

Abstract
The reference human genome sequence is inarguably the most important and widely used resource in the fields of human genetics and genomics. It has transformed the conduct of biomedical sciences and brought invaluable benefits to the understanding and improvement of human health. However, the commonly used reference sequence has profound limitations, because across much of its span, it represents the sequence of just one human haplotype. This single, monoploid reference structure presents a critical barrier to representing the broad genomic diversity in the human population. In this review, we discuss the modernization of the reference human genome sequence to a more complete reference of human genomic diversity, known as a human pangenome.
Keywords: Human Genome Project, diversity, clinical genomics, pangenome
INTRODUCTION
Over the past two decades, we have seen unprecedented advancements in DNA sequencing technologies, bioinformatics, and clinical genetics. It has never been faster, easier, or cheaper to generate and assemble a human genome sequence. An individual’s entire genetic code can be revealed in as little as an hour, allowing researchers to broaden catalogs of human genomic variation around the globe and within clinical cohorts. Additionally, with the recent technological gains of long-read DNA sequencing, we are reaching new community standards for genome sequence quality and completeness (89). In this era where entire chromosomal sequences are correctly assembled (90, 98), we are able to report genomic variation comprehensively, including differences in our genetic code that fall in large repeat-rich regions, which were previously difficult to characterize in clinical genetic and genomic studies. Throughout this period of tremendous progress, the reference human genome sequence has served as the foundation for genetic and epigenetic research and clinical genomics. Each chromosomal reference sequence offers a centralized coordinate system for systematically reporting and comparing results across a multitude of studies around the world. This universal reliance on one reference sequence, derived largely from a single individual (61), has shaped the way we design experiments, construct our bioinformatic tools, and, ultimately, interpret biomedical research results. Although this reference has been incredibly important to human genomic research, it is now clear that a single reference sequence is inadequate to fully detect and catalog sequence variants across the human population (19, 52, 64).
There is broad consensus that it is time to modernize the reference human genome sequence to include a collection of highly accurate, haplotype-phased genome sequence assemblies that better represent all common haplotypes in the human population. This diversity panel requires consistency in quality, in that each reference sequence would need to match or exceed the continuity and base-level accuracy of the most recent reference human genome sequence (GRCh38) (121), to support unbiased and comprehensive variant calling and genomic analyses. This collection of high-quality phased reference sequences would be organized into a multigenome, or pangenome, reference structure (34, 46, 138). Broadening the reference to represent genomic diversity, or allelic variation within and across human populations, will improve variant detection and functional annotation of genomic variants and, ultimately, will be transformative for biomedical research (44, 52).
Resequencing of human genomes and various functional genomics assays presently rely on second-generation sequencing technologies, which generate short sequence reads, and mapping or aligning such reads back to the reference genome sequence. Current short-read alignment and variant detection strategies that rely on a single reference genome sequence can lead to inaccuracies and incomplete assessment, and sequence reads that differ considerably from the reference result in incorrect mapping and false-negative or false-positive variant calls, presenting a common problem known as reference bias (52, 94). In alignment data sets, reference bias affects clinically significant regions of the genome. For example, due to the high levels of sequence diversity in the human leukocyte antigen (HLA) genes, genotyping has been inaccurate. HLA genotype studies from the 1000 Genomes Project previously reported that 18.6% of single-nucleotide polymorphisms (SNPs) identified were incorrect due to reference biases (3, 19). Another area of clinical research negatively affected by reference bias is structural variant discovery. Structural variants are relatively large genome differences (tens to hundreds of bases or longer) and include deletions, insertions, tandem duplications, inversions, and translocations. Algorithms to identify structural variants rely largely on detecting patterns of discordant read pairs or split (short or long) read alignments, an approach that depends on the accuracy of read mapping (8, 124). Reference bias therefore limits the detection of novel structural variants, impacting the characterization of structural variants in both short- and long-read data sets (26, 133). Improved representations of common variants within the pangenome reference will enable short- and long-read alignments directly to shared haplotypes, thereby improving the efficiency of read mapping and the accuracy of variant detection (10, 43, 117).
The development, release, and international acceptance of a new human pangenome reference resource is critical for future biomedical research. Although this goal holds great promise, it presents considerable technical and societal challenges. Similarly to the Human Genome Project, the success of developing a new human pangenome reference will require a large team of scientists with expertise in population genomics, genome sequencing technologies, computational biology, and ethics. Population sampling to support this new effort will require explicit attention to community engagement, inclusion, and fair representation, relying on careful ethical and policy guidance to ensure respectful domestic and international partnerships. Beyond sampling and representation, the pangenome infrastructure depends on the production of high-quality, phased, chromosome-level haplotype sequences that each meet or exceed the quality of the current reference human genome sequence (GRCh38). Data generation to support the assembly of hundreds of new reference genome sequences depends on the production of long-read data in a highly coordinated manner across several centers, all while navigating the ever-changing advancements in genome sequencing technologies and platform innovation. These emerging data sets will serve as a call to arms for computational scientists and software developers, leading to new established methods and paradigms to construct the pangenome infrastructure and tooling to support all downstream analyses. The release of the pangenome reference sequence will transform the way we perform basic and clinical research and lead to improved standards for genomic research, data sharing, and reproducible cloud-based workflows. Because this will not be an incremental change, outreach and training will be critical to ensuring that the pangenome reference resource is widely adopted.
Here, we provide a high-level review of the historic progress that enabled the rise of a human pangenome reference initiative (Figure 1). In particular, we discuss the influence of the Human Genome Project, previous efforts to survey human genomic diversity, and the rise of the field of computational pangenomics (34). We address the need to improve genomic diversity in biomedical research and focus on how a human pangenome reference is an important step for addressing this imbalance. Like the Human Genome Project, the human pangenome reference initiative is a big-science effort (32), whose success relies on the collaborative organization of a large, multidisciplinary team of genomic researchers, geneticists, computational biologists, policy experts, and ethicists to navigate the inherent technological and societal challenges that will be faced. Nevertheless, this important and historic initiative is expected to accelerate genotype-to-phenotype studies, drive technology innovation, and enable a new era of human biomedical research.
Figure 1.
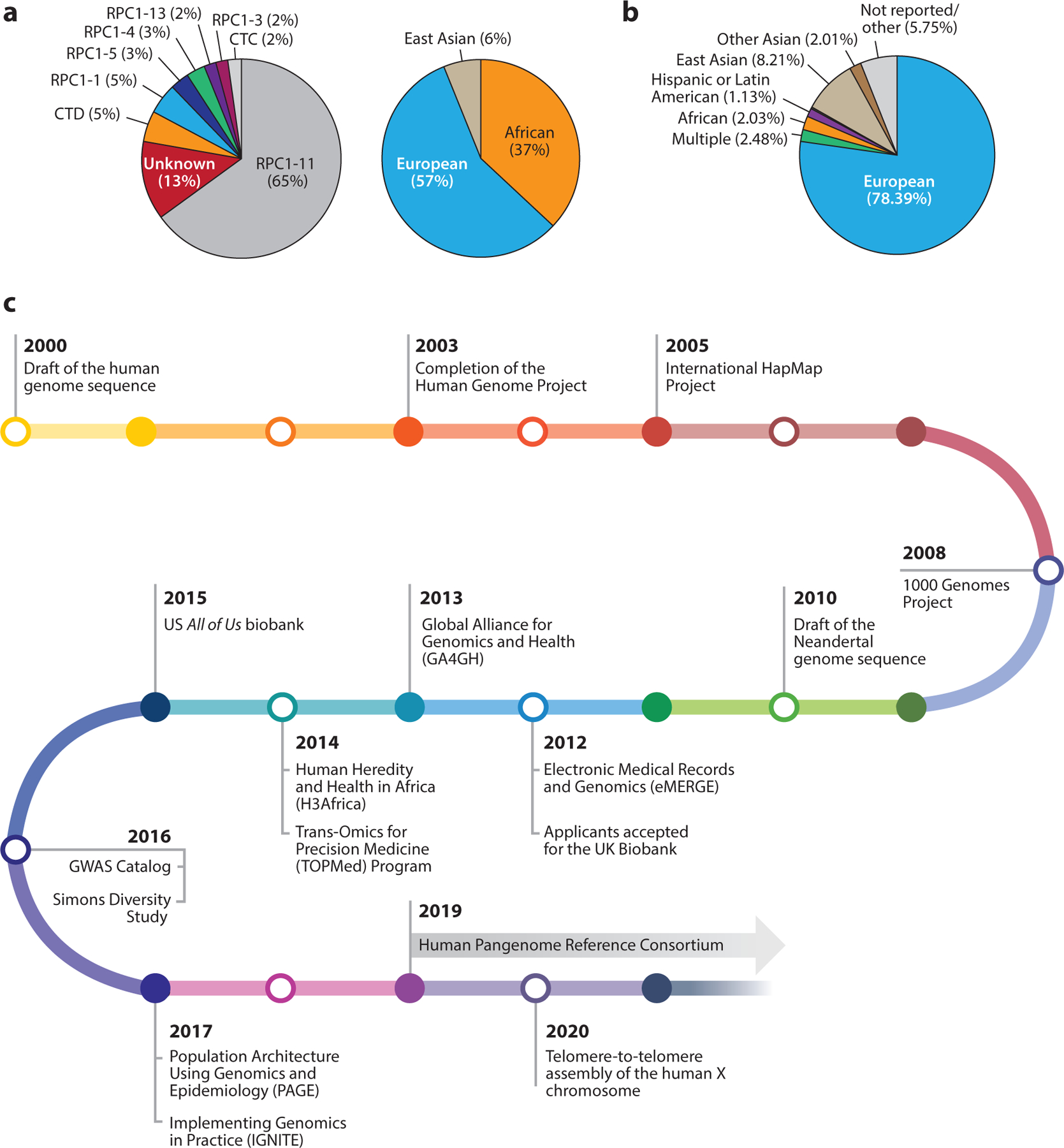
Historic progress over the last 20 years has enabled the launch of the human pangenome reference initiative. (a) Genomic representation of the current reference human genome sequence, as determined by Green et al. (61), demonstrated that the majority of the reference human genome sequence (65%) is derived from a single bacterial artificial chromosome library (RPCI-11). Further evaluation of the reference sequence (GRCh37) revealed that it reflected DNA from one male donor who was 37% African and 57% European. (b) This underrepresentation of diversity is also reported in genome-wide association databases, where the vast majority of data are from people of European ancestry (78%). (c) Following the first release of the reference human genome sequence, there have been several large initiatives to prioritize the inclusion of more diverse participants in human genomic research. In the past 20 years, several efforts have been launched to expand both domestic and international surveys of genomic diversity as well as to develop data infrastructure and governance, with the goal of improving the implementation of actionable findings. These big-science investments and initiatives have enabled the launch of the Human Pangenome Reference Consortium, a group responsible for leading a five-year initiative that aims to enhance the reference human genome sequence in a fashion that better represents common haplotypes in the human population. Panel a adapted from Reference (127); panel b adapted from Reference (100).
THE LEGACY OF THE HUMAN GENOME PROJECT
Following the historic race between the publicly funded International Human Genome Sequencing Consortium and Celera Genomics to sequence the human genome, the first draft human genome sequences were reported a little over 20 years ago in two monumental publications, one in Nature (85) and the other in Science (137). The celebrated release of the draft human genome sequence offered the first base-level resolution and order of tens of thousands of genes, organized along each chromosome. Although this early information was important for the advancement of biomedical research, arguably one of the most profound legacies of the Human Genome Project was that access to these more comprehensive maps rapidly led to the prioritization of genome-wide studies: Single-gene-based studies now faced the challenge of understanding broader genomic structure, causal alleles and disease association could now be mapped and studied across long intergenic regions, and local repeat structure and organization presented new models for gene regulation. In other words, the first release of the human genetic blueprint required a seismic shift in technology development to ensure that research could broaden in scope to operate at the genomic level. Furthermore, the ordered bases of each human chromosome offered an entirely new infrastructure—a centralized coordinate system for genomic research. Stewardship of this infrastructure through decades of work by the Genome Reference Consortium (30) ensured that discoveries from around the world were reported with base-level precision and that these results could be replicated, benchmarked, and studied in combination to advance basic and translational research. The ceremonial announcement and subsequent publications reporting a draft sequence of the human genome marked the beginning of a new age for genomic research, one that was dependent on the accuracy and representation of a single monoploid reference sequence derived primarily from one individual (61) (Figure 1a).
The Human Genome Project launched a new era of genomic research. The need to sequence one human genome quickly transformed into the need to sequence the genomes of many humans (3, 92), the genomes of many species (53), and the genomes of diseased cells (71, 113) in addition to understanding the functional meanings of the generated genome sequences (49, 50). These needs and motivations transformed an entire industry of technologies, with the last two decades bringing profound advancements in DNA sequencing technologies and bioinformatic methods to meet the demands of genome-wide analyses. High-coverage and high-quality whole-genome sequencing is now economical, with headroom for improvement in terms of read lengths, base-level accuracy, and production efficiency. The cost of sequencing a human genome went from $3 billion to less than $1,000. Second- and third-generation DNA sequencing technologies not only have provided the means to sequence a genome, but also have become common platforms for providing reads for numerous genomic assays. The rapid succession and dramatic improvement of DNA sequencing technologies resulted in the production of numerous large sequence files, which prompted the establishment of new sequence data repositories, as well as expectations of reproducible workflows and open data sharing (142). Necessity drives innovation, and with the rapid accumulation of digital sequence data came a need to develop a wide range of new bioinformatic tools to support data management and sequence analyses. Open and responsible genomic data sharing is a legacy of the Human Genome Project and is reflected across data commons ecosystems, including the National Human Genome Research Institute (NHGRI) Genomic Data Science Analysis, Visualization, and Informatics Lab-Space (AnVIL) project (https://anvilproject.org) and global collaborative initiatives, such as the Global Alliance for Genomics and Health (GA4GH) (58), that offer federated ecosystems, standards, and policies for genomic and related health data sharing.
The release of detailed genomic information had momentous societal and ethical implications. Therefore, a dedicated component of the Human Genome Project examined issues of privacy and the prevention of discrimination. The NHGRI’s investment in ethical, legal, and social implications (ELSI) research led to the world’s largest bioethics program, which provided a model for other programs worldwide (96). Overall funding and support for research and education programs aimed at health professionals and the general public resulted in a rapid increase in the understanding of genomic ethics and bioethics. ELSI scholarship, operating in parallel with scientific and technical advances, has led to a great deal of public debate, policy making, and the enactment of national legislation (129). Common infrastructures and research platforms, open access and sharing policies, and new forms of international collaborations in genomic research have led to a need for additional ELSI research to ensure that a coordinated response to societal needs can occur at the global level (21, 80, 83).
The magnitude of the technological challenge and the necessary financial investment prompted the Human Genome Project to take an integrated, large-scale, collaborative approach. In addition to researchers from the United States, the International Human Genome Sequencing Consortium comprised investigators from the United Kingdom, France, Australia, and China, as well as myriad other spontaneous relationships (31). This project led to a new era of team-oriented science; the NHGRI alone has been involved in launching more than 25 analogous projects since 2000 (60). Such an investment in big science included other large, cross-disciplinary genome-related projects—such as the International HapMap Project to study human genomic variation (72) and the Encyclopedia of DNA Elements (ENCODE) Project (49)—that maximized economies of scale by concentrating research in major centers (32). Collectively, the legacies of the Human Genome Project aim to meet the ultimate goal: to better understand how each base in the human genome influences human health and diseases.
For decades, genomic research has invested tremendous amounts of time and resources in decoding a single linear map, largely representing the genome of one individual. Now, however, even in light of the achievements of the Human Genome Project, it is time to reevaluate the capacity and utility of a single reference sequence for the human genome. Even if every chromosome reference sequence were complete from end to end and entirely error-free, it would still be challenging to identify genomic variants correctly, comprehensively, and equally due to sequence mapping inaccuracies in regions that differ in their genomic structure. In an effort to address this challenge, the current reference human genome sequence (GRCh38) included alternative assembled versions of regions with common complex variation (261 alternate loci in total, corresponding to approximately 2% of the total) (121). These alternative assemblies created multimapping challenges, in that complex regions were presented more than once in the reference sequence, which required new alternate-locus-aware variant-calling strategies (77). Additionally, the increase in the amount of genomic variation data in numerous databases [e.g., dbSNP (128)] and other high-quality reference genome sequences with diverse haplotypes made it difficult to host and build tooling to fully integrate these data relative to the reference coordinate system. These challenges motivated the development of a new human pangenome reference structure, one that was fully capable of representing diversity across a large number of genomes and was supported with the necessary bioinformatics pipelines to ensure the mapping of short sequencing reads, detection of genomic variants, and discovery of functional elements (52, 82, 110).
GLOBAL AND POPULATION-BASED SURVEYS OF GENOMIC DIVERSITY
Even before the launch of the Human Genome Project in 1990, it was proposed that global and population-based surveys of genomic diversity were necessary to understand evolution and disease [the Human Genome Diversity Project (25, 87)]. Shortly after the release of the first reference human genome sequence, the international human genomics community took the next logical step—to begin cataloging genomic differences among humans and decode their importance in health and disease. To build catalogs of common variants in the human genome, researchers began to collect DNA resources from a large number of individuals from around the world. As part of the International HapMap Project (72), investigators analyzed combinations of markers that yield mosaic-like patterns called haplotypes in order to determine the common association patterns of DNA sequence variation in the human genome. The two phases of this project (73, 74) collectively characterized more than 3.1 million SNPs in 270 individuals from four geographically diverse populations and helped launch a massive wave of genome-wide association studies (91).
The International HapMap Project was followed by the 1000 Genomes Project, which continued to establish a more detailed catalog of common and low-frequency variants from more than 2,500 individuals from 26 diverse populations (1–3, 133). At its conclusion, the latter project was credited with cataloging more than 99% of all common human genomic variants (that could be confidently mapped to the single reference human genome sequence) as well as many rare variants, which was immensely valuable for studies that relate genomic variation to health and disease. Efforts to increase sequence coverage of the 1000 Genomes Project resources (40) or broaden sequencing efforts to include more diverse populations outside of the 1000 Genomes Project collection (14, 92) and archaic human genomes (61, 97) have resulted in highly valuable short-read Illumina data sets that have established a foundation to explore how the human genome has evolved over time and how that evolution influences human health.
More recently, improvements in long-read DNA sequencing technologies have enriched our surveys to now include maps of human structural variants (26). Accurate detection of large, complex structural variants and genomic rearrangement is important in biomedical research; such events are three times as likely as single-nucleotide variants to be associated with a genome-wide significant trait locus, and structural variants are as much as 50 times as likely as single-nucleotide variants to affect gene expression (28). Importantly, expanding long-read and long-range genome sequencing studies revealed a large structural variant data set, which has enabled at least a threefold increase in structural variant detection compared with data sets from the 1000 Genomes Project and other available standard high-throughput genome sequencing studies. This would suggest that our survey of human genomic variation is not yet complete. Further improvements in DNA sequencing and variant calling technologies will undoubtedly provide a more comprehensive understanding of large genomic variation.
In addition to global sampling efforts, there has been a dramatic increase in regional and clinical biobanks (22, 101, 132). These local biobanks, which are composed of a collection of biological samples donated by thousands of individuals from the general population, offer the ability to derive a unique survey of regional population allele frequencies (e.g., (144). As such, these repositories are well positioned to link patient genotype data with their medical history and lifestyle information (6). Ultimately, these data will assist in the transition to an era of personalized and precision medicine. Such efforts have expanded on the national level, with notable examples being the UK Biobank (22), BioBank Japan (101), and the US All of Us Research Program (33, 120), which are already contributing to biomedical research and accelerating medical breakthroughs. Although the UK Biobank is restricted to national participants, the collected data are shared openly and responsibly with researchers around the world; this data sharing has enabled a wide range of studies and new discoveries, including in cancer (18), diabetes (139), and heart disease (119). Notably, the impact of this work is in both the data analyses and the establishment of new, collaborative working groups that span scientific, ethical, and legal disciplines (108, 136).
Global and domestic genomic biobanks and diversity initiatives carry enormous potential for advancing scientific discovery in biomedical and basic research. However, these studies are positioned at the crossroads of genetics and society and demand careful policy and ethical oversight to ensure community engagement and respect (59, 123). To help improve partnerships with communities where distrust in science is born out of historical exploitation, future diversity-based genomic initiatives are charged with understanding challenges faced in the past in terms of the ELSI aspects of genetic and genomic research. The 40-year Tuskegee Study of Untreated Syphilis in the African American Male, which involved the US Public Health Service and Center for Disease Control, provides an example of a major violation of ethical standards and severe erosion of public trust (116). Although egregious physical, mental, and emotional research abuses like those that occurred in the Tuskegee study have been outlawed, many individuals remain skeptical of research.
Other obvious instances of research abuse, including the expansive use of HeLa cells, pervaded medical research prior to the Tuskegee experiments, and they have significant implications for the public’s perception of genomic research as well as creating ongoing challenges to informed consent, privacy, and data sharing (16). Early efforts to survey global genomic diversity by the Human Genome Diversity Project (25, 87) were met with great opposition and outrage due to ethical concerns and power asymmetries. In 1993, the World Congress of Indigenous Peoples labeled the Human Genome Diversity Project “the Vampire Project,” saying that it was more interested in collecting blood from indigenous peoples than in their well-being (39). More recently, the Sanger Institute was accused of misusing South African DNA samples in order to commercialize a research tool based on the material, a violation of agreements with African scientists who had collected the sequences (99). Researchers at Arizona State University gathered blood samples from the Havasupai tribe to search for a link to diabetes, then also used the samples to look for other diseases and genetic markers, thereby violating the basic tenets of human subjects research and underlying principles of informed consent (35). Even ongoing efforts, like the National Institutes of Health’s All of Us Research Program, are facing tribal concerns with enrolling Native Americans and looping around tribal sovereign policies (37). Future global and domestic participant recruitment must be keenly aware of controversies that have accompanied past efforts and strive to avoid those unfortunate precedents while building on promising frameworks for collaboration that respect the rights and interests of those who contribute samples.
THE NEED TO IMPROVE GENOMIC DIVERSITY IN BIOMEDICAL RESEARCH
The need to improve the representation of diverse ancestral backgrounds in genomic research is well known (13). The majority of data analyzed in genome-wide association studies, which are used to identify connections between genomic variants and disease risk, come from people of predominantly European descent (Figure 1b). Rationales for this disparity can be attributed to the desire to avoid potential population stratification by focusing on ancestrally homogeneous populations (84). In part, such studies are expected to have a reduced number of participants of non-European ancestry due to the mistrust that has resulted from past exploitation and ethical misuses of genetic and genomic data (7). As a result, in human genomic research studies, cohorts composed almost entirely of people of European ancestry are far larger and better characterized than cohorts of people of non-European ancestry. Individuals with African ancestry harbor the greatest genomic diversity, and greater representation will undoubtedly bring novel insights into genome biology and enable improvements in clinical care. In addition to the scientific justifications for broader inclusion, increasing diversity in genomics has important ethical and public health implications (68). It is increasingly clear that a lack of genomic diversity in basic and clinical research deepens the divide in terms of health disparities.
Efforts are now ongoing to increase diversity in population-based genomic research and to advance translation for improving clinical care (Figure 1c). In particular, it is recognized that multiethnic populations are essential for replicating and refining initial results from genome-wide association studies and for identifying trait-causing genomic variants. The Population Architecture Using Genomics and Epidemiology (PAGE) program, launched in 2008, facilitated fine-mapping of several diseases of public health importance across a large cohort of approximately 100,000 participants, with an emphasis on including individuals with non-European ancestry to better characterize how genetic factors and environmental modifiers influence susceptibility to disease (24, 42, 95). Additionally, it is useful to evaluate conditions that are assumed to be pan-ethnic, as this assignment may be based on incomplete allele frequency data from global populations, and addressing this issue will require recruiting diverse participants and communities. The Human Heredity and Health in Africa (H3Africa) Consortium recently revealed more than 3 million genomic variants with 62 new chromosomal locations that are under strong selection, from the whole-genome sequences of 426 people across 50 ethnolinguistic groups (29).
Ultimately, there is a clear need not only to improve and broaden foundational genomic data resources, but also to ensure that they are accessible for analysis and interpretation and that databases and the literature are well positioned to implement precision medicine in an equitable fashion. The Clinical Sequencing Evidence-Generating Research (CSER) Consortium is a national multisite research program that studies the effectiveness of integrating genome sequencing into the clinical care of diverse and medically underserved individuals (9). Implementing Genomics in Practice (IGNITE) offers better communication strategies tailored for diverse patients in order to bring the value of genomic testing to a broader range of patients (56, 131). One of the aims of the Human Pangenome Reference Consortium is to help address the racial and ethnic biases in genomic resources, most specifically in the reference human genome sequence. Establishing such a reference that better represents human diversity is an important step in addressing the inequity and imbalance of prior human genetic and genomic research.
THE EMERGENCE OF HUMAN PANGENOMICS
The number and scope of human whole-genome sequencing initiatives have grown dramatically since the release of the first reference human genome sequence. We are entering a new era where genome sequencing technologies and computational innovation in long-read sequence assembly can support the production of reference human genome sequence assemblies that meet or exceed the quality of the single reference human genome sequence (125). With the growing collection of available diverse human cohorts, there is an urgency to move away from a single reference and develop novel, qualitatively different computational methods capable of fully integrating variation, in the form of assembled haplotypes, to be analyzed jointly or to be used as a reference.
The availability of several individual human genome sequences motivated the first human pangenome study in 2010, when Wang and colleagues (88) integrated the sequence assemblies of an Asian and an African genomes with the reference human genome sequence available at the time. This study revealed that at least 5 Mb of novel population- or individual-specific sequences in each assembly are not present in the reference human genome sequence. Subsequent studies have continued to underscore the need for and feasibility of building a human pangenome sequence (65, 70, 134). For example, in 2015, a Danish pangenome sequence was generated from 10 trios, revealing hundreds of thousands of novel single-nucleotide variants and insertions or deletions (indels) (15). In 2017, GenomeDenmark sequenced and assembled the genomes of 50 trios as a population reference (93). In 2019, Salzberg and colleagues (126) sequenced 910 individuals of African descent and constructed an African pangenome sequence. They discovered 300 Mb of novel sequences present in the populations of African descent, demonstrating that the African pangenome sequence contains approximately 10% more DNA than the GRCh38 reference human genome sequence. Most recently, in 2020, Kwok and colleagues (143) analyzed 338 high-quality human genome sequence assemblies of genetically divergent human populations to identify missing sequences in the reference human genome sequence with precise breakpoints, and demonstrated that more than 400,000 previously unmapped reads from a given genome sequenced to approximately 40× coverage can now be mapped to a reference sequence using their Human Diversity Reference.
On the forefront of this paradigm shift is the emergence of computational pangenomics (34). This new research subarea of computational biology aims to identify efficient data structures, algorithms, and statistical methods to perform bioinformatic analyses of pangenomes. The transition from a collection of linear reference sequences to a graph-based model, or genome graph, is currently showing improvements in read mapping, variant calling, and haplotype determination and gaining momentum (106). Unlike standard linear references, this structure makes use of information from an entire population to characterize genomic variants with unprecedented accuracy (41). Graph structures have the potential to learn from every new person’s sequenced genome, meaning that the graph-based reference can improve with each additional generated genome sequence. Better still, this improvement happens with only minimal increases in file size, allowing analyses of genomes at a population scale.
In a typical pangenome reference, genomic data from a population can be organized into an edge-based sequence variation graph (Figure 2). In such graphs, edges are the primary data carrier elements, and alternative haplotypes are represented as different paths through the graph. The linear reference assembly forms the graph backbone, and additional variants are added as new edges in the graph. A longer genomic haplotype can be obtained by following a path through the graph and concatenating the (sub)sequences contained by the visited edges. A graph genome reference can contain both small variants (SNPs and indels up to several dozen base pairs in length) and the larger structural variants, which are typically difficult to deal with using short-read DNA sequencing. Consequently, graph references provide the means to both accurately detect structural variation and reduce errors in small variant calls. Alternatively, a diverse collection of genomes can be represented as an acyclic or cyclic sequence graph. Individual genomes in the population can then be identified as paths in the graph. Dilthey and colleagues (38) applied this approach to the HLA region, constructing a directed acyclic graph using multiple data sources: primary and alternative sequences from GRCh37, SNPs identified by the 1000 Genomes Project, and sequences from the International Immunogenetics Information System. SNP inference using the graph reference had high accuracy, with a high concordance of SNPs identified by mapping sequence data to the graph reference and via a SNP genotyping array. This study demonstrated how the diversity of a population can be captured in a single data structure, the graph reference genome sequence.
Figure 2.
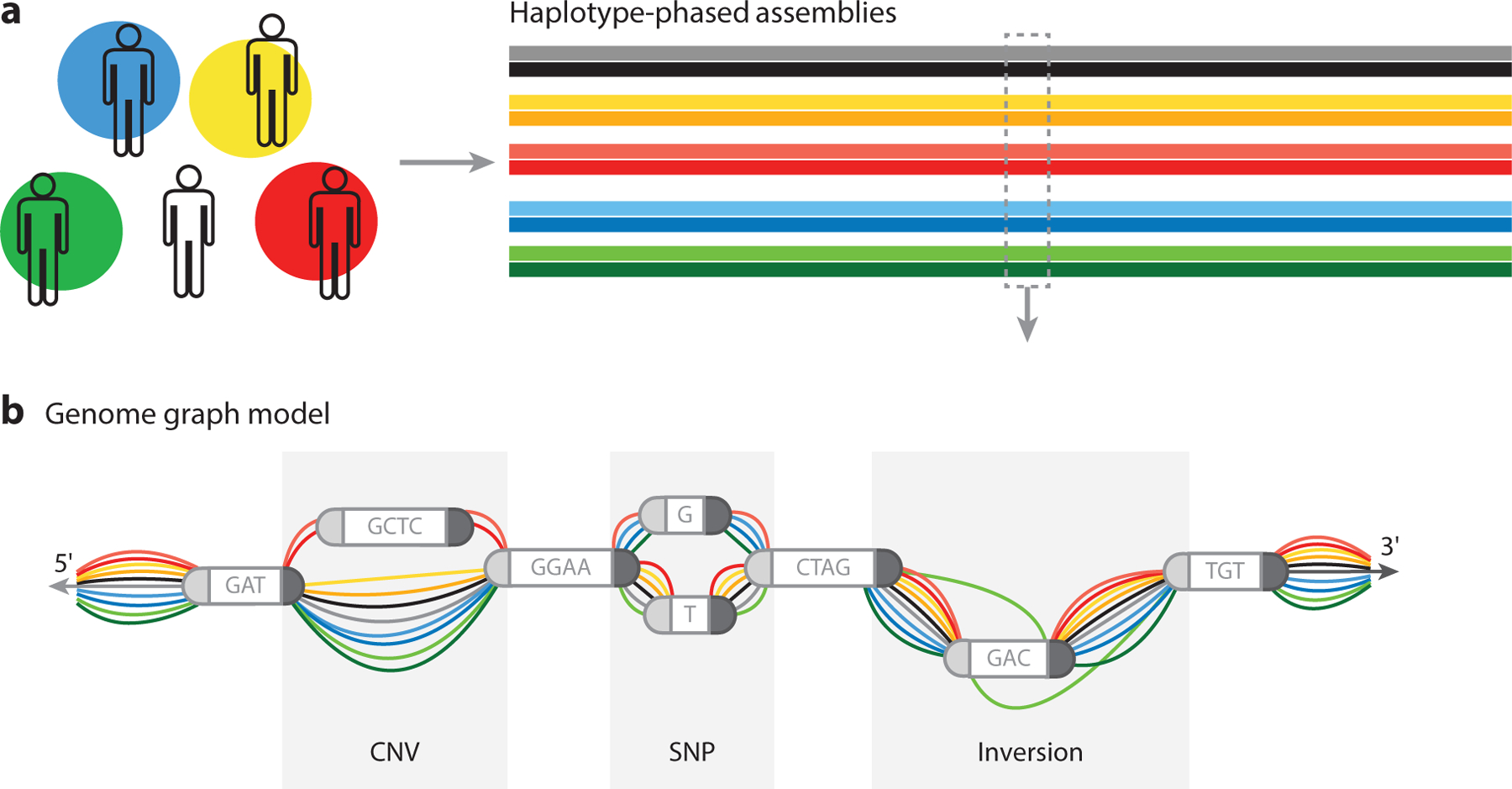
Genome graphs are useful in representing differences in genomic structure. Genomic data from a population can be organized into an edge-based sequence variation graph. (a) Consistency is important. The result of sampling from a population of diverse individuals and whole-genome sequencing is a database of haplotype-phased assemblies (ideally complete, error-free, chromosome-scale sequence assemblies) for each individual. (b) These references can be collectively studied as a graph, where nodes represent sequence information and edges describe the ordering of these sequences in each assembled haplotype. Studying these data reveals sites of copy number variants (CNVs) (regions of insertions or deletions) and sites of single-nucleotide polymorphisms (SNPs), and by creating a data structure with 5′and 3′directions, one can provide an opportunity to track inversions. In addition to reporting and representing these events, there is an opportunity to determine allelic frequency and population-based association of variants.
GENOME TECHNOLOGY AND TELOMERE-TO-TELOMERE ASSEMBLIES
The initial human genome sequence produced by the Human Genome Project provided a baseline to compare with other genomes and has been pivotal for advancing our understanding of basic genome biology, clinical genetics, and human evolution. Although foundational for genetic and genomic research, this initial reference sequence reflected only approximately 95% of the human genome, and approximately 5% remained largely unknown and uncharacterized (45). These missing sequences largely reflect long tracts of repeats, or sequences that are nearly identical and have been historically difficult to clone, sequence, and assemble. The Human Genome Project continued working on finishing the human genome sequence assembly and released the complete sequence in 2003 (75). This assembly was the highest-quality mammalian genome sequence assembly available, but it was not without shortcomings. It contained hundreds of gaps in the euchromatic portion of the genome, in addition to the large, multimegabase gaps in the satellite and duplicated regions. This original human genome reference became a work in progress as efforts started to focus on refinement, gap closure, and correction of assembly and sequencing errors. Yet these gaps were persistent, and notable large gaps on every chromosome, spanning millions of bases in centromeric and the five acrocentric short arms, inhibited comprehensive, genome-wide variant studies.
The ability to generate gapless, telomere-to-telomere, chromosome-level sequence assemblies is now a reality (90, 98). This advance is credited to tremendous improvements in long-read DNA sequencing technologies in terms of overall read length and single-read base-level accuracy. Notably, nanopore-based DNA sequencing platforms (Oxford Nanopore Technologies), where single-molecule DNA sequencing is performed by directly measuring the current changes mediated by DNA passage through the pores, can generate long-read data without a theoretical length limitation. It is possible to routinely generate moderate coverage of reads that are hundreds of kilo-bases in length [ultralong data (78)] with minimal capital cost (125), with an increasing number of reported reads that are greater than a million bases (107). These reads are capable of completely spanning and closing large and persistent assembly gaps in the GRCh38 reference and have been incredibly useful in assembling human centromeric satellite arrays (23, 78, 79, 98). Additionally, even the most difficult repeat regions can be correctly assembled using highly accurate reads (99.9% or Q30) of moderate length (e.g., 10–20 kb), or high-fidelity (HiFi) reads generated using the circular consensus method (Pacific Biosciences) (141). These data provide an opportunity to capitalize on microheterogeneity, or sparsely arranged single-nucleotide differences that alone or in combination are capable of distinguishing one repeat copy from another (103). Indeed, in September 2020, the Telomere-to-Telomere Consortium, an open, community-based effort to generate the first complete assembly of a human genome sequence, announced that they had filled in all the gaps, obtaining complete sequences for all the chromosomes (apart from the five ribosomal DNA arrays) for an effectively haploid cell line [derived from a complete hydatidiform mole (CHM13)]. With continuing gains in both HiFi read length and nanopore single-read base-level quality, it is clear that we are entering a new era of genomics. Soon, telomere-to-telomere chromosome sequence assemblies will be the new standard in population-level genomic studies, enabling the first comprehensive study of variation in the human genome.
THE HUMAN PANGENOME PROJECT
In 2019, the NHGRI funded a new initiative to generate and release a new human pangenome reference, constructed from more than 350 individuals representing highly diverse ethnic backgrounds, to serve as the foundation for future biomedical and genomic medicine research. This effort, led by the Human Pangenome Reference Consortium, falls in step with the legacy of the Human Genome Project in terms of once again relying on a big-science international production that requires working groups of multidisciplinary experts, including leading computational biologists, genomic technologists, population geneticists, and ethicists who can guide the project’s approach to community engagement and sampling (Figure 3). Like the Human Genome Project, this work will push the capabilities of current DNA sequencing technologies and assembly-based methods to reach phased and finished telomere-to-telomere diploid genome sequences for hundreds of reference samples, a goal that is well beyond what has been attempted before. The 350 genomes (700 phased chromosomal haplotypes) are not very useful as a reference on their own, requiring researchers to combine the data and create a unified genomic representation that is both usable and computable. This requires the development of innovative methods in computational pangenomics to ensure that the collection of diverse haploid reference sequences can be weaved together or studied jointly to understand where they are the same and where they differ. This herculean task will drive technology to not only enrich diversity within the reference human genome sequence but also improve common analysis workflows for comprehensive variant calling and biomedical technologies [RNA sequencing (RNA-seq) (140), chromatin immunoprecipitation followed by sequencing (ChIP-seq) (105), assay for transposase-accessible chromatin using sequencing (ATAC-seq) (20), DNase I hypersensitive site sequencing (DNase-seq) (130), micrococcal nuclease digestion with deep sequencing (MNase-seq) (66, 122), formaldehyde-assisted isolation of regulatory elements (FAIRE-seq) (57), etc.], which are driving advances in basic and clinical genomics.
Figure 3.
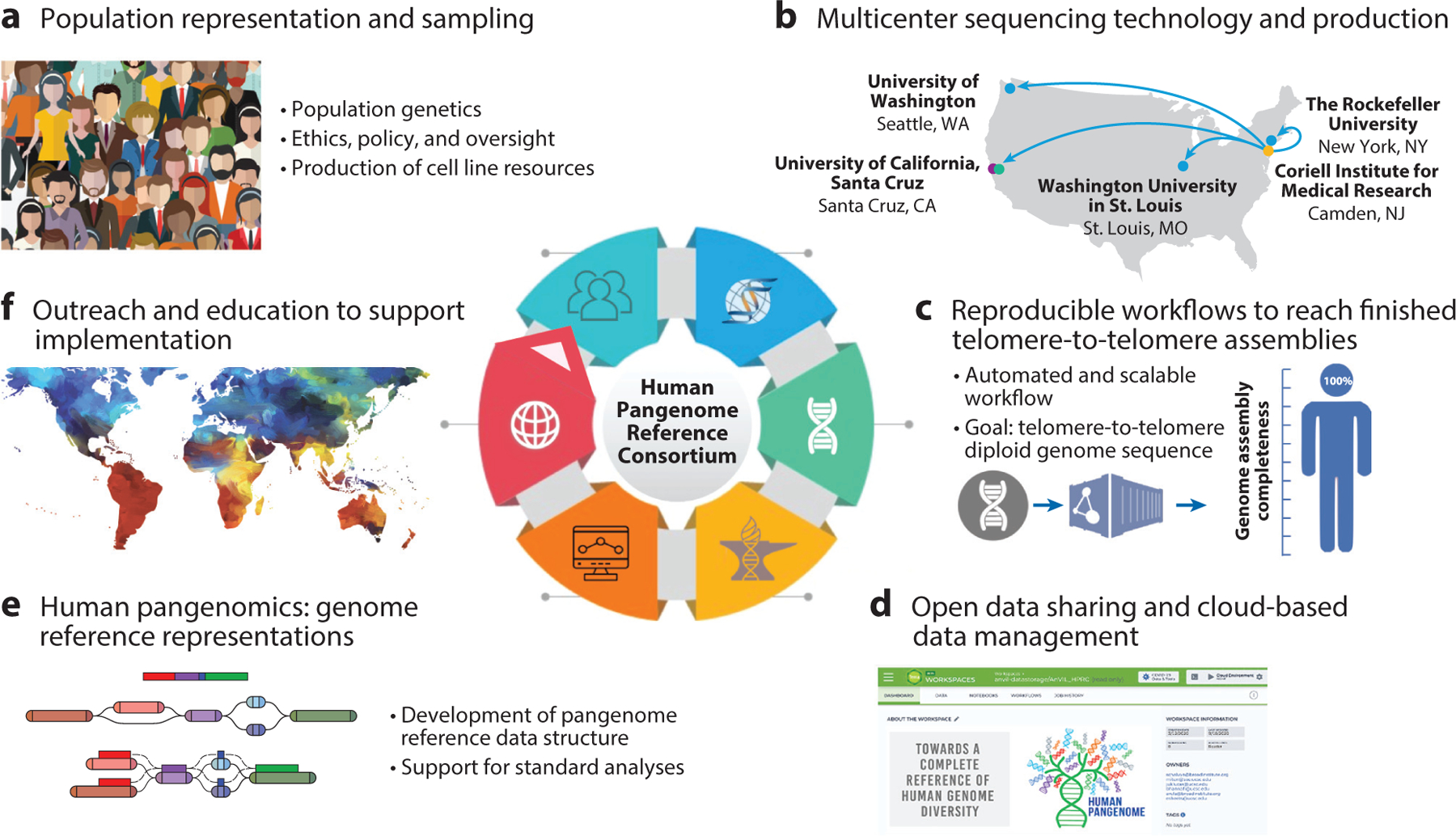
The Human Pangenome Reference Consortium is conducting a big-science initiative that relies on the collaborative organization of a large, multidisciplinary team of geneticists, computational biologists, policy experts, and ethicists. The production effort can be subdivided into six focused areas. (a) First is population representation and sampling, where guidance on participant inclusion is provided by population geneticists, ELSI oversight, and a consent model that is transparent and respectful to communities. Participants in this initiative will provide blood, which will be used to establish cell lines. (b) Cell lines will be used for data production across multiple centers and a broad range of DNA sequencing technologies. The blue circles indicate three production centers involved in the release of long-read HiFi data (Pacific Biosciences), the green circle indicates the nanopore production center for ultra-long data (Oxford Nanopore), the purple circle indicates the production center for Hi-C data (Omni-C, Dovetail Genomics, which has a company partnership with Illumina), and the yellow circle indicates the location of our cell line biorepository (the Coriell Institute for Medical Research). (c) The resulting data management will consist of open, reproducible workflows, with the goal of identifying the best combination of genomic data and computational tools to reach finished, telomere-to-telomere genome sequence assemblies (https://dockstore.org/organizations/HumanPangenome). (d) Data will be made available as soon they are as determined to be of sufficient quality and will be hosted on AnVIL (Terra workspace: https://app.terra.bio/#workspaces/anvil-datastorage/AnVILHPRC) to offer a federated data ecosystem to collaborate with other genomic resources through the adoption of FAIR principles. (e) These assemblies are used for the development of the human pangenome reference data structure (illustrated here as a graph), with tooling, benchmarks, and workflows (illustrating read alignments to graph) to ensure the support of standard analyses in human genetic and genomic research. We thank Jordan Eizenga, from the Genomics Institute at the University of California, Santa Cruz, for sharing the multipath graph illustration. (f) Finally, the consortium will need to form global partnerships, engage in outreach, and provide education to ensure that this resource directly benefits participant communities. Abbreviations: AnVIL, Genomic Data Science Analysis, Visualization, and Informatics Lab-Space; ELSI, ethical, legal, and social implications; FAIR, findable, accessible, interoperable, reusable; HiFi, high fidelity.
The Human Pangenome Project requires innovative genomic advances at a production scale. Current efforts for producing high-quality genome sequence assemblies from a small number of individuals have focused mainly on expensive high-coverage, long-read DNA sequencing and assembly protocols. While this work has been successful in delivering the necessary standards of quality, the overall cost and production time prohibit scaling to hundreds of individuals in a timely manner. A comprehensive representation of common variants will require the sequencing of hundreds of diploid genomes, which in turn will require production consistency across multiple research centers to ensure rapid data generation of consistent quality while maximizing cost-effectiveness. Furthermore, the success of the Human Pangenome Project relies on a coordinated effort to reach the best haplotype-resolved and accurate sequence assemblies using a combination of distinct technology platforms, offering long-read [e.g., Oxford Nanopore Technologies ultralong data (78) and Pacific Biosciences circular consensus sequencing (141)], Hi-C proximity ligation (12, 109), and optical mapping (17) data. Therefore, the project requires inherent flexibility and must be designed to evolve hand in hand with rapidly improving technologies to ensure use of the most powerful and cost-effective DNA sequencing and sequence assembly methods. Additionally, this effort will need to constantly adapt to improved assembly methods to reach haplotype-phased assemblies [e.g., HiFiasm (27)] and reach automated methods to evaluate assembly quality [e.g., Merqury (114) and Asset (63)].
Critical to this work is the commitment to open data sharing and the use of scalable and reproducible cloud-based workflows for reference sequence production, quality assessment, and error correction. To promote cross-system interoperability, the consortium will utilize AnVIL, a unified platform for ingestion and organization for a multitude of current and future genomic and genome-related data sets. This platform offers a federated data ecosystem to collaborate with other genomic resources through the adoption of FAIR (findable, accessible, interoperable, reusable) principles (142). Furthermore, the consortium is aligned with Docker container technology [the Dockstore Project (104)] to ensure the creation of common analytical pipelines and uniform implementation in cloud-computing environments. Importantly, the use of Docker workflows supports future federation [with support from GA4GH (58)] with other similar projects worldwide through an application programming interface (API) that makes it possible to search for containers and workflows across a global network. These collaborative cloud-based environments are useful to ensure that all resources—including study protocols, sample metadata, sequencing metrics, and bioinformatic tools—are made available to the public to broadly motivate biomedical research.
The scope of the construction and utility of a pangenome graph is quite large and is expected to offer a new paradigm for genome analysis. Building from the production of hundreds of high-quality reference genome sequences, the key goal is to create a comprehensive map of genome variation, or to revolutionize the reference data structure and coordinate system in a way that is useful. For the pangenome resource to be useful, it is critical to nucleate and foster an ecosystem of analysis tools for common applications, like read alignment and variant detection and interpretation. This includes not only algorithms and pipelines but also data standards and benchmarks for evaluating performance. For the emerging pangenome reference to be adopted by the community, it is important to include basic information used in human genetic and genomic studies, including automated annotation of genes (51), regulatory elements, and the positions and frequency of variants in the populations. One of the key deliverables of the human pangenome resource is the comprehensive and accurate reporting of genomic variation, which is expected to benefit from pairwise assembly–assembly tools that enable comparison with the reference sequence (5, 10) and pan-assembly alignment and variant-prediction tools (44, 52, 67, 76, 86, 111), with targeted local refinement of more difficult repetitive regions. As part of achieving this goal, there will be an effort to eventually reach a single human pangenome representation that describes all bases and phased haplotype samples and reach consensus new data formats for tool development to ensure broad community usage. Ultimately, the deployment of this resource in the broader community will require a road map for practical applications in real projects. Such outreach to the entire genomics community will rely on consensus regarding formats and data structure to ensure accessibility and implementation.
Studies of human sequence variation and genome structure across the globe operate at the interface of society and science, in which participants and scientists are recognized as partners in research. In diversifying the representation of humanity in the pangenome resource, it is important to establish guiding principles for future community engagement and global partnerships. In expanding partnerships in this project, researchers in the Human Pangenome Reference Consortium will need to develop an ELSI-based framework that enshrines concepts of transparency, engagement, and cultural awareness. The success of the consortium will rest on understanding what policy and ethical precedents were successful in the past and should be built upon and deciding which ones should be avoided (112). Ensuring that this scientific resource is broadly available around the world will require a consent model that facilitates open data sharing of the pangenome resource.
The first phase of the Human Pangenome Reference Consortium’s reference production will focus on existing cell lines generated by the 1000 Genomes Consortium (3), which offer a deep catalog of human genomic variation from 26 populations representing more than 2,500 participants, with a compatible consent for unrestricted (open access) data release. Cell lines from the 1000 Genomes Consortium, which are available in the NHGRI biorepository at the Coriell Institute for Medical Research, are prioritized for this project based on their potential to cover genetic and geographic diversity. These cell lines are from children of trios (to ensure the availability of parental data for the development of methods for haplotype phasing) and are limited in their growth to minimize genetic changes that may occur during extended cell culture.
Although the 1000 Genomes materials offer a powerful resource for diversity studies, they are limited by the size of the cohort and geographical sampling, making them insufficient to support the Human Pangenome Project alone. Additional participant recruitment will require new domestic and international partnerships with resources that provide greater diversity. One such resource is the BioMe BioBank Program, an ongoing, broadly consented electronic health record (EHR)–linked biobank housed in the Mount Sinai Health System that enrolls participants from the New York metropolitan area (6, 11). For hundreds of years, New York City has served as a point of entry to the United States for people from many populations, with recent origins from more than 160 countries worldwide. It will be necessary to expand the network to include a number of domestic biobanks and global partnerships and increase diversity and population representation. Such international genetic and genomic research will involve broader ethical, social, and political considerations and strategic partnerships with organizations such as GA4GH (58). This will broaden interactions in the areas of genomics and ethical oversight and potentially improve equitable educational programs to ensure consistency in training and access to the pangenome reference data.
ADVANCING FUNCTIONAL ANNOTATION OF THE HUMAN GENOME SEQUENCE
The reference human genome sequence provides a platform for annotating the functional elements encoded in the human genome. In parallel with the effort to document genomic diversity, significant efforts have been invested in comprehensively annotating the elements in the human genome that confer function, such as genes, control elements, and transcript isoforms. One representative behemoth effort is the ENCODE Project (49), which was launched in 2003 and has had four phases (47–50). In less than 20 years, the project went from analyzing 1% of the genome using array-based methods in order to map transcribed regions, open chromatin, and regions associated with transcription factors and histone modification in multiple cell lines (48), to defining more than 50,000 protein-coding and noncoding genes and close to 1 million cis-regulatory elements in the human genome, to integrating nearly 10,000 experiments in more than 500 cell and tissue types, and to functionally characterizing some of the annotated elements (50). The ENCODE Project also annotated the genomes of several model organisms, including mouse, fly, and worm (4, 54, 69, 102, 145). These efforts have provided valuable, accessible resources for the research community, and the ENCODE standards have been adopted widely by scientists when conducting functional genomics assays. The ENCODE model also stimulated several large-scale group efforts to expand the functional annotation of the human genome to different dimensions, such as the Roadmap Epigenomics Mapping Consortium (115), the 4D Nucleome Network (36), and the Human Cell Atlas (118). More recently, a new consortium, the Impact of Genomic Variation on Function Consortium, is being established to develop a framework for systematically understanding the effects of genomic variation on genome function and how these effects shape phenotypes. We are at a time when we can not only systematically record the nature of genomic variations among humans but also comprehensively functionalize genomic variants.
By design, an improved reference human genome sequence will provide a better platform for functionally annotating the human genome because it is aimed at comprehensively cataloging and presenting genomic variations relative to the diversity of the human species. At least two immediate efforts can be imagined. First, the vast amount of functional genomics data accumulated over the past decade through the ENCODE, Roadmap Epigenomics, and 4D Nucleome Projects can be reanalyzed in the context of an improved reference human genome sequence. Several studies have already demonstrated that by using more complete reference genome sequences, including in the form of a genome graph, one can readily increase the accuracy of mapping functional genomics data, detect more functional elements, and derive more value from existing data (62). Second, a complete catalog of human genomic variants in the context of their haplotype structures will help investigators better design experiments to test the potential functional consequences of these variants.
An improved reference human genome sequence will also set a new stage for the investigation of not only genomic variation but also epigenetic variation. Much like how the Roadmap Epigenomics Project followed the Human Genome Project to create a reference human epigenome sequence, a more complete and improved human epigenome map will likely follow the generation of an improved reference human genome sequence. Because the epigenome sits at the boundary between the genome and the environment, epigenome variation is both a function of genomic variation and a function of the environment, including both the cellular and developmental environment as well as how organisms interact with the world.
SUMMARY AND OUTLOOK
The Human Genome Project was an ambitious endeavor that changed many things (55). As we reflect on the impact that it has had on the field of genomics, it is apparent that the lessons we have learned are invaluable. In his 2013 State of the Union address, President Barack Obama stated, “Every dollar we invested to map the human genome returned $140 to our economy.” As we write this review, the world is in the midst of the unprecedented and ongoing COVID-19 pandemic. Also unprecedented is how much we appreciate the value of genomics and the importance of knowing our genetic code. Scientists have been able to trace the epidemiology of the SARS-CoV-2 virus using genomic data and determine both why humans are susceptible (81, 147) and why some individuals are more susceptible than others (135, 146). We are racing to develop potential vaccines and therapies using the knowledge of genomics—and we can thank the Human Genome Project for setting the stage to make all of these efforts possible.
We have also come to recognize that the single, monoploid reference structure of the current reference human genome sequence presents a critical barrier to representing the broad genomic diversity in the human population, and to appreciate that new and much-improved reference human genome sequences will further transform genomic medicine and dramatically increase the wealth and value created by the Human Genome Project. The planned human pangenome reference will include a collection of diverse and highly accurate, complete, haplotype-phased genome sequence assemblies. It will also include the algorithmic innovations to efficiently and effectively represent the pangenome, such as the genome graph, along with the software, tools, and data ecosystem for its usage and dissemination. This standardized representation of our species’ collective genome will aid the future of human genetic and genomic research and facilitate the clinical implementation of genomic medicine.
ACKNOWLEDGMENTS
K.H.M. is supported by National Institutes of Health grants R01HG011274 and U01HG010971; T.W. is supported by National Institutes of Health grants R01HG007175, U24ES026699, U01CA200060, U01HG009391, U41HG010972, and UM1HG011585. We thank all members of the Human Pangenome Reference Consortium.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.1000 Genomes Proj. Consort. 2010. A map of human genome variation from population-scale sequencing. Nature 467:1061–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.1000 Genomes Proj. Consort. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491:56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.1000 Genomes Proj. Consort. 2015. A global reference for human genetic variation. Nature 526:68–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abe M, Ishikawa O, Miyachi Y. 1998. Lupoid sycosis successfully treated with minocycline. Br. J. Dermatol 138:199–200 [DOI] [PubMed] [Google Scholar]
- 5.Abel HJ, Larson DE, Regier AA, Chiang C, Das I, et al. 2020. Mapping and characterization of structural variation in 17,795 human genomes. Nature 583:83–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abul-Husn NS, Kenny EE. 2019. Personalized medicine and the power of electronic health records. Cell 177:58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.After Havasupai litigation, Native Americans wary of genetic research. 2010. Am. J. Med. Genet. A 152A:ix [DOI] [PubMed] [Google Scholar]
- 8.Alkan C, Coe BP, Eichler EE. 2011. Genome structural variation discovery and genotyping. Nat. Rev. Genet 12:363–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amendola LM, Berg JS, Horowitz CR, Angelo F, Bensen JT, et al. 2018. The Clinical Sequencing Evidence-Generating Research Consortium: integrating genomic sequencing in diverse and medically underserved populations. Am. J. Hum. Genet 103:319–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Audano PA, Sulovari A, Graves-Lindsay TA, Cantsilieris S, Sorensen M, et al. 2019. Characterizing the major structural variant alleles of the human genome. Cell 176:663–75.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belbin GM, Odgis J, Sorokin EP, Yee M-C, Kohli S, et al. 2017. Genetic identification of a common collagen disease in Puerto Ricans via identity-by-descent mapping in a health system. eLife 6:e25060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belton J-M, McCord RP, Gibcus JH, Naumova N, Zhan Y, Dekker J. 2012. Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58:268–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bentley AR, Callier SL, Rotimi CN. 2020. Evaluating the promise of inclusion of African ancestry populations in genomics. npj Genom. Med 5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bergström A, McCarthy SA, Hui R, Almarri MA, Ayub Q, et al. 2020. Insights into human genetic variation and population history from 929 diverse genomes. Science 367:eaay5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Besenbacher S, Liu S, Izarzugaza JMG, Grove J, Belling K, et al. 2015. Novel variation and de novo mutation rates in population-wide de novo assembled Danish trios. Nat. Commun 6:5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beskow LM. 2016. Lessons from HeLa cells: the ethics and policy of biospecimens. Annu. Rev. Genom. Hum. Genet 17:395–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bocklandt S, Hastie A, Cao H. 2019. Bionano genome mapping: high-throughput, ultra-long molecule genome analysis system for precision genome assembly and haploid-resolved structural variation discovery. Adv. Exp. Med. Biol 1129:97–118 [DOI] [PubMed] [Google Scholar]
- 18.Bradbury KE, Murphy N, Key TJ. 2020. Diet and colorectal cancer in UK Biobank: a prospective study. Int. J. Epidemiol 49:246–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brandt DYC, Aguiar VRC, Bitarello BD, Nunes K, Goudet J, Meyer D. 2015. Mapping bias overestimates reference allele frequencies at the HLA genes in the 1000 Genomes Project phase I data. G3 5:931–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buenrostro JD, Wu B, Chang HY, Greenleaf WJ. 2015. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol 109:21.29.1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burke W, Appelbaum P, Dame L, Marshall P, Press N, et al. 2015. The translational potential of research on the ethical, legal, and social implications of genomics. Genet. Med 17:12–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, et al. 2018. The UK Biobank resource with deep phenotyping and genomic data. Nature 562:203–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bzikadze AV, Pevzner PA. 2020. Automated assembly of centromeres from ultra-long error-prone reads. Nat. Biotechnol 38:1309–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carlson CS, Matise TC, North KE, Haiman CA, Fesinmeyer MD, et al. 2013. Generalization and dilution of association results from European GWAS in populations of non-European ancestry: the PAGE study. PLOS Biol. 11:e1001661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cavalli-Sforza LL. 2005. The Human Genome Diversity Project: past, present and future. Nat. Rev. Genet 6:333–40 [DOI] [PubMed] [Google Scholar]
- 26.Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D, et al. 2019. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun 10:1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng H, Concepcion GT, Feng X, Zhang H, Li H. 2020. Haplotype-resolved de novo assembly with phased assembly graphs. arXiv:2008.01237 [q-bio.GN] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiang C, Scott AJ, Davis JR, Tsang EK, Li X, et al. 2017. The impact of structural variation on human gene expression. Nat. Genet 49:692–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choudhury A, Aron S, Botigué LR, Sengupta D, Botha G, et al. 2020. High-depth African genomes inform human migration and health. Nature 586:741–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Church DM, Schneider VA, Graves T, Auger K, Cunningham F, et al. 2011. Modernizing reference genome assemblies. PLOS Biol. 9:e1001091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collins FS, Galas D. 1993. A new five-year plan for the U.S. Human Genome Project. Science 262:43–46 [DOI] [PubMed] [Google Scholar]
- 32.Collins FS, Morgan M, Patrinos A. 2003. The Human Genome Project: lessons from large-scale biology. Science 300:286–90 [DOI] [PubMed] [Google Scholar]
- 33.Collins FS, Varmus H. 2015. A new initiative on precision medicine. N. Engl. J. Med 372:793–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Comput. Pan-Genomics Consort. 2018. Computational pan-genomics: status, promises and challenges. Brief. Bioinform 19:118–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Couzin-Frankel J 2010. DNA returned to tribe, raising questions about consent. Science 328:558. [DOI] [PubMed] [Google Scholar]
- 36.Dekker J, Belmont AS, Guttman M, Leshyk VO, Lis JT, et al. 2017. The 4D nucleome project. Nature 549:219–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Devaney SA, Malerba L, Manson SM. 2020. The “All of Us” program and Indigenous peoples. N. Engl. J. Med 383:1892–93 [DOI] [PubMed] [Google Scholar]
- 38.Dilthey AT, Mentzer AJ, Carapito R, Cutland C, Cereb N, et al. 2019. HLA*LA—HLA typing from linearly projected graph alignments. Bioinformatics 35:4394–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dodson M, Williamson R. 1999. Indigenous peoples and the morality of the Human Genome Diversity Project. J. Med. Ethics 25:204–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dolzhenko E, van Vugt JJFA, Shaw RJ, Bekritsky MA, van Blitterswijk M, et al. 2017. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 27:1895–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duan Z, Qiao Y, Lu J, Lu H, Zhang W, et al. 2019. HUPAN: a pan-genome analysis pipeline for human genomes. Genome Biol. 20:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dumitrescu L, Carty CL, Taylor K, Schumacher FR, Hindorff LA, et al. 2011. Genetic determinants of lipid traits in diverse populations from the population architecture using genomics and epidemiology (PAGE) study. PLOS Genet. 7:e1002138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ebler J, Clarke WE, Rausch T, Audano PA, Houwaart T, et al. 2020. Pangenome-based genome inference. bioRxiv 2020.11.11.378133. 10.1101/2020.11.11.378133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eggertsson HP, Kristmundsdottir S, Beyter D, Jonsson H, Skuladottir A, et al. 2019. GraphTyper2 enables population-scale genotyping of structural variation using pangenome graphs. Nat. Commun 10:5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eichler EE, Clark RA, She X. 2004. An assessment of the sequence gaps: unfinished business in a finished human genome. Nat. Rev. Genet 5:345–54 [DOI] [PubMed] [Google Scholar]
- 46.Eizenga JM, Novak AM, Sibbesen JA, Heumos S, Ghaffaari A, et al. 2020. Pangenome graphs. Annu. Rev. Genom. Hum. Genet 21:139–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.ENCODE Proj. Consort. 2004. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 306:636–40 [DOI] [PubMed] [Google Scholar]
- 48.ENCODE Proj. Consort. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447:799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.ENCODE Proj. Consort. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.ENCODE Proj. Consortium, Moore JE, Purcaro MJ, Pratt HE, Epstein CB, et al. 2020. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583:699–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fiddes IT, Armstrong J, Diekhans M, Nachtweide S, Kronenberg ZN, et al. 2018. Comparative Annotation Toolkit (CAT)—simultaneous clade and personal genome annotation. Genome Res. 28:1029–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garrison E, Sirén J, Novak AM, Hickey G, Eizenga JM, et al. 2018. Variation graph toolkit improves read mapping by representing genetic variation in the reference. Nat. Biotechnol 36:875–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Genome 10K Community Sci. 2009. Genome 10K: a proposal to obtain whole-genome sequence for 10,000 vertebrate species. J. Hered 100:659–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gerstein MB, Rozowsky J, Yan K-K, Wang D, Cheng C, et al. 2014. Comparative analysis of the transcriptome across distant species. Nature 512:445–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gibbs RA. 2020. The Human Genome Project changed everything. Nat. Rev. Genet 21:575–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ginsburg GS, Horowitz CR, Orlando LA. 2019. What will it take to implement genomics in practice? Lessons from the IGNITE Network. Pers. Med 16:259–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD. 2007. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 17:877–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Glob. Alliance Genom. Health. 2016. A federated ecosystem for sharing genomic, clinical data. Science 352:1278–80 [DOI] [PubMed] [Google Scholar]
- 59.Greely HT. 2007. The uneasy ethical and legal underpinnings of large-scale genomic biobanks. Annu. Rev. Genom. Hum. Genet 8:343–64 [DOI] [PubMed] [Google Scholar]
- 60.Green ED, Watson JD, Collins FS. 2015. Human Genome Project: twenty-five years of big biology. Nature 526:29–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, et al. 2010. A draft sequence of the Neandertal genome. Science 328:710–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Groza C, Kwan T, Soranzo N, Pastinen T, Bourque G. 2020. Personalized and graph genomes reveal missing signal in epigenomic data. Genome Biol. 21:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guan D 2020. Asset. Github https://github.com/dfguan/asset
- 64.Günther T, Nettelblad C. 2019. The presence and impact of reference bias on population genomic studies of prehistoric human populations. PLOS Genet. 15:e1008302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hehir-Kwa JY, Marschall T, Kloosterman WP, Francioli LC, Baaijens JA, et al. 2016. A high-quality human reference panel reveals the complexity and distribution of genomic structural variants. Nat. Commun 7:12989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Henikoff JG, Belsky JA, Krassovsky K, MacAlpine DM, Henikoff S. 2011. Epigenome characterization at single base-pair resolution. PNAS 108:18318–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hickey G, Heller D, Monlong J, Sibbesen JA, Sirén J, et al. 2020. Genotyping structural variants in pangenome graphs using the vg toolkit. Genome Biol. 21:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hindorff LA, Bonham VL, Ohno-Machado L. 2018. Enhancing diversity to reduce health information disparities and build an evidence base for genomic medicine. Pers. Med 15:403–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ho JWK, Jung YL, Liu T, Alver BH, Lee S, et al. 2014. Comparative analysis of metazoan chromatin organization. Nature 512:449–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huddleston J, Chaisson MJP, Steinberg KM, Warren W, Hoekzema K, et al. 2017. Discovery and genotyping of structural variation from long-read haploid genome sequence data. Genome Res. 27:677–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.ICGC/TCGA Pan-Cancer Anal. Whole Genomes Consort. 2020. Pan-cancer analysis of whole genomes. Nature 578:82–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Int. HapMap Consort. 2003. The International HapMap Project. Nature 426:789–96 [DOI] [PubMed] [Google Scholar]
- 73.Int. HapMap Consort. 2005. A haplotype map of the human genome. Nature 437:1299–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Int. HapMap Consort. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449:851–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Int. Hum. Genome Seq. Consort. 2004. Finishing the euchromatic sequence of the human genome. Nature 431:931–45 [DOI] [PubMed] [Google Scholar]
- 76.Iqbal Z, Caccamo M, Turner I, Flicek P, McVean G. 2012. De novo assembly and genotyping of variants using colored de Bruijn graphs. Nat. Genet 44:226–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jäger M, Schubach M, Zemojtel T, Reinert K, Church DM, Robinson PN. 2016. Alternate-locus aware variant calling in whole genome sequencing. Genome Med. 8:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jain M, Koren S, Miga KH, Quick J, Rand AC, et al. 2018. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol 36:338–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jain M, Olsen HE, Turner DJ, Stoddart D, Bulazel KV, et al. 2018. Linear assembly of a human centromere on the Y chromosome. Nat. Biotechnol 36:321–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kaye J, Meslin EM, Knoppers BM, Juengst ET, Deschênes M, et al. 2012. ELSI 2.0 for genomics and society. Science 336:673–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim D, Lee J-Y, Yang J-S, Kim JW, Kim VN, Chang H. 2020. The architecture of SARS-CoV-2 transcriptome. Cell 181:914–21.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol 37:907–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Knoppers BM, Zawati MH, Kirby ES. 2012. Sampling populations of humans across the world: ELSI issues. Annu. Rev. Genom. Hum. Genet 13:395–413 [DOI] [PubMed] [Google Scholar]
- 84.Knowler WC, Williams RC, Pettitt DJ, Steinberg AG. 1988. Gm3;5,13,14 and type 2 diabetes mellitus: an association in American Indians with genetic admixture. Am. J. Hum. Genet 43:520–26 [PMC free article] [PubMed] [Google Scholar]
- 85.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409:860–921 [DOI] [PubMed] [Google Scholar]
- 86.Li H, Feng X, Chu C. 2020. The design and construction of reference pangenome graphs with mini-graph. Genome Biol. 21:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, et al. 2008. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319:1100–4 [DOI] [PubMed] [Google Scholar]
- 88.Li R, Li Y, Zheng H, Luo R, Zhu H, et al. 2010. Building the sequence map of the human pan-genome. Nat. Biotechnol 28:57–63 [DOI] [PubMed] [Google Scholar]
- 89.Logsdon GA, Vollger MR, Eichler EE. 2020. Long-read human genome sequencing and its applications. Nat. Rev. Genet 21:597–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Logsdon GA, Vollger MR, Hsieh P, Mao Y, Liskovykh MA, et al. 2020. The structure, function, and evolution of a complete human chromosome 8. bioRxiv 2020.09.08.285395. 10.1101/2020.09.08.285395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Loos RJF. 2020. 15 years of genome-wide association studies and no signs of slowing down. Nat. Commun 11:5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, et al. 2016. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538:201–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maretty L, Jensen JM, Petersen B, Sibbesen JA, Liu S, et al. 2017. Sequencing and de novo assembly of 150 genomes from Denmark as a population reference. Nature 548:87–91 [DOI] [PubMed] [Google Scholar]
- 94.Martiniano R, Garrison E, Jones ER, Manica A, Durbin R. 2020. Removing reference bias and improving indel calling in ancient DNA data analysis by mapping to a sequence variation graph. Genome Biol. 21:250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Matise TC, Ambite JL, Buyske S, Carlson CS, Cole SA, et al. 2011. The next PAGE in understanding complex traits: design for the analysis of Population Architecture Using Genetics and Epidemiology (PAGE) Study. Am. J. Epidemiol 174:849–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McEwen JE, Boyer JT, Sun KY, Rothenberg KH, Lockhart NC, Guyer MS. 2014. The Ethical, Legal, and Social Implications Program of the National Human Genome Research Institute: reflections on an ongoing experiment. Annu. Rev. Genom. Hum. Genet 15:481–505 [DOI] [PubMed] [Google Scholar]
- 97.Meyer M, Kircher M, Gansauge M-T, Li H, Racimo F, et al. 2012. A high-coverage genome sequence from an archaic Denisovan individual. Science 338:222–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Miga KH, Koren S, Rhie A, Vollger MR, Gershman A, et al. 2020. Telomere-to-telomere assembly of a complete human × chromosome. Nature 585:79–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Moodley K, Kleinsmidt A. 2020. Allegations of misuse of African DNA in the UK: Will data protection legislation in South Africa be sufficient to prevent a recurrence? Dev. World Bioeth 10.1111/dewb.12277 [DOI] [PubMed] [Google Scholar]
- 100.Morales J, Welter D, Bowler EH, Cerezo M, Harris LW, et al. 2018. A standardized framework for representation of ancestry data in genomics studies, with application to the NHGRI-EBI GWAS Catalog. Genome Biol. 19:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nagai A, Hirata M, Kamatani Y, Muto K, Matsuda K, et al. 2017. Overview of the BioBank Japan Project: study design and profile. J. Epidemiol 27:S2–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nègre N, Brown CD, Ma L, Bristow CA, Miller SW, et al. 2011. A cis-regulatory map of the Drosophila genome. Nature 471:527–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nurk S, Walenz BP, Rhie A, Vollger MR, Logsdon GA, et al. 2020. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. 30:1291–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.O’Connor BD, Yuen D, Chung V, Duncan AG, Liu XK, et al. 2017. The Dockstore: enabling modular, community-focused sharing of Docker-based genomics tools and workflows. F1000Research 6:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Park PJ. 2009. ChIP-seq: advantages and challenges of a maturing technology. Nat. Rev. Genet 10:669–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Paten B, Novak AM, Eizenga JM, Garrison E. 2017. Genome graphs and the evolution of genome inference. Genome Res. 27:665–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Payne A, Holmes N, Rakyan V, Loose M. 2019. BulkVis: a graphical viewer for Oxford nanopore bulk FAST5 files. Bioinformatics 35:2193–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Petersen A 2005. Securing our genetic health: engendering trust in UK Biobank. Sociol. Health Illn 27:271–92 [DOI] [PubMed] [Google Scholar]
- 109.Putnam NH, O’Connell BL, Stites JC, Rice BJ, Blanchette M, et al. 2016. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 26:342–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rakocevic G, Semenyuk V, Lee W-P, Spencer J, Browning J, et al. 2019. Fast and accurate genomic analyses using genome graphs. Nat. Genet 51:354–62 [DOI] [PubMed] [Google Scholar]
- 111.Rautiainen M, Marschall T. 2020. GraphAligner: rapid and versatile sequence-to-graph alignment. Genome Biol. 21:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Reardon J 2009. Race to the Finish: Identity and Governance in an Age of Genomics. Princeton, NJ: Princeton Univ. Press [Google Scholar]
- 113.Rheinbay E, Nielsen MM, Abascal F, Wala JA, Shapira O, et al. 2020. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 578:102–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rhie A, Walenz BP, Koren S, Phillippy AM. 2020. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Roadmap Epigenom. Consort., Kundaje A, Meuleman W, Ernst J, Bilenky M, et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518:317–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rockwell DH, Yobs AR, Moore MB Jr. 1964. The Tuskegee Study of Untreated Syphilis: the 30th year of observation. Arch. Intern. Med 114:792–98 [DOI] [PubMed] [Google Scholar]
- 117.Rosen Y, Eizenga J, Paten B. 2017. Modelling haplotypes with respect to reference cohort variation graphs. Bioinformatics 33:i118–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rozenblatt-Rosen O, Stubbington MJT, Regev A, Teichmann SA. 2017. The Human Cell Atlas: from vision to reality. Nature 550:451–53 [DOI] [PubMed] [Google Scholar]
- 119.Said MA, Verweij N, van der Harst P. 2018. Associations of combined genetic and lifestyle risks with incident cardiovascular disease and diabetes in the UK Biobank study. JAMA Cardiol. 3:693–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sankar PL, Parker LS. 2017. The Precision Medicine Initiative’s All of Us Research Program: an agenda for research on its ethical, legal, and social issues. Genet. Med 19:743–50 [DOI] [PubMed] [Google Scholar]
- 121.Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen H-C, et al. 2017. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27:849–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schones DE, Cui K, Cuddapah S, Roh T-Y, Barski A, et al. 2008. Dynamic regulation of nucleosome positioning in the human genome. Cell 132:887–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Scott CT, Caulfield T, Borgelt E, Illes J. 2012. Personal medicine—the new banking crisis. Nat. Biotechnol 30:141–47 [DOI] [PubMed] [Google Scholar]
- 124.Sedlazeck FJ, Lee H, Darby CA, Schatz MC. 2018. Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nat. Rev. Genet 19:329–46 [DOI] [PubMed] [Google Scholar]
- 125.Shafin K, Pesout T, Lorig-Roach R, Haukness M, Olsen HE, et al. 2020. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol 38:1044–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sherman RM, Forman J, Antonescu V, Puiu D, Daya M, et al. 2019. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat. Genet 51:30–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sherman RM, Salzberg SL. 2020. Pan-genomics in the human genome era. Nat. Rev. Genet 21:243–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, et al. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29:308–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Skirton H, Goldsmith L, Jackson L, O’Connor A. 2012. Direct to consumer genetic testing: a systematic review of position statements, policies and recommendations. Clin. Genet 82:210–18 [DOI] [PubMed] [Google Scholar]
- 130.Song L, Crawford GE. 2010. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc 2010:db.prot5384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sperber NR, Carpenter JS, Cavallari LH, J Damschroder L, Cooper-DeHoff RM, et al. 2017. Challenges and strategies for implementing genomic services in diverse settings: experiences from the Implementing GeNomics In pracTicE (IGNITE) network. BMC Med. Genom 10:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Stark Z, Dolman L, Manolio TA, Ozenberger B, Hill SL, et al. 2019. Integrating genomics into health-care: a global responsibility. Am. J. Hum. Genet 104:13–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, et al. 2015. An integrated map of structural variation in 2,504 human genomes. Nature 526:75–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Telenti A, Pierce LCT, Biggs WH, di Iulio J, Wong EHM, et al. 2016. Deep sequencing of 10,000 human genomes. PNAS 113:11901–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Toh C, Brody JP. 2020. Evaluation of a genetic risk score for severity of COVID-19 using human chromosomal-scale length variation. Hum. Genom 14:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tutton R, Kaye J, Hoeyer K. 2004. Governing UK Biobank: the importance of ensuring public trust. Trends Biotechnol. 22:284–85 [DOI] [PubMed] [Google Scholar]
- 137.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, et al. 2001. The sequence of the human genome. Science 291:1304–51 [DOI] [PubMed] [Google Scholar]
- 138.Vernikos G, Medini D, Riley DR, Tettelin H. 2015. Ten years of pan-genome analyses. Curr. Opin. Microbiol 23:148–54 [DOI] [PubMed] [Google Scholar]
- 139.Wainberg M, Mahajan A, Kundaje A, McCarthy MI, Ingelsson E, et al. 2019. Homogeneity in the association of body mass index with type 2 diabetes across the UK Biobank: a Mendelian randomization study. PLOS Med. 16:e1002982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang Z, Gerstein M, Snyder M. 2009. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet 10:57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wenger AM, Peluso P, Rowell WJ, Chang P-C, Hall RJ, et al. 2019. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol 37: 1155–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wilkinson MD, Dumontier M, Aalbersberg IJJ, Appleton G, Axton M, et al. 2016. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 3:160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wong KHY, Ma W, Wei C-Y, Yeh E-C, Lin W-J, et al. 2020. Towards a reference genome that captures global genetic diversity. Nat. Commun 11:5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yu Y, Wei C. 2020. A powerful HUPAN on a pan-genome study: significance and perspectives. Cancer Biol. Med 17:1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, et al. 2014. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515:355–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zeberg H, Pääbo S. 2020. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 587:610–12 [DOI] [PubMed] [Google Scholar]
- 147.Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, et al. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–73 [DOI] [PMC free article] [PubMed] [Google Scholar]