

SUMMARY
yki-induced gut tumors in Drosophila are associated with host wasting, including muscle dysfunction, lipid loss, and hyperglycemia, a condition reminiscent of human cancer cachexia. We previously used this model to identify tumor-derived ligands that contribute to host wasting. To identify additional molecular networks involved in host-tumor interactions, we develop PathON, a web-based tool analyzing the major signaling pathways in Drosophila, and uncover the Upd3/Jak/Stat axis as an important modulator. We find that yki-gut tumors secrete Upd3 to promote self-overproliferation and enhance Jak/Stat signaling in host organs to cause wasting, including muscle dysfunction, lipid loss, and hyperglycemia. We further reveal that Upd3/Jak/Stat signaling in the host organs directly triggers the expression of ImpL2, an antagonistic binding protein for insulin-like peptides, to impair insulin signaling and energy balance. Altogether, our results demonstrate that yki-gut tumors produce a Jak/Stat pathway ligand, Upd3, that regulates both self-growth and host wasting.
In brief
Ding et al. show that yki3SA-gut tumors produce Upd3 as a cachectic ligand to simultaneously promote self-growth and host organ wasting via systemic activation of Jak/Stat signaling in Drosophila. The Upd3/Jak/Stat axis induces host ImpL2 production and perturbs insulin response, leading to muscle mitochondrial dysfunction, lipid loss, and carbohydrate elevation.
Graphical abstract

INTRODUCTION
Tumor-induced host organ wasting is a general phenomenon observed in both vertebrates and invertebrates. Patients with advanced cancers frequently develop severe organ wasting, referred to as “cancer cachexia,” including muscle dysfunction, lipid loss, and hyperglycemia. Organ wasting is associated with resistance to chemotherapy, reduced quality of life, and mortality among patients with cancer (Fearon et al., 2013). Recent studies based on cultured cells and tumor-bearing mice have implicated a number of tumor-secreted cachectic proteins (e.g., PTHrP, activins, LIF, IL-6, and Hsp70/90), which remotely crosstalk to muscle and adipose tissues and modulate their metabolic homeostasis. In support of their roles, antibody neutralization of some of these proteins has been shown to improve organ wasting and survival of tumor-bearing subjects (Hirata et al., 2013; Kandarian et al., 2018; Kir et al., 2014; Zhang et al., 2017; Zhou et al., 2010). Despite these advances, however, there remains a need for genetic animal models that facilitate comprehensive evaluation of tumor-secreted proteins, associated signaling pathways, and their effects in host organs.
Drosophila is emerging as an excellent model to decipher the molecular mechanisms of tumor-induced host wasting. For example, aberrant activation of yki3SA, a homolog of the human oncogene YAP1, in intestinal stem cells (ISCs) results in severe tumor cell overproliferation in the gut and subsequent muscle dysfunction, lipid loss, and hyperglycemia (Kwon et al., 2015). Similar host wasting effects were also reported in other fly tumor models (Figueroa-Clarevega and Bilder, 2015; Katheder et al., 2017; Newton et al., 2020; Nie et al., 2019). As there is no evidence of metastasis from yki3SA-gut tumors, a likely explanation is that host wasting is caused by tumor-secreted proteins that act remotely on host tissues. Previously, we identified two tumor-secreted proteins, the insulin-like polypeptide binding protein ImpL2 and the Pvr receptor tyrosine kinase ligand Pvf1, that impair the anabolism/catabolism balance of host organs (Kwon et al., 2015; Song et al., 2019). However, removal of either ImpL2 or Pvf1 from yki3SA tumors only partially alleviates host wasting, suggesting that additional secreted proteins are involved in tumor-host interactions. Moreover, despite the impacts of tumor-derived ImpL2 and Pvf1 on host organs, they fail to affect growth of the gut tumor. Whether and how tumor-secreted ligands coordinate host wasting and tumor growth remain unclear.
Hormone/ligand-induced signaling plays a major role in interorgan communication. Classically, hormones/ligands trigger specific intracellular signaling pathways, including via activation of receptors, kinases/phosphatases, and transcriptional factors, and eventually regulate downstream gene expression. For example, the Unpaired 1, 2, and 3 (Upd1/2/3) ligands bind to their common receptor Dome and activate downstream kinase Jak/Hop and transcriptional factor Stat92E to regulate expression of Stat target genes such as Socs36E and Dome (Herrera and Bach, 2019). Thus, monitoring ligand expression in a sending organ and signature target gene expression of associated signaling pathways in a receiving organ can help identify the secreted ligands and corresponding signal reception and transduction pathways involved in interorgan communication.
In this study, we developed PathON, a web-based tool for analysis of ligands, signaling components, and signature target genes for 14 common canonical Drosophila signaling pathways: EGFR/FGFR/PvR, Hedgehog, Hippo, insulin, Jak/Stat, NF-κB/Imd, and NF-κB/ToII, Notch, TGF-β/BMP and TGF-β/Activin, TNF-α, and Wnt. We found that Jak/Stat signaling participates in tumor-host interactions. Combining genetic and molecular assays, we demonstrate that yki3SA-tumor-derived Upd3 promotes both tumor growth and host wasting, including muscle dysfunction, lipid loss, and hyperglycemia, via systemic activation of Jak/Stat signaling. The results demonstrate that Upd3 acts as a tumor-secreted ligand that affects both tumor growth and host wasting.
RESULTS
Potential signaling pathways involved in tumor-host crosstalk
To develop PathON (https://www.fiyrnai.org/tools/pathon/web), we compiled a list of ligands (agonists and antagonists) and signaling components (receptors, kinases/phosphatases, adaptor proteins, and transcriptional factors) for 14 canonical Drosophila signaling pathways on the basis of the published literature (Table S1). Signature target genes for these pathways were selected if they had been previously validated by more than two of the following criteria: physiological function, gene expression, promoter activity, and direct binding to transcriptional factor(s) (Table S2).
On the basis of our hypothesis that yki-induced gut tumors produce specific ligands that activate associated signaling pathways and induce target gene expression in host organs, we applied PathON to analyze the expression levels of ligands and signature target genes of 14 signaling pathways in published RNA sequencing (RNA-seq) datasets from yki3SA-tumor guts (GSE113728) and muscles (GSE65325) of yki3SA-tumor-bearing files. Interestingly, a Jak/Stat signaling ligand (upd3, 37-fold induction) is one of the top genes upregulated in yki3SA-tumor guts (Figure 1A). Signature target genes of Jak/Stat signaling are also significantly enriched among differentially expressed genes in the muscles of yki3SA-tumor-bearing files (Figure 1A). Together, this suggests that Upd3 from yki3SA-gut tumors activates Jak/Stat signaling in muscles.
Figure 1. Upd3/Jak/Stat signaling is involved in tumor-host interactions.
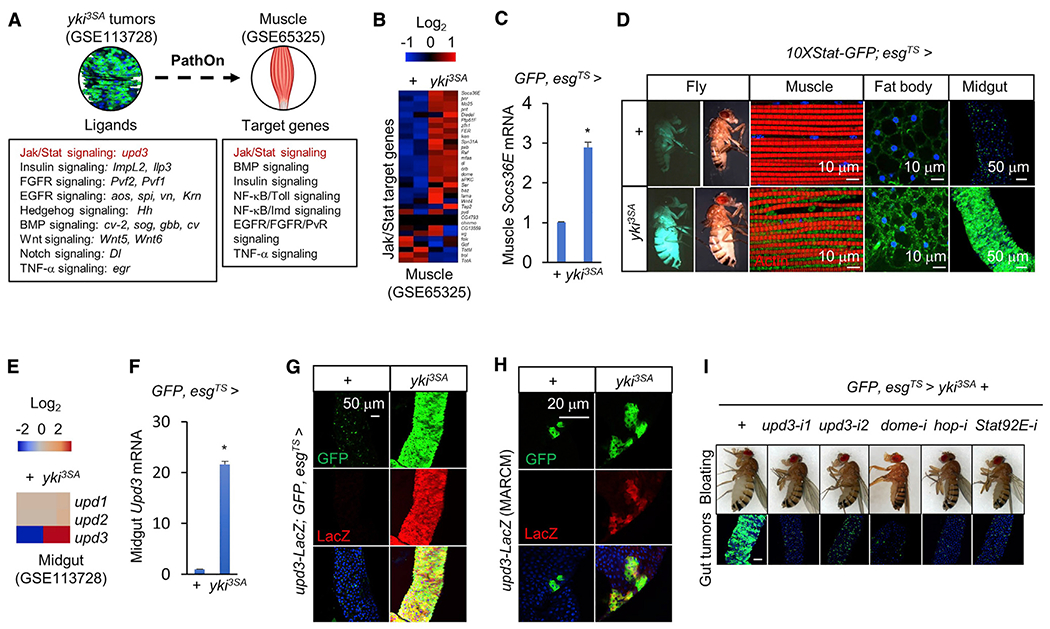
(A) Results of analysis with PathON, a software resource covering 14 canonical signaling pathways in Drosophila, reveal transcriptional changes in ligands and signature target genes of the indicated signaling pathways in gut tumors and muscles of yki3SA-tumor-bearing flies, respectively, at day 8.
(B and C) Expression levels of target genes of Jak/Stat signaling in the muscles of control flies (+: esg-GALl4, UAS-GFP, tub-GAL80TS/+) or yki3SA-tumor-bearing flies (yki3SA: esg-GAL4, UAS-GFP, tub-GAL80TS/+; UAS-yki3SA/+) at day 8. (B) Heatmap generated from GSE65325 RNA-seq data. (C) qPCR results (n = 3,5 flies/replicate).
(D) 10XStat-GFP signals in the muscle, fat body, midgut, and whole fly. Scale bars: muscle, fat body, 10 μm; midgut, 50 μm.
(E–G) Expression levels of Upd ligands in the midguts of control (+) and yki3SA-tumor-bearing flies (yki3SA) at day 8. (E) Heatmap generated from GSE113728 RNA-seq data. (F) qPCR results (n = 3, 10 flies/replicate). (G) Immunostaining indicating Upd3-LacZ expression (green) in the midgut. Scale bar: 50 μm.
(H) MARCM assays indicating that upd3-LacZ expression (red) is increased mainly in the yki-tumor cells (green) and marginally in other gut cells. Scale bar: 20 μm.
(I) Changes in the bloating phenotype (top) and gut tumors (bottom) of indicated genotypes at day 8. Scale bar: 50 μm.
Data are presented as mean ± SEM. *p < 0.05.
We note that the ligands of insulin (ImpL2, 58-fold induction; and Ilp3, 2.5-fold induction), PvR (Pvf2, 20-fold; Pvf1, 14-fold), EGFR (aos, 8-fold; spi, 4-fold; vn, 3.5-fold; Krn, 2.6-fold), Hedgehog (hh, 7-fold), TGF-β/BMP (cv-2, 6-fold; sog, 4-fold; gbb, 2.5-fold; cv, 2.2-fold), Wnt (Wnt5, 6-fold and Wnt6, 1.8-fold), Notch (Dl, 5-fold), and TNF-α (egr, 2.5-fold) signaling pathways are also significantly increased in yki3SA-tumor guts (Figure 1A). In addition, signature target genes of TGF-β/BMP, NF-κB/Imd, NF-κB/Toll, insulin, EGFR/FGFR/PvR, and TNF-α signaling pathways are significantly enriched among differentially expressed genes in the muscles of tumor-bearing fiies (Figure 1A). As we previously analyzed the roles of ImpL2 and Pvf1 in tumor-host interaction (Kwon et al., 2015; Song et al., 2019), we decided to focus on upd3, the second most increased ligand, and its associated Jak/Stat signaling pathway.
We first examined the target genes of Jak/Stat signaling and found that the transcriptional levels of most of these genes were significantly changed in the muscles of yki3SA-tumor-bearing fiies, as indicated by RNA-seq (Figure 1B; Figure S1). For example, Socs36E was robustly increased. Upregulation of Socs36E in this context was further validated by qPCR (Figure 1C). We also examined activation of Jak/Stat signaling in the muscles of yki3SA-tumor-bearing files using reporter assays. To do this, either control or yki3SA files without GFP labeling in ISCs were crossed to 10XStat-GFP reporter lines. The muscles from yki3SA files exhibited abnormal myofibril morphology, and more Stat-GFP accumulated in the space between myofibrils and nuclei compared with control (Figure 1D), confirming that muscle Jak/Stat signaling is enhanced. Stat-GFP signaling was also increased in the fat body and in tumor-bearing guts in yki3SA files (Figure 1D).
We next validated ligand induction in yki3SA-gut tumors. The transcriptional level of upd3, but not upd1 or upd2, was dramatically increased in tumors, as revealed by RNA-seq (>20-fold; Figure 1E). This result was further confirmed using qPCR, upd3-LacZ reporter, and MARCM clones (Figures 1F–1H). Altogether, the changes in transcription of the Jak/Stat ligand upd3 in yki3SA-gut tumors and of Jak/Stat target genes in muscles suggest that Upd3/Jak/Stat signaling is involved in tumor-host interactions.
Upd3 is essential for yki3SA-gut tumor growth
To test whether yki3SA-tumor-derived Upd3 directly regulates host wasting, we knocked down upd3 expression specifically in yki3SA tumors. Interestingly, the growth of yki3SA-gut tumors and Socs36E expression indicating Jak/Stat pathway activity in the muscle were potently suppressed under these conditions (Figure 1I; Figure S2A). Consistent with this result, the growth of yki3SA-gut tumors in upd3 mutant files was also diminished (Figure S2B). Previous studies have indicated that Upd3 triggers Jak/Stat signaling in ISCs to promote proliferation in response to tissue damage and infection (Shaw et al., 2010). We found that knockdown of dome, hop (Jak), or Stat92E (Stat) in yki3SA-gut tumors also suppressed tumor growth (Figure 1I). Taken together, our results show that yki3SA tumors produce Upd3 and activate Jak/Stat signaling to promote self-growth in an autocrine/paracrine manner.
Tumor-derived Upd3 is essential for host wasting
To investigate the role of Upd3 in tumor-induced host wasting, we expressed a constitutively active form of hop (hopTumL) in yki3SA-gut tumors, creating a situation in which Jak/Stat signaling and tumor growth are sustained independent of extracellular Upd3, and then removed tumor-derived upd3 (Figure 2A). Consistently, compared with flies bearing either yki3SA or yki3SA+hopTumL gut tumors, specific upd3 knockdown in yki3SA+hopTumL tumors using two different RNAi lines no longer suppressed tumor growth (Figure 2B; Figure S2C). Strikingly, removal of upd3 in yki3SA+hopTumL tumors sufficiently alleviated the effects of systemic wasting, including the bloating phenotype, reduction in TAG levels, elevation of carbohydrate levels, and decreased climbing ability (Figures 2B–2D and 3A; Figures S2C and S2D). We note that removal of upd3 from yki3SA+hopTumL tumors does not affect the expression levels of ImpL2 or Pvf1, two previously identified tumor-derived cachectic ligands (Figure 2E).
Figure 2. Tumor-produced Upd3 causes bloating and muscle dysfunction.
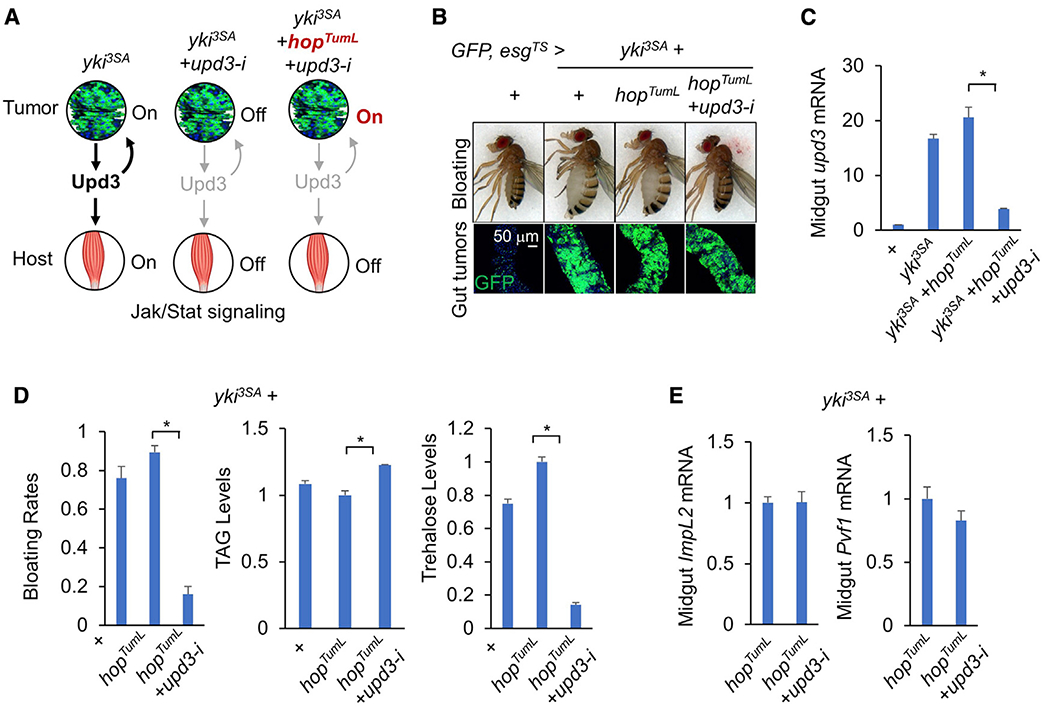
(A) Experimental strategy for uncoupling Jak/Stat signaling in the tumors and host organs.
(B–D) Wasting effects in flies bearing yki3SA+hopTumL tumors with upd3 RNAi (HMS00646) at day 6: (B) bloating phenotype (top) and gut tumors (bottom; scale bar: 50 μm), (C) midgut gene expression of upd3 (n = 3, 10 flies/replicate), and (D) bloating rates (n = 4, 20 flies/replicate) and TAG and trehalose levels (n = 3, 5 flies/replicate).
(E) Midgut gene expression of ImpL2 and Pvf1 (n = 3, 10 flies/replicate).
Data are presented as mean ± SEM. *p < 0.05.
Figure 3. Upd3/Jak/Stat signaling impairs muscle mitochondrial homeostasis.
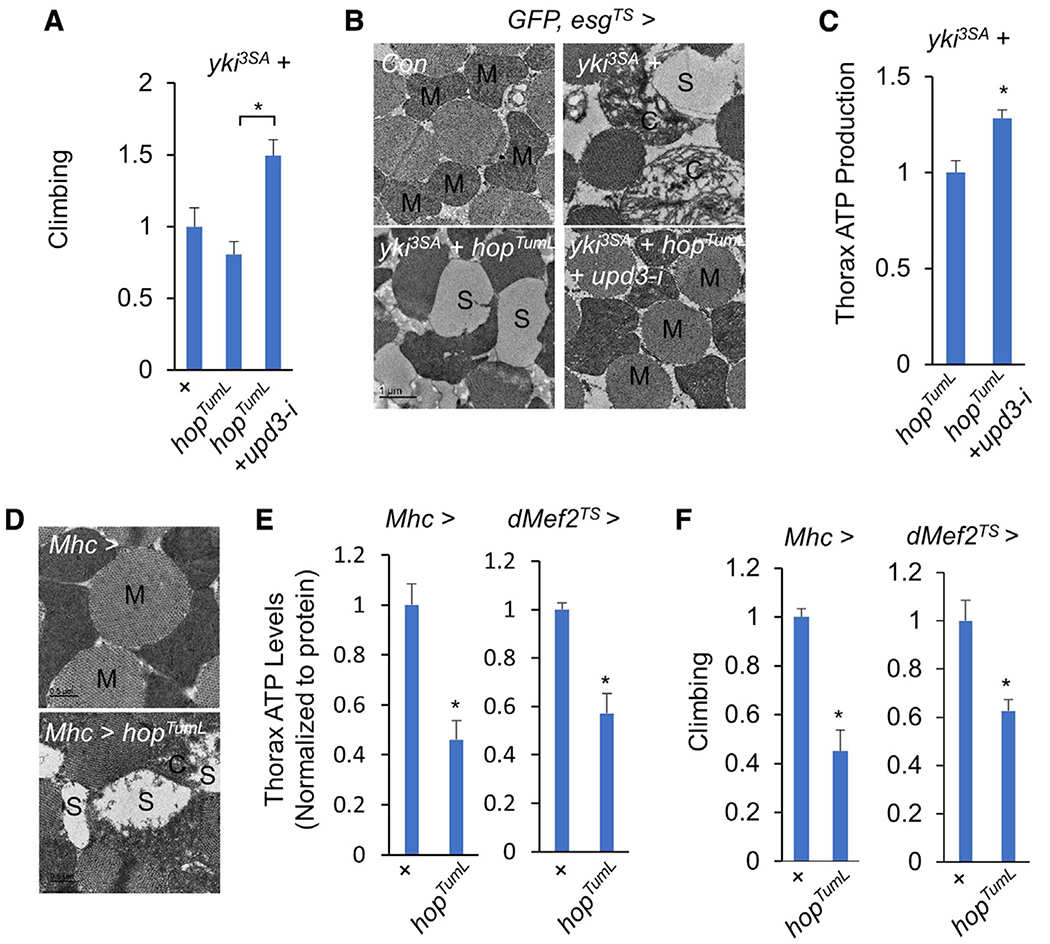
(A–C) Climbing rates (A; n = 15), muscle mitochondrial morphologies (scale bar: 1 μm) (B), and ATP production (C; n = 3, 5 flies/replicate) of flies bearing yki3SA+hopTumL tumors with or without upd3 RNAi (HMS00646) at day 6.
(D–F) Muscle mitochondrial morphologies including normal mitochondria (M) and injured mitochondria with fragmented cristae (C) or blank space (S) (D; scale bar: 0.5 μm), ATP production (E; n = 3, 5 flies/replicate), and climbing rates (F; n = 15) of flies with activation of Jak/Stat signaling in the muscle at day 4.
Data are presented as mean ± SEM. *p < 0.05.
We also asked whether inhibition of Upd3/Jak/Stat signaling in host organ tissues is sufficient to suppress tumor-induced wasting. To do this, we fed yki3SA-gut tumor flies methotrexate (MTX), a potent small-molecule inhibitor of Jak/Stat signaling (Thomas et al., 2015), simultaneously with yki3SA-tumor induction and found that treatment with 10 or 100 μM MTX strongly suppressed Jak/Stat signaling in both host organs and yki3SA-gut tumor and decreased tumor growth at day 8 of tumor induction (Figures S3A and S3B). In order to reduce the impact of the drug on tumors and investigate its roles in host organs, we constitutively enhanced Jak/Stat signaling in yki3SA tumors by inducing hopTumL and decreased the MTX dose. Interestingly, although treatment with 0.1 or 1 μM MTX rarely suppressed the growth of yki3SA+hopTumL tumors, it significantly reduced bloating and improved wasting effects, including climbing defects, lipid loss, and hyperglycemia (Figures S3C and S3D). These results collectively indicate that gut tumor-derived Upd3 acts remotely to induce Jak/Stat signaling in host organs, resulting in bloating/wasting.
Upd3/Jak/Stat signaling perturbs muscle mitochondrial homeostasis
As upd3 removal from yki3SA tumors significantly restored fly climbing rates (Figure 3A), we asked how tumor-derived Upd3 regulates muscle function. We first investigated muscle mitochondrial activity, which has been shown to be remotely impaired by yki3SA-gut tumors (Kwon et al., 2015). As expected, transmission electron microscopy (TEM) revealed normal mitochondrial integrity (M) in control muscles. In contrast, we observed swelling mitochondria with fragmented cristae (C) and low-density inner space (S) in the muscles of flies bearing yki3SA-tumors, suggesting a classic degenerative phenotype of mitochondria (Figure 3B). Flies bearing yki3SA+hopTumL tumors exhibited severer mitochondrial degeneration, as indicated by the absence of cristae and blank inner space (S) in the muscle (Figure 3B). Strikingly, upd3 removal in yki3SA+hopTumL tumors robustly eliminated mitochondrial degeneration and restored mitochondrial morphology, almost to control levels (Figure 3B). Moreover, upd3 knockdown in yki3SA+hopTumL tumors also increased muscle ATP production, another important indicator of mitochondrial dysfunction (Figure 3C).
We next wondered whether Upd3/Jak/Stat signaling directly impairs muscle function. To address this, we genetically activated Jak/Stat signaling in adult muscles via hopTumL overexpression driven by Mhc-GAL4. Interestingly, obvious morphological changes indicative of mitochondrial degeneration was observed in adult muscle (Figure 3D). Muscle ATP production was also decreased and fly climbing ability was impaired (Figures 3E and 3F). These results were further confirmed using another temperature-sensitive muscle driver tub-GAL80TS; dMef2-GAL4 (dMef2TS) (Figures 3E and 3F). Taken together, our results indicate that the Upd3/Jak/Stat cascade mediates tumor-induced muscle mitochondrial degeneration and muscle dysfunction.
Excessive ubiquitin-associated protein degradation has also been associated with tumor-induced muscle dysfunction with (Acharyya and Guttridge, 2007). In line with this observation, we observed an increase in protein ubiquitination in the muscles of yki3SA flies (Figure S4A). However, neither upd3 knockdown in the yki3SA+hopTumL tumors nor activation of Jak/Stat signaling in the wild-type adult muscle significantly affected ubiquitination of muscle proteins (Figure S4B). The expression level of CG11658, which encodes a fly homolog of Atrogin-1 E3 ubiquitin ligase, was significantly increased in the muscle of yki3SA flies but not in hopTumL-overexpressing muscle (Figure S4C), indicating that Upd3/Jak/Stat signaling does not increase ubiquitination of muscle proteins in Drosophila.
Upd3/Jak/Stat cascade impairs muscle insulin response
Insulin signaling has been shown to regulate mitochondrial homeostasis and muscle function across species (Del Campo et al., 2016). We therefore examined whether tumor-derived Upd3 remotely affects muscle through attenuation of the insulin response. We knocked down upd3 expression in yki3SA+hopTumL gut tumors and as expected, we found a significant decrease in the expression level of the Jak/Stat pathway target gene Socs36E in the fly muscle (Figure 4A). Interestingly, expression levels in the muscle of 4EBP and InR, two target genes negatively regulated by insulin signaling, were also significantly decreased (Figure 4A). Akt phosphorylation (pAkt), an important positive readout of insulin signaling, was increased in the fly muscle (Figure 4B). These results collectively indicate that upd3 removal in yki3SA+hopTumL gut tumors remotely enhances muscle insulin signaling.
Figure 4. Upd3/Jak/Stat signaling impairs muscle insulin responses.
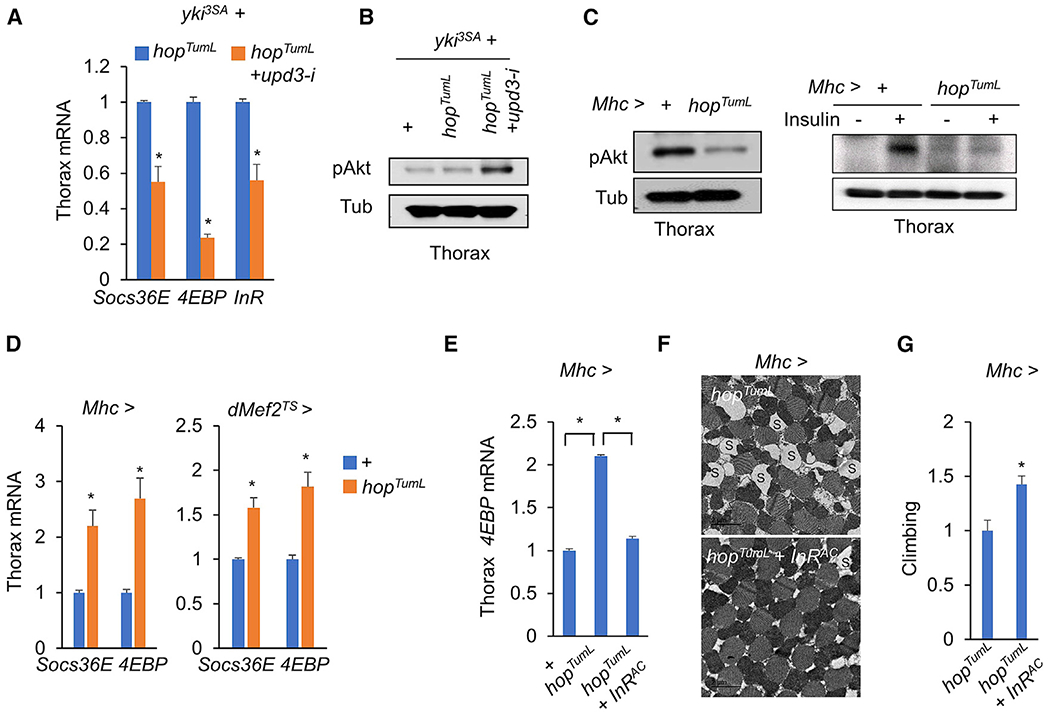
(A and B) Insulin signaling, as indicated by target gene expression (A; n = 3,5 flies/replicate) or p-Akt (B), in thethoraces of flies bearing yki3SA-gut tumors with or without upd3 RNAi (HMS00646) at day 6.
(C and D) Insulin response, as indicated by p-Akt (C; left, freshly isolated thoraces; right, isolated thoraces that were treated with 1 μg/mL insulin for 30 min) or target gene expression (D; n = 3, 5 flies/replicate), in adult thoraces with activation of Jak/Stat signaling at day 4.
(E–G) Muscle gene expression (E; n = 3, 5 flies/replicate), muscle mitochondrial morphology including injured mitochondria with blank space (S) (scale bar: 2 μm)
(F), and climbing rates (G; n = 15) of adult flies with activation of Jak/Stat and insulin signaling in the muscle at day 4.
Data are presented as mean ± SEM. *p < 0.05.
To investigate whether Jak/Stat signaling directly attenuates insulin signaling, we specifically expressed hopTumL in the muscle to activate Jak/Stat signaling and observed a significant decrease in pAkt (Figure 4C, left). This result was further validated by ex vivo assays in which adult thoracic muscles were isolated and treated with recombinant human insulin. Compared with control thoraces that exhibit a robust increase in insulin-stimulated pAkt, hopTumL overexpression in thoraces potently blunted this effect (Figure 4C, right). Consistent with pAkt changes, hopTumL overexpression increased muscle expression of Socs36E, as well as 4EBP (Figure 4D, left). Genetic manipulation using the temperature-sensitive muscle driver dMef27S also obtained similar results (Figure 4D, right), suggesting that Jak/Stat signaling autonomously suppresses insulin response in the muscle.
To investigate whether Jak/Stat signaling impairs muscle functions through attenuation of insulin response, we manually restored the insulin response by overexpressing a constitutively active form of the InR (InRAC) in the muscle in the context of hopTumL. As expected, 4EBP expression in the muscle was dramatically suppressed (Figure 4E), indicating an increase of insulin signaling. Moreover, muscle mitochondrial degeneration/swelling and fly climbing defects associated with hopTumL were also remarkably diminished (Figures 4F and 4G). Therefore, our results collectively demonstrate that production of Upd3 by yki3SA-gut tumors remotely activates Jak/Stat signaling and impairs insulin response in muscles, causing mitochondrial degeneration and muscle dysfunction.
Upd3/Jak/Stat cascade impairs insulin signaling and carbolipid metabolism in the fat body
Our results have demonstrated that tumor-derived Upd3 not only affects muscles but also leads to systemic lipid loss and carbohydrate elevation (Figure 2D), two additional major features of wasting. To investigate whether Upd3/Jak/Stat cascade directly affects the function of the fat body, the major metabolic organ in Drosophila, we activated Jak/Stat signaling specifically in the fat body. hopTumL overexpression in wild-type adult fat body using the temperature-sensitive driver Cg-GAL4, tub-GAL80TS (CgTS) for 4 days robustly increased Socs36E mRNA levels in abdomens that contain large amounts of fat body cells (Figure S5A, left). Interestingly, fat-body hopTumL overexpression significantly decreased TAG storage (Figure S5A, right). The abdominal fat layer potently disappeared in CgTS > hopTumL flies, so their abdomens appeared more translucent (Figure S5B, top). BODIPY staining also revealed that the lipid droplet mass in the fat body was dramatically reduced in CgTS > hopTumL flies (Figure S5B, bottom). Fat body hopTumL overexpression using another driver, tub-GAL80TS; Lpp-GAL4 (LppTS), phenocopied systemic lipid loss in the abdomen (Figure S5C). In addition to lipid loss, fat-body hopTumL overexpression also resulted in an elevated level of trehalose, the predominant insect circulating carbohydrate composed of two α-glucose, in CgTS > hopTumL flies (Figure S5A, right). Again, the metabolic imbalance was associated with impairment of the insulin response, which predominantly controls carbolipid metabolism in the fat body, as hopTumL overexpression significantly elevated 4EBP mRNA levels and decreased pAkt in the abdomen (Figure S5A, left; Figure S5D).
We have demonstrated that yki3SA-gut tumors also secrete ImpL2, leading to wasting via antagonization of systemic insulin signaling (Kwon et al., 2015). As upd3 removal in yki3SA+hopTumL gut tumors does not affect ImpL2 expression, we wondered whether tumor-derived ImpL2 and Upd3 are independent regulators. We knocked down both ImpL2 and upd3 in yki3SA+hopTumL gut tumors to examine potential crosstalk. Interestingly, compared with knockdown of either upd3 or ImpL2, knockdown of both ligands in yki3SA+hopTumL tumors further alleviated wasting effects, including bloating rates and hyperglycemia, without affecting tumor growth (Figures S6A–S6D). Lipid loss was also improved but not statistically significantly so (Figures S6A–S6D). Our results demonstrate that tumor-derived Upd3 and ImpL2 exhibit, at least, differential regulation of host wasting.
Upd3/Jak/Stat signaling induces ImpL2 expression in the host organ
We next dissected the molecular mechanisms by which Jak/Stat signaling impairs the insulin response. To do this, we knocked down expression of Stat92E, the transcriptional factor of Jak/Stat signaling, in the context of hopTumL overexpression in the muscle and, interestingly, observed an enhancement in pAkt levels and fly climbing ability (Figures S4D and S4E). The results suggest that Jak/Stat signaling might hamper insulin responses via Stat92E-dependent transcriptional regulation. Thus, we searched for potential Stat92E targets with the following three criteria: (1) known negative regulators of insulin signaling, (2) putative Stat-binding sites in the promoter regions, and (3) expression highly associated with Upd3/Jak/Stat signaling in the muscle. Our bioinformatic analysis identified ImpL2 as a top candidate with four putative Stat-binding sites in its promoter region (BS1, BS2, BS3, and BS4) (Figure 5A). We further transfected Stat92E and HopTumL together with BS1–4 luciferase constructs in S2R+ cells and found that BS2 luciferase activity was potently increased under these conditions (Figure 5B). We further mutated the two Stat-binding sites in BS2 region and, as expected, observed that BS2 luciferase activity was markedly blunted (Figure 5C). Chromatin immunoprecipitation (ChIP) assays also indicated that Stat92E specifically binds to the BS2 promoter region of ImpL2 (Figure 5D). There results demonstrate that Stat92E is a major regulator of ImpL2 expression that acts via a binding site in the BS2 region.
Figure 5. Upd3/Jak/Stat signaling promotes muscle ImpL2 expression.
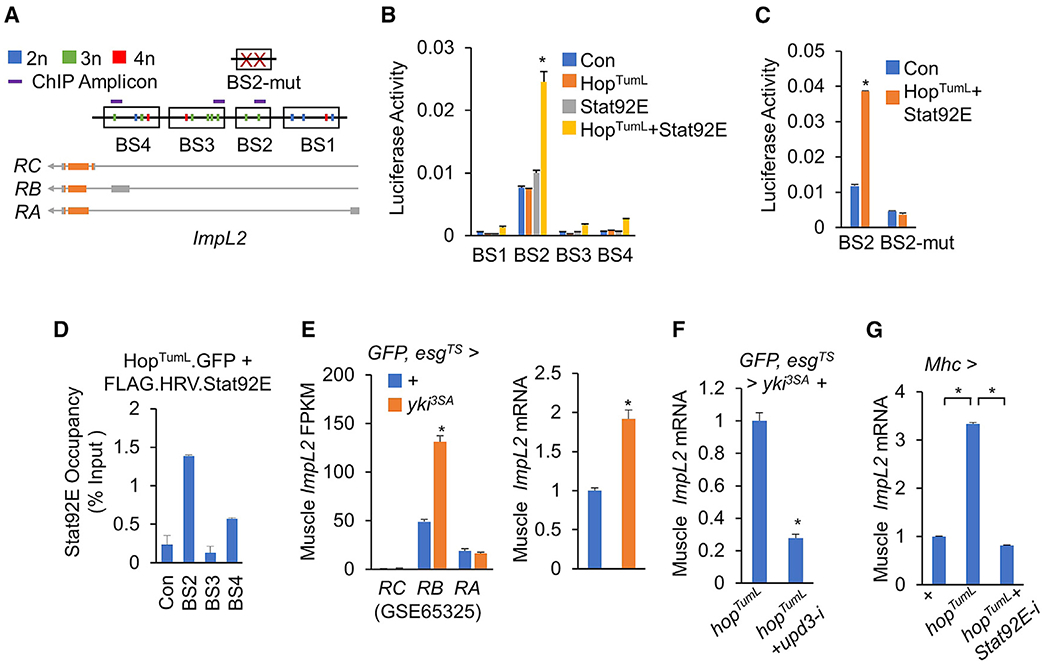
(A) Regions that contain putative Stat92E-binding sites (BS1-4) (TTCNNGAA, 2n, blue; TTCNNNGAA, 3n, green; TTCNNNNGAA, 4n, red) in the ImpL2 gene, BS2 with two mutated Stat-binding sites (BS2-mut), and ChIP amplicons are shown.
(B and C) Relative luciferase activities of the indicated vectors that contain normal (B; BS1–4) and mutated (C; BS2-mut) Stat92E binding sites with Stat92E and/or HopTumL expression in S2R+ cells (n = 3).
(D) The S2R+ cells were transfected with indicated vector were lysed and pulled down with anti-FLAG antibody to perform in vitro ChIP assays. Stat92E occupancies at promoter regions of ImpL2 as indicated relative to input were analyzed using qPCR. All ChIP-qPCR experiments were performed in duplicate.
(E) Expression levels of ImpL2 in thoraces of flies bearing yki3SA-gut tumors at day 8 from RNA-seq analysis (left; GSE65325, NCBI-GEO) and qPCR (right; n = 3, 5 flies/replicate).
(F and G) Muscle ImpL2 expression levels of flies bearing tumors with or without upd3 RNAi (HMS00646) at day 6 (F) or flies bearing hopTumL overexpression with or without Stat92E RNAi in the muscle at day 6 (G) (n = 3, 5 flies/replicate).
Data are presented as mean ± SEM. *p < 0.05.
We next asked if Upd3/Jak/Stat signaling regulates ImpL2 expression in vivo in the host organ. Our RNA-seq data (GSE65325) for yki3SA flies showed a significant increase in ImpL2 mRNA levels in the muscle (Figure 5E, left), a finding that correlates with upregulation of upd3 production in yki3SA-gut tumors and Jak/Stat signaling in the matched muscle (Figure 1). The result was also confirmed using qPCR (Figure 5E, right). Conversely, we specifically knocked down upd3 expression in yki3SA+hopTumL gut tumors, which significantly suppressed Jak/Stat signaling in the muscle (Figure 4A), and found that muscle ImpL2 mRNA levels were dramatically decreased (Figure 5F). In order to study the autonomous regulation of Jak/Stat signaling on ImpL2 expression, we activated Jak/Stat signaling by overexpressing hopTumL in the muscle and observed a 3-fold induction of ImpL2 mRNA levels in a Stat92E-dependent manner (Figure 5G). Thus, our data indicate that Upd3/Jak/Stat signaling transcriptionally regulates ImpL2 expression in the muscle.
Upd3/Jak/Stat signaling causes host wasting in part via autonomous ImpL2 expression
To examine whether Jak/Stat signaling impairs insulin response and muscle functions via autonomous ImpL2 production, we decreased ImpL2 expression in the context of muscle hopTumL overexpression. As expected, ImpL2 RNAi in hopTumL-overex pressing muscle dramatically decreased ImpL2 mRNA level and also suppressed 4EBP expression (Figure 6A), which reflects a restoration of the muscle insulin response. Furthermore, hopTumL-associated mitochondrial degeneration in the muscle and fly climbing defects were both potently improved by ImpL2 RNAi (Figures 6B and 6C).
Figure 6. Jak/Stat signaling promotes organ wasting via ImpL2.
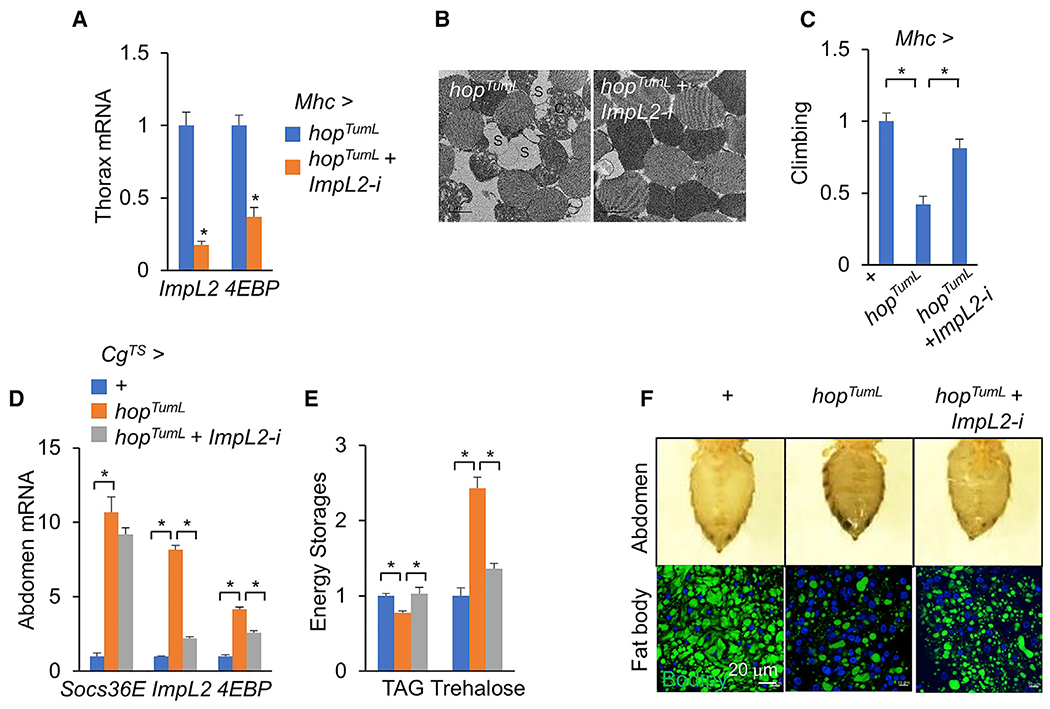
(A–C) Thorax gene expression (A; n = 3, 5 flies/replicate), muscle mitochondrial morphology including injured mitochondria with blank space (S) (scale bar: 1 μm) (B), and climbing rates (C), n = 15) of flies with activation of Jak/Stat signaling plus ImpL2 RNAi (NIG 15009R-3) in the muscle at day 6.
(D–F) Gene expression in abdomens (D; n = 3, 5 flies/replicate), whole-body TAG and trehalose levels (E; n = 3, 5 flies/replicate), abdomen appearance (F, top), and BODIPY that labels lipid droplets in the abdominal fat body (F, bottom; green, BODIPY; blue, DAPI; scale bar: 20 μm) of flies with activation of Jak/Stat signaling plus ImpL2 RNAi (HMC05809) in the fat body at day 4
Data are presented as mean ± SEM. *p < 0.05.
Similar ImpL2 regulation by Jak/Stat signaling was also observed in the fat body. Overexpression of hopTumL in the fat body resulted in an increase in ImpL2 and 4EBP mRNA levels, systemic TAG decline, and hyperglycemia (Figures 6D–6F), whereas ImpL2 RNAi in the context of hopTumL overexpression in the fat body diminished the effects associated with hopTumL (Figures 6D–6F). Taken together, our results demonstrate that Upd3/Jak/Stat signaling increases autonomous ImpL2 production in the muscle and fat body to impair the insulin response and its associated energy balance.
Upd3 overexpression alone in ISCs sufficiently results in both tumor growth and host wasting
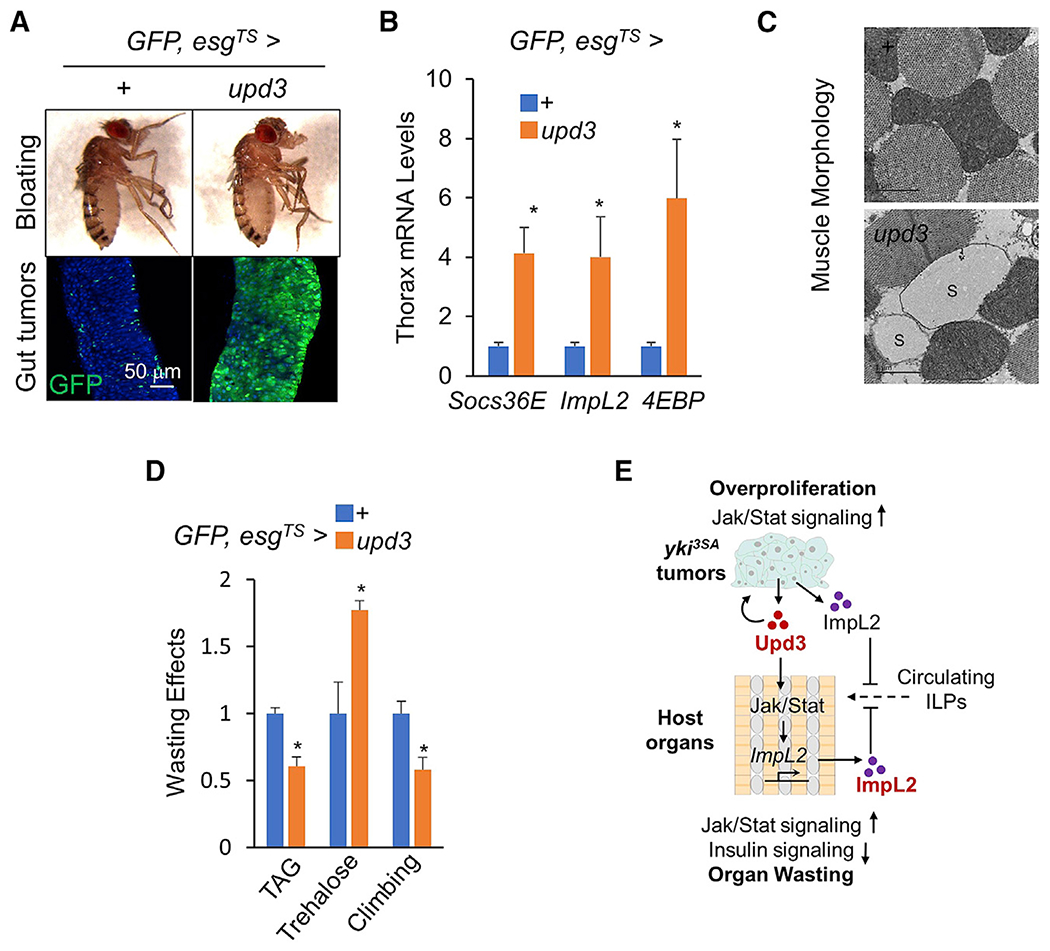
Finally, we asked whether Upd3 production alone in the gut is sufficient to cause host wasting. We overexpressed upd3 in wild-type adult enterocytes (ECs) for 8 days and observed only mild ISC overproliferation (Figure S7A). However, the Socs36E and 4EBP expression levels in the muscle were significantly increased (Figure S7B). EC-derived Upd3 production triggered host wasting, including lipid loss, carbohydrate elevation, and climbing defects (Figure S7C).
Furthermore, we overexpressed upd3 in wild-type adult ISCs and observed the dramatic overgrowth of the GFP-labeled tumor cell population, expanding to the whole gut by 8 days (Figure 7A). As expected, upd3-induced gut tumors increased the expression levels of Socs36E, ImpL2, and 4EBP in the muscle and triggered wasting effects, including muscle wasting, lipid loss, and carbohydrate elevation (Figures 7B–7D). However, we noted that Upd3 overexpression in either ECs or ISCs failed to trigger abdomen bloating and caused wasting effects not as severe as that observed with yki3SA-induced gut tumors (Figures 7A and S7A). Taken together, our results indicate that Upd3 production alone in the gut sufficiently results in host wasting.
Figure 7. Upd3 overexpression in the ISCs causes tumor formation and host wasting.
(A–D) Fly appearance (A, top), gut tumors (A bottom; scale bar: 50 μm), thoracic muscle gene expression (B; n = 3, 5 flies/replicate), and wasting effects such as muscle mitochondrial morphology including injured mitochondria with blank space (S) (C; scale bar: 1 μm), TAG and Trehalose storages (D; n = 3, 5 flies/replicate), and climbing rates (D; n = 15) of adult flies with upd3 overexpression in the ISCs at day 8. Data are presented as mean ± SEM. *p < 0.05
(E) Tumor-derived Upd3 coordinates tumor growth and host wasting via Jak/Stat signaling.
As Upd3/Jak/Stat signaling is required for ISC proliferation, we knocked down Stat92E or dome in the context of upd3 overexpression in the ISCs and found that Upd3-induced tumor growth, host Jak/Stat activation, and host organ wasting were all remarkably suppressed (Figures S7D–S7F), suggesting that the mass of Upd3-producing cells is also important for host wasting regulation. We previously identified Pvf1 as an important tumor-derived cachectic ligand and wondered whether Pvf1 would synergize with Upd3 for host wasting. We overexpressed Pvf1 in the wild-type adult ISCs and, consistent with previous studies (Bond and Foley, 2012), observed only mild ISC overproliferation (Figure S7G). Unexpectedly, we found that Pvf1 overexpression together with Upd3 antagonized Upd3-associated gut tumor formation and host wasting (Figure S7H), suggesting an unknown molecular mechanism(s) between Upd3 and Pvf1 regarding tumor-growth regulation in the gut.
DISCUSSION
The molecular mechanisms whereby malignant tumors coordinate self-growth and host organ wasting are not fully understood. In this study, which combined bioinformatics analysis and genetic validation, we identified tumor-derived Upd3 as an important mediator of tumor-host interaction. We demonstrate that tumor-derived Upd3 simultaneously activates Jak/Stat signaling in gut tumor cells, promoting overgrowth of the tumor itself and, in host organs, causing energy imbalance and wasting.
PathON for evaluating signaling pathways involved in interorgan communication
We have demonstrated that monitoring signature target genes to evaluate the activity of various signaling pathways in a receiving tissue is helpful to comprehensively understand crosstalk networks during interorgan communication (Song et al., 2017b). One potential complication of this approach is that different pathways might share a set of common target genes. For example, both insulin and TNF-α signaling directly regulate puc expression (Bai et al., 2013; McEwen and Peifer, 2005). Moreover, the targets of a given pathway might also vary in different organs or at different developmental stages. Thus, we decided to evaluate enrichment of all potential target genes rather than single ones, and we developed the web-based analysis tool PathON to facilitate this approach. Using PathON, we successfully uncovered Upd3/Jak/Stat signaling pathway as an important mediator in the tumor-host crosstalk. It will be interesting to determine whether other pathways identified using PathON, such as the BMP and TNF-α signaling pathways, also play important roles in tumor-induced host wasting. In addition, we note that because we hypothesized that tumor-host interactions were mediated by signaling ligands, we focused on 14 canonical ligand-induced signaling pathways. Target genes of other pathways and transcriptional factors, such as hypoxia/HIF and circadian clock/Period, could be worth adding to PathON in the future to facilitate assessment of a more diverse set of biological processes.
Upd3 coordinates tumor growth and host wasting
How malignant tumors coordinate selfgrowth and host wasting is not fully understood. We have previously shown that yki3SA-gut tumors produce ImpL2 to suppress systemic insulin availability and inhibit host anabolism (Kwon et al., 2015). We also found that yki3SA-gut tumors constitutively activate intracellular insulin signaling, independent of circulating insulin-like peptides (ILPs), to overcome ImpL2-associated insulin insufficiency and growth restraint (Kwon et al., 2015; Lee et al., 2021). Moreover, the yki3SA tumors also produce Vein (Vn) and Pvf1, which promote tumor growth and host wasting, respectively, through differential MEK activation in tumors and host organs (Song et al., 2019). Despite these various levels of regulation, here we document that Upd3 acts as a single tumor-derived ligand that simultaneously affects tumor growth and host wasting.
Upd3 is sufficient to mediate yki3SA-gut tumor growth via activation of local Jak/Stat signaling in a paracrine/autocrine fashion, as many studies in fly ISCs models have indicated (Amoyel et al., 2014; Jiang et al., 2009; Katheder et al., 2017; Markstein et al., 2014; Ren et al., 2010; Shaw et al., 2010; Staley and Irvine, 2010). Meanwhile, the well-expanded yki3SA tumors produce a large amount of Upd3, which has been characterized as a hormone (Huang et al., 2020; Woodcock et al., 2015), to activate Jak/Stat signaling in host organs and contributes to host wasting, including muscle dysfunction, lipid loss, and hyperglycemia. Consistent with this model, overexpression of Upd3 alone in ISCs is sufficient to cause both gut tumors and host wasting. Because Yki has been shown to bind to the upd3 promoter together with Sd to upregulate upd3 transcription (Houtz et al., 2017), upd3 induction in gut tumors is most likely directly regulated by Yki/Sd.
Upd3 has been considered a homolog of mammalian interleukins (ILs), which canonically activate Jak/Stat signaling and are highly associated with cancer cachexia and muscle wasting (Bonetto et al., 2012; Flint et al., 2016; Laird et al., 2021; Strass-mann et al., 1992). However, ILs are produced mostly in host organs, and the effects on muscle atrophy of different ILs, such as IL-1, IL-6, IL-8, and IL-12, remain controversial (Aoyagi et al., 2015), probably because of functional compensation and context-dependent differences. Using Drosophila as a conserved model organism with less gene redundancy, we demonstrate that tumor-derived Upd3 is sufficient and necessary to trigger host Jak/Stat signaling and cause wasting.
Upd3/Jak/Stat signaling impairs muscle function via mitochondrial degeneration rather than enhanced protein ubiquitination
The role of Upd3/Jak/Stat signaling in the Drosophila muscle is largely unknown. Mammalian Jak/Stat signaling has been shown to trigger muscle atrophy primarily through increased expression of E3 ubiquitin ligases (Atrogin-1 and MuRF1) and enhanced protein ubiquitination to promote protein degradation (Sala and Sacco, 2016). However, in adult fly muscles, neither tumor-derived Upd3 nor autonomous Jak/Stat signaling increases protein ubiquitination. Moreover, Jak/Stat signaling in fly muscle also fails to increase the expression of CG11658, the homolog of Atrogin-1, further suggesting differences in regulation of Jak/Stat signaling in Drosophila compared with mammals. In this study, we showed that Upd3/Jak/Stat signaling in the muscle is sufficient to cause mitochondrial degeneration and leads to muscle dysfunction. The molecular mechanisms involve, at least, an impaired muscle insulin response. Thus, our study provides a novel understanding of Jak/Stat cascade in muscle physiology in Drosophila that could be a general biological process across species.
Besides tumor-derived ImpL2 and Upd3 that antagonize host insulin signaling to perturb muscle mitochondrial activity, we have previously shown tumor-derived Pvf1 as another modulator of muscle function via activating Pvr/MEK signaling to impair protein synthesis/breakdown balance (Kwon et al., 2015; Song et al., 2019). Taken together the fact that removal of ImpL2, Pvf1 (Kwon et al., 2015; Song et al., 2019), or upd3 in the gut tumors potently but not completely diminishes host wasting, we speculate that these tumor-derived cachectic ligands synergize to regulate host energy homeostasis. In the future, it will be interesting to further investigate the crosstalk and differential effects of these ligands in the host organs.
Upd3/Jak/Stat cascade inhibits muscle insulin signaling via cell-autonomous ImpL2 induction
Several studies have indicated that activation of the Upd3/Jak/Stat cascade hampers the insulin response in various tissues (Kierdorf et al., 2020; Shin et al., 2020), although the molecular mechanisms underlying this observation are not well understood. In this study, we made the striking observation that Upd3/Jak/Stat signaling in host organs induces production of ImpL2, a well-known antagonist of both fly ILPs and human insulin/IGF (Sloth Andersen et al., 2000). This blocks extracellular ILPs and decreases the intracellular insulin response, resulting in mitochondrial degeneration and lipid loss. The results of our in vitro luciferase assays further indicate that this occurs via Stat92E-dependent transcriptional regulation of ImpL2. Interestingly, our conclusion is also supported by the results of an earlier study revealing that expression of ImpL2 is highly associated with expression of Socs36E and Upd genes in both testis and embryo (Terry et al., 2006). Mammalian Jak/Stat signaling has been reported to attenuate insulin signaling via SOCSs to degrade the insulin receptor and blunt phosphorylation of its downstream regulators (Jorgensen et al., 2013). Drosophila Socs36E functions as an important homolog of mammalian SOCSs and a negative regulator of multiple signaling pathways, including EGFR signaling, by degrading key components of these pathways (Amoyel et al., 2016; Stec et al., 2013). Thus, Socs36E might act together with ImpL2 to decrease intracellular insulin signaling.
Tumor-induced impairment of host insulin signaling is essential for systemic wasting. In our previous studies, we proposed that tumor-derived ImpL2 restrains the bioavailability of circulating ILPs and decreases muscle insulin responses (Figueroa-Clarevega and Bilder, 2015; Kwon et al., 2015). However, specific ImpL2 removal in yki3SA-gut tumors only moderately restores muscle insulin response compared with ImpL2-null mutation (Kwon et al., 2015), indicating that host organs also produce functional ImpL2 in addition to yki3SA-gut tumors. Here, we show that ImpL2 production in the host organs, which is remotely induced by tumor-derived Upd3, also contributes to the impairment of insulin response and host wasting (Figure 7E).
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Wei Song (songw@whu.edu.cn).
Materials availability
All stable reagents generated in this study are available from the lead contact without restriction.
Data and code availability
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-phosphor-Akt (Ser473) | Cell Signaling | Cat#4060, RRID:AB_2315049 |
| Mouse anti-α-tubulin | Sigma | Cat#T5168,RRID:AB_477579 |
| Mouse anti-polyubiquitin (FK2) | Enzo | Cat#BML-PW8810,RRID:AB_10541840 |
| Mouse anti-β-Gal | Promega | Cat#Z3781,RRID:AB_430877 |
| Goat Anti-Mouse IgG (H+L), HRP | ABclonal | Cat#AS003, RRID:AB_2769851 |
| Goat Anti-Rabbit IgG (H+L), HRP | ABclonal | Cat#AS014, RRID:AB_2769854 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 594 | Thermo Fisher Scientific | Cat#A32742, RRID:AB_2762825 |
| Rabbit monoclonal anti-FLAG | Genscript | Cat#A01868 |
| Bacterial and virus strains | ||
| DH5α Competent E.coli Strain | Vazyme | Cat#C502 |
| Chemicals, peptides, and recombinant proteins | ||
| Bradford Reagent | Sigma | Cat#B6916-500ML |
| Glycerol standard | Sigma | Cat#G7793-5ML |
| D-(+)-Glucose | Sigma | Cat#G7021 |
| Trehalase | Sigma | Cat#E-TREH |
| Glucose assay reagent | Megazyme | Cat#K-Gluc |
| Triglyceride reagent | Sigma | Cat#T2449-10ML |
| Free glycerol reagent | Sigma | Cat#F6428-40ML |
| Glycerol | Sigma | Cat#G7793 |
| Trizol | Thermo Fisher Scientific | Cat#15596018 |
| HiScript II Q RT Supermix | Vazyme | Cat#R222-01 |
| SYBR qPCR Master Mix | Vazyme | Cat#Q311-03 |
| DAPI | Thermo Fisher Scientific | Cat#D1306 |
| Bodipy 493/503 | Thermo Fisher Scientific | Cat#D3922 |
| Phalloidin | Thermo Fisher Scientific | Cat#A12381, |
| Insulin | Sigma | Cat#I6634 |
| Methotrexate | Selleck | Cat#S1210 |
| Critical commercial assays | ||
| CellTiter-LumiTM Plus kit | Beyotime | Cat#C0068M |
| Trehalose Assay Kit | Megazyme | Cat#K-Gluc |
| Triglyceride Assay Kit | Sigma | Cat#T2449 |
| Effectene reagent | QIAGEN | Cat#301425 |
| Pierce Protein A/G Magnetic Beads | Thermo Fisher Scientific | Cat#88803 |
| ClonExpress MultiS One Step Cloning Kit | Vazyme | Cat#C113-01 |
| HRV 3C Protease | TaKaRa | Cat#7360-1 |
| Dual-Luciferase Reporter assay kit | Promega | Cat#E1910 |
| Deposited data | ||
| RNaseq data from fly midgut | (Kwon et al., 2015) | GEO: GSE113728 |
| RNaseq data from fly muscle | (Song et al., 2019) | GEO: GSE65325 |
| Experimental models: Cell lines | ||
| D. melanogaster: Cell line S2R+ | Laboratory of Norbert Perrimon, Harvard Medical School | N/A |
| D.melanogaster: esg-GAL4, tub-GAL80TS, UAS-GFP | (Kwon et al., 2015) | N/A |
| D.melanogaster: Myo1A-GAL4, esg-GAL4, tub-GAL80TS | (Song et al., 2019) | N/A |
| D.melanogaster: CG-GAL4, tub-GAL80TS | (Song et al., 2017b) | N/A |
| D.melanogaster: tub-GAL80TS, Lpp-GAL4 | (Song et al., 2017b) | N/A |
| D.melanogaster: tub-GAL80TS, dMef2-GAL4 | (Owusu-Ansah et al., 2013) | N/A |
| D.melanogaster: tub-GAL80TS, Mhc-GAL4 | (Owusu-Ansah et al., 2013) | N/A |
| D.melanogaster: 10XStat-GFP | (Bach et al., 2007) | N/A |
| D.melanogaster: UAS-Pvf1 | (Song et al., 2019) | N/A |
| D.melanogaster: UAS-hopTumL | A gift from Dr. Erika Bach, NYU Langone Health | N/A |
| D.melanogaster: UAS-upd3 | A gift from Dr. Bruce Edgar, University of Utah | N/A |
| D.melanogaster: upd3-lacZ | A gift from Dr. Bruce Edgar, University of Utah | N/A |
| D.melanogaster: FRT19A | A gift from Dr. Zheng Guo, Huazhong University of Science and Technology | N/A |
| D.melanogaster: yw, hs-FLP, tub-GAL80, FRT19A; UAS-GFP | A gift from Dr. Zheng Guo, Huazhong University of Science and Technology | N/A |
| D.melanogaster: UAS-Stat92E-RNAi | A gift from Drs. Xinhua Lin and Yun Qi, Fudan University | N/A |
| D.melanogaster: UAS-upd3-RNAi1 | TRiP at Harvard Medical School | HMS000646 |
| D.melanogaster: UAS-upd3-RNAi2 | TRiP at Harvard Medical School | HMS05061 |
| D.melanogaster: UAS-hop-RNAi | TRiP at Harvard Medical School | HMS00761 |
| D.melanogaster: UAS-Stat92E-RNAi | TRiP at Harvard Medical School | HMS00035 |
| D.melanogaster: UAS-ImpL2-RNAi | TRiP at Harvard Medical School | HMS05891 |
| D.melanogaster: UAS-w-RNAi | TRiP at Harvard Medical School | HMS01545 |
| D.melanogaster: w1118 | Bloomington Stock Center | BDSC_3605 |
| D.melanogaster: upd3 Δ | Bloomington Stock Center | BDSC_55728 |
| D.melanogaster: USA-yki3SA | Bloomington Stock Center | BDSC_28817 |
| D.melanogaster: USA-InRAC | Bloomington Stock Center | BDSC_8248 |
| D.melanogaster: UAS-ImpL2-RNAi | National Institute of Genetics | 15009R-3 |
| D.melanogaster: UAS-dome-RNAi | Vienna Drosophila Resource Center | v19717 |
| Oligonucleotides | ||
| See Table S4 for oligonucleotide information | N/A | |
| Recombinant DNA | ||
| pAc5.1 | Thermo Fisher Scientific | Cat#V411020 |
| pGL3-Basic | Promega | Cat#E1751 |
| pAc5-Stat92E-RB | This paper | N/A |
| pAc5-hopTumL | This paper | N/A |
| pGL3-hsp70 | This paper | N/A |
| pGL3-ImpL2-BS1 | This paper | N/A |
| pGL3-ImpL2-BS2 | This paper | N/A |
| pGL3-ImpL2-BS3 | This paper | N/A |
| pGL3-ImpL2-BS4 | This paper | N/A |
| pGL3-ImpL2-BS2-mut | This paper | N/A |
| pAc-HRV.FLAG.Stat92E.RB-T2A-hopTumLGFP | This paper | N/A |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
| Microsoft Excel | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| Other | ||
| QSonica sonicator | QSonica | #Q125 system |
| Multi-sample tissuelyser-24 | Shanghai Jingxin Technology | N/A |
| Nikon SMZ18 | Nikon | N/A |
| Nikon Eclipse Ts2 | Nikon | N/A |
| Zeiss LSM880 | Zeiss | N/A |
| CFX96 Real-Time System/C1000 Thermal Cycler | Bio-Rad | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fly strains
All flies, stocks and crosses, were grown under standard laboratory conditions (25°C, 12:12 h light/dark). to induce gut tumors, we followed the experimental procedures described previously (Song et al., 2019). Briefly, different UAS insertions were crossed to esg-GAL4, tub-GAL80TS, UAS-GFP at 18°Cto inactivate GAL4, thus restricting the expression of the Gal4-induced transgenes. 4-day-old adult progenies were collected and placed at 29°C to induce the transgenes (day 0 for tumor induction). Progenies from a cross between esg-GAL4, tub-GAL80TS, UAS-GFP and w1118 or UAS-w-RNAi were used as controls. During incubation at 29°C, flies were transferred onto fresh food every 2 days.
For MARCM assays, we generated the progenies with indicated genotypes at 25°C. The 4-day old virgin adults were heat-shocked at 37°C for 30 min twice to induce gene expression and maintained back at 25°C for 6 days prior to midgut dissection and analysis.
METHOD DETAILS
Design and development of PathON
All ligands, signaling components (receptors, adaptor proteins, kinases and phosphatases, and transcriptional factors), and signature target genes for 14 signaling pathways were annotated from the published literature. Signature target genes for these pathways were selected only if they had been previously validated by more than two of the following criteria: physiological function, gene expression, promoter activity, or direct binding to transcriptional factor(s).
In many cases, ligands-induced signaling eventually leads to post-translational changes, like phosphorylation and ubiquitination, of signaling components and transcriptional changes of target genes. We integrated two RNA-seq datasets and focused on transcriptional expression of ligands in the yki3SA-tumors and signature target genes in the muscle to evaluate the tumor-muscle signal crosstalk in this study. Because expression of signaling components and signature target genes is more context-dependent (e.g., different stages and tissues) than ligands, we evaluate them as a pool using Chi-square enrichment test. We also note that most of signature target genes for the PvR, EGFR, and FGFR signaling pathways overlap, and thus we grouped them together as targets of EGFR/FGFR/PvR signaling.
Lipid and carbohydrate measurements in flies
We measured fly TAG and carbohydrates as described previously (Song et al., 2017a; Song et al., 2017b; Song et al., 2010; Song et al., 2014). Briefly, 10 flies from each group were homogenized with 1 mL PBS containing 0.2% Triton X-100 using Multi-sample tissuelyser-24 (Shanghai Jingxin Technology) and heated at 70°C for 5 min. The supernatant was collected after centrifugation at 12,000 X g for 10 min at 4°C. 10 μL of supernatant was used for protein quantification using Bradford Reagent (Sigma, B6916-500ML). Whole body trehalose levels were measured from 10 μL of supernatant treated with 0.2 μL trehalase (Megazyme, E-TREH) at 37°C for 30 min using glucose assay reagent (Megazyme, K-GLUC) following the manufacturer’s protocol. We subtracted the amount of free glucose from the measurement and then normalized the subtracted values to protein levels in the supernatant. To measure whole body triglyceride levels, we processed 10 μL of supernatant using a Serum Triglyceride Determination kit (Sigma, TR0100), subtracted the amount of free glycerol in the supernatant from the measurement, and then normalized to protein levels in the supernatant.
Climbing activity
Flies were placed in an empty vial and then tapped down to the bottom. They were allowed to climb for 3 s. Climbing was video recorded and climbing height and speed were calculated from the video. A minimum of 15 flies and 10 separate trials were performed for each condition.
Muscle ATP measurements
Five adult thoraces were freshly homogenized in 100 μL of PBS, immediately heated at 70°C for 5 min, and centrifuged at 12,000 Xg for 5 min at 4°C to remove cuticle and cell debris. The supernatants were 1:500 diluted to measure the ATP levels using a CellTiter-Lumi™ Plus kit (Beyotime, C0068M). The final ATP production levels were normalized to protein levels that were measured using Bradford Reagent (Sigma, B6916-500ML) in the supernatant.
Immunostaining
Adult midguts were dissected in PBS and fixed for 15 min in PBS containing 4% paraformaldehyde. After fixation, the samples were washed with PBS containing 0.2% Triton X-100 (PBST)and blocked with 1% BSA in PBST. After incubation with primary antibodies anti-β-Gal (1:500, Promega, Z3781) overnight at 4°C. Midguts were washed and then incubated with Alexa fluorescence secondary antibody (1:1000, Thermo Fisher, A32742) and DAPI (1:1000, ThermoFisher, D1306) for 1 h at room temperature, washed, and mounted in Vectashield (Vector, H-1000). Adult midguts, thorax muscles, as well as abdomens containing fat bodies, were dissected in PBS and fixed for 15 min in PBS containing 4% paraformaldehyde. After fixation, the samples were washed with PBST, incubated with Bodipy 493/503 (1 μg/mL, Thermo Fisher, D3922), Phalloidin (1:1000, Thermo Fisher, A12381), or DAPI (1:1000) for 1 h at room temperature, washed, and mounted in Vectashield (Vector, H-1000). Images of fly appearances were performed on a Nikon SMZ18 or Nikon Eclipse Ts2 and confocal images were obtained using a Zeiss LSM880.
Western blotting
Ten adult thoraces or abdomens without ovaries were lysed in RIPA buffer (50 mM Tris-HCl [pH 7.5], 5 mM EDTA, 10 mM Na4P2O7, 100 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3VO4, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1% Nonidet P-40). Extracts were immunoblotted with indicated antibodies: rabbit anti-phospho-Akt (S473) (1:1000, Cell Signaling, 4060), mouse anti-polyubi-quitin (FK2) (1:1000, Enzo, BML-PW8810), α-tubulin (1:5000, Sigma, T5168).
qPCR
A total of 10 adult midguts, 5 whole adult flies, or 5 adult thoraces of each genotype and were lysed with Trizol (Thermo Fisher, 15596018) for RNA extraction and cDNA was transcribed using HiScript II Q RT Supermix (Vazyme, R222-01). qPCR was then performed using ChamQ SYBR qPCR Master Mix (Vazyme, Q311-03) on a CFX96 Real-Time System/C1000 Thermal Cycler (Bio-Rad). Drosophila gene expression was normalized to RpL32.
Electron microscopy
Adult thoraces were processed and analyzed in cross-section following standard protocols (Electron Microscopy Facility at Harvard Medical School; https://electron-microscopy.hms.harvard.edu). Briefly, thoraces were fixed in 0.1 M sodium cacodylate buffer (pH 7.4) containing 2.5% glutaraldehyde, 2% paraformaldehyde overnight. The fixed samples were washed in 0.1M cacodylate buffer, fixed again with 1% osmium tetroxide (OsO4) and 1.5% potassium ferrocyanide (KFeCN6) for 1 hour, and washed 3 times in water. Samples were incubated in 1% aqueous uranyl acetate for 1 hour and followed by 2 washes in water and subsequent dehydration in grades of alcohol. The samples were then put in propylene oxide for 1 hour and embedded in TAAB Epon (Marivac Canada Inc.). Ultrathin sections (about 60 nm) were cut on a Reichert Ultracut-S microtome, moved to copper grids, and then stained with lead citrate. Sections were examined in a JEOL 1200EX transmission electron microscope, and images were recorded with an AMT 2k CCD camera.
Pharmaceutical Jak/Stat inhibition in flies
For pharmaceutical inhibition of Jak/Stat signaling, flies were transferred to food containing methotrexate (Selleckchem, S1210) simultaneously with tumor induction at 29°C.
Cell culture, constructs, luciferase, and ChIP assays
Stat92E-RB and hopTumL fragments were cloned from cDNAs of wild-type and hopTumL-overexpressing flies, respectively, into the pAc5.1 vector (ThermoFisher, V411020) at EcoRI and XhoI sites using the exonuclease-based DNA assembly method (Vazyme, C113-01). The hsp70 promoter was excised from the pUAST plasmid and inserted into the pGL3-basic Luciferase Reporter Vector (Promega, E1751) at the HindIII site. Different fragments (BS1, BS2, BS3, BS4) of ImpL2 promoter were cloned from the genome of w1118 flies and inserted into the pGL3-hsp70 vector at XhoI site.
For luciferase assays, Drosophila S2R+ cells were seeded into a 48-well plate at 25°C with Schneider’s medium supplemented with 10% fetal bovine serum, then transfected with 15ng pGL3-BS1, pGL3-BS2, pGL3-BS3, pGL3-BS4, or mutant pGL3-BS2 together with 60 ng pAc-Stat92E-RB and/or 15 ng pAc-hopTumL and 5 ng pAc-renilla for 2 days using the Effectene reagent (QIAGEN, 301425). Next, S2R+ cells were washed with PBS and lysed for measurements of firefly and Renilla luciferase activities using the Dual-Luciferase Reporter Assay Reagent (Promega, E1910). Firefly luciferase activities were normalized to Renilla luciferase activities. All of the results were obtained from at least three independent experiments.
For ChIP assays, Drosophila S2R+ cells were transiently transfected with pAc-HRV.FLAG.Stat92E.RB-T2A-hopTumL.GFP for 2 days using the Effectene and cross-linked with 1% formaldehyde for 20 min and quenched with 135 mM glycine for 15 min at 25°C. Cells were lysed and chromatins were sheared by sonication in a QSonica sonicator (QSonica, LLC, Q125) for 15 times (10 s ON and 50 s OFF, 40% Amplitude). After centrifugation at 13,200 rpm for 15 min at 4°C and lysate supernatant was adjusted into 1.3 mL TE buffer containing 1% Triton X-100, 0.1% DOC, proteinase inhibitor cocktails. 50 μL of chromatin solution was used as Input DNA. 1.2 mL chromatin solution was incubated with the beads (Pierce Protein A/G Magnetic Beads, 88803) that were preblocked using 0.2 mg/mL glycogen, 0.2 mg/mL BSA, 0.2 mg/mL tRNA for 2 hours and pretreated with 10 μL anti-FLAG antibodies (Genscript, A01868) for 2 h. The mixture was incubated with HRV 3C Protease (TaKaRa, 7360-1) at 22°C for 2 h and later incubated with 120 μL Elution Buffer overnight at 65°C. The input DNA and immunoprecipitated DNA samples were subjected to qPCR in CFX384 Touch Real-Time PCR Detection System (Bio-Rad Laboratories). A fragment in the CDS region of Sam-S that contains no Stat-binding sites was used as the negative control.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as the mean ± SEM. Unpaired Student’s t test and one-way ANOVA followed by post hoc test were performed to assess differences. A p value of < 0.05 was considered statistically significant. All of the statistical details of experiments and p values can be found in the figure legends.
Supplementary Material
Highlights.
yki3SA-gut tumors produce Upd3 to autonomously promote self-growth
yki3SA-gut tumors produce Upd3 to cause host wasting
Upd3/Jak/Stat axis impairs muscle mitochondrial functions and carbolipid metabolism
Upd3/Jak/Stat axis induces host ImpL2 production to hamper insulin response
ACKNOWLEDGMENTS
We thank the Transgenic RNAi Project (TRiP) at Harvard Medical School and the Bloomington Drosophila Stock Center for providing fly stocks; Liangyou Rui, Yong Liu, and Michele Markstein for comments and suggestions; Erika Bach for UAS-hopTumL lines; Bruce Edgarfor UAS-upd3 and upd3-LacZ lines; Zheng Guo for MARCM lines; Xinhua Lin and Yun Qi for UAS-Stat92E-i line; and Pedro Saavedra for help with the dissection of thoraces. Work in the Song lab was supported by the Chinese National Natural Science Foundation (91957118, 31800999, and 31971079) and the Fundamental Research Funds for the Central Universities. Work in the Perrimon lab is supported by NIH grants (P01CA120964, R01DK121409, and R01GM067761), the American Diabetes Association (1-16-PDF-108), and the Harvard Blavatnik Accelerator Fund (FY 2017). N.P. is an investigator of the Howard Hughes Medical Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109553
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Acharyya S, and Guttridge DC, (2007). Cancer cachexia signaling pathways continue to emerge yet much still points to the proteasome. Clin. Cancer Res 13, 1356–1361. [DOI] [PubMed] [Google Scholar]
- Amoyel M, Anderson AM, and Bach EA, (2014). JAK/STAT pathway dysregulation in tumors: a Drosophila perspective. Semin. Cell Dev. Biol 28, 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoyel M, Anderson J, Suisse A, Glasner J, and Bach EA, (2016). Socs36E controls niche competition by repressing MAPK signaling in the Drosophila testis. PLoS Genet. 12, e1005815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyagi T, Terracina KP, Raza A, Matsubara H, and Takabe K, (2015). Cancer cachexia, mechanism and treatment. World J. Gastrointest. Oncol 7, 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach EA, Ekas LA, Ayala-Camargo A, Flaherty MS, Lee H, Perrimon N, and Baeg GH, (2007). GFP reporters detect the activation of the Drosophila JAK/STAT pathway in vivo. Gene Expr. Patterns 7, 323–331. [DOI] [PubMed] [Google Scholar]
- Bai H, Kang P, Hernandez AM, and Tatar M, (2013). Activin signaling targeted by insulin/dFOXO regulates aging and muscle proteostasis in Drosophila. PLoS Genet. 9, e1003941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond D, and Foley E, (2012). Autocrine platelet-derived growth factor-vascular endothelial growth factor receptor-related (Pvr) pathway activity controls intestinal stem cell proliferation in the adult Drosophila midgut. J. Biol. Chem 287, 27359–27370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, Koniaris LG, and Zimmers TA, (2012). JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab 303, E410–E421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Campo A, Jaimovich E, and Tevy MF, (2016). Mitochondria in the aging muscles of flies and mice: new perspectives for old characters. Oxid. Med. Cell. Longev 2016, 9057593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon K, Arends J, and Baracos V, (2013). Understanding the mechanisms and treatment options in cancer cachexia. Nat. Rev. Clin. Oncol 10, 90–99. [DOI] [PubMed] [Google Scholar]
- Figueroa-Clarevega A, and Bilder D, (2015). Malignant Drosophila tumors interrupt insulin signaling to induce cachexia-like wasting. Dev. Cell 33, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint TR, Janowitz T, Connell CM, Roberts EW, Denton AE, Coll AP, Jodrell DI, and Fearon DT, (2016). Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab. 24, 672–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera SC, and Bach EA, (2019). JAK/STAT signaling in stem cells and regeneration: from Drosophila to vertebrates. Development 746, dev167643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata H, Tetsumoto S, Kijima T, Kida H, Kumagai T, Takahashi R, Otani Y, Inoue K, Kuhara H, Shimada K, et al. (2013). Favorable responses to tocilizumab in two patients with cancer-related cachexia. J. Pain Symptom Manage 46, e9–e13. [DOI] [PubMed] [Google Scholar]
- Houtz P, Bonfini A, Liu X, Revah J, Guillou A, Poidevin M, Hens K, Huang HY, Deplancke B, Tsai YC, and Buchon N, (2017). Hippo, TGF-β, and Src-MAPK pathways regulate transcription of the upd3 cytokine in Drosophila enterocytes upon bacterial infection. PLoS Genet. 73, e1007091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K, Miao T, Chang K, Kim J, Kang P, Jiang Q, Simmonds AJ, Di Cara F, and Bai H, (2020). Impaired peroxisomal import in Drosophila oenocytes causes cardiac dysfunction by inducing upd3 as a peroxikine. Nat. Commun 11, 2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Patel PH, Kohlmaier A, Grenley MO, McEwen DG, and Edgar BA, (2009). Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila midgut. Cell 137, 1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen SB, O’Neill HM, Sylow L, Honeyman J, Hewitt KA, Palanivel R, Fullerton MD, Öberg L, Balendran A, Galic S, et al. (2013). Deletion of skeletal muscle SOCS3 prevents insulin resistance in obesity. Diabetes 62, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandarian SC, Nosacka RL, Delitto AE, Judge AR, Judge SM, Ganey JD, Moreira JD, and Jackman RW, (2018). Tumour-derived leukaemia inhibitory factor is a major driver of cancer cachexia and morbidity in C26 tumour-bearing mice. J. Cachexia Sarcopenia Muscle 9, 1109–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katheder NS, Khezri R, O’Farrell F, Schultz SW, Jain A, Rahman MM, Schink KO, Theodossiou TA, Johansen T, Juhász G, et al. (2017). Microenvironmental autophagy promotes tumour growth. Nature 541, 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierdorf K, Hersperger F, Sharrock J, Vincent CM, Ustaoglu P, Dou J, Gyoergy A, Groß O, Siekhaus DE, and Dionne MS, (2020). Muscle function and homeostasis require cytokine inhibition of AKT activity in Drosophila. eLife 9, e51595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kir S, White JP, Kleiner S, Kazak L, Cohen P, Baracos VE, and Spiegelman BM, (2014).Tumour-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 573, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y, Song W, Droujinine IA, Hu Y, Asara JM, and Perrimon N, (2015). Systemic organ wasting induced by localized expression of the secreted insulin/IGF antagonist ImpL2. Dev. Cell 33, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird BJ, McMillan D, Skipworth RJE, Fallon MT, Paval DR, McNeish I, and Gallagher IJ, (2021). The emerging role of interleukin 1β (IL-1β) in cancer cachexia. Inflammation 44, 1223–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Ng KG, Dombek KM, Eom DS, and Kwon YV, (2021). Tumors overcome the action of the wasting factor ImpL2 by locally elevating Wnt/Wingless. Proc. Natl. Acad. Sci. USA 778, e2020120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markstein M, Dettorre S, Cho J, Neumüller RA, Craig-Müller S, and Perrimon N, (2014). Systematic screen of chemotherapeutics in Drosophila stem cell tumors. Proc. Natl. Acad. Sci. USA 111, 4530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen DG, and Peifer M, (2005). Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development 132, 3935–3946. [DOI] [PubMed] [Google Scholar]
- Newton H, Wang YF, Camplese L, Mokochinski JB, Kramer HB, Brown AEX, Fets L, and Hirabayashi S, (2020). Systemic muscle wasting and coordinated tumour response drive tumourigenesis. Nat. Commun 11, 4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Y, Yu S, Li Q, Nirala NK, Amcheslavsky A, Edwards YJK, Shum PW, Jiang Z, Wang W, Zhang B, et al. (2019). Oncogenic pathways and loss of the Rab11 GTPase synergize to alter metabolism in Drosophila. Genetics 212, 1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu-Ansah E, Song W, and Perrimon N, (2013). Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell 155, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren F, Wang B, Yue T, Yun EY, Ip YT, and Jiang J, (2010). Hippo signaling regulates Drosophila intestinestem cell proliferation through multiple pathways. Proc. Natl. Acad. Sci. USA 107, 21064–21069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala D, and Sacco A, (2016). Signal transducer and activator of transcription 3 signaling as a potential target to treat muscle wasting diseases. Curr. Opin. Clin. Nutr. Metab. Care 19, 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RL, Kohlmaier A, Polesello C, Veelken C, Edgar BA, and Tapon N, (2010). The Hippo pathway regulates intestinal stem cell proliferation during Drosophila adult midgut regeneration. Development 137, 4147–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M, Cha N, Koranteng F, Cho B, and Shim J, (2020). Subpopulation of macrophage-like plasmatocytes attenuates systemic growth via JAK/STAT in the Drosophila fat body. Front. Immunol 11, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloth Andersen A, Hertz Hansen P, Schaffer L, and Kristensen C, (2000). A new secreted insect protein belonging to the immunoglobulin superfamily binds insulin and related peptides and inhibits their activities. J. Biol. Chem 275, 16948–16953. [DOI] [PubMed] [Google Scholar]
- Song W, Ren D, Li W, Jiang L, Cho KW, Huang P, Fan C, Song Y, Liu Y, and Rui L, (2010). SH2B regulation of growth, metabolism, and longevity in both insects and mammals. Cell Metab. 11, 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Veenstra JA, and Perrimon N, (2014). Control of lipid metabolism by tachykinin in Drosophila. Cell Rep. 9, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Cheng D, Hong S, Sappe B, Hu Y, Wei N, Zhu C, O’Connor MB, Pissios P, and Perrimon N, (2017a). Midgut-derived activin regulates glucagon-like action in the fat body and glycemic control. Cell Metab. 25, 386–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Owusu-Ansah E, Hu Y, Cheng D, Ni X, Zirin J, and Perrimon N, (2017b). Activin signaling mediates muscle-to-adipose communication in a mitochondria dysfunction-associated obesity model. Proc. Natl. Acad. Sci. U S A 114, 8596–8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Kir S, Hong S, Hu Y, Wang X, Binari R, Tang HW, Chung V, Banks AS, Spiegelman B, and Perrimon N, (2019). Tumor-derived ligands trigger tumor growth and host wasting via differential MEK activation. Dev. Cell 48, 277–286.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley BK, and Irvine KD, (2010).Warts and Yorkie mediate intestinal regeneration by influencing stem cell proliferation. Curr. Biol 20, 1580–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stec W, Vidal O, and Zeidler MP, (2013). Drosophila SOCS36E negatively regulates JAK/STAT pathway signaling via two separable mechanisms. Mol. Biol. Cell 24, 3000–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassmann G, Fong M, Kenney JS, and Jacob CO, (1992). Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J. Clin. Invest 89, 1681–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry NA,Tulina N, Matunis E, and DiNardo S, (2006). Novel regulators revealed by profiling Drosophila testis stem cells within their niche. Dev. Biol 294, 246–257. [DOI] [PubMed] [Google Scholar]
- Thomas S, Fisher KH, Snowden JA, Danson SJ, Brown S, and Zeidler MP, (2015). Methotrexate is a JAK/STAT pathway inhibitor. PLoS ONE 10, e0130078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock KJ, Kierdorf K, Pouchelon CA, Vivancos V, Dionne MS, and Geissmann F, (2015). Macrophage-derived upd3 cytokine causes impaired glucose homeostasis and reduced lifespan in Drosophila fed a lipid-rich diet. Immunity 42, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Liu Z, Ding H, Zhou Y, Doan HA, Sin KWT, Zhu ZJ, Flores R, Wen Y, Gong X, et al. (2017). Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat. Commun 8, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, et al. (2010). Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142, 531–543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-phosphor-Akt (Ser473) | Cell Signaling | Cat#4060, RRID:AB_2315049 |
| Mouse anti-α-tubulin | Sigma | Cat#T5168,RRID:AB_477579 |
| Mouse anti-polyubiquitin (FK2) | Enzo | Cat#BML-PW8810,RRID:AB_10541840 |
| Mouse anti-β-Gal | Promega | Cat#Z3781,RRID:AB_430877 |
| Goat Anti-Mouse IgG (H+L), HRP | ABclonal | Cat#AS003, RRID:AB_2769851 |
| Goat Anti-Rabbit IgG (H+L), HRP | ABclonal | Cat#AS014, RRID:AB_2769854 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 594 | Thermo Fisher Scientific | Cat#A32742, RRID:AB_2762825 |
| Rabbit monoclonal anti-FLAG | Genscript | Cat#A01868 |
| Bacterial and virus strains | ||
| DH5α Competent E.coli Strain | Vazyme | Cat#C502 |
| Chemicals, peptides, and recombinant proteins | ||
| Bradford Reagent | Sigma | Cat#B6916-500ML |
| Glycerol standard | Sigma | Cat#G7793-5ML |
| D-(+)-Glucose | Sigma | Cat#G7021 |
| Trehalase | Sigma | Cat#E-TREH |
| Glucose assay reagent | Megazyme | Cat#K-Gluc |
| Triglyceride reagent | Sigma | Cat#T2449-10ML |
| Free glycerol reagent | Sigma | Cat#F6428-40ML |
| Glycerol | Sigma | Cat#G7793 |
| Trizol | Thermo Fisher Scientific | Cat#15596018 |
| HiScript II Q RT Supermix | Vazyme | Cat#R222-01 |
| SYBR qPCR Master Mix | Vazyme | Cat#Q311-03 |
| DAPI | Thermo Fisher Scientific | Cat#D1306 |
| Bodipy 493/503 | Thermo Fisher Scientific | Cat#D3922 |
| Phalloidin | Thermo Fisher Scientific | Cat#A12381, |
| Insulin | Sigma | Cat#I6634 |
| Methotrexate | Selleck | Cat#S1210 |
| Critical commercial assays | ||
| CellTiter-LumiTM Plus kit | Beyotime | Cat#C0068M |
| Trehalose Assay Kit | Megazyme | Cat#K-Gluc |
| Triglyceride Assay Kit | Sigma | Cat#T2449 |
| Effectene reagent | QIAGEN | Cat#301425 |
| Pierce Protein A/G Magnetic Beads | Thermo Fisher Scientific | Cat#88803 |
| ClonExpress MultiS One Step Cloning Kit | Vazyme | Cat#C113-01 |
| HRV 3C Protease | TaKaRa | Cat#7360-1 |
| Dual-Luciferase Reporter assay kit | Promega | Cat#E1910 |
| Deposited data | ||
| RNaseq data from fly midgut | (Kwon et al., 2015) | GEO: GSE113728 |
| RNaseq data from fly muscle | (Song et al., 2019) | GEO: GSE65325 |
| Experimental models: Cell lines | ||
| D. melanogaster: Cell line S2R+ | Laboratory of Norbert Perrimon, Harvard Medical School | N/A |
| D.melanogaster: esg-GAL4, tub-GAL80TS, UAS-GFP | (Kwon et al., 2015) | N/A |
| D.melanogaster: Myo1A-GAL4, esg-GAL4, tub-GAL80TS | (Song et al., 2019) | N/A |
| D.melanogaster: CG-GAL4, tub-GAL80TS | (Song et al., 2017b) | N/A |
| D.melanogaster: tub-GAL80TS, Lpp-GAL4 | (Song et al., 2017b) | N/A |
| D.melanogaster: tub-GAL80TS, dMef2-GAL4 | (Owusu-Ansah et al., 2013) | N/A |
| D.melanogaster: tub-GAL80TS, Mhc-GAL4 | (Owusu-Ansah et al., 2013) | N/A |
| D.melanogaster: 10XStat-GFP | (Bach et al., 2007) | N/A |
| D.melanogaster: UAS-Pvf1 | (Song et al., 2019) | N/A |
| D.melanogaster: UAS-hopTumL | A gift from Dr. Erika Bach, NYU Langone Health | N/A |
| D.melanogaster: UAS-upd3 | A gift from Dr. Bruce Edgar, University of Utah | N/A |
| D.melanogaster: upd3-lacZ | A gift from Dr. Bruce Edgar, University of Utah | N/A |
| D.melanogaster: FRT19A | A gift from Dr. Zheng Guo, Huazhong University of Science and Technology | N/A |
| D.melanogaster: yw, hs-FLP, tub-GAL80, FRT19A; UAS-GFP | A gift from Dr. Zheng Guo, Huazhong University of Science and Technology | N/A |
| D.melanogaster: UAS-Stat92E-RNAi | A gift from Drs. Xinhua Lin and Yun Qi, Fudan University | N/A |
| D.melanogaster: UAS-upd3-RNAi1 | TRiP at Harvard Medical School | HMS000646 |
| D.melanogaster: UAS-upd3-RNAi2 | TRiP at Harvard Medical School | HMS05061 |
| D.melanogaster: UAS-hop-RNAi | TRiP at Harvard Medical School | HMS00761 |
| D.melanogaster: UAS-Stat92E-RNAi | TRiP at Harvard Medical School | HMS00035 |
| D.melanogaster: UAS-ImpL2-RNAi | TRiP at Harvard Medical School | HMS05891 |
| D.melanogaster: UAS-w-RNAi | TRiP at Harvard Medical School | HMS01545 |
| D.melanogaster: w1118 | Bloomington Stock Center | BDSC_3605 |
| D.melanogaster: upd3 Δ | Bloomington Stock Center | BDSC_55728 |
| D.melanogaster: USA-yki3SA | Bloomington Stock Center | BDSC_28817 |
| D.melanogaster: USA-InRAC | Bloomington Stock Center | BDSC_8248 |
| D.melanogaster: UAS-ImpL2-RNAi | National Institute of Genetics | 15009R-3 |
| D.melanogaster: UAS-dome-RNAi | Vienna Drosophila Resource Center | v19717 |
| Oligonucleotides | ||
| See Table S4 for oligonucleotide information | N/A | |
| Recombinant DNA | ||
| pAc5.1 | Thermo Fisher Scientific | Cat#V411020 |
| pGL3-Basic | Promega | Cat#E1751 |
| pAc5-Stat92E-RB | This paper | N/A |
| pAc5-hopTumL | This paper | N/A |
| pGL3-hsp70 | This paper | N/A |
| pGL3-ImpL2-BS1 | This paper | N/A |
| pGL3-ImpL2-BS2 | This paper | N/A |
| pGL3-ImpL2-BS3 | This paper | N/A |
| pGL3-ImpL2-BS4 | This paper | N/A |
| pGL3-ImpL2-BS2-mut | This paper | N/A |
| pAc-HRV.FLAG.Stat92E.RB-T2A-hopTumLGFP | This paper | N/A |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/download.html |
| Microsoft Excel | Microsoft | https://www.microsoft.com/en-us/microsoft-365/excel |
| Other | ||
| QSonica sonicator | QSonica | #Q125 system |
| Multi-sample tissuelyser-24 | Shanghai Jingxin Technology | N/A |
| Nikon SMZ18 | Nikon | N/A |
| Nikon Eclipse Ts2 | Nikon | N/A |
| Zeiss LSM880 | Zeiss | N/A |
| CFX96 Real-Time System/C1000 Thermal Cycler | Bio-Rad | N/A |