

Abstract
Ischemic stroke is the most serious disease that harms human beings. In principle, its treatment is to restore blood flow supply as soon as possible. However, after the blood flow is restored, it will lead to secondary brain injury, that is, ischemia-reperfusion injury. The mechanism of ischemia-reperfusion injury is very complicated. This study showed that P2X4 receptors in the pyramidal neurons of rat hippocampus were significantly upregulated in the early stage of ischemia-reperfusion injury. Neurons with high expression of P2X4 receptors are neurons that are undergoing apoptosis. Intraventricular injection of the P2X4 receptor antagonist 5-(3-bromophenyl)-1,3-dihydro-2H-benzofuro[3,2-e]-1,4-diazepin-2-one (5-BDBD) and PSB-12062 can partially block neuronal apoptosis, to promote the survival of neurons, indicating that ATP through P2X4 receptors is involved in the process of cerebral ischemia-reperfusion injury. Therefore, identifying the mechanism of neuronal degeneration induced by extracellular ATP via P2X4 receptors after ischemia-reperfusion will likely find new targets for the treatment of ischemia-reperfusion injury, and will provide a useful theoretical basis for the treatment of ischemia-reperfusion injury.
Keywords: P2X4 purinoceptor, Ischemia-reperfusion injury, Hippocampus, Rat
Introduction
Ischemic stroke is the leading cause of death and disability in the global population [1]. The principle of treatment for ischemic stroke is generally to restore the blood supply to the ischemic area in a timely manner. However, after the ischemic area is re-perfused, the brain function and neurons of the ischemic area are further damaged. This phenomenon is called ischemia-reperfusion injury (IRI) [2]. Animal experiments have found that although IRI can reverse the death of ischemic penumbra neurons, some vulnerable areas in the central nervous system can induce delayed neuronal death (DND). The main death pattern is apoptosis, which occurs 2–4 days after ischemia-reperfusion [3–10]. Occurrence in the brain areas are mainly hippocampal CA1, cerebral cortex, and striatum. At present, the DND phenomenon presented by hippocampal CA1 pyramidal neurons has been widely recognized as an important indicator for judging IRI [5, 7, 8]. DND after cerebral ischemia-reperfusion is a multi-factor-mediated and extremely complicated mechanism. There are still many key issues to be further explored. If the occurrence of DND could be effectively prevented, it will provide some useful theoretical support for the treatment of ischemic stroke. Current research shows that DND is closely related to the following factors after ischemia-reperfusion: extracellular excitatory toxin accumulation (excitatory amino acid toxicity such as glutamic acid (Glu) and extracellular fluid ATP), intracellular calcium overload, oxygen free radicals, the generation of free radicals and lipid free radicals, the large secretion of inflammatory cytokines, the high accumulation of white blood cells, damage of the blood-brain barrier, and the lack of high-energy phosphate compounds are related [7, 8, 11–15]. There are interactions between the above factors, which makes the molecular mechanism of DND very complicated.
Previous studies have shown that one of the most critical mechanisms leading to DND is the release of large amounts of excitatory Glu, which leads to the opening of glutamate ion channels (NMDA, AMPA, and KA receptors, mainly NMDA receptors) [16, 17]. Numerous studies have shown that Glu is the central link of cerebral ischemic injury. After cerebral ischemia, the supply of oxygen and blood glucose is insufficient; energy generation is reduced, resulting in depolarization of neurons and glial cells; presynaptic membrane voltage-dependent Ca2+ ion channels are opened; and Glu is released into the synaptic cleft. In addition, due to lack of energy, reuptake of Glu by the presynaptic membrane is inhibited, further increasing the accumulation of extracellular Glu. Glu stimulates excessive excitement of NMDA receptors, leading to a large influx of Ca2+, initiating a series of pathological responses to nerve cell damage [18]. In addition to NMDA receptors, glutamate metabotropic I receptors are also involved in excitatory neurotoxicity induced by glutamate [17, 19], although II and III receptors are involved in neuroprotective effects [20].
In addition to Glu, the direct involvement of extracellular ATP and its derivatives in ischemic injury has received increasing attention [21–25]. ATP is degraded extracellularly to adenosine, which produces biological effects through its receptors. Adenosine receptors are designated P1 receptor and are all G protein–coupled receptors, which are divided into four subtypes: A1, A2A, A2B, and A3. Studies have shown that adenosine and adenosine A1 receptor agonists have important neuroprotective effects in in vivo and in vitro ischemia models, mainly by inhibiting the release of excitatory amino acid Glu. A1 receptor antagonists can aggravate ischemic nerve injury [26–28]. While the effects of A2A and A3 receptors are opposite to those of A1 receptors, blocking A2A and A3 receptors can significantly reduce the extent of ischemic damage [29, 30]. ATP mainly acts as a neurotransmitter or modulator acting via P2 receptors. The role of ATP in ischemic injury is getting more and more attention; for example, ATP can directly regulate the signaling pathway of cultured neurons during ischemia and hypoxia, which has led to further investigation of the effects of ATP in ischemic injury of the central nervous system [22, 31]. Previous experiments proved that extracellular ATP can indeed cause death of cultured neurons [32]. In fact, experiments have shown that P2 receptor antagonists can indeed block the death of cultured neurons caused by excessive Glu [33] or by serum/potassium deprivation, hypoglycemia, or chemical hypoxia [22, 31]. The above evidence indicates that extracellular ATP and its derivatives are involved in neuronal death during ischemia and hypoxia in vitro.
ATP produces biological effects through P2 receptors. P2 receptors are divided into P2X and P2Y [26]. The P2X receptor family are non-selective cation channels, which allow calcium and sodium ions to flow in and potassium ions to flow out, and the permeability of calcium ions is very high. Seven different P2X subtypes have been cloned, which are called P2X1, P2X2, P2X3, P2X4, P2X5, P2X6, and P2X7 [21]. Previous studies have shown that P2X1, P2X2, P2X4, P2X5, P2X6, and P2X7 are widely expressed in the central nervous system [21]. Morphological and functional experimental data show that neurons in the central nervous system, especially the hippocampus, mainly express P2X2, P2X4, and P2X6 receptor subtypes [34–38]; microglia express P2X4 and P2X7 receptors [33, 39–41]; oligodendrocytes express P2X7 receptors [15]; and astrocytes express all subtypes of P2X receptors (P2X1-7) [42].
Several publications have been published in recent years concerning the involvement of P2X4 in the mechanism of cerebral ischemic injury [24, 25, 43]. This study found that ATP may be involved in delayed neuronal death in cerebral ischemia-reperfusion injury through the P2X4 receptor.
Materials and methods
Animals and surgical procedures
Male Sprague–Dawley rats weighing 250–300 g were provided by the Animal Center of Second Military Medical University. All experimental procedures were approved by the Institutional Animal Care and Use Committee at Second Military Medical University and conformed to the UK Animals (Scientific Procedures) Act 1986 and associated guidelines on the ethical use of animals.
Fifteen minutes of global cerebral ischemia was induced by the four-vessel occlusion (4-VO) method, with a slight modification [44]. Briefly, rats were anesthetized with 10% (w/v) choral hydrate (400 mg/kg, intraperitoneally (i.p.)), then the bilateral common carotid arteries (CCAs) were freed, and both vertebral arteries were permanently electrocauterized. Rats were allowed to recover for 24 h after closing the surgical incisions. On the following day (0D), anesthesia was applied, the surgical incision in the neck was opened, and both CCAs were occluded with aneurysm clips to induce global cerebral ischemia for 15 min. The clips were removed for reperfusion. Rectal temperature was maintained at 36.0 to 37.0 °C throughout the procedures. Cerebral blood flow (CBF) before and after clamping the bilateral CCAs was monitored using a laser Doppler flowmetry (MBF3D, Moor Instruments, Axminster, Devon, UK), and rats with a decrease in CBF of less than 80% were excluded [44].
Experimental groups and drug administration
To study the effect of the P2X4 receptor antagonist (5-BDBD, Torcris) on delayed neuronal death in the hippocampal CA1 region after ischemia-reperfusion injury (IRI), rats were divided into three groups: sham group (sham operated), dimethyl sulfoxide (DMSO) group, 5-BDBD group, and PSB-12062 group. Rats in the sham and DMSO control groups received i.v. injection of 10 μl 20% DMSO; rats in 5-BDBD group received i.v. injection of 10 μl, 10 mM, 5-BDBD in DMSO; and rats in PSB-12062 group received i.v. injection of 10 μl, 1.0 mM, PSB-12062 in DMSO. Lateral ventricular injections were given once immediately after the injury and once 24 h later. Both 5-BDBD and PSB-12062 powder were reconstituted in DMSO and then diluted with 0.9% NaCl into a stock concentration of 10 mM and 1.0 mM, respectively.
Immunohistochemistry
After 12 h, 24 h, 48 h, 4 days, and 7 days of I/RI, rats were anesthetized and perfused intracardially with saline, followed by 4% (w/v) paraformaldehyde in 0.1 mol/L PBS, pH 7.4. Brains were removed and fixed overnight in 4% (w/v) paraformaldehyde, then transferred to 25% sucrose in PBS and kept in the solution until they sank to the bottom. Thereafter, the tissue blocks were rapidly frozen, and coronal sections (20 μm in thickness) were cut with a Leica cryostat and floated in PBS.
The following protocol was used for immunofluorescence. The sections were washed 3–5 min in PBS, and then preincubated in a blocking solution (10% normal bovine serum, 0.2% Triton X-100, 0.4% sodium azide in 0.01 mol/l PBS pH 7.2) for 30 min followed by incubation with the primary antibodies (P2X4 (1:400), Alomone, rabbit polyclonal, APR-004 and NeuN (1500), Millipore, monoclonal, clone A60. Subsequently, the sections were incubated with Cy3-conjugated donkey anti-rabbit IgG (Jackson) diluted 1:400 for P2X4 and FITC-conjugated donkey anti-mouse IgG for NeuN (Jackson). All incubations were separated by 5–10-min washes in PBS.
The following protocol was used for the double immunofluorescence of P2X4, Iba-1, NeuN, and GFAP. The sections were washed 3–5 min in PBS, and then preincubated in a blocking solution (10% normal bovine serum, 0.2% Triton X-100, 0.4% sodium azide in 0.01 mol/l PBS pH 7.2) for 30 min followed by incubation with the primary antibody mixtures. Mixture 1 was P2X4 (1:400) and Iba-1 (1:50), Abcam, goat polyclonal, ab107159; mixture 2 was P2X4 (1:400) and GFAP (1:400), Boster (1:400), monoclonal, BM0055; and mixture 3 was P2X4 (1:400) and NeuN (1:500) at room temperature overnight. Subsequently, the sections were incubated with Cy3-conjugated donkey anti-rabbit IgG (Jackson) diluted 1:200 for P2X4, FITC-conjugated donkey anti-mouse IgG (Jackson) 1:400 for NeuN and GFAP, FITC-conjugated donkey anti-goat IgG (Jackson) 1:400 for Iba-1. All incubations were separated by 5–10-min washes in PBS.
TUNEL method
In situ labeling of DNA fragmentation (terminal deoxynucleotidyl transferase–mediated UTP nick end labeling (TUNEL)) was carried out with an in situ cell death detection kit (Roche, Mannheim, Germany) according to the manufacturer’s instructions. After TUNEL detection of apoptotic cells, some sections were further immunostained to demonstrate P2X4 receptor proteins according to the above method.
Photomicroscopy
Images were taken with a Nikon digital camera DXM1200 (Nikon, Japan) attached to a Nikon Eclipse E600 microscope (Nikon). Images were imported into a graphics package (Adobe Photoshop 5.0, USA).
Western blot
For western blotting, the rats were killed after 12 h, 24 h, 48 h, 4 days, and 7 days of I/R injury, and their hippocampuses were removed immediately and lysed with 20 mM Tris–HCl buffer, pH 8.0, containing 1% NP-40, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 0.1% l-mercaptoethanol, 0.5 mM dithiothreitol, and a mixture of proteinase and phosphatase inhibitors (Sigma). Protein concentration was determined by the BCA protein assay method using bovine serum albumin (BSA) as standard. One hundred micrograms of protein samples from the hippocampus was loaded per lane, separated by SDS-PAGE (10% polyacrylamide gels), and then was electrotransferred onto nitrocellulose membranes. The membranes were blocked with 10% non-fat dry milk in Tris buffered saline for 1 h and incubated overnight at 4 °C with P2X4 (1:1000, Alomone), GAPDH (1:1000, Beyotime) diluted in 2% BSA in PBS. The membranes were then incubated with alkaline phosphatase–conjugated goat anti-rabbit IgG (Sigma) or goat anti-mouse IgG (Sigma) diluted 1:5000 in 2% BSA in PBS for 1 h at room temperature. The color development was performed with 400 μg/ml NitroBlue Tetrazolium, 200 μg/ml 5-bromo-4-chloro-3-indolyl phosphate and 100 mg/ml levamisole in TSM2 (0.1 mol/l Tris–HCl2 buffer, pH 9.5, 0.1 mol/l NaCl, and 0.05 mol/l MgCl2) in the dark. Bands were scanned using a densitometer (GS-700; Bio-Rad Laboratories).
Morris water maze test
At day 8, spatial learning and memory were tested using the Morris water maze, a circular black tank of 130 cm in diameter and 60 cm in height. The tank was filled with a depth of 30 cm water at 25 ± 1 °C. The maze was divided into four equal quadrants. The trials were performed according to Vorhees’ method [45]. Spatial acquisition: all rats received a training trial consisting of daily sessions of four consecutive trials for 5 days. The hidden platform (diameter 10 cm, 1.5 cm below the water surface) or a visible platform (diameter 10 cm, 1.5 cm above the water surface) was positioned in the middle of the southwest (SW) quadrant for all rats. The rats were released into the tank facing the maze wall at north (N), west (W), south (S), or east (E) quadrants in a predetermined pseudorandom order. A trial was terminated as soon as the rat found the platform; if the rat did not succeed within 120 s, it was guided onto the platform with a stick. The rat was allowed to stay on the platform for 20 s before being removed. Probe trial: immediately after the final training trial, the platform was removed. Rats were released into the pool at NE position and allowed to swim freely for 2 min. The time needed to find the platform (escape latency) in the training trials and time spent in the quadrants in the probe trial were recorded. The mean value of four escape latencies in the daily four training trials was taken as the escape latency for the rat. Values from eight rats in the same group were averaged to generate a mean escape latency for that day. Only these rats, which were trained to find the hidden platform, were included in the conducted probe trial.
Quantitative analysis
The apoptotic positive neurons for TUNEL in the hippocampal CA1 region at high magnification (×400) were counted. The percentage of TUNEL-positive in P2X4-positive neurons and the percentage of P2X4-positive in TUNEL-positive cells were quantified. In addition, the percentage of Iba-1-positive and the percentage of NeuN-positive cells in P2X4-positive cells in the CA1 region of the hippocampus 4 days after reperfusion injury were also quantified. Photomicrographs of the CA1 region were taken using a Nikon digital camera DXM1200 (Nikon, Japan) attached to a Nikon Eclipse E600 microscope (Nikon). The numbers of surviving/TUNEL-positive neurons and total neurons in the hippocampal CA1 region per 1-mm length were counted and analyzed blind by investigators. Six sections were used for each rat and the mean number of these six sections was calculated. Six rats were used for one group.
Results
Immunofluorescence histochemical results showed that the P2X4 receptor was normally expressed at low levels in pyramidal neurons in the hippocampal CA1 region (Fig. 1 a, b). After ischemia-reperfusion, the expression level of P2X4 receptors in the hippocampal CA1 pyramidal neurons gradually increased (Fig. 1 c, d, e, f, g, h). Peak expression of P2X4 receptors in pyramidal neurons was reached 48 h after ischemia-reperfusion (Fig. 1 g, h). Four days after IRI, P2X4 receptor expression decreased, and cells expressing P2X4 receptors were no longer neurons, but smaller cells (Fig. 1 i, j). Further verification experiments showed that these cells were mainly Iba-1-positive microglia, not astrocytes, oligodendrocytes. In total, 90% ± 5% P2X4 receptor–positive cells were also immunostained by Iba-1. Other cells positive for P2X4 receptors and negative for Iba-1 were generally NeuN-positive neurons (Fig. 2). Western blotting showed that the hippocampal P2X4 receptor showed the same trend after ischemia-reperfusion (Fig. 3).
Fig. 1.

Expression of P2X4 receptors in the CA1 region of the rat hippocampus in normal conditions (a, b) and after ischemia-reperfusion injury (c–j). After ischemia-reperfusion, the expression of P2X4 receptors gradually increased, peaked at 48 h (d), and then decreased, and the positive cells were small cells (i, j). The mean gray value of P2X4 receptor immunostaining in CA1 regions was obtained by using the ImageJ software. Data represent mean ± SEM. Statistical analysis was carried out with a Mann-Whitney U test. ∗p < 0.05, ** p < 0.01. n = 5. All scale bars = 130 μm
Fig. 2.

The identification of P2X4 receptor–positive cells 4 days after ischemia-reperfusion injury. The results show that P2X4 receptors coexist with Iba-1 (microglia marker), but not with GFAP (astrocyte marker), NeuN (neuronal marker), or Olig2 (oligodendrocyte marker). A yellow arrow indicates a cell (yellow) double-labeled by both P2X4 and Iba-1, and a red arrow indicates a neuron-like cell (red) with P2X4 receptor in A, and a yellow arrow indicates a cell with both P2X4 and NeuN in C. All scale bars = 130 μm
Fig. 3.

Western blotting showed that the hippocampal P2X4 receptor protein expression trend was similar to the immunofluorescence: it gradually increased in the early stage and decreased after 2 days of ischemic-reperfusion injury. Data represent mean ± SEM. Statistical analysis was carried out with a Mann-Whitney U test. ∗p < 0.05. n = 3
To observe whether P2X4 receptor antagonists have an effect on the activation process of microglia, we compared Iba-1 staining in the hippocampal CA1 region at 48 h of ischemia in the sham, saline, 5BDBD, and PSB-12062 groups and showed that microglia morphology was significantly altered in the saline group with a significant reduction in their protrusions and a decrease in the number of cells, but there was no significant difference between the saline, 5BDBD, and PSB-12062 groups (Fig. 4). This result indicates that the P2X4 antagonist 5BDBD and PSB-12062 did not significantly affect the activation process of microglia.
Fig. 4.

Effect of 5BDBD and PSB-12062 on microglia activation at 48 h after ischemia-reperfusion injury in the CA1 region. a, b, c, and d show Iba-1 immunostaining in the sham, saline-treated, 5BDBD-treated, and PSB-12062-treated groups respectively. e and f summarize the mean gray value and the microglial cell number in the CA1 region at 48 h after ischemia-reperfusion injury respectively. Note that the number of microglia protrusions and cell number were significantly reduced in the other three groups compared to the sham group, but there was no significant difference between those three groups. The mean gray value of P2X4 receptor immunostaining in CA1 regions was obtained by using the ImageJ software. Data represent mean ± SEM. Statistical analysis was carried out with a Mann-Whitney U test. ∗p < 0.05, **p < 0.01. n = 5
Combined immunofluorescence and apoptosis detection experiments showed that neurons in the hippocampal CA1 region that expressed high levels of P2X4 receptors were apoptotic cells (Fig. 5). A total of 85 ± 5% P2X4 receptor–positive neurons were also labeled by TUNEL assay, and 72 ± 7% cells with TUNEL signals were also immunostained by P2X4 receptor antibody.
Fig. 5.
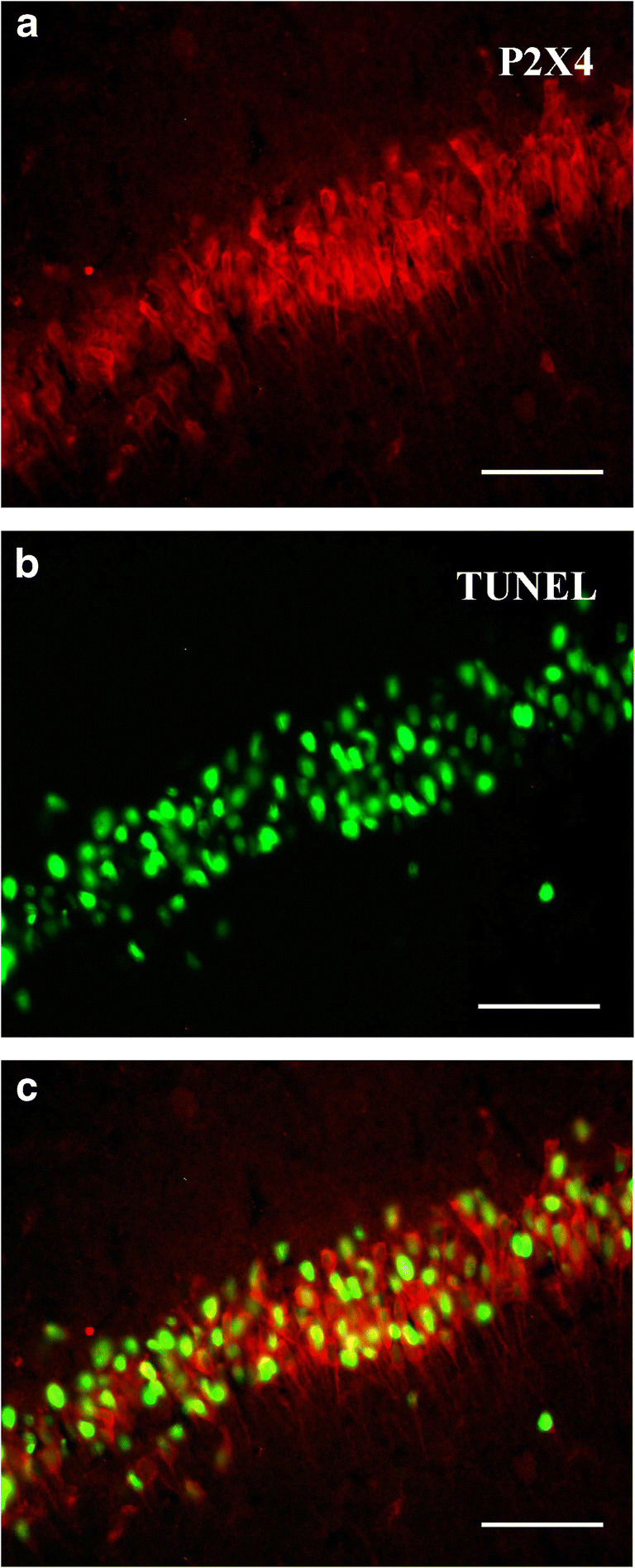
Combined immunofluorescence and TUNEL methods showed that neurons highly expressed P2X4 receptors (red) and apoptotic cell nuclei (green) in the hippocampal CA1 region. Note that majority of the neurons with P2X4 receptor immunofluorescence (red) were also labeled by TUNEL method (green). All scale bars = 65 μm
In order to further verify whether the high-level expression of P2X4 receptors in hippocampal CA1 area was involved in neuronal apoptosis after ischemia-reperfusion, we injected the lateral ventricle of ischemic animals with a P2X4 receptor–specific antagonist 5-BDBD and PSB-12062. The TUNEL method found that 5-BDBD and PSB-12062 protected neurons and reduced apoptosis of hippocampal CA1 neurons after ischemia-reperfusion (Fig. 6). Immunofluorescence of NeuN showed that the number of neurons that survived in the hippocampal CA1 area was significantly higher than that in the ischemia-reperfusion group, although there was still significant neuron loss (Fig. 7).
Fig. 6.

The TUNEL method showed apoptotic cells of the hippocampal CA1 region on the second and fourth days after ischemia-reperfusion injury in the sham (a, d, g), DMSO (b, c), 5-BDBD-treated (e, f), and PSB-12062-treated (h, i) groups. j summarizes apoptosis in the groups at the 2nd and 4th day. Note that 5-BDBD partially reduced the number of apoptotic cells. Data represent mean ± SEM. Statistical analysis was carried out with a Mann-Whitney U test. ***p < 0.001. n = 5. All scale bars = 65 μm
Fig. 7.

NeuN immunostaining of hippocampal regions in sham (a, b), DMSO-treated (c, d), 5-BDBD-treated (e, f), and PSB-12062-treated (g, h) groups after ischemic-reperfusion injury. i summarizes the number of neurons that survived in all groups at the 2nd and 4th day. Data represent mean ± SEM. Statistical analysis was carried out with a Mann-Whitney U test. ∗p < 0.05, ** p < 0.01. n = 5. All scale bars in a, c, e, g =1300 μm, in b, d, f, h = 130 μm
Spatial memory was evaluated using the Morris water maze. During the 5-day hidden platform trial, escape latency of sham, DMSO-treated, 5-BDBD-treated, and PSB-12062-treated groups decreased in a day-dependent pattern. However, the sham group took significantly less time to find the platform than the DMSO-treated group on all 5 days. In addition, the DMSO-treated group required significantly more time to find the platform than the 5-BDBD-treated and PSB-12062 groups. In the probe trial, the DMSO-treated group spent significantly less time than the 5-BDBD-treated, PSB-12062-treated, and sham groups in the SW quadrant (Fig. 8). These data suggested that both 5-BDBD and PSP-12062 could improve neurobehavioral outcomes after IRI.
Fig. 8.

Effect of 5-BDBD and PSB-12062 on spatial learning and memory in the water maze. a shows escape latency to find the hidden platform during the 5 days of the training trial from the 8th to the 12th day after IRI. b shows the escape latency to find the visible platform. c shows the distances to the hidden platform. d shows the time spent during the probe trial in the quadrants. Data represent mean ± SEM. Statistical analysis was carried out with multi-factor ANOVA. ∗p < 0.05, **p < 0.01. 5BDBD-treated group and PSB-12062 versus the relevant value of the DMSO group on the same day. n = 8
Discussion
The present study showed that P2X4 receptors are gradually upregulated during the early stage of cerebral ischemic injury in rats, and the expression peak time is the time point when neurons begin to die by apoptosis. The P2X4 receptor antagonist 5-BDBD and PSB-12062 partially blocked neuronal apoptosis, indicating that ATP through P2X4 receptors is involved in the process of cerebral ischemia-reperfusion injury.
The possible role of ATP and its derivatives in cerebral ischemia has been reported previously. Suramin, a broad-spectrum P2X receptor antagonist, can reduce the area of cerebral ischemia and the extent of cerebral edema in the early middle cerebral artery occlusion model of ischemia in rats [4]. In vitro culture of brain slices confirmed that after oxygen and glucose deprivation, the P2X2 and P2X4 receptors were significantly upregulated and neurons died [23]. Previous data showed that the P2X7 receptor is not a primary regulator of neuronal survival during cerebral ischemia [46]. These experiments indicate that ATP does not directly regulate neuron survival through the P2X7 receptor expressed by the neuron itself. Our recent experimental results show that after ischemia-reperfusion, CA1 microglial cells in the hippocampus are activated, their P2X7 receptors are significantly upregulated, and the inflammatory factor interleukin 1β (IL-β) upregulated [47]. These events occur before DND occurs. The antagonists brilliant blue G and A74003 of the P2X7 receptor can partially inhibit the occurrence of DND and IL-1β upregulation in the hippocampal CA1 region, and improve learning and memory functions of animals at the same time [47], which indicates that ATP activates microglia through microglia P2X7 receptors; thus, indirectly, they are involved in the occurrence of DND in the hippocampal CA1 region after ischemia-reperfusion, and these results are similar to those reported earlier by others [48]. This evidence suggests that ATP is indeed involved in IRI through P2X receptors.
Source of extracellular ATP
Normally, the extracellular fluid concentration of ATP is very low, at the nanomolar level but intracellular concentrations reach the millimolar level. Although physiologically, extracellular ATP can be derived from synaptic vesicle exocytosis, secretion of astrocytes and capillary endothelial cells, but it is diluted soon after release [49–52], and is hydrolyzed by the extracellular ATPase system, so ATP can be maintained at very low levels in the extracellular fluid [11, 13, 53]. Under pathological conditions, nerve cell injury can cause a large outflow of ATP, and at that time, extracellular ATPase activity is inhibited under conditions such as inflammatory response or oxidative stress. Released ATP cannot be rapidly hydrolyzed as normal and accumulates in the extracellular fluid at higher concentrations [52, 54]. A high concentration of extracellular ATP can act on neurons and glial cells through its corresponding receptors to produce a series of biological effects, which then affect their shapes and function of the local nervous system.
Because P2X receptors are highly permeable to Ca2+, Ca2+ influx induced by ATP in cortical or hippocampal pyramidal neurons is comparable to or greater than that induced by the excitatory amino acid Glu through NMDA receptors [55–57]. Based on the permeability of Ca2+ to the P2X receptor, it has been classified as a “superfamily of ligand-gated Ca2+ channels (LGCCs)” [57]. Therefore, overexpression and activation of P2X receptors is likely to be one of the causes of calcium overload during DND.
Ca2+ homeostasis is the basis for maintaining many important physiological functions of cells, including signal transmission and action potentials. Cells can regulate cytosolic Ca2+ balance and stability through the following mechanisms: calcium pumps and channels in the cell membrane; intracellular mitochondria, endoplasmic reticulum, Golgi, and other Ca2+ reservoirs precisely regulate the level of cytosolic Ca2+. After cerebral ischemia, this balance is disrupted. A large number of studies have shown that many cells rapidly and continuously increase the free Ca2+ concentration in the cytoplasm in the early stage of apoptosis [58, 59]. The excitotoxic substances Glu and ATP activate the NMDA receptor and P2X receptor, respectively, causing Ca2+ overload in the cytoplasm, activating Ca2+-dependent proteases, and inducing cell death. Ca2+ plays an important role in the death of ischemic neurons [60].
This study found that the expression of P2X4 receptors in pyramidal cells of hippocampal CA1 area was significantly upregulated, reaching a peak at 48 h, at which time TUNEL examination revealed a large number of DNDs in the CA1 region. The combination of immunohistochemistry and TUNEL showed that apoptotic hippocampal pyramidal cells expressed high levels of P2X4 receptors, suggesting that the upregulation of P2X4 receptors may be related to DND. Administration of 5-BDBD and PSB-12062 can significantly reduce the occurrence of DND in the CA1 region. These data suggest that extracellular ATP may be involved in the apoptosis of pyramidal neurons in the hippocampal CA1 region after ischemia-reperfusion through the P2X4 receptor and calcium ion overload pathway.
Researchers found in the amyotrophic lateral sclerosis animal model that motor neurons that are undergoing degenerative changes express high levels of P2X4 receptors, accompanied by abnormal changes such as the loss of NeuN or aggregation of microglia [12]. Its specific mechanism needs further study. This finding further suggests that DND in the hippocampal CA1 region during IRI is closely related to the upregulation of the P2X4 receptor.
Much of the previous data suggests that the hippocampus is involved in learning and memory processes [61]. Ischemia-reperfusion injury loses most of the pyramidal neurons in the CA1 region, so the learning and memory functions of the injured rats is definitely affected. The results of the water maze tests also confirmed the importance of pyramidal neurons of hippocampal CA1 area in learning and memory processes.
In summary, the occurrence of DND in the hippocampal CA1 region after cerebral ischemia-reperfusion is closely related to the release of extracellular ATP. It may be similar to the excitatory amino acid Glu that induces neuronal calcium overload via the NMDA receptor, and the ATP ligand–gated calcium channel superfamily member, the P2X4 receptor, induces or aggravates calcium overload in pyramidal neurons of the hippocampal CA1 region, triggering DND. Therefore, identifying the mechanism of neuronal degeneration induced by extracellular ATP via P2X4 receptors after cerebral ischemia-reperfusion will likely find new targets for the treatment of IRI, and will provide a useful theoretical basis for the treatment of cerebral ischemia-reperfusion injury.
Acknowledgements
The authors thank Dr. Gillian E. Knight for her excellent editorial assistance.
Zhenghua Xiang
His main research interests are in the role of purinoceptors in the development and injury repair of the nervous system.

Funding
This work was supported by the National Natural Science Foundation of the People’s Republic of China (81471260 to Z Xiang, 31900647 to R. Ji).
Data availability
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Compliance with ethical standards
Conflict of interest
Zhenghua Xiang declares that she has no conflict of interest.
Xin Jiang declares that she has no conflict of interest.
Ruihua Ji declares that she has no conflict of interest.
Hongbin Yuan declares that she has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Zhenghua Xiang, Xin Jiang and Rihui Ji contributed equally to this work.
References
- 1.Phipps MS, Cronin CA. Management of acute ischemic stroke. BMJ. 2020;368:l6983. doi: 10.1136/bmj.l6983. [DOI] [PubMed] [Google Scholar]
- 2.Amantea D, Nappi G, Bernardi G, Bagetta G, Corasaniti MT. Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. FEBS J. 2009;276:13–26. doi: 10.1111/j.1742-4658.2008.06766.x. [DOI] [PubMed] [Google Scholar]
- 3.Cho CH, Byun HR, Jover-Mengual T, Pontarelli F, Dejesus C, Cho AR, Zukin RS, Hwang JY. Gadd45b acts as neuroprotective effector in global ischemia-induced neuronal death. Int Neurourol J. 2019;23:S11–S21. doi: 10.5213/inj.1938040.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kharlamov A, Jones SC, Kim DK. Suramin reduces infarct volume in a model of focal brain ischemia in rats. Exp Brain Res. 2002;147:353–359. doi: 10.1007/s00221-002-1251-1. [DOI] [PubMed] [Google Scholar]
- 5.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 6.Lee TK, Kim H, Song M, Lee JC, Park JH, Ahn JH, Yang GE, Ohk TG, Shin MC, Cho JH, et al. Time-course pattern of neuronal loss and gliosis in gerbil hippocampi following mild, severe, or lethal transient global cerebral ischemia. Neural Regen Res. 2019;14:1394–1403. doi: 10.4103/1673-5374.255977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicotera P, Leist M, Ferrando-May E. Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol Lett. 1998;102-103:139–142. doi: 10.1016/S0378-4274(98)00298-7. [DOI] [PubMed] [Google Scholar]
- 8.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiong TQ, Chen LM, Gui Y, Jiang T, Tan BH, Li SL, Li YC. The effects of epothilone D on microtubule degradation and delayed neuronal death in the hippocampus following transient global ischemia. J Chem Neuroanat. 2019;98:17–26. doi: 10.1016/j.jchemneu.2019.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Zhu H, Yoshimoto T, Imajo-Ohmi S, Dazortsava M, Mathivanan A, Yamashima T. Why are hippocampal CA1 neurons vulnerable but motor cortex neurons resistant to transient ischemia? J Neurochem. 2012;120:574–585. doi: 10.1111/j.1471-4159.2011.07550.x. [DOI] [PubMed] [Google Scholar]
- 11.Agteresch HJ, Dagnelie PC, van den Berg JW, Wilson JH. Adenosine triphosphate: established and potential clinical applications. Drugs. 1999;58:211–232. doi: 10.2165/00003495-199958020-00002. [DOI] [PubMed] [Google Scholar]
- 12.Casanovas A, Hernandez S, Tarabal O, Rossello J, Esquerda JE. Strong P2X4 purinergic receptor-like immunoreactivity is selectively associated with degenerating neurons in transgenic rodent models of amyotrophic lateral sclerosis. J Comp Neurol. 2008;506:75–92. doi: 10.1002/cne.21527. [DOI] [PubMed] [Google Scholar]
- 13.Flavin MP, Zhao G, Ho LT. Microglial tissue plasminogen activator (tPA) triggers neuronal apoptosis in vitro. Glia. 2000;29:347–354. doi: 10.1002/(SICI)1098-1136(20000215)29:4<347::AID-GLIA5>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 14.Wang JY, Xia Q, Chu KT, Pan J, Sun LN, Zeng B, Zhu YJ, Wang Q, Wang K, Luo BY. Severe global cerebral ischemia-induced programmed necrosis of hippocampal CA1 neurons in rat is prevented by 3-methyladenine: a widely used inhibitor of autophagy. J Neuropathol Exp Neurol. 2011;70:314–322. doi: 10.1097/NEN.0b013e31821352bd. [DOI] [PubMed] [Google Scholar]
- 15.Yan BC, Park JH, Lee CH, Yoo KY, Choi JH, Lee YJ, Cho JH, Baek YY, Kim YM, Won MH. Increases of antioxidants are related to more delayed neuronal death in the hippocampal CA1 region of the young gerbil induced by transient cerebral ischemia. Brain Res. 2011;1425:142–154. doi: 10.1016/j.brainres.2011.09.063. [DOI] [PubMed] [Google Scholar]
- 16.Michaelis EK. Molecular biology of glutamate receptors in the central nervous system and their role in excitotoxicity, oxidative stress and aging. Prog Neurobiol. 1998;54:369–415. doi: 10.1016/S0301-0082(97)00055-5. [DOI] [PubMed] [Google Scholar]
- 17.Park JH, Kim YH, Ahn JH, Choi SY, Hong S, Kim SK, Kang IJ, Kim YM, Lee TK, Won MH, Lee CH. Atomoxetine protects against NMDA receptor-mediated hippocampal neuronal death following transient global cerebral ischemia. Curr Neurovasc Res. 2017;14:158–168. doi: 10.2174/1567202614666170328094042. [DOI] [PubMed] [Google Scholar]
- 18.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/S0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 19.Nicoletti F, Bruno V, Copani A, Casabona G, Knopfel T. Metabotropic glutamate receptors: a new target for the therapy of neurodegenerative disorders? Trends Neurosci. 1996;19:267–271. doi: 10.1016/S0166-2236(96)20019-0. [DOI] [PubMed] [Google Scholar]
- 20.Henrich-Noack P, Flor PJ, Sabelhaus CF, Prass K, Dirnagl U, Gasparini F, Sauter A, Rudin M, Reymann KG. Distinct influence of the group III metabotropic glutamate receptor agonist (R,S)-4-phosphonophenylglycine [(R,S)-PPG] on different forms of neuronal damage. Neuropharmacology. 2000;39:911–917. doi: 10.1016/S0028-3908(99)00256-7. [DOI] [PubMed] [Google Scholar]
- 21.Burnstock G. Purinergic signalling and disorders of the central nervous system. Nat Rev Drug Discov. 2008;7:575–590. doi: 10.1038/nrd2605. [DOI] [PubMed] [Google Scholar]
- 22.Cavaliere F, D’Ambrosi N, Ciotti MT, Mancino G, Sancesario G, Bernardi G, Volonte C. Glucose deprivation and chemical hypoxia: neuroprotection by P2 receptor antagonists. Neurochem Int. 2001;38:189–197. doi: 10.1016/S0197-0186(00)00088-7. [DOI] [PubMed] [Google Scholar]
- 23.Juranyi Z, Sperlagh B, Vizi ES. Involvement of P2 purinoceptors and the nitric oxide pathway in [3H] purine outflow evoked by short-term hypoxia and hypoglycemia in rat hippocampal slices. Brain Res. 1999;823:183–190. doi: 10.1016/S0006-8993(99)01169-5. [DOI] [PubMed] [Google Scholar]
- 24.Srivastava P, Cronin CG, Scranton VL, Jacobson KA, Liang BT, Verma R. Neuroprotective and neuro-rehabilitative effects of acute purinergic receptor P2X4 (P2X4R) blockade after ischemic stroke. Exp Neurol. 2020;329:113308. doi: 10.1016/j.expneurol.2020.113308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verma R, Cronin CG, Hudobenko J, Venna VR, McCullough LD, Liang BT. Deletion of the P2X4 receptor is neuroprotective acutely, but induces a depressive phenotype during recovery from ischemic stroke. Brain Behav Immun. 2017;66:302–312. doi: 10.1016/j.bbi.2017.07.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H. Purinergic signalling in the nervous system: an overview. Trends Neurosci. 2009;32:19–29. doi: 10.1016/j.tins.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 27.Cunha RA. Neuroprotection by adenosine in the brain: from A(1) receptor activation to A (2A) receptor blockade. Purinergic Signal. 2005;1:111–134. doi: 10.1007/s11302-005-0649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jurkowitz MS, Litsky ML, Browning MJ, Hohl CM. Adenosine, inosine, and guanosine protect glial cells during glucose deprivation and mitochondrial inhibition: correlation between protection and ATP preservation. J Neurochem. 1998;71:535–548. doi: 10.1046/j.1471-4159.1998.71020535.x. [DOI] [PubMed] [Google Scholar]
- 29.Chen GJ, Harvey BK, Shen H, Chou J, Victor A, Wang Y. Activation of adenosine A3 receptors reduces ischemic brain injury in rodents. J Neurosci Res. 2006;84:1848–1855. doi: 10.1002/jnr.21071. [DOI] [PubMed] [Google Scholar]
- 30.Chen JF, Huang Z, Ma J, Zhu J, Moratalla R, Standaert D, Moskowitz MA, Fink JS, Schwarzschild MA. A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci. 1999;19:9192–9200. doi: 10.1523/JNEUROSCI.19-21-09192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cavaliere F, D’Ambrosi N, Sancesario G, Bernardi G, Volonte C. Hypoglycaemia-induced cell death: features of neuroprotection by the P2 receptor antagonist basilen blue. Neurochem Int. 2001;38:199–207. doi: 10.1016/S0197-0186(00)00087-5. [DOI] [PubMed] [Google Scholar]
- 32.Amadio S, D’Ambrosi N, Cavaliere F, Murra B, Sancesario G, Bernardi G, Burnstock G, Volonte C. P2 receptor modulation and cytotoxic function in cultured CNS neurons. Neuropharmacology. 2002;42:489–501. doi: 10.1016/S0028-3908(01)00197-6. [DOI] [PubMed] [Google Scholar]
- 33.Volonte C, Merlo D. Selected P2 purinoceptor modulators prevent glutamate-evoked cytotoxicity in cultured cerebellar granule neurons. J Neurosci Res. 1996;45:183–193. doi: 10.1002/(SICI)1097-4547(19960715)45:2<183::AID-JNR10>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 34.Bo X, Zhang Y, Nassar M, Burnstock G, Schoepfer R. A P2X purinoceptor cDNA conferring a novel pharmacological profile. FEBS Lett. 1995;375:129–133. doi: 10.1016/0014-5793(95)01203-Q. [DOI] [PubMed] [Google Scholar]
- 35.Collo G, North RA, Kawashima E, Merlo-Pich E, Neidhart S, Surprenant A, Buell G. Cloning OF P2X5 and P2X6 receptors and the distribution and properties of an extended family of ATP-gated ion channels. J Neurosci. 1996;16:2495–2507. doi: 10.1523/JNEUROSCI.16-08-02495.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo W, Xu X, Gao X, Burnstock G, He C, Xiang Z. Expression of P2X5 receptors in the mouse CNS. Neuroscience. 2008;156:673–692. doi: 10.1016/j.neuroscience.2008.07.062. [DOI] [PubMed] [Google Scholar]
- 37.Kanjhan R, Housley GD, Burton LD, Christie DL, Kippenberger A, Thorne PR, Luo L, Ryan AF. Distribution of the P2X2 receptor subunit of the ATP-gated ion channels in the rat central nervous system. J Comp Neurol. 1999;407:11–32. doi: 10.1002/(SICI)1096-9861(19990428)407:1<11::AID-CNE2>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 38.Soto F, Garcia-Guzman M, Karschin C, Stuhmer W. Cloning and tissue distribution of a novel P2X receptor from rat brain. Biochem Biophys Res Commun. 1996;223:456–460. doi: 10.1006/bbrc.1996.0915. [DOI] [PubMed] [Google Scholar]
- 39.Collo G, Neidhart S, Kawashima E, Kosco-Vilbois M, North RA, Buell G. Tissue distribution of the P2X7 receptor. Neuropharmacology. 1997;36:1277–1283. doi: 10.1016/S0028-3908(97)00140-8. [DOI] [PubMed] [Google Scholar]
- 40.Xiang Z, Burnstock G. Expression of P2X receptors on rat microglial cells during early development. Glia. 2005;52:119–126. doi: 10.1002/glia.20227. [DOI] [PubMed] [Google Scholar]
- 41.Xiang Z, Chen M, Ping J, Dunn P, Lv J, Jiao B, Burnstock G. Microglial morphology and its transformation after challenge by extracellular ATP in vitro. J Neurosci Res. 2006;83:91–101. doi: 10.1002/jnr.20709. [DOI] [PubMed] [Google Scholar]
- 42.Kukley M, Barden JA, Steinhauser C, Jabs R. Distribution of P2X receptors on astrocytes in juvenile rat hippocampus. Glia. 2001;36:11–21. doi: 10.1002/glia.1091. [DOI] [PubMed] [Google Scholar]
- 43.Ozaki T, Muramatsu R, Sasai M, Yamamoto M, Kubota Y, Fujinaka T, Yoshimine T, Yamashita T. The P2X4 receptor is required for neuroprotection via ischemic preconditioning. Sci Rep. 2016;6:25893. doi: 10.1038/srep25893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Q, Xu J, Rottinghaus GE, Simonyi A, Lubahn D, Sun GY, Sun AY. Resveratrol protects against global cerebral ischemic injury in gerbils. Brain Res. 2002;958:439–447. doi: 10.1016/S0006-8993(02)03543-6. [DOI] [PubMed] [Google Scholar]
- 45.Vorhees CV, Williams MT (2006) Morris water maze: procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1:848–858 [DOI] [PMC free article] [PubMed]
- 46.Lee JM, Zipfel GJ, Choi DW. The changing landscape of ischaemic brain injury mechanisms. Nature. 1999;399:A7-14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- 47.Yu Q, Guo Z, Liu X, Ouyang Q, He C, Burnstock G, Yuan H, Xiang Z. Block of P2X7 receptors could partly reverse the delayed neuronal death in area CA1 of the hippocampus after transient global cerebral ischemia. Purinergic Signal. 2013;9:663–675. doi: 10.1007/s11302-013-9379-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chu K, Yin B, Wang J, Peng G, Liang H, Xu Z, Du Y, Fang M, Xia Q, Luo B. Inhibition of P2X7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J Neuroinflammation. 2012;9:69. doi: 10.1186/1742-2094-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beaudoin AR, Grondin G, Gendron FP. Immunolocalization of ATP diphosphohydrolase in pig and mouse brains, and sensory organs of the mouse. Prog Brain Res. 1999;120:387–395. doi: 10.1016/S0079-6123(08)63571-2. [DOI] [PubMed] [Google Scholar]
- 50.Kaczmarek E, Koziak K, Sevigny J, Siegel JB, Anrather J, Beaudoin AR, Bach FH, Robson SC. Identification and characterization of CD39/vascular ATP diphosphohydrolase. J Biol Chem. 1996;271:33116–33122. doi: 10.1074/jbc.271.51.33116. [DOI] [PubMed] [Google Scholar]
- 51.Wang TF, Guidotti G. CD39 is an ecto-(Ca2+,Mg2+)-apyrase. J Biol Chem. 1996;271:9898–9901. doi: 10.1074/jbc.271.17.9898. [DOI] [PubMed] [Google Scholar]
- 52.Zimmermann H, Braun N. Ecto-nucleotidases--molecular structures, catalytic properties, and functional roles in the nervous system. Prog Brain Res. 1999;120:371–385. doi: 10.1016/S0079-6123(08)63570-0. [DOI] [PubMed] [Google Scholar]
- 53.Schwiebert EM (2000) Extracellular ATP-mediated propagation of Ca(2+) waves. Focus on “mechanical strain-induced Ca(2+) waves are propagated via ATP release and purinergic receptor activation”. Am J Phys Cell Phys 279:C281–C283 [DOI] [PubMed]
- 54.Robson SC, Kaczmarek E, Siegel JB, Candinas D, Koziak K, Millan M, Hancock WW, Bach FH. Loss of ATP diphosphohydrolase activity with endothelial cell activation. J Exp Med. 1997;185:153–163. doi: 10.1084/jem.185.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abbracchio MP, Cattabeni F. Brain adenosine receptors as targets for therapeutic intervention in neurodegenerative diseases. Ann N Y Acad Sci. 1999;890:79–92. doi: 10.1111/j.1749-6632.1999.tb07983.x. [DOI] [PubMed] [Google Scholar]
- 56.Egan TM, Khakh BS. Contribution of calcium ions to P2X channel responses. J Neurosci. 2004;24:3413–3420. doi: 10.1523/JNEUROSCI.5429-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pankratov Y, Lalo U. Calcium permeability of ligand-gated Ca2+ channels. Eur J Pharmacol. 2014;739:60–73. doi: 10.1016/j.ejphar.2013.11.017. [DOI] [PubMed] [Google Scholar]
- 58.Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene. 2008;27:6407–6418. doi: 10.1038/onc.2008.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–129. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 60.Kristian T, Siesjo BK. Calcium-related damage in ischemia. Life Sci. 1996;59:357–367. doi: 10.1016/0024-3205(96)00314-1. [DOI] [PubMed] [Google Scholar]
- 61.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361(6407):31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.