

Abstract
Introduction
Sporadic Alzheimer's disease (AD) is strongly correlated with impaired brain glucose metabolism, which may affect AD onset and progression. Ketolysis has been suggested as an alternative pathway to fuel the brain.
Methods
RNA‐seq profiles of post mortem AD brains were used to determine whether dysfunctional AD brain metabolism can be determined by impairments in glycolytic and ketolytic gene expression. Data were obtained from the Knight Alzheimer's Disease Research Center (62 cases; 13 controls), Mount Sinai Brain Bank (110 cases; 44 controls), and the Mayo Clinic Brain Bank (80 cases; 76 controls), and were normalized to cell type: astrocytes, microglia, neurons, oligodendrocytes.
Results
In oligodendrocytes, both glycolytic and ketolytic pathways were significantly impaired in AD brains. Ketolytic gene expression was not significantly altered in neurons, astrocytes, and microglia.
Discussion
Oligodendrocytes may contribute to brain hypometabolism observed in AD. These results are suggestive of a potential link between hypometabolism and dysmyelination in disease physiology. Additionally, ketones may be therapeutic in AD due to their ability to fuel neurons despite impaired glycolytic metabolism.
Keywords: Alzheimer's disease, astrocytes, glycolysis, ketolysis, metabolic RNA‐seq profiles, microglia, neurons, oligodendrocytes
1. BACKGROUND
Sporadic Alzheimer's disease (AD) is the most common form of senescence‐related dementia, and is clinically characterized by neurodegeneration that progressively impairs memory and behavior. Amyloid and tau buildup in the brain have been the dominant hypotheses of AD etiology and the target of numerous AD treatments. However, amyloid‐targeted treatments have been largely unsuccessful, 1 contributing to controversy about its role in disease etiology. 2 , 3 Whether these classic phenotypes are the primary causes of AD or simply consequential of other mechanisms remains largely unknown.
In early 2‐Fluoro‐2‐deoxy‐D‐glucose positron emission tomography (FDG‐PET) studies, it was observed that AD patients presented with regional reductions in cerebral rate of glucose (CMRg) use. 4 , 5 It is now widely accepted that impaired brain glucose metabolism is strongly correlated with disease severity 6 and that regional CMRg declines throughout the progression from normal cognition to clinically relevant AD. 7 Therefore, it has been suggested that AD‐associated neurodegeneration may be the result of a starving brain. 8 Although these studies categorize AD in a metabolic framework and are not specific to cell type, the interpretations have been largely neurocentric and have not accounted for glial cells, which make up a large portion of metabolically active cells in the brain. 9
Glial cells (i.e., astrocytes, oligodendrocytes, microglia), in addition to their numerous functions in regulating blood‐brain barrier integrity and permeability, recycling neurotransmitters, 10 producing insulating myelin to maintain structural axonal integrity, 11 and providing immune support, 12 also play metabolic roles in sustaining neuronal function. Glia are capable of supplying neurons with aerobic glycolysis‐produced lactate. 13 Although glucose is considered the major energy fuel for neuronal activity, glia‐derived lactate provides neuronal fuel under prolonged stimulation. 14 Astrocytes can use fatty acids to produce ketones, which have also been hypothesized to fuel neurons. 15 Additionally, astrocytes and oligodendrocytes are connected to each other via gap junctions, 16 positioning them to sense bioenergetic changes and respond accordingly due to their physical proximity to neurons and their connections to the blood‐brain barrier. As essential contributors to neuronal metabolism, 17 it is likely that they contribute to impaired brain glucose metabolism in AD brains.
The efficacy of metabolic strategies in AD, including diets, have been explored in both animal AD models and human AD patients. In both rodents and humans, exogenous ketone administration and ketogenic diets are effective at improving AD pathology, 18 , 19 likely because brain ketone (acetoacetate) uptake is normal in AD patients. 20 Many studies have hypothesized that ketones fuel the brain under AD‐induced impairments in brain glucose metabolism. 20 , 21 , 22 While impaired brain glucose metabolism has been demonstrated by downregulated expression of glycolytic genes in the hippocampus, 23 ketolytic gene expression has not been previously evaluated. Our study focused on whether dysfunctional AD brain glucose metabolism could be identified by impairments in glycolytic gene expression in neurons, astrocytes, oligodendrocytes, and microglia, and explored the extent to which ketolytic gene expression is altered in AD patients. RNA‐seq profiles of post mortem AD brains from several available datasets were used to compare glycolytic and ketolytic gene expression between AD patients and cognitively normal (CN) controls. We provide evidence that AD affects both glycolytic and ketolytic metabolism within regions of the brain relevant to AD. Our findings suggest that ketone‐induced alleviation of AD symptoms may be due to their ability to fuel neurons under glycolytic dysfunction, and support investigation of glia‐targeted therapies.
HIGHLIGHTS
Glycolytic genes were impaired in Alzheimer's disease (AD) oligodendrocytes.
Ketolytic genes were significantly downregulated in AD oligodendrocytes.
Ketolytic gene expression was unchanged in AD neurons, astrocytes, and microglia.
Both ketolytic and glycolytic pathways may be therapeutic targets for treating AD.
RESEARCH IN CONTEXT
Systematic review: A growing body of evidence demonstrates that Alzheimer's disease (AD) is strongly correlated with impaired glucose metabolism and implicates metabolic dysfunction in AD onset and progression. However, the degree to which glia contribute to impaired glucose metabolism and whether ketone metabolism is also impaired in AD is unknown.
Interpretation: RNA‐seq profiles of post mortem brains illustrate widespread glycolytic and ketolytic dysfunction in oligodendrocytes in brain regions relevant to AD pathology. They also demonstrate impairments in glycolytic but not ketolytic gene expression in neurons. These findings suggest that oligodendrocytes play a larger role in AD metabolic dysfunction than previously thought and that ketones may be therapeutic in AD due to their ability to fuel the brain.
Future directions: We propose further analyses of the contributions of glial cells in AD energy metabolism, and further investigation of potential links between oligodendrocyte hypometabolism, dysmyelination, and AD pathology. These results implicate both glycolytic and ketolytic dysregulation in AD pathology, which may provide future therapeutic targets for treating AD.
2. METHODS
RNA‐seq data were analyzed from three available datasets: Knight Alzheimer's Disease Research Center (Knight‐ADRC), Mayo Clinic Brain Bank (Mayo), and Mount Sinai Brain Bank (MSBB). RNA‐seq data were normalized to four cell types using cell‐specific markers (Table S1 in supporting information). 24 Analysis of variance (ANOVA) P‐values and Cohen's d effect size were reported for neurons, astrocytes, oligodendrocytes, and microglia. Each dataset was filtered to include only AD cases and CN controls. Table S2 in supporting information contains Clinical Dementia Ratings (CDR) and Braak scoring for AD cases and controls from the Knight‐ADRC and MSBB datasets. CDR scores were unavailable from the Mayo dataset. A chi‐squared test showed that AD cases were more likely to have CDR scores ≥1.0 in both the Knight‐ADRC and MSBB datasets (P‐values = 5.944 × 10–13 and 9.308 × 10–93, respectively), indicating that CDR and Braak staging were congruent. We recognize that slight variations in diagnostic criteria among the three datasets, as well as inherent limitations of using neuropathology to classify AD (e.g., variation in clinical presentation) 25 may introduce some differences in the case selection process. We also recognize that the lack of co‐morbidities, especially diabetes, in these datasets may limit the implications of some of analyses. However, the criteria used by each dataset are consistent with current diagnostic criteria, 26 and both CDR and Braak staging indicate that AD cases were selected.
Individuals were excluded from each dataset if their sex was missing. Genes were required to have expression levels > 10 (transcript counts produced by Salmon 27 ) in at least 85% of the samples. Genes were also excluded when the ANOVA of RNA expression between males and females in either AD cases or CN controls had a P‐value < .05, excluding sex‐specific differences in gene expression. The number of genes that had sufficient support but were removed from the analysis due to differential expression based on sex in AD cases but not CN controls for each cell type is shown in Table S3 in supporting information. Table S4 in supporting information shows the number of genes, number of cases, and number of controls that passed our filtering criteria for each brain region. Although the number of genes passing all filters varied by cell type, the number of cases and controls passing all filters for each cell type was the same across a brain region.
Bonferroni corrections 28 were used to establish α values for each dataset using the remaining genes (Tables S5 and S6 in supporting information). Additionally, we determined the empirical distribution of ANOVA P‐values across all genes, both metabolic and non‐metabolic, by calculating the ANOVA P‐values for each gene and determining the top 5% of differentially expressed genes in each brain region for each cell type (i.e., empirical P‐value of .05; Table S7 in supporting information). While the Bonferroni correction provides a statistical cutoff of significance, the empirical P‐value provides a distribution of the observed gene expressions regardless of a global skew in differential gene expression (i.e., all genes may exceed a Bonferroni correction, but the empirical P‐value will cap the results at the top 5% of differentially expressed genes across the entire dataset). Therefore, glycolytic or ketolytic genes with ANOVA P‐values less than or equal to the Bonferroni α value and less than or equal to the empirical P‐value threshold indicated the strongest support for differential expression between AD and CN controls and were classified as significant. Genes that met either the Bonferroni‐corrected significance threshold or the empirical P‐value suggested significance but were not included in our prioritized list of genes.
We selected 20 genes associated with glycolysis 29 , 30 and other aspects of brain glucose metabolism 31 (Figure 1A,B, Table S7) to characterize the glycolytic expression profiles of AD patients and CN controls. We selected genes that encode the insulin receptor (INSR) and insulin receptor substrates (IRS1, IRS2); glucose transporters (SLC2A1, SLC2A3, SLC2A4); and enzymes of glycolysis, which include hexokinase (HK1, HK2), glucose‐6‐phosphate isomerase (GPI), phosphofructokinase (PFKM, PFKL, PFKP), aldolase (ALDOA), triose phosphate isomerase (TPI1), glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH), phosphoglycerate kinase (PGK1), phosphoglycerate mutase (PGAM1), enolase (ENO2), pyruvate kinase (PKM), and pyruvate dehydrogenase (PDHA1).
FIGURE 1.
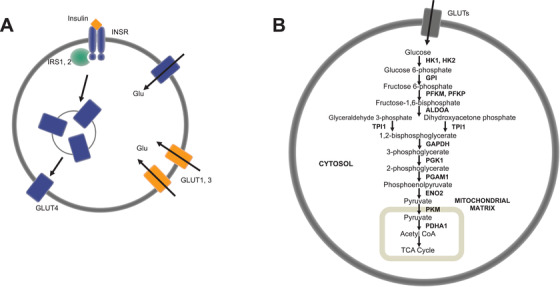
Glycolytic genes analyzed and their roles in brain glucose metabolism—insulin signaling (A) and glycolysis (B). Insulin controls the uptake of glucose via the insulin signaling pathway, which involves activation of the insulin receptor (INSR), phosphorylation of insulin receptor substrates (IRS1, IRS2), and the subsequent translocation of insulin‐sensitive GLUT4 transporters to the plasma membrane (A). Glucose is transported into blood‐brain barrier epithelia, neurons, and glia through GLUT transporters (GLUT1, GLUT2, GLUT4) into the cytoplasm, where it is metabolized to pyruvate in glycolysis, transported into the mitochondrial matrix, and converted to acetyl coenzyme A for tricarboxylic acid cycle oxidation (B)
We also selected six genes specific to ketone transport (i.e., facilitate ketone transport across the blood‐brain barrier into neurons and astrocytes) and metabolism to acetyl coenzyme A (CoA; Figure 2) 32 , 33 to determine whether ketolytic genes are affected by AD. These include monocarboxylate transporters (MCT) 1, 2, and 4 (encoded by SLC16A1, SLC16A7, SLC16A3, respectively), beta‐hydroxybutyrate dehydrogenase (BDH1), 3‐oxoacid CoA transferase 1 (OXCT1/SCOT, succinyl‐CoA:3‐ketoacid coenzyme A transferase 1), and acetoacetyl‐CoA thiolase/thiolase (ACAT1). Although ketone production has been detected in astrocytes in regions of the brain involved in regulating food intake (e.g., ventromedial hypothalamus), 34 the extent to which astrocytic ketone production in other regions of the brain contribute to fueling neurons is largely unknown. Therefore, because our main objective for this study was to determine the extent to which ketolytic metabolism machinery was intact in AD to allow ketones to fuel the brain, we focused on ketone catabolism.
FIGURE 2.
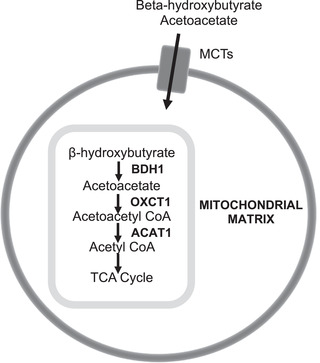
Ketolytic genes analyzed and their roles in brain ketone metabolism. Ketone bodies, beta‐hydroxybutyrate and acetoacetate, are transported through the plasma membrane of blood‐brain barrier epithelia, neurons, and glia through monocarboxylate transporters (MCT1, MCT2, MCT4). They diffuse into the mitochondrial matrix, where they are metabolized by BDH1, OXCT1, and ACAT1 to yield acetyl coenzyme A for oxidation in the tricarboxylic acid cycle
2.1. Knight‐ADRC
Washington University School of Medicine in St. Louis obtained parietal lobe tissue of post mortem brains for research purposes, as approved by their institutional review board. Tissue Lyser LT and RNeasy Mini Kit (Qiagen) were used to extract RNA from the frozen brains for RNA‐seq. Illumina HiSeq 4000 sequencing generated paired‐end read lengths of 2 × 150 base pairs with a mean coverage of 58.14 ± 8.62 million reads per sample. CDR scores 35 were calculated regularly pre mortem, and other measurements, including Braak staging, 36 were collected post mortem during the autopsy. Additional details on sequencing protocols are reported by Li et al. 24 We used the neuropathology diagnosis for each sample to separate AD cases from controls. In total, we analyzed 86 AD cases (33 males and 53 females) and 15 controls (5 males and 10 females) from the Knight‐ADRC dataset.
2.2. Mayo Clinic Brain Bank
We used the Advanced Medicines Partnership—Alzheimer's Disease (AMP‐AD) portal (synapse ID = 5,550,404; accessed January 2017) to download the RNA‐seq data from the Mayo Clinic Brain Bank. Illumina HiSeq 2000 sequencing was conducted on each sample and produced 2 × 101 paired end reads, averaging 134.9 million reads per sample. Additional details on sequencing, quality control procedures, and neuropathology criteria are reported by Allen et al. 37 Only definite AD diagnoses and CN controls were included in our analyses. A definite AD diagnosis was made according to National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (NINCDS‐ADRDA) criteria, 38 and had a Braak neurofibrillary tangles (NFT) stage of IV or greater. Controls had a Braak NFT stage of III or less, Consortium to Establish a Registry for Alzheimer's Disease (CERAD) 39 , 40 , 41 neuritic and cortical plaque densities of zero or one, and lacked any additional neuropathologic diagnoses. Two brain regions are included in the Mayo Clinic Brain Bank: cerebellum and temporal cortex. In the cerebellum, we analyzed 82 AD cases (34 males and 48 females) and 77 controls (40 males and 37 females). In the temporal cortex, we analyzed 82 AD cases (33 males and 49 females) and 78 controls (41 males and 37 females).
2.3. Mount Sinai Brain Bank
Mount Sinai Brain Bank RNA‐seq data were downloaded from the AMP‐AD portal (synapse ID = 3,157,743; accessed January 2017). Illumina HiSeq 2500 generated single‐end reads of 100 nucleotides, averaging 38.7 million reads per sample. RNA‐seq data from the MSBB include four brain regions: anterior prefrontal cortex, superior temporal gyrus, parahippocampal gyrus, and inferior frontal gyrus. Using CERAD‐defined neuropathology, we limited our analyses to samples that were definite AD (cases) and CN (controls). For the anterior prefrontal cortex, we included 139 AD cases (52 males and 87 females) and 93 controls (42 males and 51 females) in our analyses. For the superior temporal gyrus, we analyzed 148 AD cases (59 males and 89 females) and 86 controls (43 males and 43 females). For the parahippocampal gyrus, we analyzed 146 AD cases (50 males and 96 females) and 83 controls (39 males and 44 females). For the inferior frontal gyrus, we analyzed 130 AD cases (49 males and 81 females) and 91 controls (47 males and 44 females).
3. RESULTS
3.1. Glycolysis
The number of genes that met the criteria outlined above varied among datasets, brain regions, and cell types. Genes included in glycolytic analyses for each cell type and brain region are included in Tables S8‐11 in supporting information. P‐values and Cohen's d of glycolytic genes that met the Bonferroni‐corrected α value and empirical P‐value cutoffs for significance are included in Table 1.
TABLE 1.
P‐values and Cohen's d of glycolytic genes that met the Bonferroni‐corrected α value and empirical P‐value cutoffs for significance in neurons, astrocytes, and oligodendrocytes, and microglia
| Glycolytic P‐values and Cohen's d‐values | ||||
|---|---|---|---|---|
| Dataset | Gene | P | Cohen's d‐value | |
| Neuron | MSBB parahippocampal gyrus | PFKL | 8.70 × 10‐13 | 1.515373 |
| MSBB superior temporal gyrus | PFKL | 5.91 × 10‐6 | 0.900013 | |
| Astrocyte | Mayo cerebellum | HK1 | 1.07 × 10‐14 | 1.554021 |
| IRS1 | 4.60 × 10‐15 | 1.581132 | ||
| SLC2A3 | 1.00 × 10‐14 | 1.556237 | ||
| Oligodendrocyte | Mayo temporal cortex | TPI1 | 5.48 × 10‐9 | 0.996358377 |
| MSBB frontal pole | TPI1 | .000716 | 0.621992169 | |
| PFKM | .000118 | 0.760133881 | ||
| PGAM1 | .00021 | 0.730112972 | ||
| MSBB inferior frontal gyrus | PKM | .000258 | 0.719315744 | |
| TPI1 | .000178 | 0.738811918 | ||
| ENO2 | 7.13 × 10‐12 | 1.441230355 | ||
| PFKM | 7.63 × 10‐10 | 1.27078558 | ||
| MSBB parahippocampal gyrus | PGAM1 | 3.02 × 10‐9 | 1.21863907 | |
| PGK1 | 5.89 × 10‐10 | 1.280517083 | ||
| PKM | 4.41 × 10‐9 | 1.204070491 | ||
| TPI1 | 5.19 × 10‐11 | 1.369950693 | ||
| ENO2 | .00017 | 0.738741297 | ||
| Microglia | MSBB superior temporal gyrus | GPI | .000386 | 0.695532656 |
| PFKM | .000154 | 0.743899193 | ||
| PKM | .000386 | 0.695485281 | ||
| Mayo cerebellum | HK1 | .001678 | 0.569799 | |
| PFKP | 2.80 x 10‐6 | 0.870544 | ||
| Mayo temporal cortex | SLC2A4 | .000261 | 0.602561 | |
| MSBB parahippocampal gyrus | ENO2 | 7.29 x 10‐5 | 0.785553 | |
Abbreviation: MSBB, Mount Sinai Brain Bank.
3.1.1. Neurons
After correcting for cell type, AD neurons displayed few significant changes in glycolytic gene expression (Figure 3A). Differentially expressed genes were downregulated in AD compared to controls, which is consistent with previous studies documenting glucose hypometabolism in AD brains. 42 PFKL met both the Bonferroni α value and empirical P‐value cutoffs for significance (Tables S5 and S7) in the parahippocampal gyrus (MSBB; P = 8.70 × 10–13; d = 1.525) and in the superior temporal lobe (MSBB; P = 5.91 × 10–6; d = 0.9). Both genes had large Cohen's d effect sizes, indicating a strong association between impaired PFKL expression and AD.
FIGURE 3.
Volcano plots of glycolytic gene expression from Alzheimer's disease patients in neurons (A) and oligodendrocytes (B) from each dataset. The ‐log10 P‐values of genes are plotted versus the log2FoldChange along the X‐axis. Bonferroni‐corrected α‐values are indicated by the black dashed line and the empirical P‐values are indicated by the blue dashed line. Genes with P‐values below either cutoff are plotted above their respective cutoff lines, marked in red, and labeled
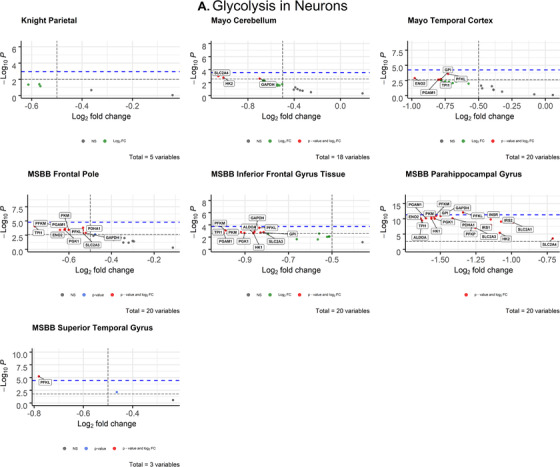
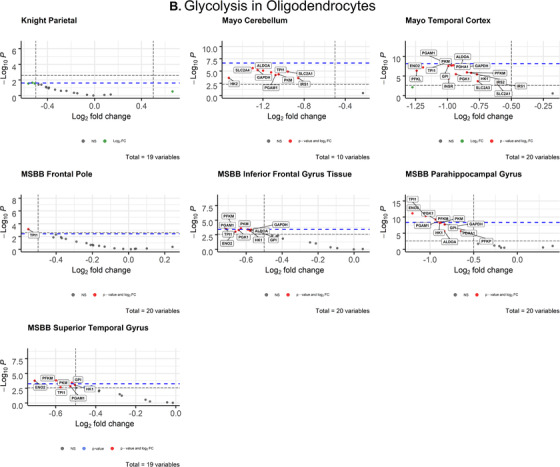
3.1.2. Astrocytes
After correcting for cell type, astrocytes also displayed few significant changes in glycolytic gene expression between AD brains and CN controls (Figure S1 in supporting information). Significant astrocytic genes were upregulated in AD brains compared to controls. These included cerebellar HK1 (P = 1.07 × 10–14; d = 1.554), IRS1 (P = 4.60 × 10–15; d = 1.581), and SLC2A3 (P = 1.00 × 10–14; d = 1.556; Figure S1, Table 1), which exceeded the Bonferroni‐adjusted α value (α = 0.0025) and empirical P‐value (P = 2.618 × 10–14) cutoffs. The Cohen's d of these genes (Table 1) indicates a strong effect size and association between the upregulated HK1, IRS1, and SLC2A3 expression in cerebellar astrocytes and AD.
3.1.3. Oligodendrocytes
After correcting for cell type, oligodendrocytes displayed widespread impairment of glycolytic gene expression (Figure 3B) that exceeded all other cell types. All differentially expressed oligodendrocytic genes were downregulated in AD brains compared to controls. The parietal lobe (Knight‐ADRC) and cerebellum (Mayo) were the only brain regions that contained no significantly impaired expression. In the temporal lobe, TPI1 (P = 5.48 × 10–9; d = 0.996) met both the Bonferroni α value (α = 0.0025) and the more stringent empirical p‐value (P = 7.233 × 10–9) cutoffs. In the frontal pole (MSBB), TPI1 (P = 0.000718; d = 0.621) was the only gene that exceeded both Bonferroni‐corrected (α = 0.0025) and empirical P‐value (P = .003687) significance. In the inferior frontal gyrus (MSBB), four genes met both the Bonferroni α value (α = 0.0025) and empirical p‐value (P = .0003743) cutoffs for significance: PFKM (P = .000118; d = 0.760), PGAM1 (P = .00021; d = 0.730), PKM (P = .000258; d = 0.719), TPI1 (P = .000178; d = 0.739). In the parahippocampal gyrus (MSBB), six genes met the Bonferroni α value (α = 0.0025) and empirical P‐value (P = 4.462 × 10–9) cutoffs: ENO2, PFKM, PGAM1, PGK1, PKM, TPI (Figure 3B, Table 1). In the superior temporal gyrus (MSBB) four genes met the Bonferroni α value (α = 0.002632) and empirical P‐value (P = .0005621) cutoffs: ENO2 (P = .00017; d = 0.738), GPI (P = .000386; d = 0.695), PFKM (P = .000154, d = 0.744), PKM (P = .000386, d = 0.695).
3.1.4. Microglia
After correcting for cell type, microglia displayed few significant changes in AD brains (Figure S2 in supporting information). In the cerebellum (Mayo), HK1 (P = .001678; d = 0.57) and PFKP (P = 2.80 × 10–6; d = 0.871) were upregulated, meeting both Bonferroni α value (α = 0.0025) and empirical P‐value (P = .002181) cutoffs. In the temporal cortex (Mayo), SLC2A4 (P = .000261; d = 0.603) was significantly upregulated exceeding both the Bonferroni (α = 0.0025) and empirical P‐value (P = .00382) cutoffs. In the parahippocampal gyrus (MSBB), ENO2 (P = 7.29 × 10–5; d = 0.786) was significantly downregulated and met both Bonferroni α value (α = 0.0025) and empirical P‐value (P = .00537) cutoffs.
3.2. Ketolysis
The number of ketolytic genes that met the inclusion criteria outlined above also varied among datasets, brain regions, and cell types (see Tables S12‐15 in supporting information). P‐values and the Cohen's d of ketolytic genes that met the Bonferroni‐corrected α value and empirical P‐value cutoffs for significance are included in Table 2.
TABLE 2.
P‐values and Cohen's d of ketolytic genes that met the Bonferroni‐corrected α value and empirical P‐value cutoffs for significance in neurons and oligodendrocytes, and microglia
| Ketolytic P‐values and Cohen's d‐values: neurons | ||||
|---|---|---|---|---|
| Dataset | Gene | P | Cohen's d | |
| Neuron | MSBB inferior frontal gyrus | SLC16A1 | 6.50 × 10‐5 | 0.789925 |
| Oligodendrocyte | Mayo temporal cortex | BDH1 | 9.85 × 10‐10 | 1.05008 |
| MSBB parahippocampal gyrus | OXCT1 | 8.69 × 10‐11 | 1.351187 | |
| MSBB superior temporal gyrus | BDH1 | .000191 | 0.732589 | |
Abbreviation: MSBB, Mount Sinai Brain Bank.
3.2.1. Neurons
After correcting for cell type, neuronal ketolytic gene expression (Figure 4A) mimicked the pattern of downregulation observed in neuronal glycolytic genes (Figure 3A). In the inferior frontal gyrus (MSBB), SLC16A1 (P = .006371; d = 0.79) met both the Bonferroni α value (α = 0.00833) and empirical P‐value (P = .0001757) cutoffs for significance (Table 2). The Cohen's d for SLC16A1 indicates a medium effect size and association with AD.
FIGURE 4.
Volcano plots of ketolytic gene expression from Alzheimer's disease patients in neurons (A) and oligodendrocytes (B) from each dataset. The ‐log10 P‐values of genes are plotted versus the log2FoldChange along the X‐axis. Bonferroni‐corrected α‐values are indicated by the black dashed line and the empirical P‐values are indicated by the blue dashed line. Genes with P‐values below either cutoff are plotted above their respective cutoff lines, marked in red, and labeled
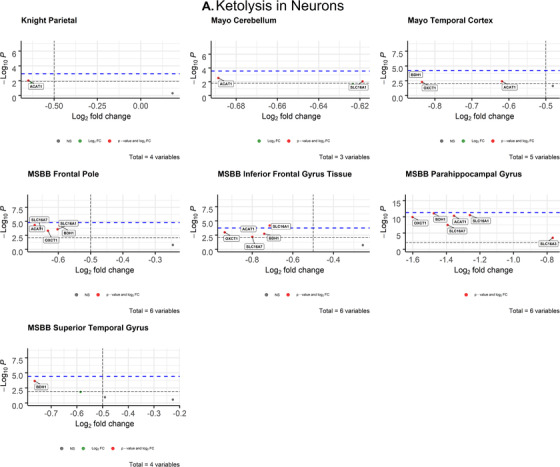
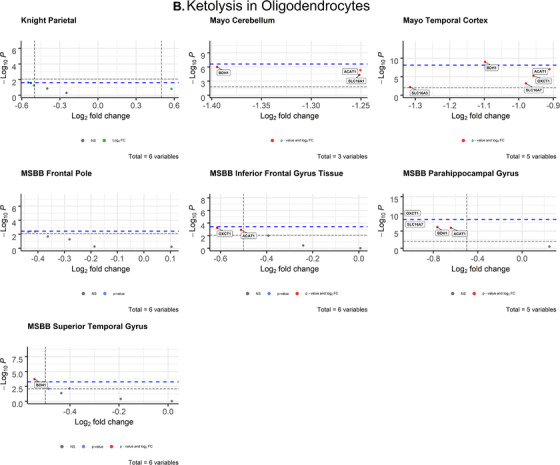
3.2.2. Astrocytes
There were no astrocytic ketolytic genes that met both the Bonferroni‐corrected α value and empirical P‐value cutoffs for significance (Figure S3 in supporting information), indicating no significant disruption to astrocytic ketolytic gene expression in AD brains.
3.2.3. Oligodendrocytes
After correcting for cell type, ketolytic gene expression in oligodendrocytes was significantly impaired in AD brains compared to CN controls (Figure 4B), similar to glycolytic gene expression (Figure 3B). Two genes met both the Bonferroni α value and empirical P‐value cutoffs for significance across three brain regions: BDH1 (P = 9.85 × 10–10; d = 1.05) in the temporal cortex (Mayo), OXCT (P = 8.69 × 10–11, d = 1.35) in the parahippocampal gyrus (MSBB), and BDH1 (P = .000191; d = 0.732) in the superior temporal gyrus (MSBB; Table 2). These analyses show that both brain glucose and ketone metabolism in oligodendrocytes are impaired in AD and highly associated with disease.
3.2.4. Microglia
There were no microglial ketolytic genes that met both cutoffs for significance (Figure S4 in supporting information), indicating no significant disruption to microglial ketolytic gene expression in AD brains.
4. DISCUSSION
This study provides further evidence that energy substrate metabolism is significantly disrupted in AD. We observed widespread impairments in glycolytic gene expression and disruption of ketolytic gene expression in oligodendrocytes (Figure 3B). Oligodendrocytes are responsible for producing myelin, which characterizes white matter in the brain. 43 While AD has historically been classified as a gray matter disease due to the distribution of amyloid beta (Aβ) and tau NFT, and gray matter atrophy, 44 recent evidence has shown that white matter abnormalities are prevalent in AD brains and are good predictors of cognitive decline in individuals with mild cognitive impairment. 45 Studies in AD mouse models have shown that AD alters myelination patterns and oligodendrocyte function before Aβ and tau pathologies are detectable. 46 Although we lack longitudinal evidence, our data suggest oligodendrocyte dysfunction in AD. Our findings add support to McKenzie et al., 47 which assessed oligodendrocytic gene expression in human post mortem AD brains and revealed key oligodendrocytic genes involved in myelin regulation that were highly associated with AD. Their findings suggested that dysfunctional gene expression in oligodendrocytes plays a causal role in downstream mitochondrial and ribosomal defects that contribute to AD pathology. The data presented here fits within this proposed framework because oligodendrocytic gene expression was highly dysregulated across various datasets and concentrated in regions of the brain relevant to AD pathology.
Interestingly, donepezil, an acetylcholinesterase inhibitor that is commonly prescribed to AD patients, has been shown to promote oligodendrocyte differentiation and remyelination. 48 While our findings further implicate impaired oligodendrocyte function in AD pathology and support targeting oligodendrocytes in AD therapies, additional research will be necessary to determine whether impaired oligodendrocytic gene expression is a major contributor to regional hypometabolism observed in AD FDG‐PET studies, 49 and the extent to which dysfunctional oligodendrocyte gene expression and downstream effects contribute to the pathogenesis and progression of AD.
Consistent with our initial hypothesis, neuronal ketolytic gene expression was not significantly altered in AD brains, with the exception of one gene (Figure 4A). In the inferior frontal gyrus (MSBB), monocarboxylate transporter 1 (MCT1, encoded by SLC16A1) was significantly downregulated in AD neurons. However, MCT1 is primarily expressed in astrocytes and endothelial cells of the blood‐brain barrier, while MCT2 (encoded by SLC16A7) is primarily expressed in neurons. 50 Neuronal SLC16A7 expression was not significantly altered (Figure 3A), which indicates that ketone transport into neurons is intact in the AD brain. Therefore, it is possible that cognitive improvements observed in AD patients treated with ketones and ketogenic diets 51 , 52 , 53 are due to the ability of ketones to fuel the brain despite glycolytic impairments. We observed significant deficits in ketolytic gene expression in oligodendrocytes that mirrored the patterns observed in glycolytic gene expression, which demonstrates that impaired brain metabolism in AD is not exclusive to neuronal glucose metabolism. Together, these findings suggest that oligodendrocytes may play a larger role in the energetic deficiency of AD pathology than previously thought. They also suggest that improvements in AD symptoms observed with exogenous ketone administration and ketogenic diets 54 may be due to adequately fueling neurons.
While ketolytic pathways appeared intact in neurons and astrocytes, changes in glycolytic gene expression were present in both populations. We observed significant downregulation of neuronal phosphofructokinase, the rate‐limiting enzyme of glycolysis, in two regions of the brain within the temporal lobe (Figure 3A), which is consistent with findings from previous studies. 7 , 55 , 56 AD‐specific impairments in PFKP expression likely contribute to impaired neuronal glucose metabolism. Conversely, astrocytic glycolytic gene expression was significantly upregulated in the cerebellum (Figure S1), possibly as a compensatory mechanism for impaired neuronal glucose metabolism. The observed widespread disruption of both ketolytic and glycolytic genes in neurons suggests that current studies using the cerebellum as a reference region in AD PET studies, 57 might require reevaluation.
Although the mechanisms are not well established, ketones have demonstrated therapeutic potential in improving cognition in both mouse AD models 18 and human AD patients. 53 Several recent studies have demonstrated that ketones enhance fuel availability to the brain despite pathological impairments in brain glucose metabolism. 49 , 52 , 58 , 59 One PET‐based analysis of brain fuel metabolism in AD showed that the cerebral metabolic rate of glucose was impaired in AD patients, but the cerebral metabolic rate of acetoacetate was unimpaired. 20 Our analyses are consistent with these findings. We show that glycolytic gene expression was significantly reduced in AD neurons and oligodendrocytes (Figure 3), while ketolytic gene expression was not significantly altered in neurons, astrocytes, and microglia (Figure 4A, S2). Although ketolytic gene expression was downregulated in oligodendrocytes, unimpaired ketolytic gene expression in neurons, astrocytes, and microglia may be sufficient to enable ketones to fuel essential neuronal processes despite impairments in oligodendrocytic ketone metabolism.
Regional brain atrophy is an AD phenotype observable later in AD progression, in which areas of the brain associated with memory are most severely affected. 60 It is possible that the extensively downregulated gene expression we observed in oligodendrocytes could simply be due to general regional brain atrophy. However, we determined this to be unlikely because similar patterns of widespread downregulation were not observed in neurons, astrocytes, and microglia, which demonstrates that these impairments are oligodendrocyte‐specific. It is important to acknowledge that we were not able to analyze the gene expression data with finer resolution (i.e., parse out hippocampal gene expression from the temporal lobe, etc.) due to limitations of the datasets, which likely skewed some analyses toward insignificance. Additionally, as these data were collected from post mortem brains, they provide no insight into correlations between disease progression and changes in gene expression. Therefore, further research with finer resolution of gene expression data as well as longitudinal studies performed in animal models of AD will be important for assessing regional effects of AD on metabolic gene expression and their correlations with disease progression.
Collectively, these findings support the characterization of AD as a metabolic disorder. Our data demonstrate impairments in glycolytic gene expression in neurons and oligodendrocytes that may be responsible for the decrease in brain glucose uptake and metabolism observed in AD patients in FDG‐PET studies. These impairments were highly concentrated in regions of the brain involved in cognition and behavior (e.g., medial temporal lobe structures, frontal lobe), which is consistent with AD pathology. These RNA‐seq profiles revealed that neuronal ketolytic gene expression was not significantly altered in AD brains, indicating that AD‐induced impairments in neural and white matter metabolism are specific to glucose, which confirms previous reports of ketone and FDG‐PET imaging. 49 , 52 , 59 , 60 Therefore, ketones may have therapeutic potential in treating AD due to their ability to fuel the brain despite impairments in brain glucose metabolism.
Overall, these data are clinically relevant in several aspects. They provide evidence that impairments in oligodendrocyte metabolism exist and may contribute to brain hypometabolism characteristic of AD. In conjunction with findings by McKenzie et al., 47 these data suggest a potential link between brain hypometabolism and dysmyelination observed in AD. Moreover, these data contribute to the growing body of evidence demonstrating therapeutic potential of ketones and intermediary metabolites in various neuropathological conditions. Recent studies have indicated clinical benefit of betahydroxybutyrate in AD 61 and Parkinson's disease, 62 sodium phenylbutyrate‐taurursodiol in amyotrophic lateral sclerosis (ALS), 61 , 63 4‐phenylbutyrate in Down's syndrome, 64 and sodium butyrate in spinal cord injury. 65 Additionally, clinical studies assessing the effects of ketosis in Parkinson's disease (NCT04477161, NCT03421899), multiple sclerosis (NCT03718247), and other neurologic conditions are ongoing.
While the current view of AD is highly neurocentric, these findings support further investigation of oligodendrocyte function in AD, specifically in the regulation of myelin and energy metabolism, and how they are potentially interlinked. Our findings suggest that future studies investigating metabolic dysfunction in AD brains should target oligodendrocytes because genes involved in ketolysis and glycolysis are both differentially expressed in that cell type in AD brains.
Supporting information
Supplementary Information
ACKNOWLEDGMENTS
We recognize the contributions of Brigham Young University, including the Office of Research Computing, for supporting our research. This work was supported by the following grants from the National Institutes of Health: RF1AG054052 (JK), R01AG044546 (CC), P01AG003991 (CC), RF1AG053303 (CC), RF1AG058501 (CC), U01AG058922 (CC), and R01AG057777 (OH). This work was also supported by the BrightFocus Foundation grant A2020118F (JM). Study data were provided by the following sources: The Mayo Clinic Alzheimer's Disease Genetic Studies, led by Dr. Nilufer Taner and Dr. Steven G. Younkin, Mayo Clinic, Jacksonville, FL, using samples from the Mayo Clinic Study of Aging, the Mayo Clinic Alzheimer's Disease Research Center, and the Mayo Clinic Brain Bank. Data collection was supported through funding by National Institute on Aging grants P50 AG016574, R01 AG032990, U01 AG046139, R01 AG018023, U01 AG006576, U01 AG006786, R01 AG025711, R01 AG017216, R01 AG003949; NINDS grant R01 NS080820; CurePSP Foundation; and support from Mayo Foundation. Study data includes samples collected through the Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson's Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer's Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer's Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05‐901 and 1001 to the Arizona Parkinson's Disease Consortium), and the Michael J. Fox Foundation for Parkinson's Research. BTB receives royalties from the sale of a book about insulin resistance.
Saito ER, Miller JB, Harari O, et al. Alzheimer's disease alters oligodendrocytic glycolytic and ketolytic gene expression. Alzheimer's Dement. 2021;17:1474–1486. 10.1002/alz.12310
Erin R. Saito and Justin B. Miller wish it to be known that, in their opinion, they contributed equally to this study.
REFERENCES
- 1. Knopman DS. Lowering of Amyloid‐Beta by β‐Secretase Inhibitors—Some Informative Failures. N Engl J Med. 2019;380:1476‐1478. [DOI] [PubMed] [Google Scholar]
- 2. Aisen PS. Failure after failure. What next in AD drug development?. J Prev Alzheimers Dis. 2019;6(3):150‐150. [DOI] [PubMed] [Google Scholar]
- 3. Kametani F, Hasegawa M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer's disease. Front Neurosci. 2018;12:25‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meltzer C, Zubieta J, Brandt J, Tune L, Mayberg H, Frost J. Regional hypometabolism in Alzheimer's disease as measured by positron emission tomography after correction for effects of partial volume averaging. Neurology. 1996;47(2):454‐461. [DOI] [PubMed] [Google Scholar]
- 5. Ishii K, Imamura T, Sasaki M, et al. Regional cerebral glucose metabolism in dementia with lewy bodies and Alzheimer's disease. Neurology. 1998;51(1):125‐130. [DOI] [PubMed] [Google Scholar]
- 6. Diehl‐Schmid J, Grimmer T, Drzezga A, et al. Decline of cerebral glucose metabolism in frontotemporal dementia: a longitudinal 18F‐FDG‐PET‐study. Neurobiol Aging. 2007;28(1):42‐50. [DOI] [PubMed] [Google Scholar]
- 7. Mosconi L, Mistur R, Switalski R, et al. FDG‐PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2009;36(5):811‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mamelak M. Sporadic Alzheimer's disease: the starving brain. J Alzheimers Dis. 2012;31(3):459‐474. [DOI] [PubMed] [Google Scholar]
- 9. von Bartheld CS, Bahney J, Herculano‐Houzel S. The search for true numbers of neurons and glial cells in the human brain: a review of 150 years of cell counting. J Comp Neurol. 2016;524(18):3865‐3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Benarroch EE. Neuron‐astrocyte interactions: Partnerships for normal function and disease in the central nervous system. 2005;80(10):1326‐1338. [DOI] [PubMed] [Google Scholar]
- 11. Saab AS, Tzvetanova ID, Nave K‐A. The role of myelin and oligodendrocytes in axonal energy metabolism. Curr Opin Neurobiol. 2013;23(6):1065‐1072. [DOI] [PubMed] [Google Scholar]
- 12. Barron KD. The microglial cell. A historical review. J Neurol Sci. 1995;134:57‐68. [DOI] [PubMed] [Google Scholar]
- 13. Chih C‐P, Roberts Jr EL. Energy substrates for neurons during neural activity: a critical review of the astrocyte‐neuron lactate shuttle hypothesis. J Cereb Blood Flow Metab. 2003;23(11):1263‐1281. [DOI] [PubMed] [Google Scholar]
- 14. Chih C‐P, Lipton P, Roberts EL. Do active cerebral neurons really use lactate rather than glucose?. Trends Neurosci. 2001;24(10):573‐578. [DOI] [PubMed] [Google Scholar]
- 15. Le Foll C, Levin BE. Fatty acid‐induced astrocyte ketone production and the control of food intake. Am J Physiol Regul Integr Comp Physiol. 2016;310(11):R1186‐R1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tress O, Maglione M, May D, et al. Panglial gap junctional communication is essential for maintenance of myelin in the CNS. J Neurosci. 2012;32(22):7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jha MK, Morrison BM. Glia‐neuron energy metabolism in health and diseases: new insights into the role of nervous system metabolic transporters. Exp Neurol. 2018;309:23‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yin JX, Maalouf M, Han P, et al. Ketones block amyloid entry and improve cognition in an Alzheimer's model. Neurobiol Aging. 2016;39:25‐37. [DOI] [PubMed] [Google Scholar]
- 19. Reger MA, Henderson ST, Hale C, et al. Effects of β‐hydroxybutyrate on cognition in memory‐impaired adults. Neurobiol Aging. 2004;25(3):311‐314. [DOI] [PubMed] [Google Scholar]
- 20. Castellano C‐A, Nugent S, Paquet N, et al. Lower brain 18F‐fluorodeoxyglucose uptake but normal 11C‐acetoacetate metabolism in mild Alzheimer's disease dementia. J Alzheimers Dis. 2015;43(4):1343‐1353. [DOI] [PubMed] [Google Scholar]
- 21. Cunnane SC, Courchesne‐Loyer A, St‐Pierre V, et al. Can ketones compensate for deteriorating brain glucose uptake during aging? Implications for the risk and treatment of Alzheimer's disease. Ann N Y Acad Sci. 2016;1367(1):12‐20. [DOI] [PubMed] [Google Scholar]
- 22. Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech RL. d‐β‐Hydroxybutyrate protects neurons in models of Alzheimer's and Parkinson's disease. Proc Natl Acad Sci. 2000;97(10):5440‐5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brooks WM, Lynch PJ, Ingle CC, et al. Gene expression profiles of metabolic enzyme transcripts in Alzheimer's disease. Brain Res. 2007;1127:127‐135. [DOI] [PubMed] [Google Scholar]
- 24. Li Z, Del‐Aguila JL, Dube U, et al. Genetic variants associated with Alzheimer's disease confer different cerebral cortex cell‐type population structure. Genome Med. 2018;10(1):43‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener. 2019;14(1):1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jack CR Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National institute on aging‐Alzheimer's association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):257‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias‐aware quantification of transcript expression. Nat Methods. 2017;14(4):417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bonferroni C. Teoria statistica delle classi e calcolo delle probabilita. Pubblicazioni del R Istituto Superiore di Scienze Economiche e Commericiali di Firenze. 1936;8:3‐62. [Google Scholar]
- 29. Gibbs M, Turner JF. Enzymes of glycolysis. Modern Methods of Plant Analysis/Moderne Methoden der Pflanzenanalyse. Springer; 1964:520‐545. [Google Scholar]
- 30. Scrutton MC, Utter MF. The regulation of glycolysis and gluconeogenesis in animal tissues. Annu Rev Biochem. 1968;37(1):249‐302. [Google Scholar]
- 31. Taha C, Klip A. The insulin signaling pathway. J Membr Biol. 1999;169(1):1‐12. [DOI] [PubMed] [Google Scholar]
- 32. Fukao T, Lopaschuk GD, Mitchell GA. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukotrienes Essent Fatty Acids. 2004;70(3):243‐251. [DOI] [PubMed] [Google Scholar]
- 33. Morris AAM. Cerebral ketone body metabolism. J Inherit Metab Dis. 2005;28(2):109‐121. [DOI] [PubMed] [Google Scholar]
- 34. Le Foll C, Dunn‐Meynell AA, Miziorko HM, Levin BE. Regulation of hypothalamic neuronal sensing and food intake by ketone bodies and fatty acids. Diabetes. 2014;63(4):1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morris JC. Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr. 1997;9(Suppl 1):173‐176.discussion 177‐178. [DOI] [PubMed] [Google Scholar]
- 36. Braak H, Braak E. Staging of Alzheimer's disease‐related neurofibrillary changes. Neurobiol Aging. 1995;16(3):271‐278.discussion 278‐284. [DOI] [PubMed] [Google Scholar]
- 37. Allen M, Carrasquillo MM, Funk C, et al. Human whole genome genotype and transcriptome data for Alzheimer's and other neurodegenerative diseases. Sci Data. 2016;3:160089‐160089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease. Neurology. 1984;34(7):939. [DOI] [PubMed] [Google Scholar]
- 39. Fillenbaum GG, van Belle G, Morris JC, et al. Consortium to Establish a Registry for Alzheimer's Disease (CERAD): the first twenty years. Alzheimers Dement. 2008;4(2):96‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41(4):479‐486. [DOI] [PubMed] [Google Scholar]
- 41. Morris JC, Heyman A, Mohs RC, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part I. clinical and neuropsychological assessment of Alzheimer's disease. Neurology. 1989;39(9):1159‐1165. [DOI] [PubMed] [Google Scholar]
- 42. Jagust WJ, Eberling JL, Richardson BC, et al. The cortical topography of temporal lobe hypometabolism in early Alzheimer's disease. Brain Res. 1993;629(2):189‐198. [DOI] [PubMed] [Google Scholar]
- 43. Hamanaka G, Ohtomo R, Takase H, Lok J, Arai K. White‐matter repair: interaction between oligodendrocytes and the neurovascular unit. Brain Circ. 2018;4(3):118‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jang H, Kwon H, Yang J‐J, et al. Correlations between gray matter and white matter degeneration in pure Alzheimer's disease, pure subcortical vascular dementia, and mixed dementia. Sci Rep. 2017;7(1):1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tosto G, Zimmerman ME, Carmichael OT, Brickman AM, ftAsDN Initiative. Predicting aggressive decline in mild cognitive impairment: the importance of white matter hyperintensities. JAMA Neurol. 2014;71(7):872‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Desai MK, Sudol KL, Janelsins MC, Mastrangelo MA, Frazer ME, Bowers WJ. Triple‐transgenic Alzheimer's disease mice exhibit region‐specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia. 2009;57(1):54‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McKenzie AT, Moyon S, Wang M, et al. Multiscale network modeling of oligodendrocytes reveals molecular components of myelin dysregulation in Alzheimer's disease. Mol Neurodegener. 2017;12(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cui X, Guo Y‐e, Fang J‐h, et al. Donepezil, a drug for Alzheimer's disease, promotes oligodendrocyte generation and remyelination. Acta Pharmacol Sin. 2019;40(11):1386‐1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roy M, Rheault F, Croteau E, et al. Fascicle‐and glucose‐specific deterioration in white matter energy supply in Alzheimer's disease. J Alzheimers Dis. 2020(Preprint):1‐19. [DOI] [PubMed] [Google Scholar]
- 50. Pierre K, Pellerin L. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem. 2005;94(1):1‐14. [DOI] [PubMed] [Google Scholar]
- 51. Krikorian R, Shidler MD, Dangelo K, Couch SC, Benoit SC, Clegg DJ. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol Aging. 2012;33(2):425. e419‐425. e427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fortier M, Castellano CA, St‐Pierre V, et al. A ketogenic drink improves cognition in mild cognitive impairment: results of a 6‐month RCT. Alzheimers Dement. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Taylor MK, Sullivan DK, Mahnken JD, Burns JM, Swerdlow RH. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer's disease. Alzheimers Dement. 2018;4:28‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Neth BJ, Mintz A, Whitlow C, et al. Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer's disease: a pilot study. Neurobiol Aging. 2020;86:54‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Van Hoesen GW, Hyman BT. Hippocampal formation: anatomy and the patterns of pathology in Alzheimer's disease. Prog Brain Res. Elsevier; 1990:445‐457. [DOI] [PubMed] [Google Scholar]
- 56. Piert M, Koeppe RA, Giordani B, Berent S, Kuhl DE. Diminished glucose transport andphosphorylation in Alzheimer's disease determined by dynamic FDG‐PET. J Nucl Med. 1996;37:201‐208. [PubMed] [Google Scholar]
- 57. Nugent S, Croteau E, Potvin O, et al. Selection of the optimal intensity normalization region for FDG‐PET studies of normal aging and Alzheimer's disease. Sci Rep. 2020;10(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cunnane SC, Trushina E, Morland C, et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discovery. 2020:1‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Croteau E, Castellano C, Fortier M, et al. A cross‐sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer's disease. Exp Gerontol. 2018;107:18‐26. [DOI] [PubMed] [Google Scholar]
- 60. Fox N, Warrington E, Freeborough P, et al. Presymptomatic hippocampal atrophy in Alzheimer's disease: a longitudinal MRI study. Brain. 1996;119(6):2001‐2007. [DOI] [PubMed] [Google Scholar]
- 61. Paganoni S, Macklin EA, Hendrix S, et al. Trial of sodium phenylbutyrate–taurursodiol for amyotrophic lateral sclerosis. N Engl J Med. 2020;383(10):919‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Norwitz NG, Dearlove DJ, Lu M, Clarke K, Dawes H, Hu MT. A ketone ester drink enhances endurance exercise performance in parkinson's disease. Front Neurosci. 2020;14(1009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Benatar M, McDermott MP. Incremental gains in the battle against ALS. N Engl J Med. 2020;383(10):979‐980. [DOI] [PubMed] [Google Scholar]
- 64. Hirata K, Nambara T, Kawatani K, et al. 4‐Phenylbutyrate ameliorates apoptotic neural cell death in down syndrome by reducing protein aggregates. Sci Rep. 2020;10(1):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lanza M, Campolo M, Casili G, et al. Sodium butyrate exerts neuroprotective effects in spinal cord injury. Mol Neurobiol. 2019;56(6):3937‐3947. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information