OMIM.
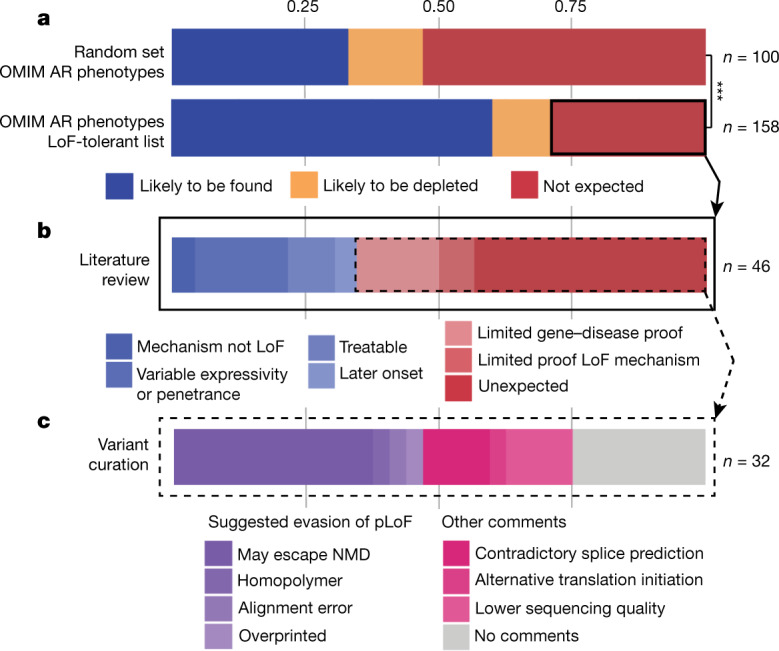
a, Autosomal recessive and X-linked (AR) OMIM phenotypes: likely to be found (blue), likely to be depleted (yellow) or not expected (red) to be found in gnomAD, for the 158 phenotypes associated with LoF-tolerant genes in gnomAD and a set of 100 randomly selected AR and X-linked OMIM traits. ***P = 3.0 × 10−5, Fisher’s exact test. b, Extended literature review of the 46 out of 158 OMIM phenotypes not expected to be found in gnomAD. c, Extended variant curation of 32 pLoF variants in 30 LoF-tolerant genes beyond criteria presented in our original Article revealed pLoF with suggested evasion of pLoF (purple), and pLoF with no conclusive (pink) or no evidence (grey) contradicting pLoF in these genes. NMD, nonsense-mediated decay. Further details are provided in Supplementary Table 1.
In contrast to what is expected to be found in gnomAD, 32 pLoF variants are in 30 genes for which homozygous LoF has been associated with severe or lethal phenotypes in OMIM. However, 10 of these 30 genes had a limited number of cases reported (n = 7) or no reported biallelic LoF variants in humans (n = 3) (Fig. 1b, light red) and only 5 genes meet current ClinGen standards for a known LoF mechanism2. We evaluated the 32 variants by applying more stringent criteria, and identified several cases in which a variety of mechanisms may result in an evasion of true loss of gene function. For 15 variants, we found evidence that disputed our previous prediction (Fig. 1c, purple), including variants that are suspected to escape nonsense-mediated decay but that did not meet the criteria for rescue applied in our original Article (n = 12), one variant that was within a small homopolymer and thus is more likely to represent a sequencing error, one alignment error, and one variant that is in an overprinted transcript and is more probably a synonymous variant in the most biologically relevant transcript. For the 17 variants for which we cannot identify conclusive (n = 9) (Fig. 1c, pink) or any (n = 8) (Fig. 1c, grey) evidence for evasion of pLoF, there are several explanations that even our stringent curation cannot confidently exclude: for example, sample swaps, a variety of residual sequencing and annotation artefact classes, the presence of an individual in gnomAD who does actually have the expected phenotype, or simply variable expressivity, late age of onset or reduced penetrance of the disease phenotype itself. Further details regarding variant curation are are available in Supplementary Table 1 and from https://gnomad.broadinstitute.org/downloads, or the curation data can be viewed at the respective gene page at https://gnomad.broadinstitute.org.
In summary, this result emphasizes the well-established need for extremely careful curation of any pLoF variant observed in a population database such as gnomAD, especially for genes for which such variants are expected to be deleterious. The variants curated here are found at low frequency and are enriched for both sequencing and annotation errors3,4. This enrichment is expected to be even larger in genes for which inactivation is associated with severe disease, because sequencing and annotation artefacts are distributed approximately uniformly across the genome, whereas true LoF variation is depleted in genes in which it results in a more detrimental effect. Although the pLoF variants found in the gnomAD dataset have been subjected to thorough quality control, any filtration process other than comprehensive experimental validation is insufficient to remove all artefacts.
In conclusion, population databases such as gnomAD are a powerful source of information when predicting human tolerance towards gene disruption. The list of LoF-tolerant genes identified in gnomAD is a useful class for downstream analysis that appears to largely comprise genes for which true homozygous disruption does not cause severe early-onset disease.
Authors S.G. and M.S.-B. carried out the analysis described in this Addendum. K.J.K., A.O.-L. and D.G.M. contributed to the experimental design, and A.O.-L. and D.G.M. supervised the work. S.G., M.S.-B., K.J.K., A.O.-L. and D.G.M. wrote the Addendum. A.O.-L. and D.G.M. contributed equally to this work.
We thank C. Arnoult, P. Ray and N. Thierry-Mieg for presenting the opportunity to further clarify the term LoF tolerance.
Supplementary Information is available in the online version of this Amendment.
Supplementary information
Homozygous pLoF variants in genes associated with autosomal recessive and X-linked phenotypes in OMIM.
Footnotes
Deceased: Pamela Sklar
Lists of authors and their affiliations appear online
Contributor Information
Konrad J. Karczewski, Email: konradk@broadinstitute.org
Daniel G. MacArthur, Email: d.macarthur@garvan.org.au
Genome Aggregation Database Consortium:
Carlos A. Aguilar Salinas, Tariq Ahmad, Christine M. Albert, Diego Ardissino, Gil Atzmon, John Barnard, Laurent Beaugerie, Emelia J. Benjamin, Michael Boehnke, Lori L. Bonnycastle, Erwin P. Bottinger, Donald W. Bowden, Matthew J. Bown, John C. Chambers, Juliana C. Chan, Daniel Chasman, Judy Cho, Mina K. Chung, Bruce Cohen, Adolfo Correa, Dana Dabelea, Mark J. Daly, Dawood Darbar, Ravindranath Duggirala, Josée Dupuis, Patrick T. Ellinor, Roberto Elosua, Jeanette Erdmann, Tõnu Esko, Martti Färkkilä, Jose Florez, Andre Franke, Gad Getz, Benjamin Glaser, Stephen J. Glatt, David Goldstein, Clicerio Gonzalez, Leif Groop, Christopher Haiman, Craig Hanis, Matthew Harms, Mikko Hiltunen, Matti M. Holi, Christina M. Hultman, Mikko Kallela, Jaakko Kaprio, Sekar Kathiresan, Bong-Jo Kim, Young Jin Kim, George Kirov, Jaspal Kooner, Seppo Koskinen, Harlan M. Krumholz, Subra Kugathasan, Soo Heon Kwak, Markku Laakso, Terho Lehtimäki, Ruth J. F. Loos, Steven A. Lubitz, Ronald C. W. Ma, Daniel G. MacArthur, Jaume Marrugat, Kari M. Mattila, Steven McCarroll, Mark I. McCarthy, Dermot McGovern, Ruth McPherson, James B. Meigs, Olle Melander, Andres Metspalu, Benjamin M. Neale, Peter M. Nilsson, Michael C. O’Donovan, Dost Ongur, Lorena Orozco, Michael J. Owen, Colin N. A. Palmer, Aarno Palotie, Kyong Soo Park, Carlos Pato, Ann E. Pulver, Nazneen Rahman, Anne M. Remes, John D. Rioux, Samuli Ripatti, Dan M. Roden, Danish Saleheen, Veikko Salomaa, Nilesh J. Samani, Jeremiah Scharf, Heribert Schunkert, Moore B. Shoemaker, Pamela Sklar, Hilkka Soininen, Harry Sokol, Tim Spector, Patrick F. Sullivan, Jaana Suvisaari, E. Shyong Tai, Yik Ying Teo, Tuomi Tiinamaija, Ming Tsuang, Dan Turner, Teresa Tusie-Luna, Erkki Vartiainen, Marquis P. Vawter, James S. Ware, Hugh Watkins, Rinse K. Weersma, Maija Wessman, James G. Wilson, and Ramnik J. Xavier
Supplementary information
The online version contains supplementary material available at 10.1038/s41586-021-03758-y.
References
- 1.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abou Tayoun AN, et al. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018;39:1517–1524. doi: 10.1002/humu.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacArthur DG, Tyler-Smith C. Loss-of-function variants in the genomes of healthy humans. Hum. Mol. Genet. 2010;19:R125–R130. doi: 10.1093/hmg/ddq365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacArthur DG, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Homozygous pLoF variants in genes associated with autosomal recessive and X-linked phenotypes in OMIM.