

Abstract
Fluorescent dyes attached to kinase inhibitors (KIs) can be used to probe kinases in vitro, in cells, and in vivo. Ideal characteristics of the dyes vary with their intended applications. Fluorophores used in vitro may inform on kinase active site environments, hence the dyes used should be small and have minimal impact on modes of binding. These probes may have short wavelength emissions since blue fluorophores are perfectly adequate in this context. Thus, for instance, KI fragments that mimic nucleobases may be modified to be fluorescent with minimal perturbation to the kinase inhibitor structure. However, progressively larger dyes, that emit at longer wavelengths, are required for cellular and in vivo work. In cells, it is necessary to have emissions above autofluorescence of biomolecules, and near infrared dyes are needed to enable excitation and observation through tissue in vivo. This review is organized to describe probes intended for applications in vitro, in cells, then in vivo and readers will see that the probes featured tend to become larger and responsive to the red end of the spectrum as the review progresses. Readers may also be surprised to realize that relatively few dyes have been used for fluorophore-kinase inhibitor conjugates, and the area is open for innovations in the types of fluorophores used.
A. Introduction
There are more than 518 kinases in the human kinome,1 most having homologous kinase domains and structurally similar ATP binding sites.2 Various kinases are overexpressed or mutated in cancers to promote cell survival and propagation. Kinase inhibitors (KIs) are designed to inhibit these kinases. Of the >60 KIs clinically approved, most (~42) are for cancer,3 and these are intended to target those specific pathways that are upregulated in tumor cells, over healthy tissue, hence to slow propagation and metastatic spread, while incurring less side-effects than more traditional, broadly cytotoxic, chemotherapeutic drugs.
As the field progresses, more clinical candidates will be approved, and off-label applications will be found revealing mechanisms of action that were perhaps unanticipated. Consequently, there is considerable interest in understanding aspects of the cell biology, pharmacokinetics (PK), and pharmacodynamics (PD) of KIs; one way of doing this is using fluorescently labelled KIs (fluor-KIs), and that is the focus of this review. Throughout this article, the word “fluorophores” is sometimes abbreviated to “fluors”.
Optimal fluor designs for labelling KIs are different for in vitro, cellular, and in vivo applications: there cannot be a single type of fluorophore-KI probe for all purposes. For instance, studies of modes of binding must prioritize minimal perturbation to the KI structure, hence features small fluorophores. Small fluors, however, usually have short wavelength absorption and emission maxima, and that renders them non-ideal for intracellular imaging and inappropriate for in vivo studies. Probes that absorb and emit beyond the range of intracellular autofluorescence (i.e., >550 nm) are most suitable for confocal imaging (including fluorescence and life-time measurements) where autofluorescence is minimal. Extended near-infrared (NIR) fluorophores are favored for in vivo imaging because dyes that absorb at less than ~750 nm are hard to excite when shielded by more than a few millimeters of tissue, even if powerful lasers are used. To calibrate, penetration of 800 nm wavelength light is twice that of 630 nm.4 In vivo imaging of fluor-KI probes requires larger dyes unless two-photon excitation5, 6 is used, and, as far as we are aware, no one has yet reported this for fluor-KI probes. Consequently, properties of KIs tend to be perturbed by the larger dyes used in cellular and in vivo studies, than by the smaller ones that may be used in vitro.
This review deals with fluor-KI probes applied in vitro, cellular, then in vivo. Consequently, readers will observe an emphasis shift in the fluors commonly used (Fig 1a, Table 1) from green BODIPY dyes to red rhodamines, to the deep red cyanines, as this review progresses. Throughout this article the KIs (Fig 1b, Table 2) are shown in magenta.
Fig 1.
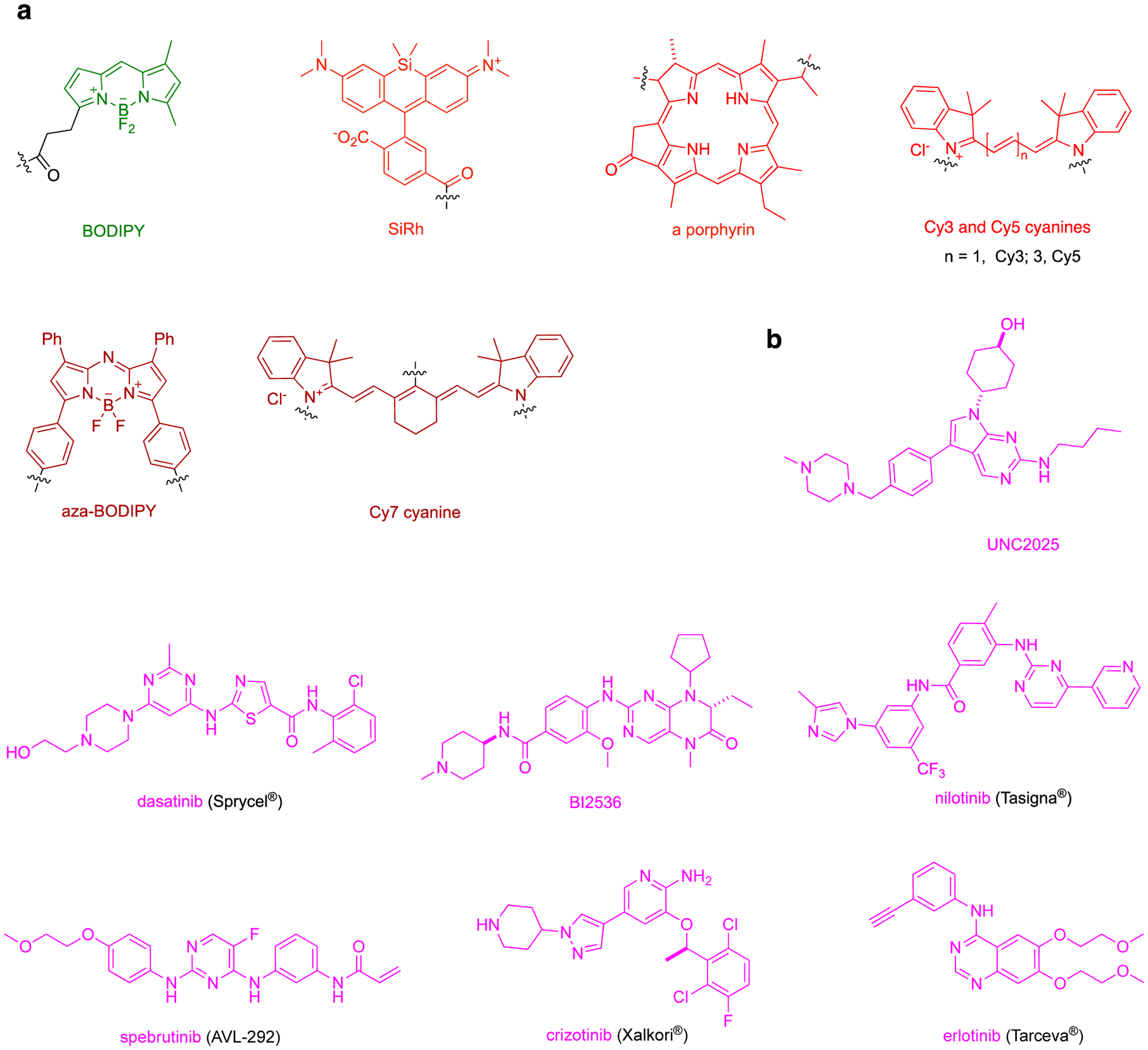
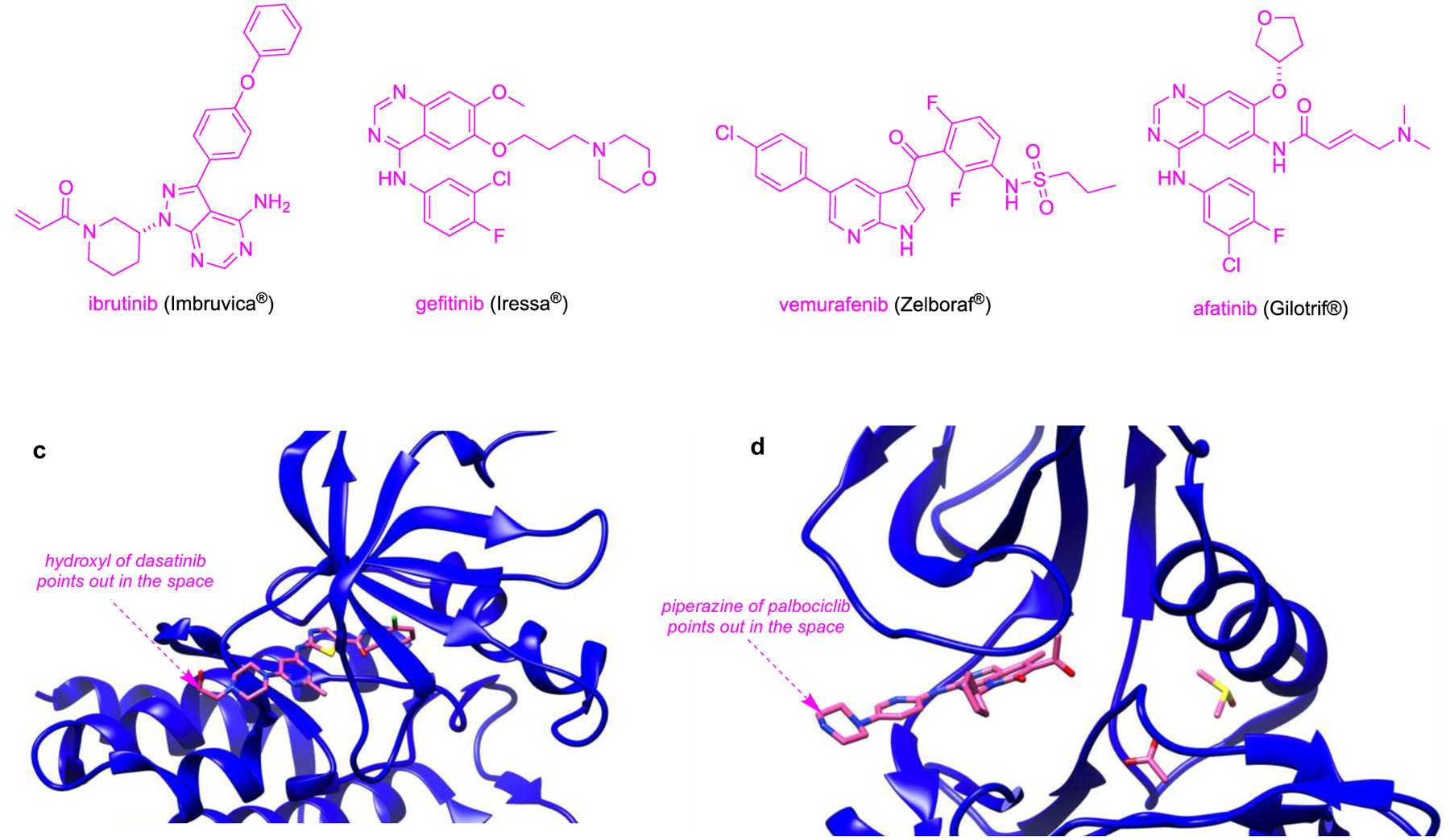
a Different type of fluorophores discussed in this review. Throughout the manuscript, the fluorophores will be colored green – red as depicted here to highlight their absorption of light. b Parent structures of kinase inhibitors. From here on, modifications made to kinase inhibitor for fluorophore conjugate will be referred as kinase inhibitor analog and shown in magenta. Modifications to the kinase inhibitors will be depicted in blue or purple. c Dasatinib binding to Abl (blue; PBD: 2GQG). d Palbociclib bound to CDK6 (blue; PBD: 5L2T).
Table 1.
Photophysical properties of the fluorophores discussed in this review.
| Fluorophore | absorbance (λmax, nm) | fluorescence (λmax, nm) | extinction co-efficient (M−1 cm−1) | Quantum Yield (%) |
|---|---|---|---|---|
| BODIPY | 503 | 509 | 92,000 | 0.97 |
| SiRh | 651 | 671 | 100,000 | 0.39 |
| porphyrin | 665 | 670 | 100,000 | 0.20 |
| Cy3 | 550 | 570 | 150,000 | 0.04 |
| Cy5 | 650 | 670 | 250,000 | 0.27 |
| aza-BODIPY | 706 | 730 | 80,000 | 0.14 |
| Cy7 | 750 | 770 | 200,000 | 0.3 |
Table 2.
The biological properties, specificities and potencies of kinase inhibitors featured in this review.
| Kinase Inhibitor | target | binding and kinase inhibition (nM, cell-free unless otherwise noted) |
|---|---|---|
| UNC202511 | MERTK | Kd: 0.74 IC50 (in cell): 1 μM |
| dasatinib23,12 | BCR-Abl | Kd: Src 0.07; Abl 0.03 IC50 (μM): Src; 0.2, Abl 0.03, Lyn 0.6, Btk 1.5 |
| BI253613 | PLK1–3 | IC50: PLK1 5.01; PLK2 19.4; PLK3 9.37 |
| nilotinib14 | BCR-Abl | IC50 (in cell): 20 – 60 |
| spebrutinib15 | Btk | IC50: < 0.5 |
| crizotinib16 | ALK | IC50 (in cells): 24 |
| erlotinib17 | EGFR | IC50: 2 |
| Ibrutinib7 | Btk | IC50: 68 |
| gefitinib18 | EGFR | IC50 (in cells): 33 |
| vemurafenib19 | BRAFV600E | IC50 (in cells): 64 – 110 |
| afatinib20 | EGFR, HER2 | IC50 (in cell): 0.35 – 0.34 μM |
Effective fluor-KI conjugates must be cell permeable, and have functionality to facilitate fluor attachment. Non-charged and hydrophilic dyes usually can permeate the cell membrane faster (through passive transport) than their charged counterparts. These non-charged dyes diffuse evenly in the cells, hence provide optimal chance for fluor-KI interaction, making them ideal for conjugation with KIs. If the parent KI does not have an appropriate attachment point, then the structure must be modified to incorporate a functional group without drastically altering its mode of action. For example, a sprebutinib analog was synthesized7 to introduce an amine for amide coupling attachment. That work is typical because the attachment point is an amine, but there are instances where KI has been coupled using methods such as substitution of meso-Cl of heptamethine cyanine (Cy7)8 or metal catalyzed cross couplings.9 Modified KIs, however, may have different activities and selectivities; consequently, to avoid synthetic work and compound excessive validation, there is a bias towards probes from KIs that have some intrinsic point of attachment that projects into solvent when bound to the target enzyme by orienting into solvent and away from the binding pocket of kinase (Fig 1c and d).
This article excludes peptidic chemosensors that, in many cases, change fluorescence on phosphorylation, without necessarily being kinase inhibitors; those were reviewed in 2012.10 Moreover, this review only covers applications of fluor-KIs related to cancer. In any case, to the best of our knowledge, so far there are no fluor-KI probes for use outside that area.
B. In Vitro Probes
Fluor-KI probes can be used in studies of kinases in vitro to report on binding constants, on and off rates, and conformational characteristics of the enzyme active sites. To understand how, however, the next section overviews the nomenclature used to describe kinase conformations (Fig 2a). Unfortunately, that nomenclature is largely not self-explanatory (type I, II etc.), and sometimes confusing (e.g., type I½).
Fig 2.
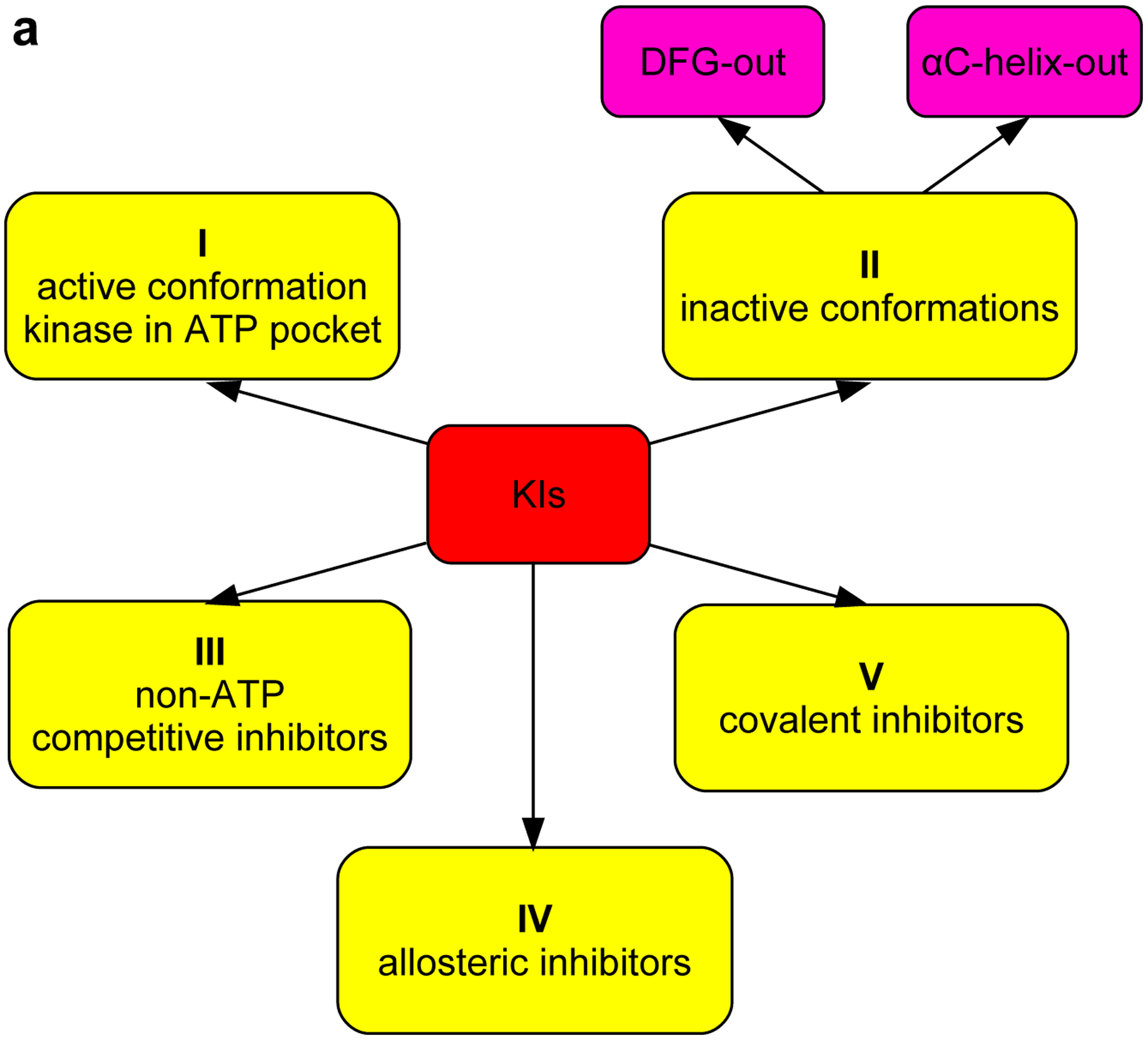
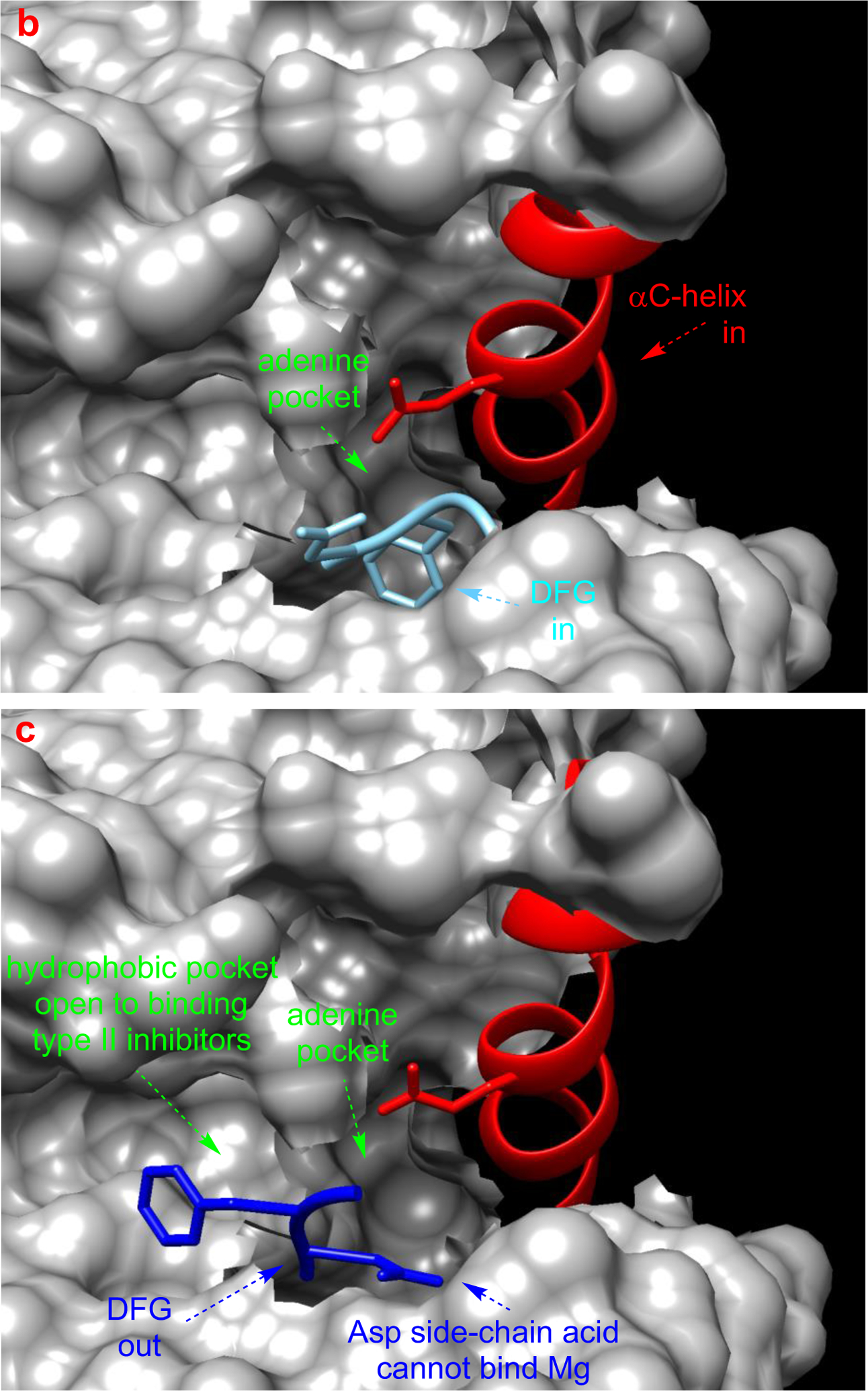
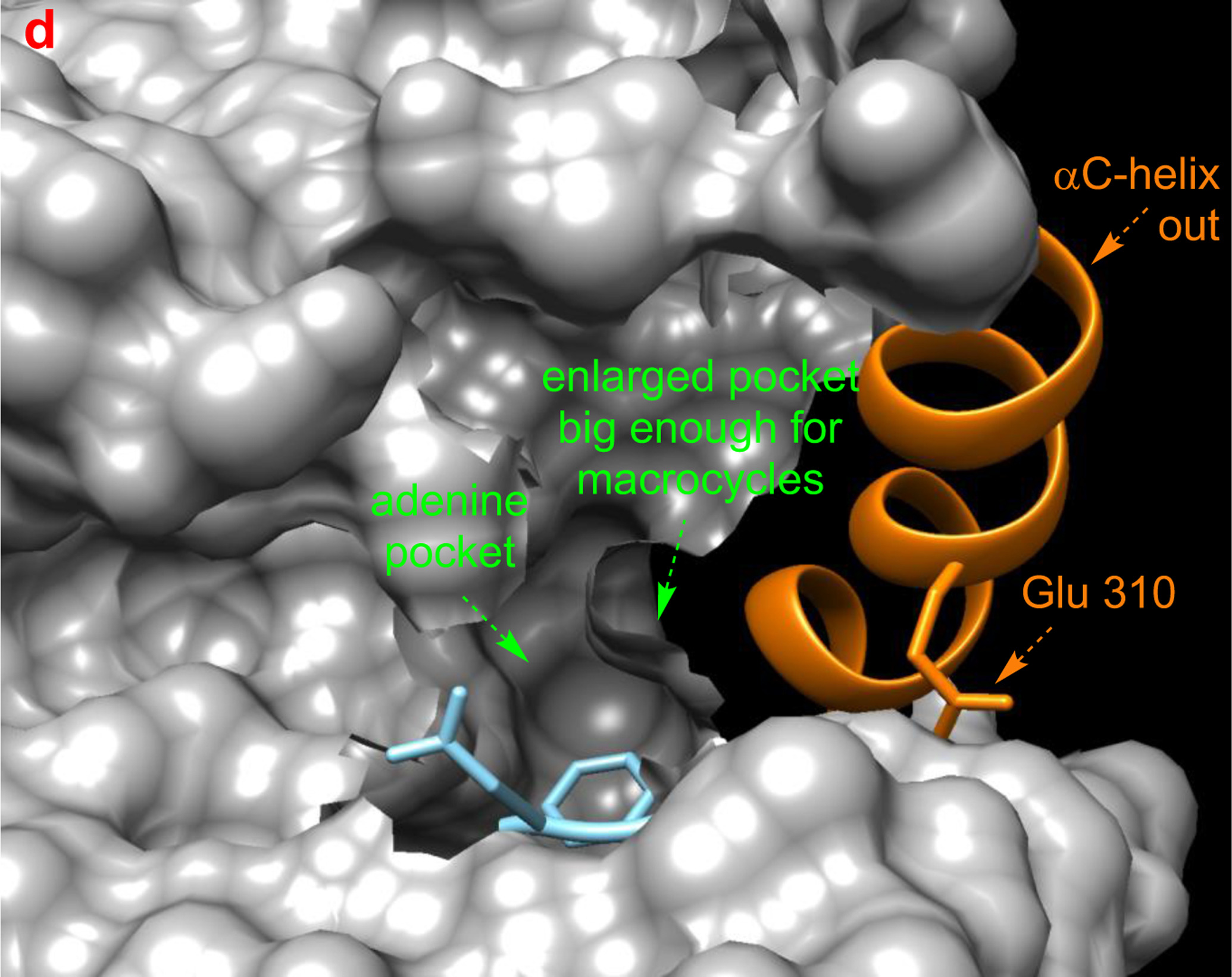
a Different type of KIs. Structure of Src with kinase inhibitors in active (b, PDB: 3G5D), DFG-out (c, PDB: 4YBJ) and C-helix-out conformation (d, PDB: 4YBK). Type II inhibitors lead to DFG-out (dark blue) in contrast to DFG-in (light blue) conformation. c-Helix-out inhibitors lead to αC-helix-out (orange) in contrast to αC-helix in (red) conformation. Small molecule ligands are excluded for simplicity.
Essential Facets Of Kinase Conformations
Kinase inhibitors can be classified in terms of conformations of kinases they bind to, and where on the protein surface they bind those enzymes. By definition, type I inhibitors bind active kinases in the ATP pocket. Active conformations of kinases generally have a DFG (Asp-Phe-Gly) motif pointing into the site for Mg2+/ATP coordination, and a salt bridge between the catalytic lysine and a glutamate in the αC-helix. Type I inhibitors tend to be less kinase selective because of the close similarities of active conformations in these enzymes. Dasatinib complexed with Src is an example of a type I inhibitor (Fig 2b).
Type II inhibitors bind various inactive conformations, including ones described as “DFG-out” and “αC-helix-out” (Fig 2c and d). Typically, type II inhibitors cause displacement resulting in DFG-out conformations, and this leads to filling of a hydrophobic site adjacent to ATP binding pocket. Type II inhibitors tend to be more selective than type I due to uniqueness of inactive protein kinase conformations.21,22
Little is said in this article about inhibitors other than type I or II, but for completeness a few more are briefly described here. Type I½ inhibitors are hybrids of I and II that bind the inactive “DFG-in” and “αC-helix-out” conformations. Type III inhibitors are non-ATP competitive and bind sites adjacent to the ATP binding pocket in ways that allow ATP and the KI to associate with the kinase simultaneously. Type IV inhibitors are allosteric ones that dock on sites distant from the ATP binding site and covalent inhibitors, sometimes also referred to as type V inhibitors bind irreversibly (Fig 2a).23, 24
DFG-out inactive conformations feature the DFG motif distal to the active site such that Mg2+ coordination to ATP and the nearby Asp of the kinase is prevented, thus suppressing capture and reactivity of ATP in the enzyme active site.23,24 Transition of the DFG motif into this “out conformation” also exposes a hydrophobic pocket that may be occupied by the KI fragments. Enhanced selectivity of binding of type II KIs in many cases can be attributed to that hydrophobic pocket. For instance, preclinical inhibitor 2-H (based on the type I inhibitor 1) binds Src by placing the 3-trifluoromethylbenzamide fragment (blue below) into the newly exposed hydrophobic pocket giving a type II inhibitor. This strategy is discussed again around Fig 4 below, where it is referred to as “Gray’s hybridization method”.
Fig 4.
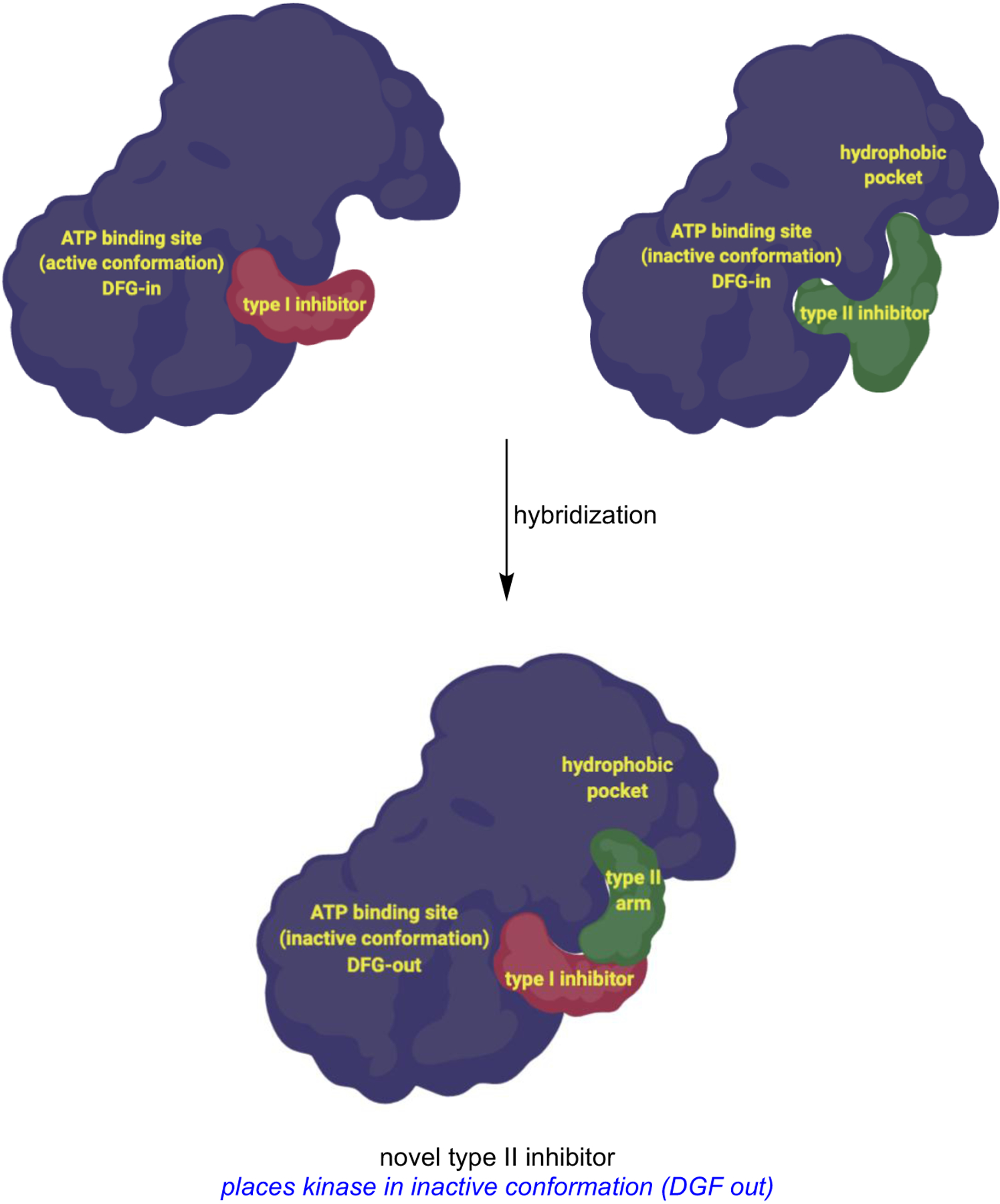
Gray’s hybridization concept to convert type I inhibitors into type II by conjugating ATP-binding active site fragment with a fragment with complementary hydrophobic pocket on inactive states.
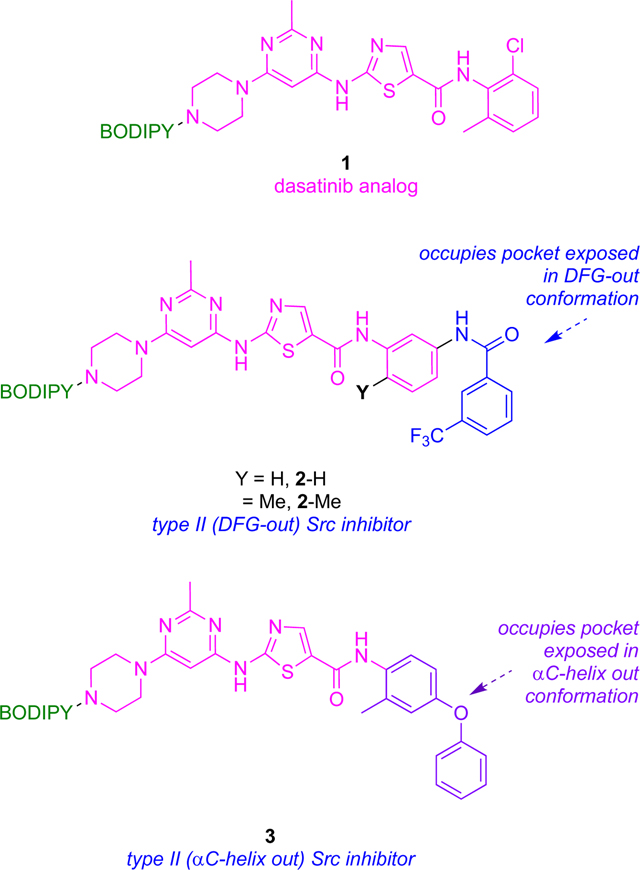
The αC-helix-out subset of type II inactive conformations have the helix rotated away from the kinase core, reorienting a key glutamate to point away from active site, breaking the salt bridge between glutamate and the catalytic lysine critical for kinase activity (Fig 2d). Twisting the αC-helix in this way extends the active site and creates a hydrophobic binding pocket large enough to favor docking of macrocyclic compounds; inhibition via this type of binding is illustrated by 3 with Src.
Type II KIs tend to have longer kinase binding residence times relative to type I inhibitors. This is important because examples covered in the next section feature KI derivatives for which fluorescence changes can be linked with residence times, and therefore to modes of binding.
All inhibitor types are somewhat arbitrary milestones on a continuum of possibilities reflecting kinase plasticity. Thus, type II inhibitors bind inactive conformations that vary between kinases, and this variability tends to engender enhanced selectivities. DFG-out conformations are not uniformly favored over “-in ones” for inactive kinases, and DFG-out orientations are sometimes accessible to active kinases. Type III (non-ATP competitive), IV (allosteric) and V (covalent) situations are more unique24 and cannot be represented by generalized conformations. These types of inhibitors are not illustrated in Fig 2.
Competitive Binding Of Labeled And Unlabeled Kinases To Infer Binding Modes
Determination Of Binding And Inhibition Constants At Equilibrium
Early steps in probe-validation for fluor-KI conjugates tend to involve measurements of dissociation constants, and/or inhibition of kinase activities. Dissociation constants can be measured by several methods, including isothermal calorimetry (ITC) and fluorescence polarization (FP, based on changes in fluorescence anisotropies). FP assays are robust, convenient, versatile and cost-effective; they feature free fluorophores excited with plane polarized light losing polarization due to fast tumbling, but their polarization is retained when they are protein-bound due to slow tumbling giving higher FP signals.25
It should be possible to use FP to deduce Kd values for fluor-KI •kinase complexes; this might be called direct FP. Direct FP apparently was not explored in any papers covered for this review, perhaps because comparative Kd measurements for the parent KIs could not be obtained in the same way (though competitive experiments are possible), and perhaps because of inadequate instrument sensitivity for detection of anisotropy when low nanomolar concentrations of KIs are involved. Instead, researchers using fluor-KI probes have tended to turn to commercial Kd determination methods, like the KINOMEscan™. KINOMEscan™ features combinations of a DNA-tagged kinase, and an immobilized, active-site directed ligand; abilities of test samples to compete with the immobilized ligand are measured via quantitative polymerase chain reaction (PCR) of the DNA-tagged kinase.
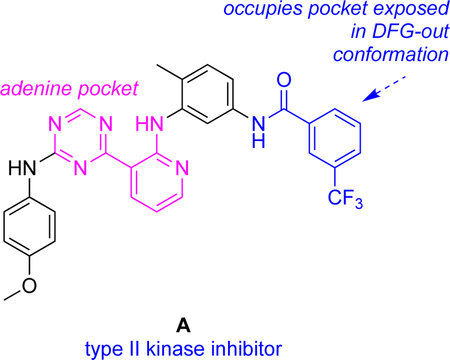
IC50’s for enzyme inhibition by KIs can reveal biochemical effects of KIs that may contrast with binding constants. For instance, the KI A was shown to have IC50’s <25 nM for seven kinases known to adopt DFG-out conformations, but for kinases that appear not to adopt DFG-out conformations, the measured IC50 values were >1000 nM.26
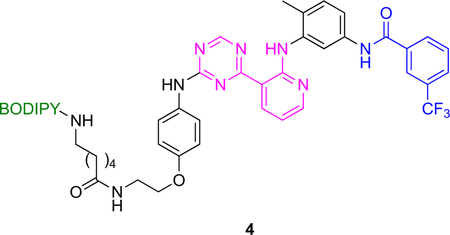
Determination Of On- And Off-rates
Use of fluorescent kinase inhibitor probes in end point fluorescence assays to measure kinetic binding parameters is illustrated in Fig 3. KI A has fragments that bind to the hydrophobic pocket exposed in DFG-out conformations and to the ATP binding site. It tends to bind DFG-out conformations of many kinases hence can be called a general type II inhibitor.26
Fig 3.
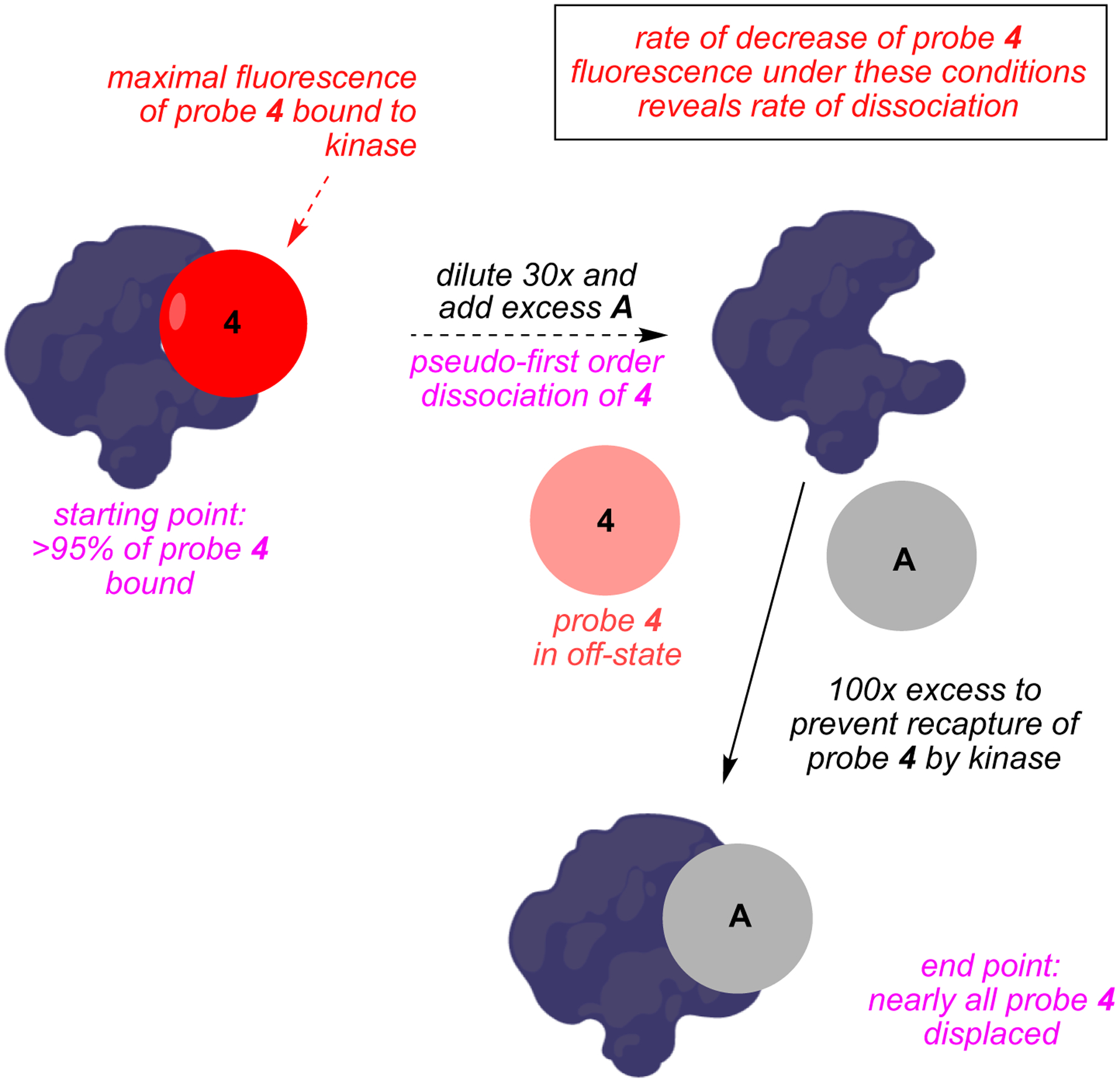
Principle of end point fluorescence assay applied to kinase inhibitors.
Probe 4 (BODIPY functionalized A) binds kinases with similar affinities to A. The brilliance (fluorescence) of 4 increases linearly with probe-to-kinase ratio (provided the kinase is not saturated); predictably, this brilliance increase is competitively suppressed by the parent non-labeled inhibitor A. Consequently, when the kinase is treated with A then 4, time dependent fluorescence changes can be used to reveal koff values for A.
Inhibitors that bind kinases in DFG-out conformations tend to have slow koff rates. Consequently, end point fluorescence assays for probe 4 binding p38 and Abl should give slow off rates because the parent probe A is known to bind those kinases in DFG-out conformations; in fact, they did (koff = 1.6 × 10−4 and 5.2 × 10−5 s−1 respectively). Kinases for which DFG-out conformations are unfavorable (e.g., CLK1 and MAP3K5) did not bind this probe with high affinity.
Recall from the discussion of compounds 1 – 3 above that Gray’s hybridization method (Fig 4)27, 28 features fragments which bind the ATP-binding active site combined with others that bind a complementary hydrophobic pocket on inactive states, thus forcing the kinase into a DFG-out inactive conformation.25 Thus, this strategy guides modifications of type I inhibitors that transform them into type II. A similar approach can be applied to enforce αC-helix-out inactive conformation. Thus, inhibitors have been made23 to amalgamate elements of dasatinib (type I) with imatinib (type II, DFG loop out) or a para-phenoxy group (known to induce type II αC-helix-out inactive conformers by displacing the 310Glu residue of the helix), as already illustrated in Fig 2. Thus, inhibitors that bind the ATP binding site in either the DFG-out or αC-helix-out inactive conformers, respectively (Fig 5a), were then modified to include a fluorescent dye to give 1 – 3. Crystallographic analyses confirmed 1 bound Src in its active conformation, 2 bound the DFG-out inactive form, and 3 bound the αC-helix-out inactive shape (Fig 5b – d).
Fig 5.
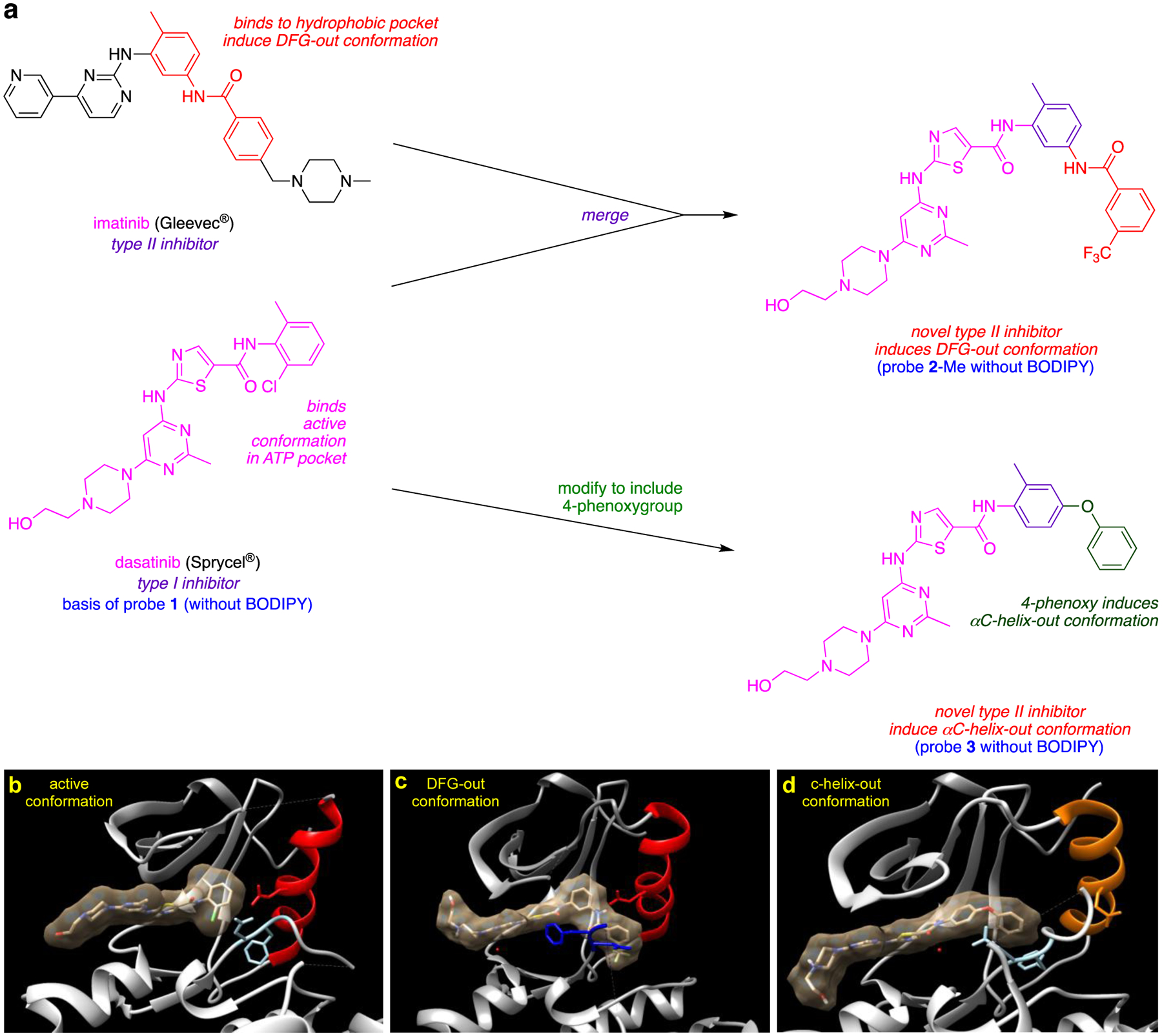
a Design of novel type II inhibitors inducing DFG-out and αC-helix-out inactive conformations. Crystal structures of dasatinib (b) or its analogs (c, d) with Src in active (b, PDB: 3G5D), DFG-out (c, PDB: 4YBJ) and αC-helix-out (d, PDB: 4YBK) conformation. c Incorporation of trifluoromethyl benzamide group to dasatinib analog II inhibitors lead to DFG-out (dark blue) in contrast to DFG-in (light blue) conformation. d Incorporation of para-phenoxy group to dasatinib analog leads to αC-helix-out (orange) in contrast to αC-helix in (red) conformation.
Probe 2 (the one giving the type II DFG-out conformation) in fluorescent end point assays with Src and Abl showed slower off-rates than expected for type I bound conformations, so it was inferred that both kinases bound to 2 are in the type II conformation. Conversely, 1 and 3 (for the active and αC-helix-out conformations, respectively) had fast, dissociation kinetics, unlike type II DFG-out inhibitors. Off-rates for compounds that bind the αC-helix-out conformation were hitherto largely unknown; the inference of this study is that they are fast.
Imatinib is a DFG-out type II inhibitor which binds Abl in preference to Src, and the reasons for this were at first unclear. Suggestions were made that imatinib preferentially binds Abl because there may be an energetic penalty for Src to adopt the DFG-out conformation. To test this hypothesis, kon, koff, and Kd parameters were compared for kinase domains of Abl and Src interacting with 1 and 2-Me; no significant kinetic or thermodynamic differences were observed which undermines the hypothesis that imatinib preferentially binds Abl because there may be an energetic penalty for Src to adopt the DFG-out conformation. Moreover, no significant difference was observed between Src kinase domain and full-length construct. There were also no binding differences for probes 1 or 2-Me to kinase domains of Src and to activation-loop-phosphorylated Src proving these conformation-selective inhibitors did not exhibit an activation state bias. In summary, probes 1 and 2 were used to test, and disprove, various notions about differences in the way imatinib binds Abl in preference to Src. Ultimately, after these fluorescent studies, NMR and fast kinetics were used to show Abl’s high affinity can be attributed mostly to an induced fit conformational change after binding that does not occur for Src.29
Environments Elucidated Via Solvatochromatic, Intrinsically Fluorescent KIs
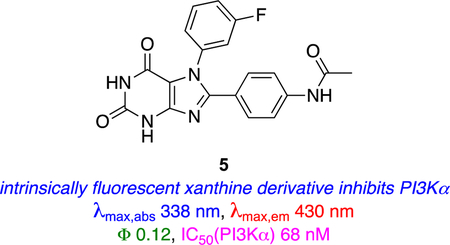
Xanthine 5
If a KI is intrinsically fluorescent and solvatochromatic, as distinct from one attached to a solvatochromatic dye, then changes in its electronic spectra on interacting with a protein can be used to infer the environment of the target KI. Intrinsically fluorescent xanthine derivatives including 5,30–32 has been developed to target the kinase ATP binding site. Probe 5 has undesirably short absorption and emission wavelength maxima (λmax,abs 338 nm, λmax,em 430 nm) for cell studies, but could be observed via confocal microscopy after deconvoluting from cellular autofluorescence. In fact, 5 was also observed, albeit extremely faintly, after direct injection into subcutaneous tumors in a murine model where the need for long wavelength emitters is even more acute.
Gefitinib Hybrids 6 – 8
Hybrids of gefitinib and a fluorescent push-pull stilbene derivative have been used to study ERBB (human epidermal growth factor receptor) kinases.33,34 Like 5, probes 6 and 7 can be excited at wavelengths used for common nuclear stains like DAPI (4′,6-diamidine-2′-phenylindole), i.e. at wavelengths available on nearly all confocal microscopes. Molar absorbances of the dyes increase with the number of double bonds in conjugation, hence probes 7 tend to be brighter than 5. Even though probe 7a absorbs at a relatively short wavelength, it has a large Stokes’ shift (absorbs at 391 nm, emits at 561 nm, in chloroform) and this characteristic compensates for the short λmax abs when the probes are applied for in intracellular imaging.
Probes 6 – 8 have high “turn-on” ratios meaning their fluorescence is greatly enhanced in the constrained, hydrophobic pockets of the ERBB receptors. Ranges of intracellular turn-on ratios on kinase binding can be estimated from enhancements measured for the probes in octanol over water (Ioctanol/Iwater) in vitro. Probe 7a possesses an on/off >50-fold ratio so it is highly responsive to changes molecular environment.
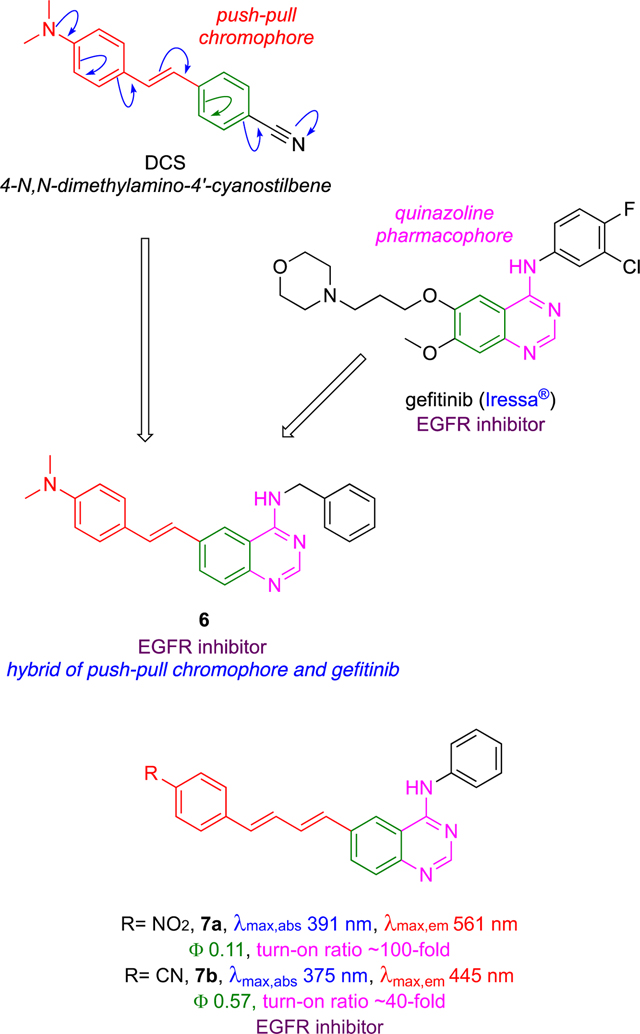
Activation of fluorescence when 6 – 8 bound to ERBB2 receptors was used to infer their modes of binding.33 For example, enhanced fluorescence of 6 correlated with inhibition of ERBB2 autophosphorylation in ERBB2+ cells (quenched in a competition experiment by an inhibitor that binds the ERBB2 active site). Most of the probes at 10 μM showed complete inhibition of phosphorylation of ERBB2 in vitro. However, in cells the probe with the longest emission maxima, 7b, did not completely inhibit ERBB2 phosphorylation at <100 μM implying it is vulnerable to alternative modes of interaction with the kinase, low cellular uptake, weak binding at the target site, aggregation, binding to other proteins, and/or displacement from the kinase via competitive binding of endogenous ATP. It is also possible that double bond electrophilicity in 7 could lead to formation of conjugate addition products with nucleophiles like glutathione causing loss of kinase affinity.35, 36
Modifications of KIs 6 and 7 led to improved systems 8;37,38 these incorporate N-methyl piperazines in place of aryl amine moieties to give more water-soluble probes that aggregate less, leading to increased brightness in cells. The pharmacophore in 8b was modified as shown below to include a hydrophobic group hence favor type II inhibition, while 8a only binds the active conformation (type I).
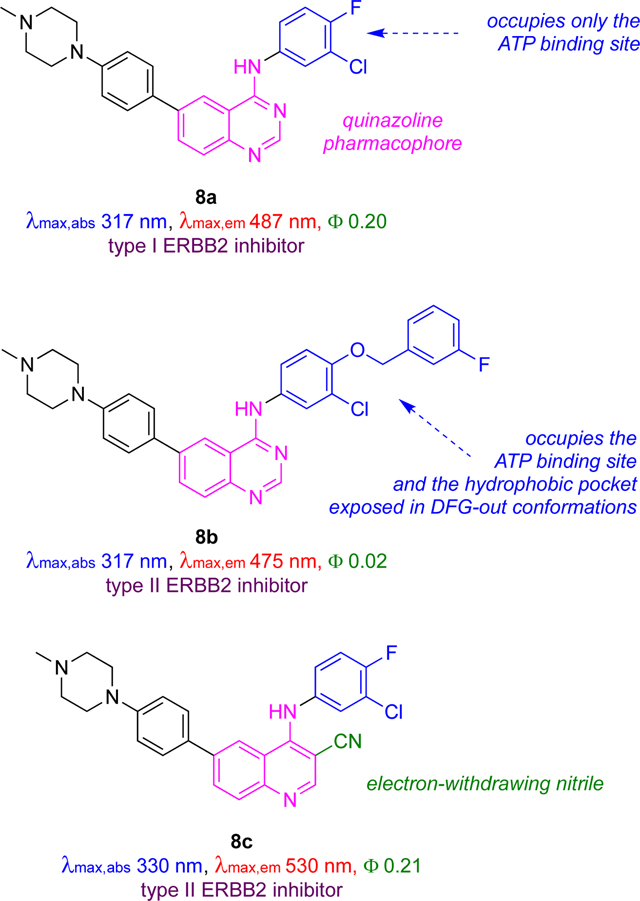
Overall, the optimized ligands type I and II ligands 8a and 8b have Ki values of 71 and 27 nM, which are comparable to those for the FDA-approved type II inhibitor lapatinib (20 nM). Both probes afforded significant turn-on emission responses in the kinase domain (11x for 8a, and 4x for 8b) and both were blue shifted by ~40 nm on binding. However, enhancement was not exclusively due to solvatochromic effects as seen by recording fluorescence intensities as a function of excitation wavelength, i.e., via excitation spectra. These spectra showed significant emission for excitation at 280 nm which does not correspond to significant absorption by the dye, but instead may be dye to energy transfer from Trp and Tyr residues in the enzyme pocket to the probe.
Access to probes 8a and 8b facilitated detailed studies of the cancer cell biology of ERBB2 inhibition.37 For instance, immunohistochemistry with an anti-ERBB2 mAb proved the probe colocalized with the ERBB2 receptor within the complex cell environment. Off rates for 8a and 8b in these experiments correlate with expectations for type I (fast) and type II (slow) inhibitors, respectively.
Heterogeneity between fluorescence levels was observed for ERBB2+ cells treated with 8a. This fluorescence heterogeneity could be due to different levels of receptor expression or to fundamental differences in levels of kinase activation. To differentiate between these possibilities, fluorescent activated cell sorting (FACS) was used to determine cell populations displaying high and low probe signals. ERBB2 receptor phosphorylation levels then were allowed to recover for 24 h prior to Western blot analysis. Even after recovery, cells with high probe signal showed 25 % higher phosphorylation signal than the ones with low probe signal, suggesting the fluorescent probes detect stable differences in kinase activation states inherent to different cells.
Further modification of structures 8 led to the cyano derivative 8c.38 Like 8a, 8c absorbed at about the same wavelength as the parent inhibitors gefitinib and lapatinib. However, unlike 8a, 8c has a huge (200 nm) Stokes shift giving a longer emission max (520 nm) in the bound form which is easily distinguishable from gefitinib and lapatinib (440 nm).
Probe 8c was used in a clever way to differentiate type I and II inhibitors and their relative binding affinities. Briefly, cells were first incubated with 8c to saturate internal ERBB2, hence the probe showed an increased fluorescent signal which plateaued. To illustrate the outcome with type I inhibitors, addition of gefitinib, a type I inhibitor with modest affinity (5 μM) towards ERBB2, induced the active ERBB2 conformation, caused dimerization, and more ERBB2 to be internalized. Thus, an increase in fluorescence was observed that peaked as 8c (Ki 390 nM) competed with gefitinib for the binding site, but fluorescence intensity did not increase drastically when a KI with higher ERBB2 affinity, canertinib, was added. For lapatinib and neratinib, however, there was no change in emission when cells were treated with 8c because these type II inhibitors bind in ways that favor inactive conformations of ERBB2, which tend not to dimerize or be internalized. Thus, 8c reports on the dynamics of internalization ERBB2 activation states because the probe senses binding modes that correlate with endocytosis and also with type I or II conformations.
Prodan Derivatives 9
An interesting combinatorial approach has been used to find fluor-KIs against initially undefined kinases. Prodan (red), an uncharged small molecule fluorescent group, was attached to pyrazolo{3,4-d}pyrimidine, a generic scaffold commonly used in competitive ATP kinase inhibitors.39 A series of compounds were synthesized by modifying the 3-position of the heterocycle, and photophysical properties were calculated in different organic and aqueous solvents. Probes 9a and 9b displayed good fluorescence in aqueous conditions, hence they were evaluated further in kinome screens against 65 kinases at 1 μM to assess kinase selectivity. Probe 9a did not inhibit any of the kinases significantly, but 9b had a significant effect on Aurora-A, Blk and LCK. Cell free assay IC50 values revealed 9b as nanomolar inhibitor of Aurora-A (222), Blk (554) and LCK (124 nM).
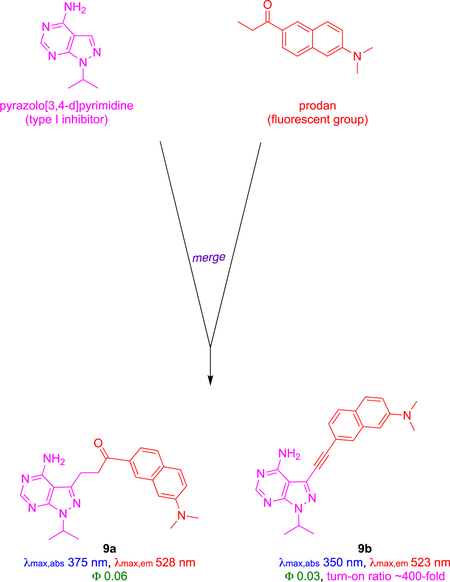
Compound 9b was used to study activity of LCK in live cells. Titration showed the fluorescence of 9b increased by 400x relative to free probe when one equivalent of LCK was added, presumably due to enhancement in the hydrophobic pocket of LCK. Fluorescence emission of 9b decreased in the presence of the high affinity LCK inhibitor TC-S7003 (IC50 = 7 nM). These in vitro experiments were augmented with cellular assays that confirmed the role of the probe. Thus, confocal imaging showed intracellular fluorescence correlated with increased concentration of 9b when the probe was incubated with LCK-positive Jurkat cells. Partial co-localization of fluorescence signal of 9b and LCK-specific antibody (anti-LCK-AF647) confirmed intracellular localization of 9b and LCK.
C. Cellular Probes
Fluor-KIs have two main applications in cellular assays. First, they can be used to report on localization of the probe-kinase complex, particularly if the kinase is labeled with a complementary fluorescent dye (usually a protein like mCherry) so that two channel observations (KI and kinase probe) can be used to check for co-localization. These studies reveal if the probe has selectivity for the targeted kinases in cells, and where in the cell those complexes concentrate.
A Src Probe
Dasatinib-BODIPY derivative 1 has been investigated for intracellular imaging of its binding with Src.12 As a prelude to those experiments, kinase binding and inhibition properties of 1 were validated via a kinase assay, specifically a Z’-Lyte assay.
Z’-Lyte methodology is based on differential rates of proteolytic cleavage for peptides in phosphorylated and non-phosphorylated states. Peptides modified to include donor-acceptor or donor-quencher FRET (Förster Resonance Energy Transfer) systems are cleaved rapidly in non-phosphorylated state, i.e., when the featured kinase is inhibited, disrupting energy transfer, and giving a conspicuous spectroscopic change. Phosphorylation of the peptide impedes this process; hence the level of kinase inhibition can be quantitated.13
A pan-SRC KI can inhibit several SRC kinases. Dasatinib, for instance, has nanomolar potencies against nine SRC family kinases including Src, Yes, Lck, Fyn, Blk, Frk, Fgr and Hck, and inhibits kinases in the Ephrin and Tec family.40–42 Formation of fluor-KI conjugates frequently results in reduced kinase inhibition and less potent effects on cell proliferation.12 Z’-Lyte assays against ten known kinase targets of dasatinib showed 1 exhibited single-digit micromolar IC50s against seven (including several Src family kinases), representing two- to ten-fold decreased activities compared to dasatinib. For instance, 1 exhibited a 40-fold decreased activity against Abl vs dasatinib, and it was 23x less active (IC50 = 47 nM) in a cell proliferation assay featuring BCR-Abl-transformed cells than the parent KI (IC50 2.1 nM). Conjugate 1 was not cytotoxic at <5 μM towards parental cells that do not express BCR-Abl, excluding the possibility that cytotoxicity was unrelated to inhibition of that kinase.
Uptake of 1 was determined by probing its interaction with Src in live cells. Hepatoma cells were engineered to express a Src-mCherry fusion protein (ex/em 587/610 nm) to facilitate this study by searching for co-localization of the mCherry and fluor-KI fluorescence. Flow cytometry showed high correlation of signals in the mCherry(Src) and BODIPY channels when the cells were treated with 1, suggesting the probe bound Src in cells. This experiment does not exclude the possibility that 1 bind more than one intracellular protein; consequently, a control was necessary. Thus, incubation and counting were repeated using the parent, unlabeled, inhibitor in competition with the probe. That experiment showed significant reduction of the colocalized fluorescence implying dasatinib and 1 compete for the same protein target (Fig 6).
Fig 6.
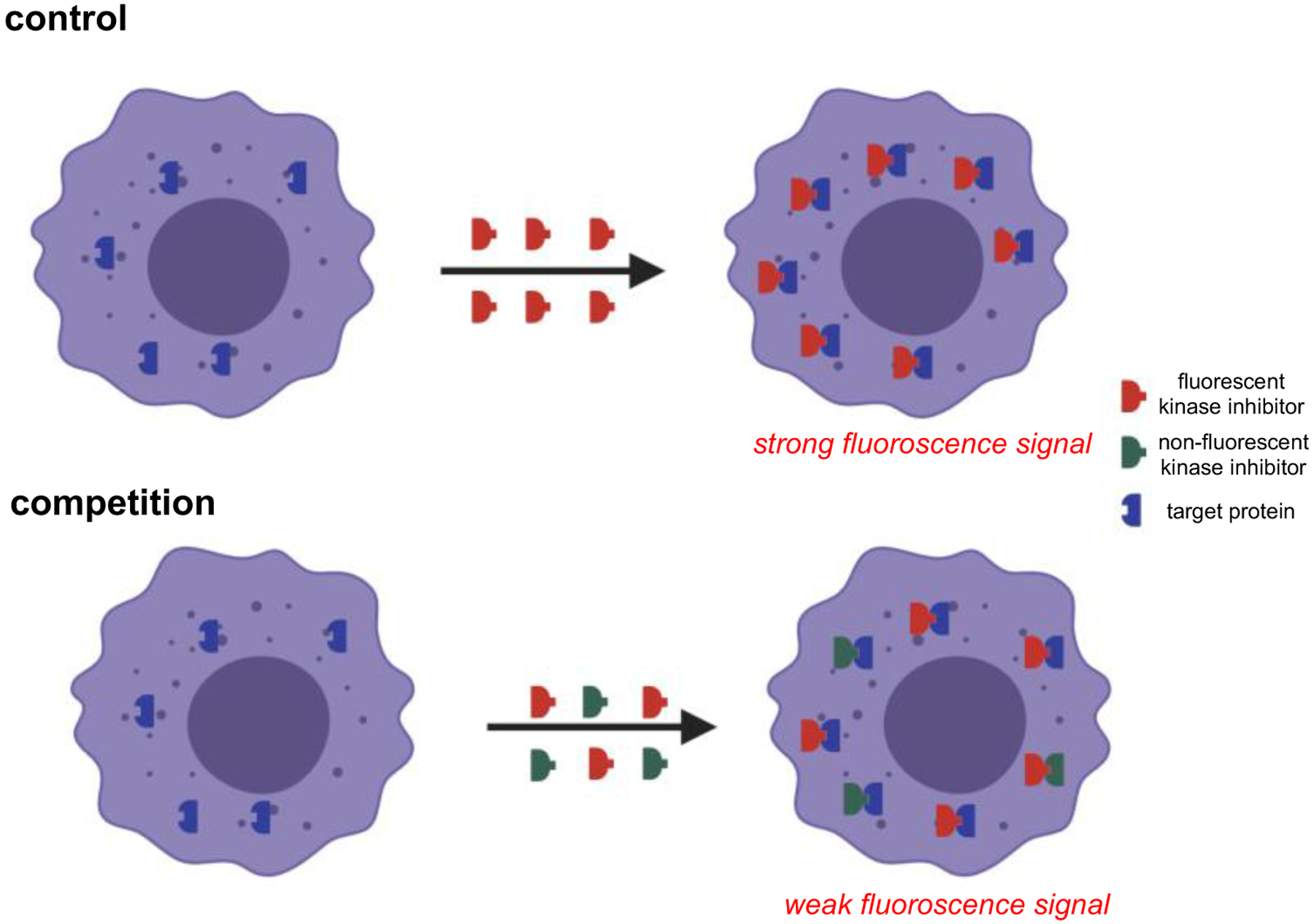
Cellular based assay to show competitive binding of a fluor-KI to probe target selectivity.
Bruton’s Tyrosine Kinase (Btk) Probes Form Covalently Bound KIs
KIs that bind non-covalently and reversibly are prone to be effluxed from cells faster than ones that become covalently attached to the enzyme. Consequently, probes based on irreversible kinase inhibitors resist wash out from cells, have long residence times in vivo, and data obtained from these are easier to interpret than for non-covalent probes which are necessarily more dynamic.
Ibrutinib (FDA approved for treatment of various forms of myeloma, leukemia, and non-Hodgkin’s lymphoma) is an irreversible KI that inhibits Bruton’s tyrosine kinase (Btk) by covalently binding a free Cys residue. Probe 10a links ibrutinib and a BODIPY.43 The purified kinase, and cell lysates containing it, were reacted with 10a, and fluorescence labeling of Btk was observed via gel analyses. Btk-10a immunoprecipitation was shown to deplete that protein from a cell lysate.
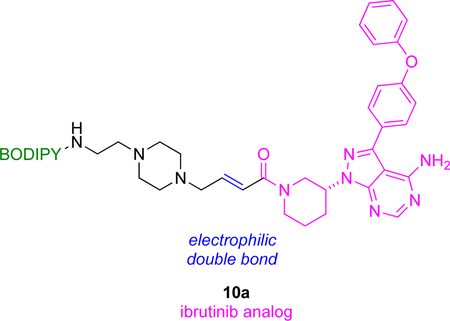
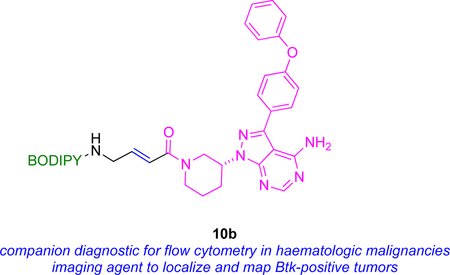
Other work, but from another group, has featured 10b, very similar to probe 10a, on the same kinase44 The apparent IC50 of 10b (~200 nM) was 100x greater than that of the parent drug, but it still labeled Btk with high selectivity, presumably due to covalent binding. In any case, IC50 data is not a reliable indicator of inhibition by covalently binding KIs because of the time-dependent and irreversible interactions involved.45 Flow cytometry was used to measure uptake of 10b; only cells that express this kinase were labeled, ones containing a Btk-mCherry fusion (cf Fig 6) colocalized the fluorescent protein and BODIPY fluorescence, and that staining was blocked by the parent drug. Consequently, there is potential to use probes 10 as a diagnostic agent for hematologic malignancies via flow cytometry.
Probes For Polo-like Kinases 1 – 3 (PLK1 – 3)
Polo-like kinases (PLK1 – 3) are concentrated in subcellular structures, mainly in the cytoplasm, microtubules and interphase-centrosomes, during mitosis,46 hence regulate cell cycle progression.47 This motivated studies of the inhibition of PLK1 – 3 using a BODIPY-functionalized derivative of BI2536 (inhibitor of PLK kinases),13 i.e. probe 11.
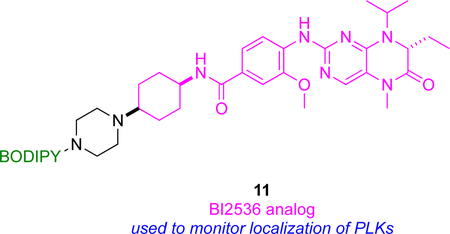
Compound 11 was first validated as a PLK probe by using the Z’-Lyte kinase assay, and western blots to detect inhibition of the PLKs. High and sustained cyclin B levels were observed when cells were treated with 11. Cyclin B is a mitotic marker, so this observation suggests conjugating this fluor-KI potently induces mitotic arrest.
The same fluor-KI, 11, was used to monitor localization of PLKs. Confocal microscopy showed localization of 11 in centrosomes (organelle involved in organizing microtubules), kinetochores (disc shaped protein assemblies where spindle fibers attach during cell division), the mid-body of cells during telophase, and in tubulin. In a control competitive experiment, tubulin staining by probe 11 was disrupted in cells treated with excess unlabeled KI, supporting the assertion that 11 selectively binds PLK kinases.
Probes To Measure The Effects Of A KI On Cellular Efflux
Small molecules can be internalized into cells by passive diffusion, endocytosis, or active transport, and pumped out via pathways which may overlap but are not necessarily the same as the import mechanisms. For instance, KIs may passively diffuse into cells, and be effluxed via ATP-binding cassette (ABC) transporters,48 which include P-glycoprotein (Pgp) and ABCG2.48 Thus, observation of labeled fluor-KIs at any instant represents a net effect snap-shot of opposing dynamic processes which can provide information on KI influx-efflux dynamics that would be difficult to obtain otherwise.
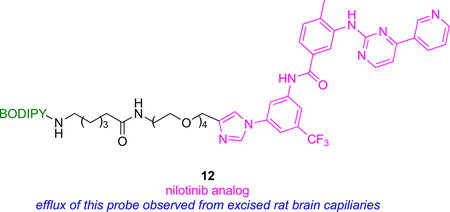
The BODIPY derivative 12 was made from the BCR-Abl inhibitor nilotinib (FDA approved to treat drug-resistant chronic myelogenous leukemia CML).49 In flow cytometry, cells that express Pgp or ABCG2 retained significantly less 12 than ones that have neither of these receptors, while retention of 12 in Pgp+ and ABCG2+ cells was enhanced using inhibitors of these transporters. Together these two observations indicate 12 is pumped out via Pgp or ABCG2.
An important function of Pgp and ABCG2 is to maintain homeostasis across blood brain barrier (BBB), so an ex vivo model featuring expressed Pgp and ABCG2 was used to mimic efflux of 12 across the BBB. Freshly isolated rat brain capillaries incubated with 12 effluxed its fluorescence via pathways that were inhibited by nilotinib and by Pgp and ABCG2 specific inhibitors. It was also shown that nilotinib and its fluorescent derivative bind at substrate binding site of Pgp and ABCG2.50 Thus, nilotinib inhibits Pgp and ABCG2 but, surprisingly, not at the ATP-binding site of these transporter proteins.
Incidentally, attempts to simulate physiochemical properties of the probes above failed because most computer programs used to do this are not parameterized for the B-F bonds of BODIPY dyes. Lack of parameterization is a trap that is relevant to simulations of physiochemical properties for other KI-BODIPY probes, and potentially for other containing molecular fragments not found in pharmaceuticals (e.g., C – Si bonds in silyl rhodamine, SiRh dyes).
D. In Vivo Probes
The structure of the fluorescent reporting group is critical for in vivo imaging of fluor-KI probes, the ideal being one that allows permeation of small molecules into cells and does not interfere with the kinase binding. Fluor-KIs designed to report on the location of a kinase in vivo are critically dependent on the dye used. Consequently, the following section is organized in terms of those dyes, then sub-divided into applications based on the dyes used, then the kinases targeted, or where the fluorophore binds, if that is a dominant factor.
Probes Containing Silyl-rhodamines Or Aza-BODIPYs
Fluor-KIs Comprising Bruton’s Tyrosine Kinase (Btk) Inhibitors
KIs that become covalently attached to kinases, like the Bruton’s tyrosine kinase (Btk) inhibitors, ibrutinib (FDA approved) and spebrutinib (in clinical trials) should probe for the KI and the kinase interactions in vivo. Thus, ibrutinib and spebrutinib have been modified to incorporate an amine handle, then functionalized with four different dye cores (silyl rhodamine, rhodamine, fluorescein and BODIPY) to give six different conjugates.7 Conjugates 13a and 13b formed from these dyes were shown to have good cellular uptake, binding selectivity, and inhibit Btk. In cell assays, these probes colocalized with Btk-mCherry proved that the silicon rhodamine carboxylate conjugates 13a and 13b were most closely associated with the kinase. Fluorescent gel scanning confirmed 13a covalently and selectively bound Btk in Btk+ lymphocytes but not in Btk− lymphocytes.
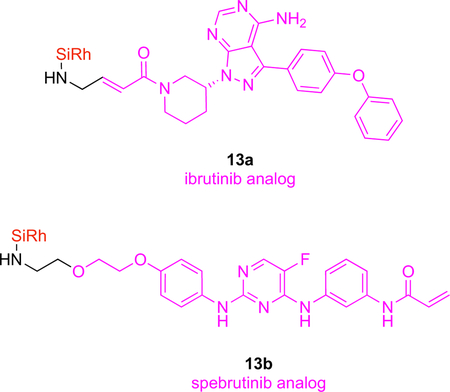
Absorption and emission wavelength maxima of silyl-rhodamines dyes are not ideal for in vivo studies, but data from mice can be collected. Experiments with more than one tumor type, each intrinsically labeled (e.g., with mCherry or GFP), can be used to show the fluor-KI probes preferentially accumulate in tumors that express the kinases they are targeted to. For instance, 13a only collected in Btk+ tumors when injected into mice implanted in experiments featuring tumors formed from Btk-mCherry or Btk−-GFP cells. Another advantage of fluor-KIs in vivo is that measurement of blood half-lives for the probes is relatively easy by fluorescence (provided the probe is not degraded to a nonfluorescent metabolite); in this case 13a had a vascular t1/2 of ~18 min.
Fluor-KIs Comprising Inhibitors Of B-Raf Kinase
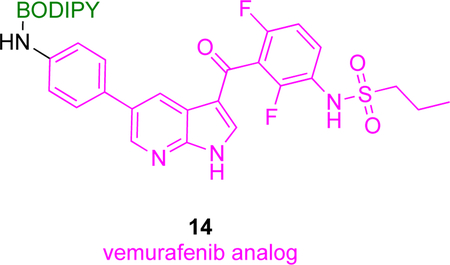
Another in vivo application of fluor-KI probes features BODIPY and silicon rhodamine derivatives19 and vemurafenib, a non-covalent serine/threonine B-Raf kinase inhibitor directed to the BRAFV600E mutation. BRAFV600E is an oncogenic driver of several cancer types, particularly melanoma.51 Structural analyses show the p-chlorophenyl substituent of vemurafenib points into solvent and away from the kinase binding pocket, hence modifications at that position were prepared because they are relatively unlikely to compromise the KI’s binding. Six fluorescent conjugates were synthesized based on BODIPY and silicon rhodamine dyes and tested for inhibition of proliferation of melanoma cancer cells, and for binding affinities to BRAFV600E. The top three conjugates showed EC50 in the range of 95–570 nM against melanoma cancer cells and 280–380 nM binding affinities against BRAFV600E (cf vemurafenib had an EC50 (64–160 nM) and a binding affinity of 110 nM in the corresponding assays). These three compounds were further screened for penetration and retention in melanoma cancer cells, which revealed 14 had the most favorable properties. Moreover, confocal imaging showed 14 localized in cellular cytoplasm, the putative site for accumulation of BRAFV600E, and the fluorescent signal lingered there for an extended period consistent with covalent binding.
In vivo imaging in mice models developed from vemurafenib-sensitive and -resistant cancer cell lines revealed 14 preferentially localized in tumors that responded to vemurafenib, and its fluorescence was retained there even after 24 h.
Fluor-KIs Comprising Inhibitors Of Mer Tyrosine Kinase
In vivo studies have also been performed for a fluorescent conjugate of UNC2025, a preclinical inhibitor of tyrosine kinase Mer (MERTK).11 MERTK is a transmembrane protein implicated in various cancers, wherein it plays a role in metastatic spread.52 Crystallographic analyses of Mer complexed with UNC2025 and with a closely related structure (UNC569; PDB ID: 3TCP) indicate modifications to the methyl piperazine of the KI should not significantly impact complexation, so analogs 15 with that replaced by a piperazine conjugated to a zwitterionic silicon rhodamine carboxylate were prepared.53
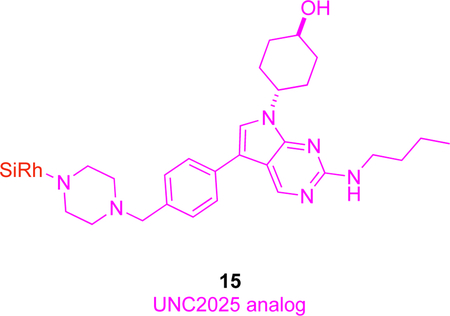
In vitro imaging of 15 on cells which overexpress Mer (SK-MEL-3 melanoma), revealed that it accumulates in the cytoplasm near the nucleus. In blocking experiments, uptake of 15 was suppressed by excess UNC2025, and in SK-MEL-3 cells in which Mer had been knocked down via siRNA; both experiments indicate uptake of 15 is mediated by that kinase. Conjugate 15 was inferior to the parent KI with respect to cytotoxicity and phosphorylation activity, but, nevertheless, colocalized with Mer in vivo, as shown using a mouse model in which the endogenous expression of the kinase was engineered to be coupled with GFP expression. Probe 15 was uptaken more in tumor-associated macrophages than in tumor cells, i.e., in the inflammation surrounding the cancer more than the tumor itself.
Fluor-KI Probes Targeting the Polyamine Uptake System
A targeted prodrug approach has been used to deliver gefitinib to non-small cell lung cancer (NSCLC),54 wherein the 16a consists of a targeting ligand, cleavable linker, and gefitinib. The targeting ligand used is a polyamine mimic which is endocytosed through polyamine uptake system (PUS);55 that PUS-system is overexpressed in NSCLC.56 A cleavable disulfide linker was used to connect gefitinib with the polyamine-mimic targeting group (green) and an aza-BODIPY fluorophore. Glutathione (GSH), an abundant antioxidant in cancer cells compared to normal ones (up to 10 times),57, 58 cleaves the disulfide bond resulting in selective release of gefitinib in the cancer cells.
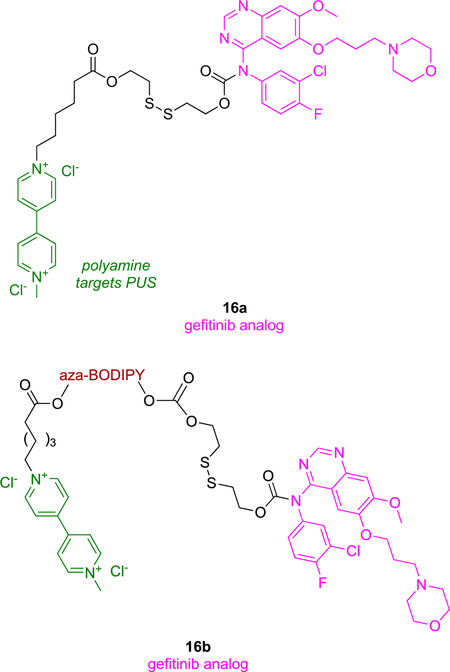
Polyamine ligands used in the study were tested for phosphorylation inhibition of Akt-Ser473; the Akt pathway is downstream of EGFR (epidermal growth factor receptor) stimulation, and plays an integral role in cell survival.59 Flow cytometry and western blotting indicated that the polyamine mimics inhibited pAkt resulting in apoptotic cell death. Non-fluorescent (a) and fluorescent (b) forms of 16 were tested in two NSCLC cell lines, and both conjugates were more cytotoxic than the parent KI, possibly due to polyamine inhibition of pAkt.60 Annexin V/PI (propidium iodide) staining in flow cytometry indicated compounds 16 had higher tendency to kill cells by apoptosis than necrosis, implying cytotoxicity was induced by disruption of cell signaling pathways by the KI.
In two parallel experiments, probe 16b was administered iv via the tail of mice impregnated with subcutaneous tumors, some from cells that are sensitive, and others that are resistant, to gefitinib. In both cases this probe selectively localized in the tumors, and robust fluorescence persisted for up to 24 h post injection. An attribute of 16b is that it gains fluorescence after the disulfide bond is cleaved inside the tumor microenvironment. Both 16b and gefitinib inhibited the growth of tumors from gefitinib-sensitive cells to about the same degree, but the impact of 16b on gefitinib-resistant cells was greater than gefitinib, demonstrating the effect of the PUS targeting group. Conjugate 16b performed better in detecting samples from patient derived NSCLC than commercially available serum biomarkers (carcinoembryonic antigen and cytokeratin 19).
Probes Containing “Tumor-seeking” Heptamethine Cyanines
Recent work from our laboratories61 and others8, 62 showed some fluor-KI conjugates limit cell proliferation more effectively than the parent KI.61 All these conjugates feature a “tumor-seeking dye” like the cyanine-7 dye MHI-148 A’ (Fig 7a). The special characteristics of MHI-148 A’ are: first that it accumulates in tumor tissues after iv injection, and second that it is retained there for several days.63, 64 Amazingly, this characteristic is independent of the type of implanted solid tumor.
Fig 7.
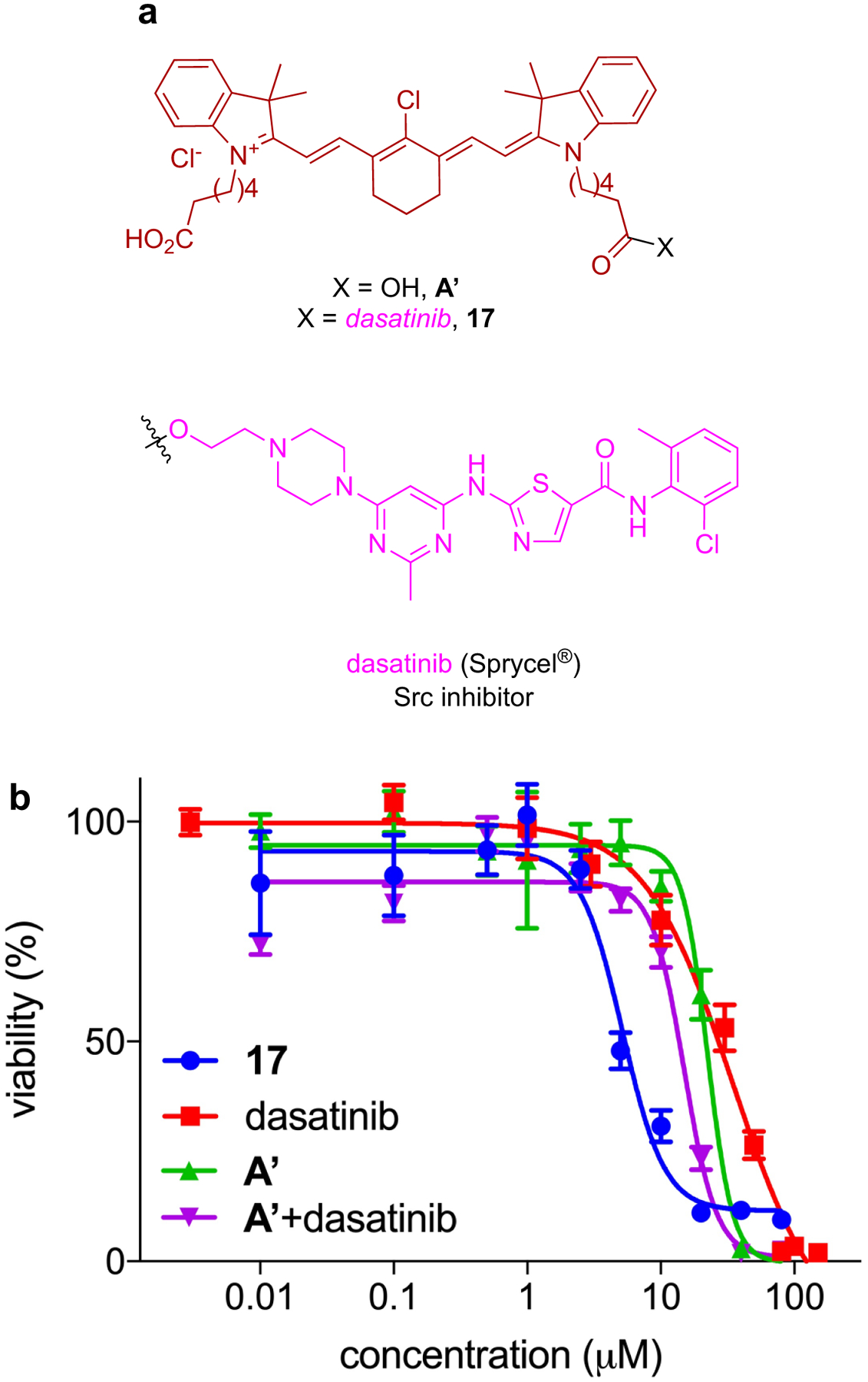
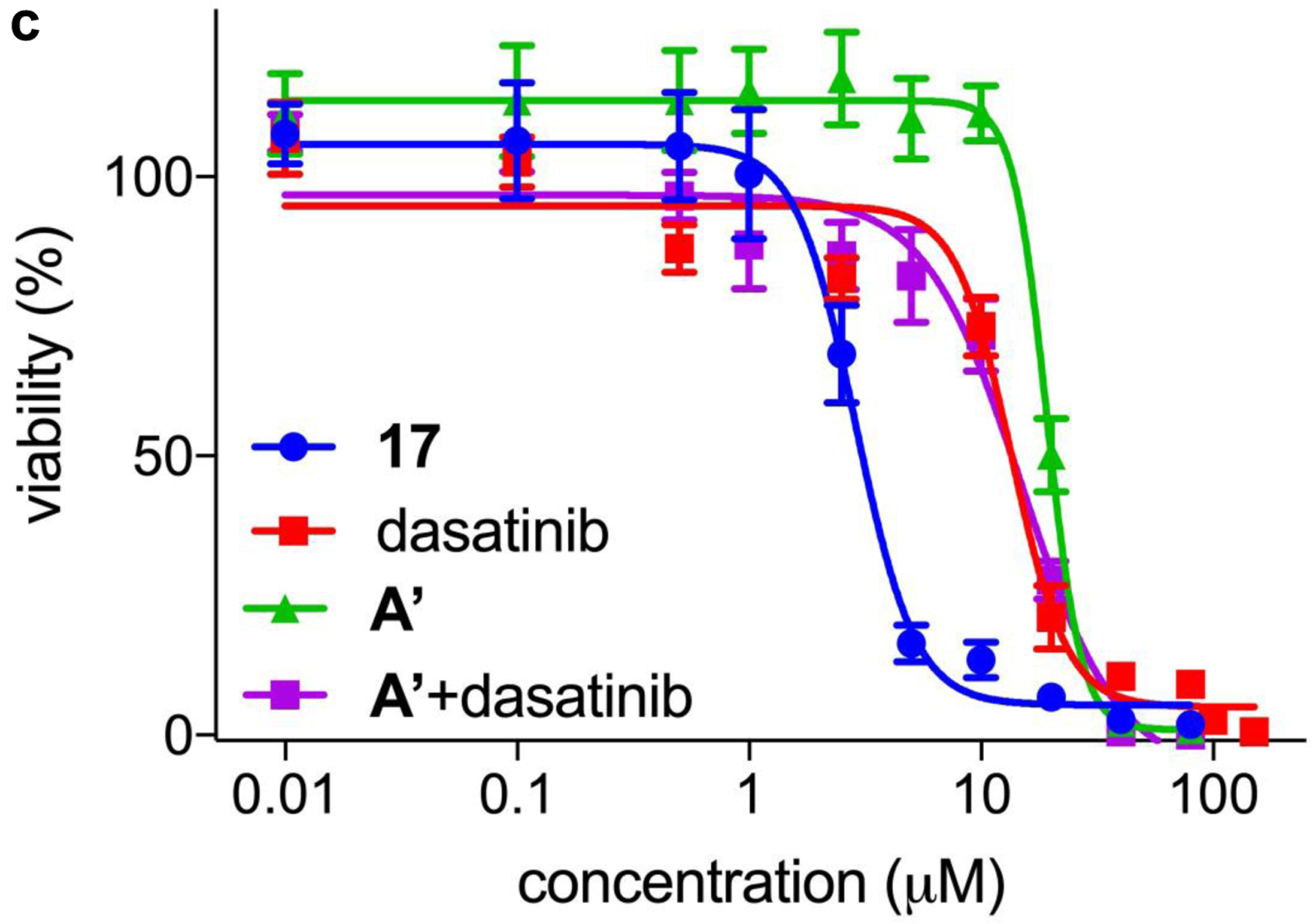
a Conjugate 17, from a tumor seeking NIR dye and dasatinib, gives enhanced impact on the viability of: b HepG2 liver cancer cells; and c U87-MG glioblastoma cancer cells.
We reasoned conjugates of KIs with MHI-148 could be used to target solid tumors and cause the kinases are overexpressed there. Unfortunately, but as expected, probe 17, conjugate of dasatinib with MHI-148, binds weakly to two illustrative kinases than the parent KI (Kd for 17: 2.4 nM with Src, and 17 nM with Lyn; literature Kd values for dasatinib are 0.07 nM with Src61 and 0.57 nM with Lyn61). Moreover, in assays to gauge enzyme activity the conjugate was also less active than the parent KI (18.4 nM to Src, and 55.6 nM to Lyn: while the corresponding IC50 values for dasatinib were 1.2 nM for Src, and 1.8 nM for Lyn). However, 17 is cell permeable (localizes in the mitochondria and lysosome) and has a greater effect on cell proliferation than the parent KI and MHI-148 mixed at equimolar concentrations (Fig 7b and c).
In other work we have established that thiols (but not amines, alcohols or phenols) under physiological conditions react with A’ via displacement of its meso-chloride.65 Only free thiol in albumin is 34Cys. It transpired that the 34Cys of albumin forms a covalent conjugate via binding to the cyanine at the meso-position.66 Independent studies by Goncalves and co-workers were published almost simultaneously, and confirmed these observations.67
The inference of the observations above is that the effects of KIs on cell viability sometimes can be increased by binding to albumin. Consistent with this, it has been reported that erlotinib analogues conjugated to albumin slowed proliferation of NSCLC cell lines more than erlotinib itself.68
For nearly a decade, literature reports consistently claimed MHI-148 A’ (and related dyes) were transported into cells via Organic Anion Transporting Polypeptides (OATPs). However, we showed evidence for OATP transport in cell studies is only seen if serum free media are used, i.e. an environment without albumin.69 Subsequently, we showed MHI-148 A’ binds albumin non-covalently then slowly transforms to the covalent adduct by displacement of the meso-Cl of MHI-148 by 34Cys of the protein. Both the covalent and the non-covalent adducts were uptaken into tumor cells selectively over non-tumorigenic cells. Moreover, MHI-148 A’ non-covalently bound to albumin-FITC (i.e., albumin-fluorescein isothiocyanate conjugate) was uptaken into tumor cells and the two labels colocalized. Based on these experiments, it would appear MHI-148 A’ is imported via albumin receptors. There are several albumin receptors (e.g. SPARC and gp60), and they tend to be overexpressed on cancer cells,70 probably because of their enhanced metabolism rates. On the basis of those data, based exclusively on cellular assays, one might conclude that uptake into tumors were mediated exclusively by albumin interacting with its receptor. However, if the uptake into cancer cells could be mediated via both non-covalent and covalent binding to albumin, then covalent binding might not be necessary for persistence of the dye in tumor tissue over several days. Research to establish if covalent binding is required, or if non-covalent binding alone is sufficient, is now described, because their KI conjugates will presumably behave in the same way.
A series of in vivo experiments were planned since accumulation of MHI-148 A’ into tumor tissue could be explained via albumin receptors, but reasons for persistence of the tumor fluorescence were unclear. These in vivo studies featured four dyes with complementary properties which include A’. Fluor A’-Ph forms a non-covalent adduct with albumin and cannot form a covalent one. “QuatCy” B’ does not form a non-covalent albumin adduct, and only slowly forms a covalent one, again by displacement of meso-Cl by 34Cys of albumin. Finally, B’-Ph does not form a non-covalent albumin adduct and cannot form a covalent one.
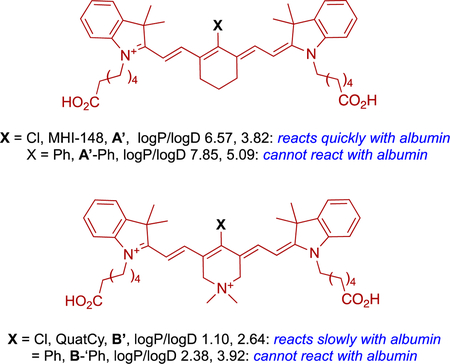
Complimentary characteristics of A’, A’-Ph, B’, and B’-Ph facilitated deduction of structural features that determine in vivo accumulation and persistence. These dyes were injected intravenously into healthy mice, and into others implanted with subcutaneous triple negative breast tumors (E0771). The passage of fluors were monitored via near IR fluorescence in the living animals, and in their organs post mortem.69
Experiments with the healthy mice revealed A’ was predominantly cleared via the liver, while B’ cleared more quickly via both renal and hepatic pathways. Fluor B’ was detected in the gastrointestinal tract 1 h post-injection, indicating fast excretion from liver and bile duct, while A’ gave signal in the liver and duodenum after 4 h. Insignificant urinary excretion was observed for A’, and this dye had a blood plasma fluorescence half-life of 195.2 min. Compound B’ dosed intravenously at 25 nmol (17 μg/kg) had a plasma elimination half-life of 100 min and 32% of injected dose was excreted through the urine.
For the tumor-bearing mice, the blocked compounds A’-Ph and B’-Ph had poor preferential tumor accumulation and no significant persistence, hence tumor-to-background ratios (TBRs) did not deviate significantly from unity, i.e., there was little contrast between the tumor and the background. Conversely, the fluors that form albumin adducts, A’ and B’, displayed high tumor accumulation and prolonged retention; TBR after 48 h post injection was 3.5 and 10, respectively. Accumulation of A’ and B’ into tumor and background tissues was rapid, but B’ cleared faster from the healthy tissue, giving comparatively high TBR values for each time point at which the tumor could be discerned; high contrast was observed 4 h post-injection of B’ while 48 h was needed for A’. At 48 h post-injection, when the meso-phenyl dyes (A’-Ph and B’-Ph) were almost completely cleared from the body, but the meso-chloride dyes (A’ and B’) could be clearly seen in the tumors.69
Studies outlined above indicate non-covalent binding is insufficient for retention of tumor seeking dyes. Both non-covalent and covalent albumin adducts might get internalized into tumors but only conjugates that will form covalent complex with albumin give tumor fluorescence that persists for extended periods. Moreover, over a relatively short time (4 – 6 h) in the body, the dyes which could not form covalent adducts did not appear to accumulate in tumors selectively over healthy tissue and were mostly excreted. This observation is unexpected from the cell data where non-covalent adduct formation did result in dye uptake. In vivo, only covalent adduct formation resulted in accumulation and persistence in tumors.69
It is unwise to extrapolate too much from cellular assays to predict in vivo behavior, and the featured experiments illustrate this nicely. Cellular data alone might lead to the prediction that uptake into tumor tissues in vivo was due to uptake via albumin receptors, but in vivo studies indicate this is not necessarily the sole mechanism. The enhanced permeability effect (EPR) effect, originally postulated based on labeled-albumin in tumors,71–75 may play a role in the accumulation of tumor seeking dyes, and might even be the predominant reason for persistence. EPR effects arise from differences in healthy and tumor tissue vasculatures. Large molecules and particles can traverse blood vessels in healthy tissues, but tumor environments have vasculature compressed enough that large molecules and particles tend to be retained.76, 77 In either case, it seems the root cause of accumulation and persistence of tumor seeking dyes is the extremely long residence time of albumin in the body coupled with the high abundance of that carrier-protein (in human blood, turn-over >20 d and concentration 0.53–0.73 mM78).
An inference of the studies above is that tumor seeking dyes require a leaving group, typically a meso-chloride, on the Cy7 system. Fluor-KI conjugates that have no meso-leaving group will not endow the KI with preferential accumulation and persistence. Beyond that, the PK behavior of A’ and B’ brings to the fore a variety of important unanswered questions, prevalent among them being the unresolved impacts of fluor-KI adducts on tumors in vivo. Imaging data8, 79 shows localization of 17 in tumors in vivo (Fig 8) but a therapeutic study has not been performed.79 Fluorescence from 17 localized and persisted for 72 h in mice impregnated with subcutaneous U87-MG tumors. This observation does not mean the intact fluor-KIs accumulated and persisted in the tumors, but it leaves open the possibility that these conjugates may drastically increase the residence time of a KI in a tumor, and, simultaneously, provide a NIR-probe for optical imaging in vivo.
Fig 8.
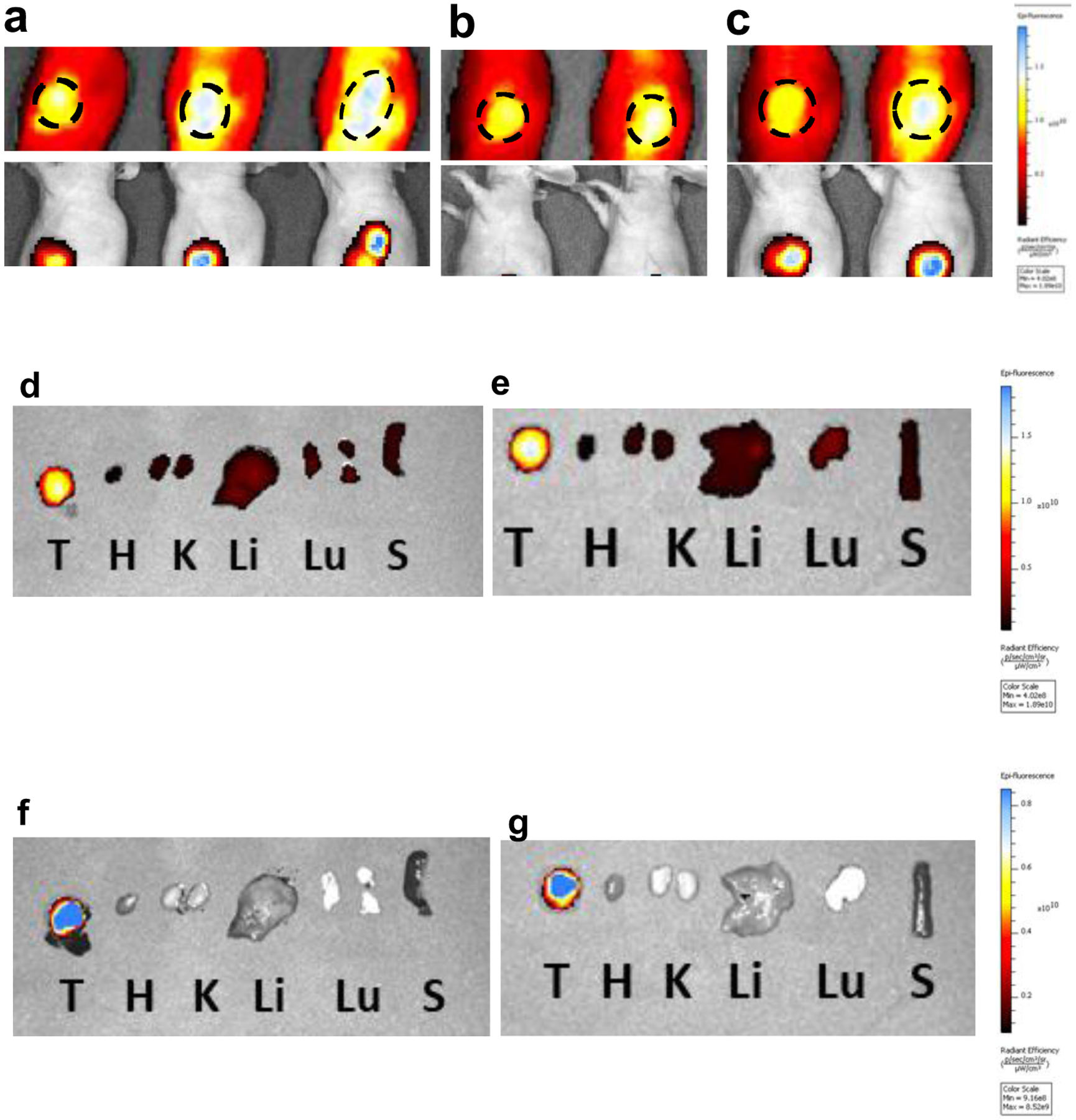
Localization of 17 and fluorescence of RFP in the tumor tissue showing the region of interest after retroorbital iv injections at 10 mg/Kg, at: a 24; b 48; and c 72 h. Post mortem fluorescence of organs after sacrifice at 72 h: d NIR for mouse 1; e NIR for mouse 2; f RFP mouse 1; and, g RFP mouse 2. T = tumor, H = heart, K = kidney, Li = liver, Lu = lung, S = spleen
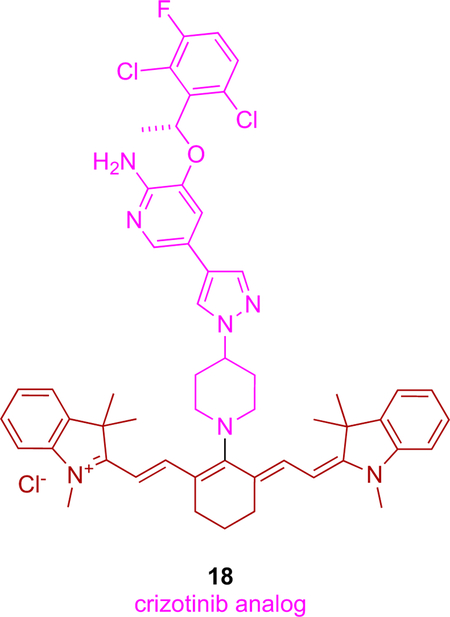
Following our work, a conjugate of crizotinib with a NIR cyanine dye (IR-786) has been reported.8 This conjugate (18) was prepared by allowing the piperazine of crizotinib to displace the meso-Cl of IR-786. Crizotinib targets ALK, a kinase overexpressed in glioblastoma multiforme (GBM),80, 81 hence this KI, free dye and conjugate 18 were tested against three patient-derived GBM cell lines. Remarkably, the cytotoxicity of 18 was 100x more than the free dye or KI. All three patient-derived cells were resistant to temozolomide (TMZ; FDA approved chemotherapeutic for glioblastoma), but synergistic effects were observed when equimolar concentrations of TMZ and 18 were added to the cells.
Drugs conjugated to tumor-seeking cyanine dyes have been reviewed,62 and these include derivatives of poly (ADP-ribose) polymerase {PARP} inhibitors82 that are similar to tumor seeking dyes with KIs.
Probes Containing Other Cyanines
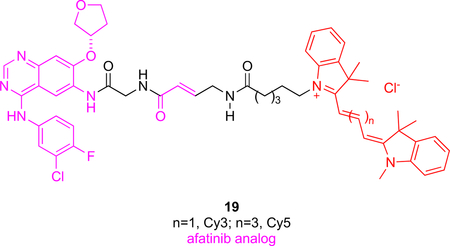
Afatinib, a dual inhibitor of HER1 and HER2 is associated with multiple different cancers such as NSCLC, breast, and ovarian cancer. Probes 19 are unique insofar as it contains fragments of the KI afatinib separated by a glycine. Trimethine (Cy3) and pentamethine (Cy5) cyanine dyes were bound at the other terminus of probes 19.20 The Michael receptor of afatinib combines with the cysteine residue near the enzyme active site to irreversibly form a covalent adduct, but there was no evidence this occurs for 19. On the contrary, flow cytometry of cells treated with 19 then with afatinib showed decreased signal intensity indicative of reversible binding to the kinase. It appears that 19 is stereochemically unable to undergo Michael additions, unlike the parent KI. These probes could be used to design competitive assays to find stronger inhibitors of HER1/HER2 or for detection of HER1/HER2 in tumors.
The Cy3-functionalized version of probe 19 was administered iv in an immune compromised murine model featuring subcutaneous tumors xenografted with NSCLC cancer cells. Fluorescence in the tumor was discerned from 12 – 48 h post injection in live mice, and this was confirmed via ex vivo imaging post mortem. It is unclear why the Cy5 modification of 19 was not used in this experiment because it should be more easily visualized in vivo than Cy3-labeled compounds.
Multimodal Probes For In Vivo Studies
Optical imaging is compatible with technologies like positron emission tomography (PET), computer tomography (CT), and magnetic resonance imaging (MRI); consequently, NIR dyes can be combined with diagnostic probes like those to provide holistic visualizations of tumor microenvironments. For instance, optical imaging and PET complement each other because fluorescence spectroscopy provides higher resolution, whereas radiolabel-based tomography can image deep tissue environments.
Multimodal strategies are most conveniently implemented in proof-of-principle studies by using nanoparticles wherein components can be added to inert matrices. This approach circumvents synthetic effort to form covalent connections in small molecules, and the rigorous characterization criteria that is often not adhered to for polymer formulations. In the long term, however, batch-to-batch variability of nanoparticles is a serious problem in clinical studies, so this review is restricted to discrete small molecules of defined structures. Most multimodal probes are based on nanoparticles, and we are only aware of two involving KIs and discrete molecules.
One example of KI-based multimodal probes explored83 convection enhanced delivery (CED), an invasive method in which catheters are inserted in and around the tumor to bypass the blood brain barrier (Fig 9).84 A probe was formed by conjugating dasatinib (SRC promiscuous KI), a Cy3 dye (useful, but not the best cyanine for NIR imaging), and a 18F capture agent (purple) in the small molecule “package” 20.85, 86 Conjugate 20 proved to be less potent than the parent KI in cellular assays, but still effective at nanomolar concentrations. Predictably, delivery to tumor region was shown to be more effective via CED than iv.
Fig 9.
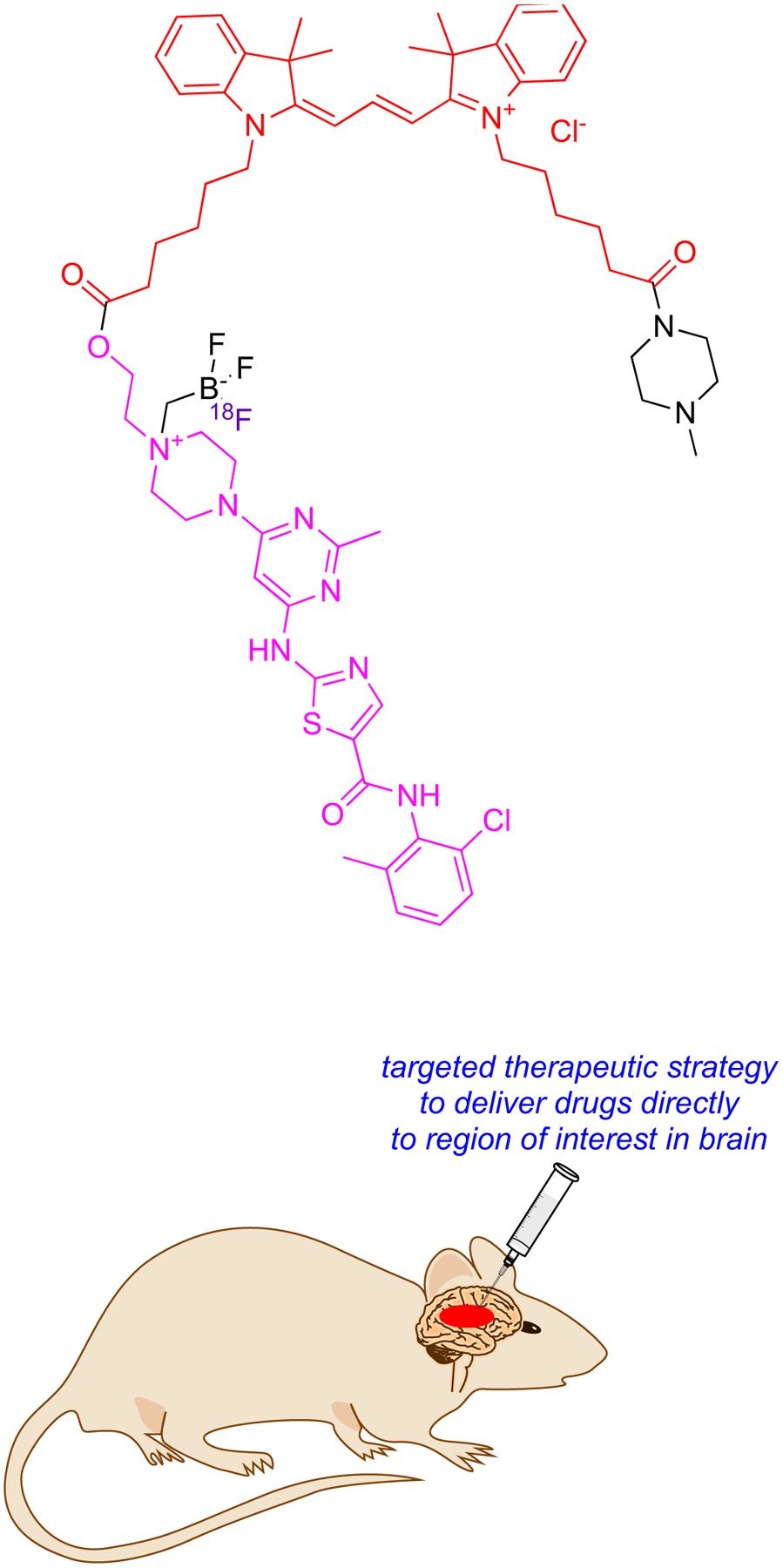
Convention Enhanced Delivery (CED) in which thin tubes penetrate the brain directly and bypass the BBB.
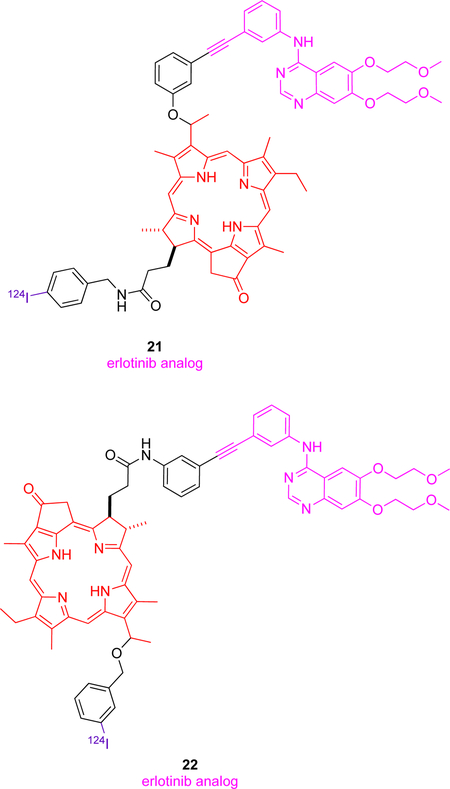
Stimulation of EGFR, a receptor tyrosine kinase overexpressed in many different cancers,87 results in cellular proliferation via inhibition of apoptosis, metastases and angiogenesis.88 One report,9 describes several theranostic molecules combining PET, PDT (photodynamic therapy) and fluorescence imaging agents. Erlotinib, a specific inhibitor of EGFR was conjugated with chlorin (a porphyrin-based imaging and PDT moiety) and 124I (purple) for PET was introduced at two different positions (3 and 17) giving conjugates 21 and 22. Their choice of attachment point for conjugation was surprising because the alkyne position on erlotinib points inside the binding pocket where this KI docks with EGFR, so modifying that alkyne might be expected to drastically reduce binding to EGFR, while crystallography indicates it is the other end of the molecule, the ethylene glycol fragment, that protrudes into solvent.68, 89 Surfactants (Tween 20 and Pluronic F-127) were required to solubilize 21 and 22 because the chlorin is hydrophobic. PET imaging using 124I is interesting; this radionuclide is harder to produce than 18F but its long half-life (4 d) facilitates shipment to regions where there is no local cyclotron. Both conjugates showed good tumor-to-background ratios for EGFR+ tissue over EGFR− (T24 and UMUC3 cells, respectively) hence these conjugates could serve as imaging agents. Conjugate 22 showed much better singlet oxygen generation as compared to chlorin structure, and on this basis, it is a better theranostic candidate of the two choices.
E. Conclusions
Overall, surprisingly few fluorophores have been used in fluorophore-KI probes; specifically, a commercially available BODIPY, an aza-BODIPY, a silyl rhodamine, a porphyrin, and a few cyanine dyes (Fig 1). However, those dyes cover a broad spectrum from short (<400 nm) to relatively long (780 nm or more) wavelength absorption and fluorescence maxima hence the range of applications covered in this review is correspondingly large.
Table 3 lists all the probes reviewed here, their primary kinase targets, the dyes used (approximately from shorter to longer wavelength absorption maxima), their binding and kinase inhibition properties (where available), and their potencies relative to the parent KIs.
Table 3.
Fluor-KI conjugates in this review, their targets, specificities, potencies, cell membrane permeability and whether the probe was applied in vivo.
| conjugate | KI | target | dye | binding and kinase inhibition (cell-free, nM, unless otherwise noted) | potencies relative to parent KI | cell membrane permeable | applied in vivo |
|---|---|---|---|---|---|---|---|
| 1 23 | dasatinib | Src, Abl | BODIPY | Kd: Src 0.5; Abl 0.15 | N/A | yes | no |
| 2 23 | dasatinib (DFG-out) | Kd: Src 2.8; Abl 0.75 | ligand induced different binding conformation | N/A | N/A | ||
| 3 23 | dasatinib (αC-helix-out) | Kd: Src 4.6; Abl 0.58. | N/A | N/A | |||
| 4 26 | a type II KI | Src, p38 | IC50:Src 25.4; p38 25.5 | N/A | N/A | N/A | |
| 5 30–32 | HS-133 | PI3Kα | intrinsically fluorescent | IC50: 68 nM | N/A | yes | yes |
| 6 33 | gefitinib | ERBB2 | Ki (in cell): 3,100 | N/A | yes | no | |
| 7a-b 34 | Ki (in cell): 7a N/A, 7b 9 μM | reporter of type I or II binding mode | yes | no | |||
| 8a 37 , 8c 38 | Ki (in cell): 8a 71; 8c N/A | yes | no | ||||
| 8b 37 | lapatinib | Ki (in cell): 27 | yes | no | |||
| 9b 39 | type I KI | Aurora-A, Blk, LCK | intrinsically fluorescent | IC50:222 nM (Arora-A), 554 nM (Blk), 124 nM (LCK) | N/A | yes | no |
| 10a43, 10b44 | ibrutinib | Btk | BODIPY | IC50: 10a N/A: 10b ~200 | 100x inferior | yes | yes |
| 11 13 | BI2536 | PLK1–3 | IC50: PLK1 5.01; PLK2 19.4; PLK3 9.37 | comparable | yes | no | |
| 12 49 | nilotinib | BCR-Abl | N/A | inferior | yes | no | |
| 13a 7 | ibrutinib | Btk | SiRh | IC50: 122 | 200~300x inferior | yes | yes |
| 13b 7 | spebrutinib | Btk | SiRh | IC50: 283 | yes | no | |
| 14 19 | vemurafenib | BRAFV600E | BODIPY | EC50: 380 | 3x inferior | yes | yes |
| 15 11 | UNC2025 | MERTK | SiRh | IC50 (in cell): 19.7 μM | inferior | yes | yes |
| 16 54 | gefitinib | EGFR | aza-BODIPY | IC50 (in cell): PC9, 50 H1650, 780 |
superior | yes | yes |
| 17 61,79 | dasatinib | Src, Lyn | NIR Cy7 (MHI-148) | IC50 (Src, cell-free): 184 IC50 (in cell): 5380 |
less potent in cell-free than in cell-based assays | yes | yes |
| 18 8 | crizotinib | ALK | NIR Cy7 (IR-786) | EC50: (average of 3 GBM cell lines): 50 ± 20 | 100x superior | yes | no |
| 19 20 | afatinib | EGFR, HER2 | Cy3 and Cy5 | IC50 (three cell lines average): Cy3-AFTN 1480 Cy5-AFTN 2460 |
inferior | yes | yes |
| 20 83 | dasatinib | Src | Cy5 | IC50 (in cell): 46 ± 30 nM | similar but inferior (used for PET) | yes | yes |
| 21, 22 9 | erlotinib | EGFR | porphyrin | N/A | used as PDT photosensitizer | yes | yes |
Probes Containing Blue-fluors
Simple BODIPY dyes tend to absorb UV and fluoresce in the 500 – 550 nm range, ideal for many in vitro assays. This spectral window overlaps with background intracellular autofluorescence, but the brilliance of BODIPY fluors compensates for this shortcoming. All the BODIPY-KI probes reported to date, 1 – 4 and 10 – 12, feature the same dye, presumably because it is commercially available (“BODIPY-FL” from Thermofisher and Lumiprobe). Practitioners studying KIs are, understandably, reluctant to spend time preparing custom BODIPY-dyes but it would be preferable to use more hydrophilic fluors to avoid non-specifically binding of fluor-KI conjugates to biomacromolecules.
Most KIs are not fluors; in fact, only KI 5 in this review is intrinsically fluorescent without modification. Consequently, tension in molecular designs is inevitable; changes to produce rigid, extended, chromophores with large cross sections (to enhance quantum yields, wavelengths, and extinction coefficients, respectively) alter the KIs-heterocyclic core and invariably result in diminished binding affinities and kinase inhibition activities. KIs with heterocyclic cores that have been modified to be fluorescent tend to comprise aromatic systems become part of “push-pull” chromophores loosely based on DCS (4-N,N-dimethylamino-4’-cyanostilbene). More specifically, modifications that conjugate an aryl alkene to the KI core (e.g., 6 – 8) are the only ones tested so far. There is some potential for building KIs with better affinity around intrinsically fluorescent chromophore that cause minimal structural perturbation, but, overall, to modify KIs to become fluorescent and retain their binding is a molecular design challenge.
Conceivably, fluorescent push-pull systems could feature alkenes electrophilic enough to covalently bind kinases, simultaneously breaking the conjugations leading to blue-shifted fluorescence. Screening these self-indicating KIs would be fast and informative, especially when paired with counter-screens to measure inhibition independent of covalent binding.
When considering kinase inhibition, binding affinities and inhibition IC50 values for isolated kinases in vitro, may not represent events in cells and in vivo90 because many kinases in cells function as quaternary complexes with other proteins; assays without those cooperative proteins therefore can be over-simplistic, and this uncertainty surrounds much of the data presented in the Table 3. In cases where this is a concern, modifying KIs to include fluorescent dyes is more likely to impact interactions with quaternary protein complexes than with isolated kinases because inclusion of large dyes may have more of a disruptive effect.
Probes Containing Dyes That Absorb In 600 – 700 nm
The silyl rhodamine featured in this review (SiRh) has an absorbance wavelength significantly above cellular autofluorescence, but shorter than ideal for imaging through tissue. The size of the probes containing are larger than necessary for in vitro assays, but not extended enough for maximal contrast in vivo. However, they are just as good, if not better, than BODIPYs for intracellular imaging, and the SiRh-probes 13 and 15 also have superior water solubility.
In contrast to SiRh fluors, aza-BODIPYs, as in 16, are poorly soluble in aqueous media hence they aggregate. This probably accounts for the literature bias towards SiRh dyes in this wavelength range for forming fluor-KI conjugates. In fact, the sole aza-BODIPY probe featured in this review, 16, has a polyamine mimic group for PUS uptake91, 92 which conveniently enhances the water solubility of the system overall. However, it would be interesting to check how the absorption/fluorescence properties of this fluor-KI probe change with concentration over the assayed range; if they do, that will indicate kinase inhibition might be influenced by formation of particles or micelles in solution.
Probe 19 is the unique one in which the key structural features of afatinib separated with a spacer (Gly). Compounds 19 feature a Cy5 (absorbs and emits above 600 nm) or a Cy3 (absorbs at 554 nm and emits at 570 nm).20 There probes emit in similar regions to r SiRh but have been used less for fluor-KI probes, due to non-specific binding of cyanine systems to biomacromolecules.
Probes Containing Tumor-seeking Dyes
Tumor-seeking characteristics of some Cy7 dyes are associated with leaving groups at the meso-position that are displaced by albumin-34Cys in vivo (and in some cell assays, depending on the conditions). Fluor-KI albumin adducts will assume the PK properties of albumin.
Probe 17 (and the parent dyes MHI-148 A’ and QuatCy B’) can form albumin adducts by displacement of their meso-Cl functionalities with the free 34Cys of albumin, but the other fluor-KI in this class, 18, cannot. Consequently, we predict the behavior of 17 and 18 will be different in vivo because 18 probably will not accumulate and persist in tumors. Contrasting PK behavior for meso-Cl and covalently bound systems has been demonstrated for molecules that do not contain KIs,93 but the experiments have not yet been reported for fluor-KI conjugates like 17 and 18. Thus there a routes to probes that do and do not bind albumin, and these may complement each other for different applications.
Multimodal Probes
Three multimodal dyes 20 – 22 are featured in this review; one of them, 20, contains a Cy3 dye attached to one of Perrin’s 18F-capture agents,85, 94–98 while the other two have porphyrin derivatives with a radioactive 124I. All these probes are designed to combine fluorescence indicators with labels for PET.99, 100 Porphyrin rings in 21 and 22 endow them with fluorescence and singlet-oxygen generation properties, hence they can be used in experimental approaches to PDT.101, 102
Combining NIR fluorescence, PET, MRI, photodynamic or photothermal characteristics are obvious combinations for futuristic KI multimodal probes. Discrete molecular systems are generally more likely to reach clinical trials than ones based on nanoparticles that cannot be reproducibly prepared to the high level of consistency required for clinical work.
Generalities Regarding Fluor-KI Conjugates As Research Probes
Fluor-KI conjugates can be used as research tools for studying kinases in vitro, in cells, and in vivo. For intracellular imaging and some animal studies, fluor-KI probes can complement strategies featuring genetically engineered fusions between fluorescence and target proteins. Fluor-KI probes can be useful provided the fluorophore does not interfere prohibitively with kinase binding, or non-selectively bind proteins in general. Thus, the selection of dyes is important, and there is room for improvement because, so few dyes have been explored (see above). For instance, one area for future exploration may involve pH- or metal-sensitive-fluors attached to KIs.
In vivo studies of fluorophore-KI probes are limited by the photophysical properties of the fluorophore. None of the studies featured above, except those with conjugates 17 and 18, involve fluors that absorb in the NIR region where penetration of light through tissue is maximal. Development of NIR fluor-KI conjugates is an area with a research future; for instance, it is not yet known how tumor-seeking fluor-KIs impact tumor growth in animal models. NIR dyes are necessarily larger than shorter wavelength probes, and endow conjugates with significantly different kinase binding properties, physiochemical profiles, and PK characteristics. In many cases these properties will compromise the activity of the KI, but in rare cases, like for tumor seeking dyes, they might enhance it because of PK factors. Almost invariably, the behavior of these systems will differ more from the properties of the KI than labelling strategies featuring small blue fluors.
A flow of routine validation steps in development of fluor-KI probes has become apparent in this review, as summarized in Fig 10.
Fig 10.
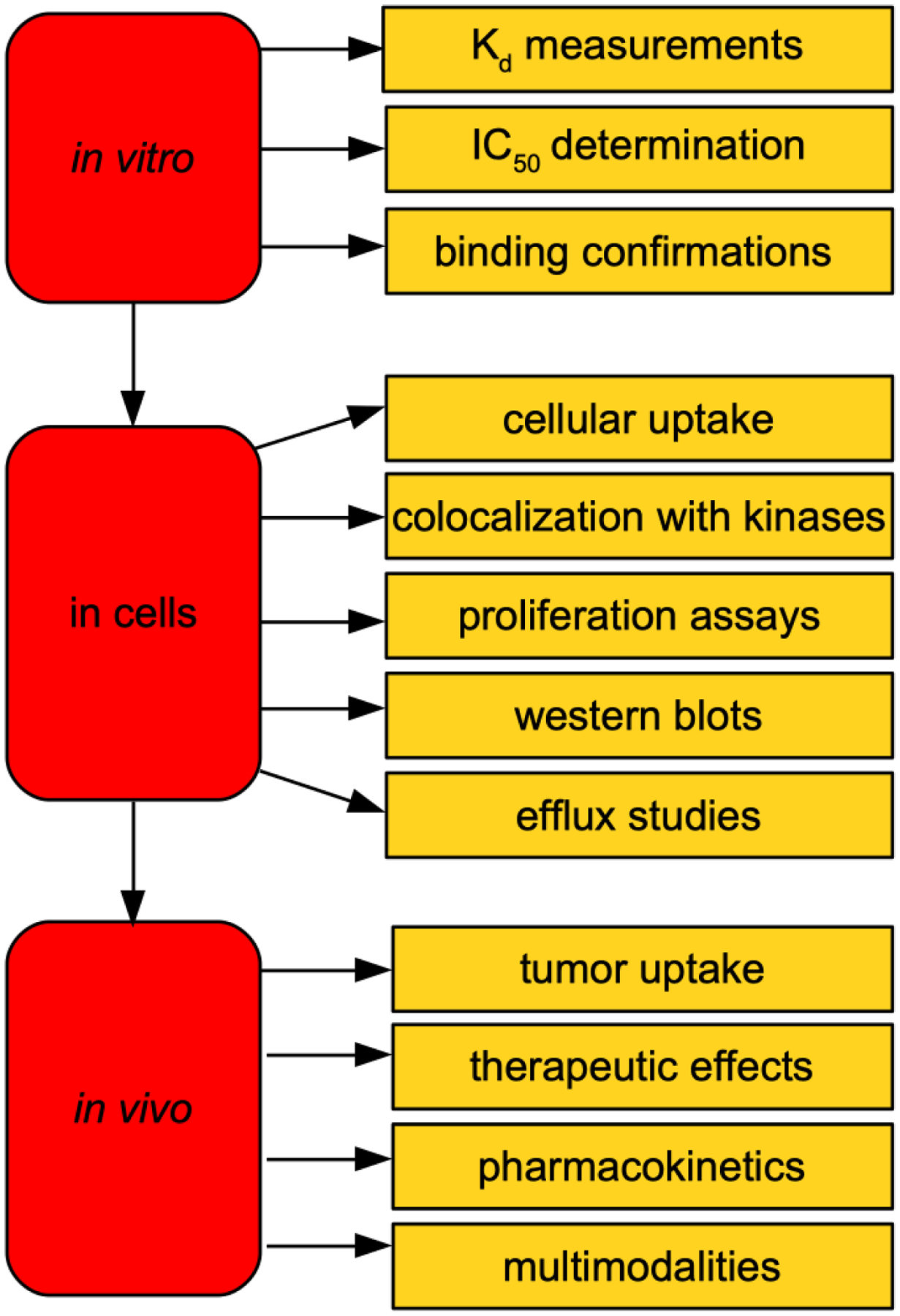
Steps in development and validation of fluor-KI probes for in vitro to in vivo application.
Outlook For Practical Applications Of Fluor-KI Conjugates
Kinase inhibitor fluorophore conjugates have potential for practical applications beyond research probes. Probably the most imminent real-world use of fluor-KI conjugates is as diagnostic markers, for instance, in flow cytometry to track certain kinases in diseased cells like in cancers of the blood, macrophages during infection, or in histological fixed tissue imaging. Labelled KIs that become covalently bound to their targets should be particularly informative and easy to apply in histology because they are impervious to wash out.
There are also opportunities for using fluor-kinase inhibitors bound to kinases expressed as luciferase fusion proteins in live cells. Displacement of a fluor-KI by the parent KI in this system would cause loss of bioluminescence energy transfer (BRET). Multiplexed parallel detection of BRET changes is convenient, so this strategy could be used in high throughput assays.
It is possible that tumor seeking dyes may be used in theranostics that deliver KIs, and simultaneously for NIR imaging during surgeries. It is more likely that tumor seeking fluors without KI cargoes will be pursued as surgical staining agents, and KI’s attached to albumin will be explored separately as potential therapeutics, but these two applications could be guided or even inspired by research on fluor-KI probes.
Finally, KIs are useful for therapeutic areas other than cancer. As far as we are aware there have been no fluor-KIs applied to studies of other diseases,103 but this is an obvious extrapolation of the research strategies described here.
ACKNOWLEDGEMENTS
We thank the following grant agencies for funding: DoD BCRP Breakthrough Award (BC141561), and CPRIT (RP170144 and RP180875), and the NIH (R01EY029645). Figures 3, 4 and 6 were created with BioRender.com
Footnotes
The authors declare no competing financial interests.
References
- 1.Wu P, Nielsen TE and Clausen MH, Trends Pharmacol. Sci, 2015, 36, 422–439. [DOI] [PubMed] [Google Scholar]
- 2.Wu P, Nielsen TE and Clausen MH, Drug Discov. Today, 2016, 21, 5–10. [DOI] [PubMed] [Google Scholar]
- 3.https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm).
- 4.Ethirajan M, Chen Y, Joshi P and Pandey RK, Chem. Soc. Rev, 2011, 40, 340–362. [DOI] [PubMed] [Google Scholar]
- 5.Xu C and Webb WW, J. Opt. Soc. Am. B, 1996, 13, 481–491. [Google Scholar]
- 6.Sonnleitner M, Schutz GJ and Schmidt T, Chem. Phys. Lett, 1999, 300, 221–226. [Google Scholar]
- 7.Kim E, Yang KS, Kohler RH, Dubach JM, Mikula H and Weissleder R, Bioconjugate Chem, 2015, 26, 1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi PJ, Denny WA, Jose J, Cooper E, Schweder P, Mee E, Faull R, Dragunow M and Park TIH, Bioorg. Med. Chem. Lett, 2019, 29, 2617–2621. [DOI] [PubMed] [Google Scholar]
- 9.Cheruku RR, Cacaccio J, Durrani FA, Tabaczynski WA, Watson R, Marko A, Kumar R, El-Khouly ME, Fukuzumi S, Missert JR, Yao R, Sajjad M, Chandra D, Guru K and Pandey RK, J. Med. Chem, 2019, 62, 2598–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez-Vera JA, Chem. Soc. Rev, 2012, 41, 1652–1664. [DOI] [PubMed] [Google Scholar]
- 11.Miller MA, Kim E, Cuccarese MF, Plotkin AL, Prytyskach M, Kohler RH, Pittet MJ and Weissleder R, Chem. Commun, 2018, 54, 42–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vetter ML, Zhang Z, Liu S, Wang J, Cho H, Zhang J, Zhang W, Gray NS and Yang PL, Chembiochem : a European journal of chemical biology, 2014, 15, 1317–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Z, Kwiatkowski N, Zeng H, Lim SM, Gray NS, Zhang W and Yang PL, Molecular BioSystems, 2012, 8, 2523–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E, Kung AL, Mestan J, Daley GQ, Callahan L, Catley L, Cavazza C, Azam M, Neuberg D, Wright RD, Gilliland DG and Griffin JD, Cancer Cell, 2005, 7, 129–141. [DOI] [PubMed] [Google Scholar]
- 15.Evans EK, Tester R, Aslanian S, Karp R, Sheets M, Labenski MT, Witowski SR, Lounsbury H, Chaturvedi P, Mazdiyasni H, Zhu Z, Nacht M, Freed MI, Petter RC, Dubrovskiy A, Singh J and Westlin WF, J Pharmacol Exp Ther, 2013, 346, 219–228. [DOI] [PubMed] [Google Scholar]
- 16.Zou HY, Li Q, Lee JH, Arango ME, McDonnell SR, Yamazaki S, Koudriakova TB, Alton G, Cui JJ, Kung PP, Nambu MD, Los G, Bender SL, Mroczkowski B and Christensen JG, Cancer Res, 2007, 67, 4408–4417. [DOI] [PubMed] [Google Scholar]
- 17.Moyer JD, Barbacci EG, Iwata KK, Arnold L, Boman B, Cunningham A, DiOrio C, Doty J, Morin MJ, Moyer MP, Neveu M, Pollack VA, Pustilnik LR, Reynolds MM, Sloan D, Theleman A and Miller P, Cancer Res, 1997, 57, 4838–4848. [PubMed] [Google Scholar]
- 18.Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ and Gibson KH, Cancer Res, 2002, 62, 5749–5754. [PubMed] [Google Scholar]
- 19.Mikula H, Stapleton S, Kohler RH, Vinegoni C and Weissleder R, Theranostics, 2017, 7, 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S, Song W, Gao X, Su Y, Gao E and Gao Q, Anal. Chem, 2019, 91, 1507–1515. [DOI] [PubMed] [Google Scholar]
- 21.Namboodiri HV, Bukhtiyarova M, Ramcharan J, Karpusas M, Lee Y and Springman EB, Biochemistry, 2010, 49, 3611–3618. [DOI] [PubMed] [Google Scholar]
- 22.Zhao Z, Wu H, Wang L, Liu Y, Knapp S, Liu Q and Gray NS, ACS Chem Biol, 2014, 9, 1230–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwarcinski FE, Brandvold KR, Phadke S, Beleh OM, Johnson TK, Meagher JL, Seeliger MA, Stuckey JA and Soellner MB, ACS Chem. Biol, 2016, 11, 1296–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhullar KS, Lagaron NO, McGowan EM, Parmar I, Jha A, Hubbard BP and Rupasinghe HPV, Molecular Cancer, 2018, 17, 48/41–48/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ansideri F, Lange A, El-Gokha A, Boeckler FM and Koch P, Anal Biochem, 2016, 503, 28–40. [DOI] [PubMed] [Google Scholar]
- 26.Ranjitkar P, Brock AM and Maly DJ, Chem. Biol, 2010, 17, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okram B, Nagle A, Adrian FJ, Lee C, Ren P, Wang X, Sim T, Xie Y, Wang X, Xia G, Spraggon G, Warmuth M, Liu Y and Gray NS, Chem Biol, 2006, 13, 779–786. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y and Gray NS, Nat Chem Biol, 2006, 2, 358–364. [DOI] [PubMed] [Google Scholar]
- 29.Agafonov RV, Wilson C, Otten R, Buosi V and Kern D, Nat Struct Mol Biol, 2014, 21, 848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim D, Jun H, Lee H, Hong S-S and Hong S, Org. Lett, 2010, 12, 1212–1215. [DOI] [PubMed] [Google Scholar]
- 31.Lee J-H, Jung Kyung H, Lee H, Son Mi K, Yun S-M, Hong S-S, Ahn S-H, Lee K-R, Lee S, Kim D and Hong S, Oncotarget, 2014, 5, 10180–10197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim D, Lee H, Jun H, Hong S-S and Hong S, Bioorg. Med. Chem, 2011, 19, 2508–2516. [DOI] [PubMed] [Google Scholar]
- 33.Sicard R, Dhuguru J, Liu W, Patel N, Landgraf R and Wilson JN, Bioorg Med Chem Lett, 2012, 22, 5532–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dhuguru J, Liu W, Gonzalez WG, Babinchak WM, Miksovska J, Landgraf R and Wilson JN, J. Org. Chem, 2014, 79, 4940–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hughes TB, Miller GP and Swamidass SJ, Chem Res Toxicol, 2015, 28, 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cee VJ, Volak LP, Chen Y, Bartberger MD, Tegley C, Arvedson T, McCarter J, Tasker AS and Fotsch C, J Med Chem, 2015, 58, 9171–9178. [DOI] [PubMed] [Google Scholar]
- 37.Lee H, Liu W, Brown AS, Landgraf R and Wilson JN, Anal. Chem, 2016, 88, 11310–11313. [DOI] [PubMed] [Google Scholar]
- 38.Lee H, Landgraf R and Wilson JN, Bioorg. Med. Chem, 2017, 25, 6016–6023. [DOI] [PubMed] [Google Scholar]
- 39.Fleming CL, Sandoz PA, Inghardt T, Onfelt B, Grotli M and Andreasson J, Angew Chem Int Ed Engl, 2019, 58, 15000–15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Das J, Chen P, Norris D, Padmanabha R, Lin J, Moquin RV, Shen Z, Cook LS, Doweyko AM, Pitt S, Pang S, Shen DR, Fang Q, de Fex HF, McIntyre KW, Shuster DJ, Gillooly KM, Behnia K, Schieven GL, Wityak J and Barrish JC, J Med Chem, 2006, 49, 6819–6832. [DOI] [PubMed] [Google Scholar]
- 41.Hantschel O, Rix U, Schmidt U, Burckstummer T, Kneidinger M, Schutze G, Colinge J, Bennett KL, Ellmeier W, Valent P and Superti-Furga G, Proc Natl Acad Sci U S A, 2007, 104, 13283–13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Z, Lee FY, Bhalla KN and Wu J, Mol Pharmacol, 2006, 69, 1527–1533. [DOI] [PubMed] [Google Scholar]
- 43.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA and Buggy JJ, Proc. Natl. Acad. Sci. U. S. A, 2010, 107, 13075–13080, S13075/13071–S13075/13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Turetsky A, Kim E, Kohler RH, Miller MA and Weissleder R, Scientific Reports, 2014, 4, 4782/4781–4782/4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harris CM, Foley SE, Goedken ER, Michalak M, Murdock S and Wilson NS, SLAS Discov, 2018, 23, 1040–1050. [DOI] [PubMed] [Google Scholar]
- 46.Zitouni S, Nabais C, Jana SC, Guerrero A and Bettencourt-Dias M, Nat Rev Mol Cell Biol, 2014, 15, 433–452. [DOI] [PubMed] [Google Scholar]
- 47.Lee SY, Jang C and Lee KA, Dev Reprod, 2014, 18, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng J, Shao J, Markowitz JS and An G, Pharm. Res, 2014, 31, 2237–2255. [DOI] [PubMed] [Google Scholar]
- 49.Shukla S, Skoumbourdis AP, Walsh MJ, Hartz AMS, Fung KL, Wu C-P, Gottesman MM, Bauer B, Thomas CJ and Ambudkar SV, Molecular Pharmaceutics, 2011, 8, 1292–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shukla S, Sauna ZE and Ambudkar SV, Leukemia, 2008, 22, 445–447. [DOI] [PubMed] [Google Scholar]
- 51.Zambon A, Niculescu-Duvaz D, Niculescu-Duvaz I, Marais R and Springer CJ, Expert Opin Ther Pat, 2013, 23, 155–164. [DOI] [PubMed] [Google Scholar]
- 52.Cummings CT, Deryckere D, Earp HS and Graham DK, Clin Cancer Res, 2013, 19, 5275–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu J, Yang C, Simpson C, DeRyckere D, Van Deusen A, Miley MJ, Kireev D, Norris-Drouin J, Sather S, Hunter D, Korboukh VK, Patel HS, Janzen WP, Machius M, Johnson GL, Earp HS, Graham DK, Frye SV and Wang X, ACS Medicinal Chemistry Letters, 2012, 3, 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song X, Han X, Yu F, Zhang X, Chen L and Lv C, Theranostics, 2018, 8, 2217–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoet PH and Nemery B, Am J Physiol Lung Cell Mol Physiol, 2000, 278, L417–433. [DOI] [PubMed] [Google Scholar]
- 56.Gerner EW and Meyskens FL Jr., Nat Rev Cancer, 2004, 4, 781–792. [DOI] [PubMed] [Google Scholar]
- 57.Gamcsik MP, Kasibhatla MS, Teeter SD and Colvin OM, Biomarkers, 2012, 17, 671–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perry RR, Mazetta JA, Levin M and Barranco SC, Cancer, 1993, 72, 783–787. [DOI] [PubMed] [Google Scholar]
- 59.Zhang HM, Rao JN, Guo X, Liu L, Zou T, Turner DJ and Wang JY, J Biol Chem, 2004, 279, 22539–22547. [DOI] [PubMed] [Google Scholar]
- 60.Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P, Michel K, Peifer M, Mermel C, Girard L, Peyton M, Gazdar AF, Minna JD, Garraway LA, Kashkar H, Pao W, Meyerson M and Thomas RK, Cancer Research, 2009, 69, 3256–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Usama SM, Zhao B and Burgess K, Bioconjugate Chem, 2019, 30, 1175–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choi PJ, Park TIH, Cooper E, Dragunow M, Denny WA and Jose J, Bioconjug. Chem, 2020, 31, 1724–1739. [DOI] [PubMed] [Google Scholar]
- 63.Yang X, Shi C, Tong R, Qian W, Zhau HE, Wang R, Zhu G, Cheng J, Yang VW, Cheng T, Henary M, Strekowski L and Chung LWK, Clinical Cancer Research, 2010, 16, 2833–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zang C, Long L and Shi C, Adv. Ther, 2018, 1, 180069.180061 – 180069.180022. [Google Scholar]
- 65.Lin C-M, Usama SM and Burgess K, Molecules, 2018, 23, 2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Usama SM, Lin C-M and Burgess K, Bioconjugate Chem, 2018, 29, 3886–3895. [DOI] [PubMed] [Google Scholar]
- 67.Canovas C, Bellaye P-S, Moreau M, Romieu A, Denat F and Goncalves V, Org. Biomol. Chem, 2018, 16, 8831–8836. [DOI] [PubMed] [Google Scholar]
- 68.Boobalan R, Liu K-K, Chao J-I and Chen C, Bioorg. Med. Chem. Lett, 2017, 27, 1784–1788. [DOI] [PubMed] [Google Scholar]
- 69.Usama SM, Park GK, Nomura S, Baek Y, Choi H-S and Burgess K, Bioconjug. Chem, 2020, 31, 248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu Z and Chen X, Chem. Soc. Rev, 2016, 45, 1432–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stehle G, Sinn H, Wunder A, Schrenk HH, Schutt S, Maier-Borst W and Heene DL, Anti-Cancer Drugs, 1997, 8, 677–685. [PubMed] [Google Scholar]
- 72.Noguchi Y, Wu J, Duncan R, Strohalm J, Ulbrich K, Akaike T and Maeda H, Japanese Journal of Cancer Research, 1998, 89, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Torchilin Vladimir P, Handbook of experimental pharmacology, 2010, 197, 3–53. [DOI] [PubMed] [Google Scholar]
- 74.Heneweer C, Holland JP, Divilov V, Carlin S and Lewis JS, Journal of Nuclear Medicine, 2011, 52, 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lammers T, Kiessling F, Hennink WE and Storm G, J. Controlled Release, 2012, 161, 175–187. [DOI] [PubMed] [Google Scholar]
- 76.Golombek SK, May JN, Theek B, Appold L, Drude N, Kiessling F and Lammers T, Adv Drug Deliv Rev, 2018, 130, 17–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matsumura Y and Maeda H, Cancer Res, 1986, 46, 6387–6392. [PubMed] [Google Scholar]
- 78.Levitt DG and Levitt MD, International Journal of General Medicine, 2016, 9, 229–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Usama SM, Jiang Z and Burgess K, ChemMedChem, 2019, 14, 1575–1579. [DOI] [PubMed] [Google Scholar]
- 80.Jorge SE, Schulman S, Freed JA, Rangachari D, Kobayashi SS, Huberman MS, VanderLaan PA and Costa DB, Lung cancer (Amsterdam, Netherlands), 2015, 90, 369–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nakada M, Kita D, Watanabe T, Hayashi Y, Teng L, Pyko IV and Hamada J.-i., Cancers, 2011, 3, 3242–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Choi PJ, Cooper E, Schweder P, Mee E, Turner C, Faull R, Denny WA, Dragunow M, Park TIH and Jose J, Bioorg. Med. Chem. Lett, 2020, 30, 127252. [DOI] [PubMed] [Google Scholar]
- 83.Wang M, Tosi U, Wu LY, Singh R, Kommidi H, Guo H, Law B, Ting R, Zhou Z, Schweitzer ME, Souweidane MM and Hou S, Mol. Cancer. Ther, 2017, 16, 2902–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL and Oldfield EH, Proc Natl Acad Sci U S A, 1994, 91, 2076–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu Z, Pourghiasian M, Radtke MA, Lau J, Pan J, Dias GM, Yapp D, Lin K-S, Benard F and Perrin DM, Angew. Chem. Int. Ed, 2014, 53, 11876–11880. [DOI] [PubMed] [Google Scholar]
- 86.Perrin DM, Acc. Chem. Res, 2016, 49, 1333–1343. [DOI] [PubMed] [Google Scholar]
- 87.Tomas A, Futter CE and Eden ER, Trends Cell Biol, 2014, 24, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yewale C, Baradia D, Vhora I, Patil S and Misra A, Biomaterials, 2013, 34, 8690–8707. [DOI] [PubMed] [Google Scholar]
- 89.Zhang Y, Tortorella MD, Liao J, Qin X, Chen T, Luo J, Guan J, Talley JJ and Tu Z, ACS Medicinal Chemistry Letters, 2015, 6, 1086–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guiley KZ, Stevenson JW, Lou K, Barkovich KJ, Kumarasamy V, Wijeratne TU, Bunch KL, Tripathi S, Knudsen ES, Witkiewicz AK, Shokat KM and Rubin SM, Science, 2019, 366, eaaw2016, 2011–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hoet PHM and Nemery B, Am. J. Physiol, 2000, 278, L417–L433. [DOI] [PubMed] [Google Scholar]
- 92.Gerner EW and Meyskens FL Jr., Nat. Rev. Cancer, 2004, 4, 781–792. [DOI] [PubMed] [Google Scholar]
- 93.Jiang Z, Pflug K, Usama SM, Kuai D, Yan X, Sitcheran R and Burgess K, J. Med. Chem, 2019, 62, 9236–9245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.auf dem Keller U, Bellac CL, Li Y, Lou Y, Lange PF, Ting R, Harwig C, Kappelhoff R, Dedhar S, Adam MJ, Ruth TJ, Benard F, Perrin DM and Overall CM, Cancer Research, 2010, 70, 7562–7569. [DOI] [PubMed] [Google Scholar]
- 95.Li Y, Ting R, Harwig CW, auf dem Keller U, Bellac CL, Lange PF, Inkster JAH, Schaffer P, Adam MJ, Ruth TJ, Overall CM and Perrin DM, MedChemComm, 2011, 2, 942–949. [Google Scholar]
- 96.Li Y, Liu Z, Harwig CW, Pourghiasian M, Lau J, Lin K-S, Schaffer P, Benard F and Perrin DM, American Journal of Nuclear Medicine and Molecular Imaging, 2013, 3, 57–70. [PMC free article] [PubMed] [Google Scholar]
- 97.Liu Z, Hundal-Jabal N, Wong M, Yapp D, Lin KS, Benard F and Perrin DM, MedChemComm, 2014, 5, 171–179. [Google Scholar]
- 98.Liu Z, Lin K-S, Benard F, Pourghiasian M, Kiesewetter DO, Perrin DM and Chen X, Nat. Protoc, 2015, 10, 1423–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ametamey SM, Honer M and Schubiger PA, Chem. Rev, 2008, 108, 1501–1516. [DOI] [PubMed] [Google Scholar]
- 100.Pandey SK, Gryshuk AL, Sajjad M, Zheng X, Chen Y, Abouzeid MM, Morgan J, Charamisinau I, Nabi HA, Oseroff A and Pandey RK, J. Med. Chem, 2005, 48, 6286–6295. [DOI] [PubMed] [Google Scholar]
- 101.Abrahamse H and Hamblin MR, Biochem. J, 2016, 473, 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mehraban N and Freeman HS, Materials, 2015, 8, 4421–4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Grimminger F, Schermuly RT and Ghofrani HA, Nat Rev Drug Discov, 2010, 9, 956–970. [DOI] [PubMed] [Google Scholar]