

Abstract
Gene therapy is at the forefront of the drive to bring the potential of cure to patients with genetic diseases. Multiple mechanisms of effective and efficient gene therapy delivery (eg, lentiviral, adeno‐associated) for transgene expression as well as gene editing have been explored to improve vector and construct attributes and achieve therapeutic success. Recent clinical research has focused on recombinant adeno‐associated viral (rAAV) vectors as a preferred method owing to their naturally occurring vector biology characteristics, such as serotypes with specific tissue tropisms, facilitated in vivo delivery, and stable physicochemical properties. For those living with hereditary diseases like hemophilia, this potential curative approach is balanced against the need to provide safe, predictable, effective, and durable factor expression. While in vivo studies of rAAV gene therapy have demonstrated amelioration of the bleeding phenotype in adults, long‐term safety and effectiveness remain to be established. This review discusses vector biology in the context of rAAV‐based liver‐directed gene therapy for hemophilia and provides an overview of the types of viral vectors and vector components that are under investigation, as well as an assessment of the challenges associated with gene therapy delivery and durability of expression.
Keywords: gene therapy, hemophilia, recombinant adeno‐associated viral (rAAV) vectors, vector
Essentials.
Viral vectors are the most commonly used gene therapy modality.
The liver is increasingly recognized as the primary natural target for all known AAV serotypes.
The goals of gene therapy in hemophilia are a “functional cure” and “health equity.”
The ideal gene therapy will provide safe, predictable, and durable factor expression.
1. OVERVIEW OF GENE THERAPY
Gene therapy research has progressed over the past 30 years, with the aim of treating, and potentially curing, genetic diseases. Multiple approaches have been explored to achieve these goals, with viral vectors commonly used to deliver the therapeutic gene to target cells. This review explores different aspects of vector biology and technology advancements, as well as the advantages and challenges of designing a gene therapy strategy, with a particular focus on recombinant adeno‐associated viral (rAAV) vectors in therapies for hemophilia.
1.1. Viral vector terminology
Gene therapy requires a vehicle to effectively deliver genetic material to a target cell. Multiple technologies to achieve this have been developed; however, viral vectors are the most commonly used because they are highly efficient, owing to their evolutionary adaptation to deliver DNA or RNA to mammalian cells.1 Viral vectors are designed to preferentially transduce a specific target cell type (in vivo); alternatively, cells may be removed from the body for genetic manipulation and expansion and then reintroduced into the original donor (ex vivo).1, 2 The process of delivery and expression of a therapeutic gene using a viral vector is termed transduction. Unlike wild‐type (WT) viruses found in nature, a viral vector cannot replicate. It delivers its payload to the nucleus, enabling expression of the therapeutic protein; once the payload is delivered, the viral “shell” or capsid is degraded.
Based on the relationship between the vector‐delivered transgene and target cell genome, vectors can be divided broadly into integrating and nonintegrating subtypes.2
1.1.1. Vectors that are designed to integrate into the host genome
Vectors based on retroviruses integrate the expression cassette (the therapeutic transgene and its regulatory components) into the target cell chromosome, allowing the transgene to be passed to daughter cells.3 These vectors are typically used for ex vivo delivery of a transgene into a stem or precursor target cell type and involves removing cells from the body, transducing them using an integrating vector and, following expansion, reintroducing the genetically modified cells into the original donor (autologous).2 However, such integrating vectors can also be delivered in vivo.4 The requirements for ex vivo gene delivery include a vector encoding the therapeutic transgene and a manufacturing facility for purification, transduction, and expansion of the primary cells.
1.1.2. Vectors designed not to integrate into the host genome
Other viruses, such as genetically modified rAAVs, introduce their transgene into the nucleus of the cell, but the delivered DNA has a very low frequency of integration5 and remains in an episomal form.3 AAV‐based hemophilia gene therapy studies in large animal models reported that some random integration events occurred but did not result in any deleterious events.6 However, the long‐term safety of AAV‐based gene therapy remains to be determined with continued monitoring to fully understand the risk of carcinogenesis.
Typically, rAAV vectors are used to deliver a transgene to a long‐lived, postmitotic, or slowly dividing cell, in vivo, with the aim to achieve long‐term expression of that gene. To the degree possible, all potentially immunogenic attributes of viral vectors are removed (Table 1). Features of a well‐designed viral vector include avoidance and/or removal of elements that may activate innate immune pathways, such as toll‐like receptor (TLR) ligands (eg, TLR2, TLR9),7 interleukins 1 and 6,8 complement proteins,9, 10, 11 and use of a manufacturing process to enhance vector quality and reduce immunogenic impurities (eg, host cell contaminants).
TABLE 1.
Considerations for vector design
| Goal | Ideal Properties |
|---|---|
| Target tissues for optimal therapeutic benefit | |
| Achieve optimal therapeutic transgene expression levels |
|
| Limit or control host immune response to the vector |
|
|
Minimize the risk of vector‐associated genotoxicity |
|
| Achieve therapeutic safety |
|
| Optimize CMC |
|
Abbreviations: CMC, chemistry, manufacturing, and controls; NAbs, neutralizing antibodies; QC, quality control.
1.2. Vector‐associated immune responses may limit efficacy
The development of viral vectors is based on the modification of viruses that the human immune system is naturally able to detect and eliminate. Therefore, immune responses can limit the therapeutic effect of viral vector–based products and include humoral (antibody) and cellular responses directed at the viral capsid proteins and the therapeutic product. Because long‐term expression is a fundamental requirement for most gene therapies, strategies to minimize innate and adaptive immune responses are important.
1.3. Gene therapy for hemophilia
Hemophilia was one of the earliest diseases considered for gene therapy due to its well‐understood disease pathology and the validation of protein replacement therapy (Table 2). A rare, X‐linked recessive bleeding disorder, hemophilia is typically caused by mutations in F8 or F9, coding for factor VIII (FVIII) and factor IX (FIX) proteins, respectively. Cloning of the F8 and F9 genes, a turning point in hemophilia care, ushered in controlled industrial production of recombinant proteins for clinical use and also led to the consideration of gene therapy as a potential cure.3
TABLE 2.
Hemophilia is an optimal candidate for gene therapy
| Rationale | Description |
|---|---|
| Monogenic inheritance | Correction in a single gene provides long‐term symptom relief and is potentially curative110 |
| Gene addition is sufficient for clinical benefit | Mutations that cause hemophilia are not dominant‐negative, and thus gene addition is sufficient to correct the phenotype |
| Cargo capacity for efficient transduction | The coding region of the F9 gene fits into AAV vectors; the F8 gene can be modified to fit by deleting the B‐domain, which does not affect FVIII activity79 |
| Target tissue is well defined and accessible with current gene delivery methods | Hepatocytes can produce active FVIII, are the natural production site of FIX, and are the natural targets for many AAV vectors; expression is driven by liver‐specific promotors |
| Even minimal increases in clotting factor activity can significantly improve symptoms/QOL |
|
| Well‐studied clinical readout/benefit |
|
| Animal models of hemophilia A and B are available |
|
Abbreviations: AAV, adeno‐associated virus; ABRs, annualized bleeding rates; FDA, US Food and Drug Administration; FIX, factor IX; FVIII, factor VIII; QOL, quality of life.
The current standard of care for hemophilia is the prophylactic use of FVIII or FIX concentrates,12 but this requires frequent intravenous (IV) administration. In addition, lack of adherence to IV therapy has resulted in suboptimal patient outcomes.13 While extended half‐life recombinant proteins and novel alternative solutions, such as bispecific antibodies (eg, emicizumab),14 have decreased dosing frequency, chronic administration is required.14 Coupled with the need to manage breakthrough bleeding and the ongoing adherence challenges,15 there remains a need for more convenient and effective therapies.
Thus, the goals of gene therapy in hemophilia are a “functional cure” and “health equity,” defined as optimized health and well‐being, which is attained only with normal hemostasis.13
2. VIRAL VECTORS IN GENE THERAPY
2.1. Lentiviral vectors improve on earlier retroviruses
Lentiviruses (LVs), a type of retrovirus, are single‐stranded RNA viruses containing a reverse transcriptase to allow the viral RNA genome to be converted into double‐stranded DNA, which then integrates into the host genome via a virus‐encoded integrase.3, 16 The most commonly used recombinant LV (rLV) vectors are derived from HIV‐1. In these rLVs, the transgene expression cassette replaces most viral genes and regulatory sequences, resulting in a replication‐deficient vector.17 Benefits of rLVs are that they transduce nondividing cells3 and can be used ex vivo or in vivo.2, 3, 16 rLVs used for gene therapy have been optimized for efficient manufacturing, are free of potential contamination with replication competent species,16, 18, 19 and boast improved transduction of target cells.20 Although current data indicate no causal association between rLV gene therapy and cancer, monitoring for this potential adverse outcome is ongoing.
2.1.1. In vivo rLV vectors
The feasibility of in vivo gene therapy using rLV vectors has been explored to avoid the complicated protocols and safety issues associated with ex vivo delivery. With in vivo rLV delivery there is the advantage that the vector can be handled like other pharmaceutical agents, that is, stored frozen and administered in an outpatient setting. However, for in vivo applications of rLVs to be successful, improvements in tissue targeting and vector manufacturing technologies are still needed.
2.1.2. Ex vivo rLV vectors
In addition to their use in two licensed chimeric antigen receptor T cells (CAR‐T) products and numerous clinical stage programs, rLVs are being studied for the treatment of primary immunodeficiencies, metabolic diseases, and genetic blood disorders, including sickle cell anemia. A recent report documented successful ex vivo rLV therapy in patients with transfusion‐dependent β‐thalassemia.2, 21 Gene therapy using an ex vivo rLV vector for patients ≥12 years of age with transfusion‐dependent β‐thalassemia was approved by the European Medicines Agency (EMA) in 2019 based on clinical trial data demonstrating durable transfusion independence of up to 57 months.22
2.1.3. LV gene therapy for hemophilia
Ex vivo LV gene therapy has been investigated in animal models of hemophilia, using lineage restricted and unrestricted hematopoietic stem cells (HSCs).3, 23 For ex vivo LV gene therapy to be successful, the vector must integrate into dividing HSCs. Animal models are also being used to explore approaches to simplify ex vivo regimens, such as avoiding the need for bone marrow transplantation and other invasive procedures. Currently, one ongoing clinical trial is using YUVA‐GT‐F901 LV‐transduced autologous HSCs and mesenchymal stem cells (MSCs) in people with hemophilia B, although the use of some (partially) myeloablative regimens is required.24 In addition, three trials of LV gene therapies for hemophilia A are enrolling participants, including CD68‐ET3 LV‐transduced high‐expressing B‐domain–deleted factor VIII (BDD‐FVIII) transgene (Expression Therapeutics, Atlanta, GA, USA) in HSCs,17 Pleightlet (MUT6) LV‐transduced BDD‐FVIII in CD34+ peripheral blood stem cells (PBSCs) (Medical College of Wisconsin, Milwaukee, WI, USA),25 and YUVA‐GT‐F801 (hemophilia A) and YUVA‐GT‐F901 (hemophilia B) LV‐transduced autologous HSCs and MSCs (NCT03217032 and NCT03961243) (Shenzhen Geno‐Immune Medical Institute, Shenzen, China) (Table 3).
TABLE 3.
Clinical trials of gene therapy for hemophilia A and B
| Sponsor | Serotype | Transgene | Enrollment | Start Date | % of Normal | ABR | Duration | ALT ↑ | ↓Expr With ALT↑ |
Statusb Phase (*abstract form) |
|---|---|---|---|---|---|---|---|---|---|---|
| Hemophilia B Trials: rAAV Vector | ||||||||||
| Avigen/CHOP62 | rAAV2 | FIX‐WT | 4 | 08/07 | 12a | 1/2 | 1/2 | NCT00515710; completed; LTFU | ||
| UCL/SJCRH87 | rAAV2/8 | FIX‐WT | 14 | 02/22/10 | 5.1 | 1.5 (−90%) | >3 y | 4/6 | 4/6 | NCT00979238; active (NR); Ph1 |
| Takeda (Shire)47, 90 | rAAV8 (BAX 335) | FIX‐Padua | 8 | 02/11/13 | 2.8–45.3 | N/A | >4 y | 8/8 | 7/8 | NCT01687608; active (NR); Ph1, 2 |
| Spark/Pfizer117 | Spark100 (SPK9001) | FIX‐Padua | 15 | 11/15 | 33 | 0.4 (−96%) | 2/10 | 2/10 | NCT02484092; completed; Ph2 | |
| Pfizer | Spark100 (SPK9001) | hFIX‐Padua | 20 (est) | 06/22/17 | NCT03307980; recruiting; LTFU of NCT02484092 | |||||
| Pfizer | Spark100 (SPK9001) | hFIX‐Padua | 55 (est) | 07/29/19 | NCT03861273; recruiting; Ph3 | |||||
| uniQure110, 118 | rAAV5 (AMT−160) | FIX‐WT | 10 | 06/10/15 | 5.1–7.5 | 0‐3.3 (−77%–100%) | >4.5 y | 3/10 | 0/10 | NCT02396342; completed; Ph1, 2* |
| uniQure119 | rAAV5 (AMT−061) | hFIX‐coPadua | 54 | 06/27/18 | 37.2 | (−83%) | 26 wk | 7/54 | N/A | NCT03569891; active (NR); Ph3* |
| uniQure120 | rAAV5 (AMT−061) | hFIX‐coPadua | 3 | 07/24/18 | 47 | 0 (−100%) | 26 wk | 3/3 | 0/3 | NCT03489291; active (NR); Ph2, dose confirmation |
| Dimension/Ultragenyx93 | rAAV‐rh10 | co‐hFIX WT | 6 | 01/17 contin | 6.7 | 3/3 | 3/3 | NCT02971969; active (NR); LTFU | ||
| UCL/Freeline94, 95 | AAVS3 (FLT180a) | FIX‐(R338L) Padua | 24 (est) | 12/05/17 | 44‐190 (n = 6, 26 wk) | 0 | 26 wk−3 y | 2/8 | NCT03369444; recruiting; Ph1, 2* | |
| Freeline | AAVS3 (FLT180a) | FIX‐(R338L) Padua | 10 | 07/10/18 | NCT03641703; recruiting; Ph1, 2; LTFU | |||||
| China IHBDHT | rAAV/BBM‐H901 | FIX | 9 (est) | 10/16/19 | NCT04135300; recruiting; N/A | |||||
| Takeda (Shire) | rAAV8 (SHP648) | FIX‐Padua | 21 (est) | 05/13/20 | NCT04394286; suspended; Ph1, 2 | |||||
| Hemophilia B Trials: Lentiviral Vector | ||||||||||
| SGIMI | LV‐FIX (YUVA‐GT‐F901) | FIX | 10 (est) | 06/01/20 | NCT03961243; not yet recruiting; Ph1 | |||||
| Hemophilia A Trials: rAAV Vector | ||||||||||
| BioMarin96, 121 | rAAV5 (BMN270) | H(BDD)‐FVIII‐SQ | 15 | 08/01/2015 | 4‐100 (cohort 3, 3y)121 | 0.8 (−95%)98 | 4 y | 7/7 | 1/7 | NCT02576795; active (NR); Ph1, 2 |
| BioMarin122 | rAAV5 (BMN270) | H(BDD)‐FVIII‐SQ | 134 | 12/19/17 | 43 | 0.8 (n=112) | 52 wk | NCT03370913; active (NR); Ph3 | ||
| BioMarin | rAAV5 (BMN270) | H(BDD)‐FVIII‐SQ | 1 | 03/14/18 | NCT03392974; active (NR); Ph3, 1‐arm dose | |||||
| BioMarin | rAAV5 (BMN270) | H(BDD)‐FVIII‐SQ | 10 (est) | 04/03/18 | NCT03520712; enrolling by invitation; with anti‐AAV5 antibodies; Ph1, 2 | |||||
| BioMarin | rAAV5 (BMN270) | H(BDD)‐FVIII‐SQ | 20 (est) | 11/10/20 | NCT04323098; active; Ph3 | |||||
| BioMarin | rAAV5 (BMN270) | H(BDD)‐FVIII‐SQ | 20 (est) | 12/25/20 | NCT04684940; active; Ph1, 2 | |||||
| Spark |
Spark200/ LK03 (SPK−8011) |
BDD‐FVIII | 30 (est) | 01/26/17 |
5.2–19.8 (n=5)99 (5 × 1011/1 × 1012 vg/kg) |
0.4, 0, 0.4, 3.6, 0.5 (−91%)99 |
>2 y99 | 3/14 | NCT03003533; recruiting; Ph1, 2, dose finding* | |
| Spark |
Spark200/ LK03 (SPK−8011) |
BDD‐FVIII | 100 (est) | 08/14/18 | NCT03432520; enrolling by invitation; LTFU inhibitors | |||||
| Spark123 | (SPK−8016) | BDD‐FVIII | 30 (est) | 01/30/19 | 6.2‐21.8 (n=4) | 0‐2.4 (−85%) | 1–1.5 y | 0/4 | N/A | NCT03734588; active (NR); Ph1, 2, dose finding; inhibitors and non‐inhibitors* |
| Bayer/Ultragenyx124 | rAAV hu37 | BDD‐FVIII | 30 (est) | 11/07/18 | 1‐70 (n=6) | N/A | 30 wk−1.5 y | 3/6 | NCT03588299; recruiting; Ph1, 2* | |
| Pfizer | rAAV2/6 (PF−07055480/SB−525) | BDD‐FVIII | 63 (est) | 08/18/20 | NCT04370054; recruiting; Ph3 | |||||
| Pfizer101 | rAAV2/6 (SB−525) | BDD‐FVIII | 11 | 06/26/17 | 70.4 (n=5; cohort 4) | N/A | 1 y | 10/11 | 1/5 (cohort 4) | NCT03061201; active (NR); Ph2* |
| Takeda (Shire)125 | rAAV8 (TAK−754) | BDD‐FVIII | 12 (est) | 3/31/18 | 1–70 (n=4) | 1.25 (n=4) | 1 y | 4/4 | N/A | NCT03370172; active (NR); Ph1, 2* |
| ASC Therapeutics | rAAV | BDD‐FVIII | 12 (est) | 07/21 (est) | NCT04676048; not yet recruiting; Ph1, 2 | |||||
| UCL | rAAV2/8 | FVIII‐V3 | 18 (est) | 06/14/17 | NCT03001830; recruiting; Ph1 | |||||
| Hemophilia A Trials: Lentiviral Vector | ||||||||||
| Expression26 | CD68‐ET3 | BDD‐hFVIII | 7 (est) | 02/21 (est) | NCT04418414; not yet recruiting; Ph1 | |||||
| MCW/Hari | Pleightlet (MUT6) | BDD‐FVIII | 5 (est) | 04/29/20 | NCT03818763; recruiting; Ph 1 | |||||
| SGIMI | NHP/TYF | FVIII (YUVA‐GT‐F801) | 10 (est) | 06/01/20 | NCT03217032; not yet recruiting; Ph 1 | |||||
Abbreviations: AAV, adeno‐associated virus; ABR, annualized bleed rate; ALT, alanine transaminase; BDD‐FVIII, B‐domain–deleted–factor VIII; CHOP, Children’s Hospital of Philadelphia; contin, continuing; engi, engineered; est, estimate; expr, expression; FIX‐WT, factor IX wild type; IHBDHT, Institute of Hematology & Blood Diseases Hospital Tianjin; LTFU, long‐term follow‐up; MCW, Medical College of Wisconsin; N/A, not applicable; NR, not recruiting; Ph, phase; rAAV, recombinant adeno‐associated virus; SGIMI, Shenzhen Geno‐Immune Medical Institute; SJCRH, St. Jude Children’s Research Hospital; UCL, University College London; ZFN, zinc finger nuclease.
One patient had 12% peak activity that declined to <1% after immune response.
Status determined by ClinicalTrials.gov as of June 2020.
Preliminary data, not peer‐reviewed; to be interpreted with caution.
In both hemophilia A and B, the use of LV in vivo has been explored in animal models, including in nonhuman primates (NHPs).26, 27, 28 and demonstrated efficient targeting of hepatocytes and reduced acute inflammation for IV‐administrated rLV.27
2.1.4. Challenges to the use of rLVs
A potential risk associated with rLVs is insertional mutagenesis, a safety concern that may be more likely with transduction of dividing cells.16, 27, 29 Newer‐generation rLV designs have greatly reduced the risk of insertional mutagenesis, and no cases of leukemic transformation have been reported in human gene therapy trials.16, 18 Previously reported cases of genotoxicity associated with retroviral vectors may be due to the vector‐encoded endogenous promoter in activating oncogenes. Subsequently, the genome of rLV has been modified to enable deletion of the viral promoter during the reverse‐transcription process—so‐called self‐inactivating rLVs—which has substantially diminished the potential for genotoxicity.
2.2. AAV vectors
AAVs are small, nonenveloped, ≈4.7‐Kb DNA genome, replication‐defective members of the parvovirus family.3 rAAV vectors are generally delivered in vivo, either by injection into a specific tissue site or by IV infusion (Figure 1). Upon transduction of a target cell, multiple copies of the rAAV vector genome are established as stable circular concatemers (multimers of the expression cassette linked via inverted terminal repeats [ITRs]) outside of the chromosomal DNA within the nucleus of the transduced cell.3, 30 rAAV is derived from a WT parent virus that is common in the human population and not associated with any known disease. AAV vectors exploit this refined evolutionary fitness to efficiently transduce human cells.30, 31
FIGURE 1.
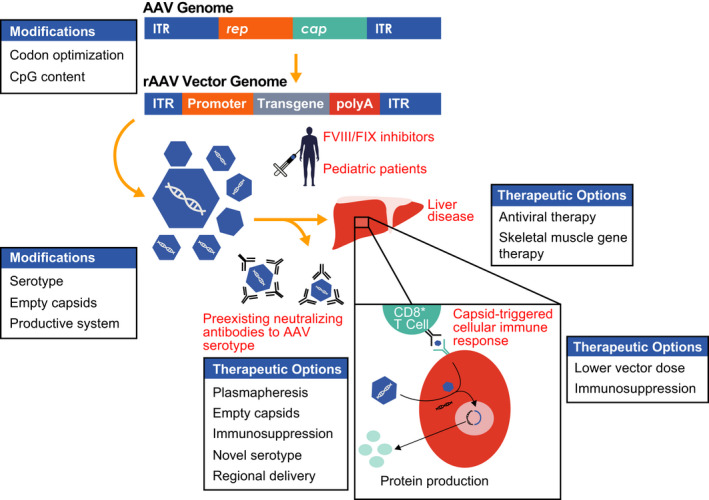
Overview of rAAV‐mediated liver‐directed gene therapy for hemophilia. The wild‐type adeno‐associated virus (AAV) genome consists of two inverted tandem repeat (ITR) regions flanking the rep (replication) and cap (capsid) genes. These genes are replaced by a tissue‐specific promoter with enhancer, intron, and transgene of interest in the recombinant adeno‐associated viral (rAAV) vector transgene expression cassette, which is packaged into capsids and injected into subjects via an intravenous infusion. Once infused, rAAV vector can be neutralized by preexisting antibodies in a serotype‐specific manner or transduce hepatocytes. The capsid is degraded and the genetic material maintained as an episome in the nucleus to produce the transgene product. Capsid peptides can be presented on the surface of hepatocytes to CD8+ T cells, thought to lead to a cellular immune response coinciding with loss of transgene and a rise in liver transaminases in some clinical trials. Modifications in the transgene, serotype, infusion of empty capsids, and production process may all affect efficacy. Options to bypass the preexisting humoral response or liver disease are listed. Additional hurdles to general application of liver‐directed AAV gene therapy include inhibitors to factor VIII (FVIII) and factor IX (FIX) as well as infusion in young people with hemophilia. (Reproduced, with permission, from Doshi et al, p 275, Figure 1)
2.2.1. In vivo rAAV vectors
Alipogene tiparvovec was the first AAV‐based gene therapy commercially approved in the European Union in 2012 for an ultra‐rare condition, hereditary lipoprotein lipase deficiency.32 Alipogene tiparvovec utilized rAAV serotype 1 (rAAV1) to deliver the expression cassette to myocytes following direct intramuscular injection. The next commercially approved therapeutic was voretigene neparvovec‐rzyl (Spark Therapeutics, Philadelphia, PA, USA), an rAAV2 serotype–based vector carrying the RPE65 transgene that is used to treat RPE65 −/−‐associated retinal dystrophy. Clinical trials led to US Food and Drug Administration (FDA) approval of this gene therapy in 2017.33
More recently, onasemnogene abeparvovec‐xioi (AveXis) was approved by the FDA in 2019 and by the EMA in 2020 for spinal muscular atrophy, a degenerative neuromuscular disease.34, 35 Onasemnogene abeparvovec‐xioi is an rAAV9‐based gene therapy administered IV with the intent of delivering a copy of the gene encoding the human SMN1 protein to the central nervous system (CNS).
2.2.2. Advantages and preferred use of AAV‐based gene therapy
rAAV vectors have emerged as the preferred tools for in vivo gene therapy due to their relative safety and ability to transduce a variety of tissue and cell types.31 The cloning steps needed to generate novel AAV vectors are well established, and the vector itself, while complex to manufacture, is stable, relatively homogeneous, and well defined biochemically.30 Based on their high physicochemical stability, rAAV vectors can be handled like many other biologics, that is, either frozen for long‐term storage (years) and kept at 4°C (days) or maintained at room temperature (hours) without detectable loss of functional activity. These properties facilitate storage, transport, and administration to patients.2, 36
2.2.3. Immunologic challenges of rAAV vector gene therapy
Immune responses, reported in both animal and human studies, remain important challenges to optimal, broad implementation of rAAV‐mediated gene therapies.11, 37, 38
One potential immunogenic target is the therapeutic transgene product. In hemophilia, antibodies, generally termed inhibitors, occur following protein replacement and are a key concern for health care providers who treat individuals with hemophilia. There have not been reports of the development of inhibitors in hemophilia AAV gene therapy trials. Although to date a limited number of adult subjects have received rAAV‐based investigational products, it is also believed that liver‐directed rAAV administration may reduce such immune response through induction of tolerance.39
A second and well‐established immunogenic target is the AAV capsid. Antibodies to the AAV capsid already exist in many people because of prior exposure to the common WT virus. AAV capsid antibodies may preclude transduction and readministration of AAV vectors.40, 41 An ongoing phase 3 study in individuals with hemophilia B included participants with modest levels of preexisting neutralizing antibodies (NAbs) in whom neither safety signals nor an impact on transgene expressions was observed.42 At the very high rAAV doses that have been required for certain disease indications, including Duchenne muscular dystrophy, anti‐AAV antibodies that were preexisting and/or rapidly formed after systemic vector administration have been proposed to form immune complexes with AAV that can activate complement and adverse events.9, 10, 11, 43, 44 It is encouraging that serial administration of AAV vectors to immunologically protected tissues and compartments (eg, eye and brain) is possible.45
A third immunogenic risk in using AAV vectors is the triggering of a cellular immune response to AAV capsid peptides expressed on the surface of the transduced cells. This response has often been observed in clinical settings and can lead to loss of transduced cells and therapeutic benefit. Administration of immunomodulatory agents such as corticosteroids ameliorates this unwanted immune response but is not always effective.3 Recent evaluation of AAV features in constructs used for hemophilia B studies support the notion that pathogen‐associated molecular patterns can contribute to the formation of capsid‐specific cytotoxic T lymphocytes (CTLs). Specifically, unmethylated cytosine‐guanine dinucleotides (CpG) motifs that are the known ligands involved in activation of TLR9, when present at sufficient density in an AAV expression cassette, may trigger CTLs, leading to the elimination of transduced hepatocytes and loss of transgene expression.46 This possibility is supported by a follow‐up analysis of a phase 1/2 study of an AAV8‐based hemophilia B gene therapy, BAX 335. The study investigators suggested that the loss of expression seen in seven of the eight participants resulted from innate immune responses triggered by the vector genome, more specifically, by the presence of unmethylated CpG motifs,47 which triggered activation of a cytotoxic T‐cell response against AAV‐transduced hepatocytes.
3. AAV BIOLOGY AND MECHANISM OF ACTION
The AAV genome consists of two ITR sequences of 145 nucleotides flanking open reading frames that encode four nonstructural replicases (Rep78/68/52/40), three structural (capsid) proteins (VP1/2/3), and additional proteins involved in capsid assembly.48, 49 The ITRs contain cis‐acting sequences required for genome replication and encapsidation.50 A critical advance in the AAV field was the discovery that the AAV2 genome could be cross‐packaged (pseudo‐serotyped) into capsids of other natural AAVs51 and bioengineered capsid variants.52 This discovery allowed for alterations of vector tropism, immunobiology, kinetics of transgene expression, and intracellular trafficking, all of which have dramatically improved clinical applicability.52, 53, 54 Additional critical AAV vector advances include the development of scalable high‐titer production strategies55 and the demonstration of rAAV usefulness in gene addition and targeted gene correction by homologous recombination.56
Although individual AAV serotypes can efficiently transduce multiple tissues,57 the liver is increasingly recognized as the primary natural target for all known AAV serotypes, as evidenced by the strong evolutionary relationship between the AAV life cycle and the host liver.58 AAV2, a human isolate from which prototypic AAV vectors were first derived, is endemic in the human population, with serologic evidence supporting lifetime infection rates of 35% to 80%, depending on geographic location.59, 60 When AAV virions encounter target cells in the absence of a helper virus, the viral genome can become latent. Single‐ or double‐stranded episomal forms of the AAV genome also can support latent infection; the mechanisms for this are less well understood but may be linked to the AAV capsid.61 In rAAV, the entire viral coding region, including rep and cap genes, is replaced with the exogenous DNA of interest or “transgene,” such as F8 or F9, and a promoter (Figure 1).3, 30 This rAAV genome is subsequently packaged into a human liver‐tropic capsid to preferentially deliver the therapeutic cargo to the liver following systemic delivery.62, 63
The infused rAAV predominantly transduces hepatocytes and travels to the cell nucleus, where the payload is released. rAAVs exhibit different physical characteristics than their WT AAV precursors and no longer maintain genetic instructions for site‐preferential integration64, 65 (Figure 1). Random integration events of rAAV genomes have been observed at a very low frequency at high vector doses (5%‐10% of hepatocyte transduction events).5, 6, 64, 66 Although such rare integrations of rAAV do not appear to have been associated with safety issues in clinical studies, the large number of vector genomes (vgs) delivered during a typical gene therapy treatment (typically >1011 vg/kg), relative to the number of all hepatocytes (139 × 109 cells/g of liver),67 suggests that there could be the potential for a high number of random integration events.1
3.1. AAVs serotypes with different tissue tropisms
There are at least 13 WT AAV serotypes51, 68 (Table 4), each with somewhat unique tissue tropisms (prevalence for CNS, liver, lung, and/or muscle),69 which have been “vectorized” for use as rAAVs in gene therapy approaches. These different tropisms are tied, in part, to the presence of preferential receptors on the preferred cell type but stem from the tissue‐specific promotor in the vector cassette.69 Results of early clinical studies of rAAV hemophilia gene therapy demonstrated that none of the existing natural AAV serotypes had a high transduction efficiency for human liver cells. These results prompted efforts to bioengineer new, highly functional human liver‐tropic capsids that could evade the immune system,52, 70 potentially allowing for efficacy at a lower dose with fewer adverse events.71 Capsid diversification strategies were thus developed; these ranged from rational design, in which specific capsid residues are modified to display random peptides (ie, ligands) on the surface‐exposed capsid variable regions, to random diversification methods, such as error‐prone polymerase chain reaction, used to amplify and introduce random point mutations into the AAV cap sequences “by chance.”72 However, a key milestone outside of these AAV bioengineered technologies was the description of directed evolution.
TABLE 4.
Examples of receptors and preferential tissue tropism of natural AAV vectors (Reproduced, with permission, from Costa Verdera, p 3, Table 1)
| Serotype | Source | Glycan Receptor | Co‐Receptor/Other | Examples of Tissue Tropism |
|---|---|---|---|---|
| AAV1 | Nonhuman primate | N‐linked sialic acid | Unknown | Skeletal muscle, lung, CNS, retina, pancreas |
| AAV2 | Human | HSPG | FGFR1, HGFR, LamR, CD9, tetraspanin | Smooth muscle, skeletal muscle, CNS, liver, kidney |
| AAV3 | Nonhuman primate | HSPG | FGFR1, HGFR, LamR | Hepatocarcinoma, skeletal muscle, inner ear |
| AAV4 | Nonhuman primate | O‐linked sialic acid | Unknown | CNS, retina |
| AAV5 | Human | N‐linked sialic acid | PDGFR | Skeletal muscle, CNS, lung, retina, liver |
| AAV6 | Human | N‐linked sialic acid, HSPG | EGFR | Skeletal muscle, heart, lung, bone marrow |
| AAV7 | Nonhuman primate | Unknown | Unknown | Skeletal muscle, retina, CNS |
| AAV8 | Nonhuman primate | Unknown | LamR | Liver, skeletal muscle, CNS, retina, pancreas, heart |
| AAV9 | Nonhuman primate | N‐linked galactose | LamR | Liver, heart, brain, skeletal muscle, lung, pancreas, kidney |
| AAV10 | Nonhuman primate | Unknown | Unknown | Liver |
Abbreviations: AAV, adeno‐associated virus; CNS, central nervous system; EGFR, epidermal growth factor receptor; FGFR1, fibroblast growth factor receptor 1; HGFR, hepatocyte growth factor; HSPG, heparan sulfate proteoglycans; LamR, laminin receptor; PDGFR, platelet‐derived growth factor receptor.
The directed evolution approach mimics natural evolutionary selection under controlled laboratory settings. Specifically, a selection pressure, such as the ability to transduce primary human hepatocytes or resistance to neutralization by preexisting human NAbs, is applied to a large AAV variant library.52, 73 Importantly, this process is highly flexible, and the selection can be performed either in vitro73 or in vivo.52 The initial library can be generated by such methods as shuffling capsid genes from genetically and functionally diverse parental AAV serotypes through enzymatic fragmentation, followed by assembly of shuffled full‐length capsid genes. Novel capsid optimization technologies were developed to improve shuffling efficiency and to enable contribution from highly diverse parental AAVs.74
3.2. Insights from novel AAV vector studies
Studies of novel bioengineered AAV vectors have led to interesting AAV vectorology insights and have provided a potential explanation for the unexpected natural AAV variant data. For example, studies of the FRG murine model (Fah−/−/Rag2−/−/Il2rg−/−)75 repopulated with primary human hepatocytes suggested that rAAV8, previously considered to have strong liver tropism, is a poor functional transducer of human hepatocytes in vivo.52, 76, 77 A more recent study using novel bioengineered AAV variants as a genetic tool to elucidate the interaction between AAV and human primary hepatocytes showed that strong binding to heparan sulfate proteoglycan, the first described AAV cellular receptor, is actually detrimental to AAV function in vivo.4, 78 These insights partially explain the lower‐than‐anticipated performance of AAV2 in the first hemophilia clinical study.62
3.3. rAAVs in hemophilia clinical trials
rAAVs used as vectors for hemophilia gene therapy in clinical trials are serotypes specific for liver tissue. The FVIII complementary DNA (cDNA), at 7 kb, is large and exceeds the capacity of AAV; however, the F8 transgene has been reduced in size by deleting the B domain of the F8 gene (≈2.6 kb), which is not required for coagulation. The resulting products are derivatives of the BDD‐FVIII transgene.3, 79 The 1.6‐kb coding region for factor IX (FIX) is much smaller and easier to package in rAAV. Therefore, despite the lower prevalence of hemophilia B, FIX was the first target for hemophilia gene therapy studied in clinical trials using rAAV vectors.3
4. RATIONALE FOR LIVER‐DIRECTED AAV FOR HEMOPHILIA
FVIII and FIX are secreted proteins and can be expressed and released into the bloodstream from various cell types, whereas the liver is the preferred target for hemophilia gene therapy due to its physiologic and functional properties that favor high vector transduction and systemic protein distribution. Furthermore, hepatocytes naturally produce FIX, which may provide additional benefits for people with hemophilia B. The liver plays key roles in metabolism, accounts for 10% to 15% of overall blood volume, and secretes many proteins into the circulation. In addition, the liver is highly vascularized, facilitating AAV transduction, ensuring that the majority of the IV‐administered AAV vector reaches its target cells and the subsequent dissemination of the transgene product. The liver can provide a “tolerizing” effect for “nonself” proteins expressed therein,39 which hypothetically may prevent activation of the immune response against the therapeutic protein. In small and large animal models, liver‐directed gene transfer with AAVs and LVs shows that expression of an antigen in hepatocytes can promote robust antigen‐specific immune tolerance.30, 41 Several studies have documented induction of antigen‐specific T‐regulatory cells (Tregs) and expression of antigen‐specific T‐cell exhaustion markers at inflammatory sites of rAAV delivery.80 Animal models of AAV vector–mediated gene transfer have confirmed the crucial role of Tregs in liver‐mediated tolerance induction; in these studies, pharmacologic blockade or depletion of Tregs resulted in an immune response against the transgene.30, 41 However, clinical studies have reported significant immunogenicity to rAAV vector capsid antigens. Although this immune response is typically treated by broad immunosuppression with steroids, alternative approaches are being developed, such as stimulation of Treg activity.30, 41
The small diameter of rAAV vectors enables easy passage through fenestrated endothelium to reach hepatocytes.81 The diameter of endothelial fenestrae in healthy humans ranges from ≈50 to 250 nm, with a mean diameter slightly >100 nm, whereas the diameters of AAV vectors are typically ≈25 nm.81, 82 Thanks to these physiologic factors, any infused liver‐targeted AAV vectors accumulate rapidly within the liver, a property that is critical to the success of liver‐mediated gene therapy83 (Figure 2).
FIGURE 2.
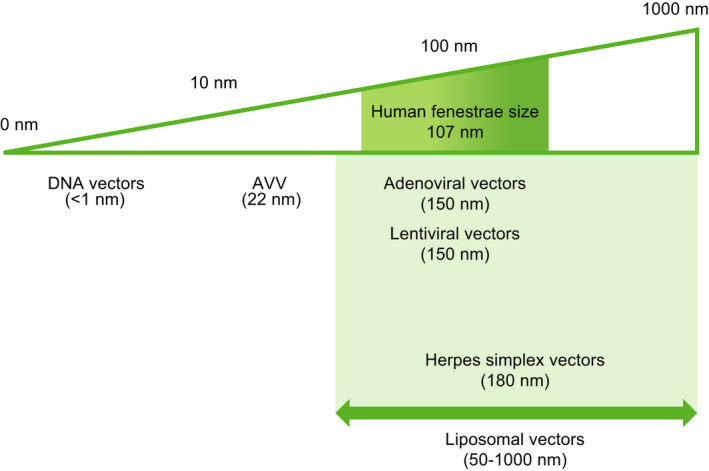
Relevance of gene therapy vector dimensions for gene transfer efficiency into hepatocytes. Human liver sinusoidal fenestrae show a Gaussian size distribution (shaded box) and have an average diameter of 107 nm. To ensure efficient transendothelial passage of gene transfer vectors, their size should be small enough to pass through the fenestrae into the space of Disse. (Reproduced, with permission, from Jacobs et al, p 1375)
In the mature adult liver, <2% of hepatocytes are actively dividing; due to this low cell turnover, any therapeutic effect achieved following AAV transduction is expected to be long lasting.81 However, this does not apply to the pediatric liver, which undergoes three doublings in the first 10 years of life due to natural organ growth.84 Moreover, the average life span of nonquiescent hepatocytes (<1%‐2% of hepatocytes) is estimated to be 200 to 300 days.81 Liver growth should be considered when contemplating the application of liver‐directed rAAV‐mediated hemophilia gene therapy in children but is less important for other tissue‐directed gene therapies (for example, onasemnogene abeparvovec‐xioi), which targets neurons.34 Data from several rAAV‐mediated gene therapy trials in adults with hemophilia B (Table 3) have shown durable transgene expression beyond 4 years. The finding that a sufficient fraction of transduced hepatocytes continues to express the transgene may indicate that the rate of vector integration may be higher than anticipated or that potentially some other longer‐lived cell type is functionally transduced with the therapeutic vector.
4.1. Clinical development of rAAV vector–mediated FIX gene therapy
The first hemophilia gene therapy studies were carried out with the F9 gene, owing to its small size.3 An early gene therapy clinical study in which skeletal muscle of participants with severe hemophilia B was injected with rAAV‐F9 demonstrated safety up to 40 months after injection but showed insufficient expression levels.62, 85 It should be noted, however, that in one participant from this study local transgene expression in skeletal muscle was detected up to 3.7 years following vector administration.86
In the first dose‐escalation AAV2‐F9 clinical trial using systemic delivery, those with severe hemophilia B who received the highest vector dose (2 × 1012 vg/kg) initially achieved up to 11% of normal FIX expression, which decreased to <1% within weeks. The drop in expression was accompanied by a transient elevation in liver transaminases. All participants demonstrated an increase in AAV2 NAbs and enzyme‐linked immune absorbent spot (ELISpot), and one participant showed interferon gamma (IFN‐γ) secretion detected 2 weeks after vector administration but showed no evidence of NAbs to FIX or ELISpot positivity to FIX peptides.3, 62
Systemic infusion of rAAV8 in a hemophilia B trial resulted in sustained, 2% to 5% FIX levels extending >8 years following treatment (University College London/St. Jude Children’s Research Hospital; NCT00979238; Table 3).87, 88 Even with prophylactic corticosteroids, participants in this trial demonstrated an immune response to the capsid, including capsid‐reactive T cells and anti‐AAV8 antibodies, but no immune response to the F9 transgene.87 Comparison of higher‐ versus lower‐dose cohorts suggested the possibility of increased capsid immunogenicity with increased vector dose. This finding prompted the development of gene therapy employing a type of naturally occurring FIX variant, FIX‐Padua, which has an 8‐fold higher specific FIX activity compared with FIX‐WT.3, 89 An rAAV8 vector with the FIX‐R338L/Padua transgene (Table 3) provided sustained FIX expression at 20% in a single participant receiving 1 × 1012 vg/kg with no observed toxicity.90 However, in participants treated with higher doses, FIX expression decreased due to a capsid immune response, even with corticosteroid treatment.3 A trial using Spark100 (Table 3), a modified rAAV variant, at a fixed dose of 5 × 1011 vg/kg, resulted in 22.9% physiologic FIX expression at 1 year following vector infusion and a lower rate of immune response.91
AMT‐061, an AAV5‐based FIX‐Padua gene therapy, is currently being evaluated in a phase 3 study enrolling participants without prior FIX inhibitors but does not exclude individuals with preexisting NAbs (NCT03569891). Recently presented data from 54 participants with hemophilia B who received 2.0 × 1013 gc/kg of AMT‐061, showed that 37.2% of participants had steady FIX expression 6 months after vector administration.42 Importantly, no relationship between corticosteroid treatment for elevated transaminases and FIX expression level or immunity was observed42, 92 (Figure 3).
FIGURE 3.
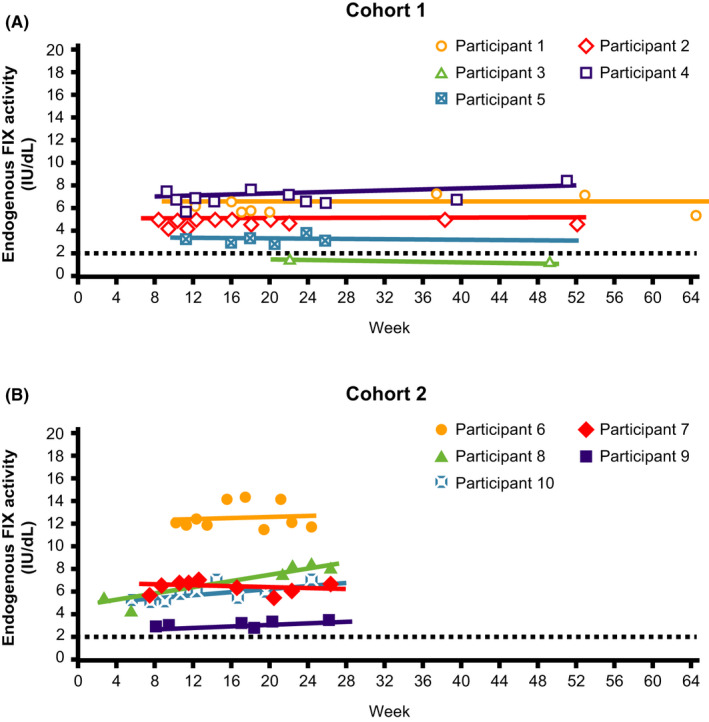
Factor IX (FIX) activity across time (uniQure FIX clinical trial data). (A) Cohort 1; (B) Cohort 2. Only values at least 10 days after the preceding FIX concentrate administration, so that they are uncontaminated by exogenous FIX, are included. Participant 3 continued with prophylaxis after AMT‐060 treatment; as a result, only limited samples uncontaminated by exogenous FIX were available. The dotted line at FIX activity of 2 IU/dL indicates the threshold required for ceasing prophylaxis per protocol. FIX prophylaxis was continued after AMT‐060 and tapered between week 6 and week 12. Participant 4 had a moderate hemophilia B phenotype at baseline (FIX activity, 1.5 IU/dL). (Reproduced, with permission, from Miesbach et al, p 1027, Figure 2)
An rAAV serotype rh10 (AAVrh10) vector containing a human F9 gene that was codon‐modified to increase expression efficiency but also contained elevated CpG dinucleotides relative to WT, with expression driven by both a liver‐specific enhancer and promoter; use of this vector achieved FIX levels of 5% to 20%.93 Therapeutic FIX expression was observed across two dose cohorts, but the levels declined to baseline, coincident with elevated transaminase levels, despite corticosteroid treatment. The loss of FIX expression is thought to potentially be related to AAVrh10 capsid immune response, and the study was subsequently terminated (Dimension Therapeutics; NCT02971969; Table 3).
FLT180a, an rAAV vector with a novel human liver‐tropic bioengineered capsid (AAVS3), was developed to express FIX‐Padua under the control of a liver‐specific promoter (Freeline Therapeutics; NCT03369444, NCT03641703, NCT04394286; Table 3). All participants treated with this therapy received prophylactic steroids. Those receiving 3.8 × 1011 vg/kg of R338L/Padua (n = 2) achieved steady‐state FIX expression in the 40% range, the highest FIX levels obtained at that dose without elevation of liver enzymes. Of the two participants who received 1.28 × 1012vg/kg, one showed supraphysiologic FIX levels and the other achieved FIX activity in the normal range. One participant in each of the other cohorts (6.4 × 1011vg/kg and 8.32 × 1011vg/kg) experienced loss of FIX expression due to transaminitis.94, 95
4.2. Clinical development of rAAV vector–mediated FVIII gene therapy
In preclinical studies, use of dual AAV canine FVIII heavy‐ and light‐chain vectors achieved long‐term success lasting >10 years in nine dogs with hemophilia A.6
Based on these animal studies, a phase 1/2 clinical trial using an rAAV5 serotype at doses ranging from 2 × 1012 vg/kg to 6 × 1013 vg/kg was initiated to evaluate the incidence of treatment‐related adverse events and to determine the dose required to achieve FVIII activity ≥5% of normal (BioMarin; NCT02576795; Table 3).96 A wide range of FVIII expression was observed in this study, with no evidence of capsid‐mediated immune response. Increased factor expression in the first participant was accompanied by a moderate increase in alanine transaminase (ALT) levels, prompting preemptive corticosteroid use in all other participants to forestall a drop in expression levels. To date, no clear connection among increased ALT, anticapsid T‐cell response, steroid use, and FVIII activity has been demonstrated.3 A 4‐year follow‐up analysis showed no ALT elevations or inhibitor development beyond year 1.97, 98 This has led to several clinical trials with rAAV5, including three phase 3 studies (NCT03370913, NCT03392974, NCT04323098; Table 3).
Results from a phase 1/2 dose‐finding trial using a novel recombinant AAV serotype, Spark200/LK03 (SPK‐8011), carrying a BDD‐FVIII (Spark Therapeutics; NCT03003533; Table 3) demonstrated that doses of 5 × 1011 and 1 × 1012 vg/kg led to increased expression levels ranging from 5.2% to 19.8% in the first two dose cohorts. In two participants, reactive corticosteroids were administered for approximately 7 weeks in response to declining FVIII levels without ALT elevation, likely due to a capsid‐based immune response, as well as loss of FVII expression. Steady‐state FVIII expression was achieved by 8 to 12 weeks in seven of nine subjects in the 2 × 1012vg/kg cohort, and at >2‐year follow‐up there was neither a change in FVIII levels nor elevations in ALT and no evidence of immune response to capsid antigens99 (NCT03432520; Table 3).
A dose‐finding trial of another novel vector, an rAAV‐hu37 serotype with a liver‐specific promotor/enhancer combination optimized for transgenic expression, has also demonstrated some success (Bayer/Ultragenyx; NCT03588299; Table 3). Of the six evaluable subjects who received this therapy, five achieved and maintained clinically meaningful FVIII levels, and one resumed prophylaxis; however, at least four subjects experienced bleeding after vector administration.100
Results of a phase 1/2 FVIII gene therapy study using an AAV6 vector serotype (SB‐525/PF‐07055480) demonstrated steady FVIII activity by week 9 following vector administration in four participants. Mean FVIII activity from week 9 to week 52 was 70.4%.101 A phase 3 study evaluating this FVIII gene therapy is ongoing (NCT04370054).
The most significant limitations of these FVIII gene therapy studies include their short‐term follow‐up and the reduction in expression seen in one study. It remains to be determined whether this is true for all FVIII gene therapy products.
5. CHALLENGES ASSOCIATED WITH GENE THERAPY DELIVERY AND EXPRESSION IN THE CONTEXT OF VECTOR SCIENCE
rAAV vectors can preferentially integrate at chromosome breaks at the location of DNA repair. Thus, in order for gene therapy to be appropriate for use in children, the possibility of insertional mutagenesis in growing pediatric livers with rapidly dividing cells remains to be resolved.3
Currently, people with preexisting NAbs to rAAVs are generally excluded from clinical trials. For this reason, as part of the Biologics License Application for an investigational AAV5 gene therapy, a companion diagnostic that tests for preexisting anti‐rAAV5 NAbs was also submitted for FDA approval.15
There is no agreement yet on which vector properties may account for the differences in factor expression. Do the differences stem from DNA conformation, from the presence of particular nucleotide sequences, vector capsid identity, product‐related impurities acquired during manufacture (such as excess noninfectious capsids, DNA contaminants, and/or other proteins), the content of the empty capsid in the final formulation, or from a combination of all these factors?37, 41
A follow‐up to the original rAAV2‐FIX study62, 102 demonstrated the persistence of high‐titer AAV NAbs for up to 15 years following vector administration. NAbs against AAV5 and AAV8 were also detected, likely reflecting cross‐reactivity of AAV antibodies. The significance of these results to vector capsid selection and engineering for lower immunogenicity and higher transduction rates is unknown.102
5.1. Approaches to reducing the effects of preexisting NAbs
An immunoadsorption procedure to remove preexisting AAV NAbs before infusion is being explored and could enable gene delivery to individuals with anti‐AAV NAbs. In an rAAV5‐F9 study using NHPs, the group that underwent immunoadsorption demonstrated lower levels of circulating NAbs (mean NAb titer decreased by >1 log after three consecutive cycles, with an average 2.3‐fold reduction per cycle) and higher factor expression than a control group. Proof of concept in humans was demonstrated in four subjects with autoimmune diseases who underwent testing for reductions in various serotypes of AAVs (the procedure resulted in an average 1.8‐fold reduction in immunoglobulin G (IgG) levels per cycle across all four subjects).103
Another strategy being explored is the use of an endopeptidase that degrades circulating IgG to eliminate circulating anti‐rAAV NAbs before rAAV gene therapy. Imlifidase, an immunoglobulin G–degrading enzyme of Streptococcus pyogenes (IdeS), is currently being evaluated in subjects who have received a solid‐organ transplant. In a mouse model of rAAV8‐mediated F9 gene therapy, imlifidase administration decreased anti‐AAV antibodies and enabled efficient liver F9 transgene expression. The results were confirmed in NHPs, a natural host for WT AAV8. Imlifidase is derived from Streptococcus pyogenes, and natural humoral immunity against Streptococcus pyogenes may represent an obstacle for human use. However, preliminary results suggest that even in the presence of anti‐imlifidase antibodies this enzyme can be effective.104 Human proof‐of‐concept data have not been published and are needed for a better understanding. Another study in animal models analyzed IdeZ, a homolog of IdeS, which efficiently cleaves IgG in a similar manner to IdeS105 and may increase the number of potential eligible individuals for clinical trials. An important caveat to these studies is the inclusion of models with only modest titers of NAbs. Additional analysis is needed in samples with high antibody titers, which are more reflective of potential participants in gene therapy redosing studies.
Although no hemophilia gene therapy has delivered predictable and durable physiologic levels of factor expression to date, there is broad agreement that factor expression can be increased to eliminate spontaneous bleeding.106 Improvements in vector design and delivery are needed to ensure consistently high, durable expression that can achieve long‐term therapeutic success.2 Ongoing work is aimed at developing new vector types with properties that optimize transduction and minimize immunogenicity via capsid bioengineering and isolation of natural liver‐tropic variants.4, 52, 70, 78
Insertional mutagenesis and risk of tumor development, while rare and more often associated with retroviral than with rAAV therapy, remain a potential risk of rAAV vectors. Even at low rates, random integration of vector genomes into the host DNA may lead to deleterious mutations that may alter cell functionality and homeostasis.6, 65
In a long‐term study of rAAV‐FVIII gene therapy in dogs with 10 years of data, AAV gene integration and clonal expansion occurred; however, no instances of malignancy have been reported to date.6, 65 Similar data were obtained in hemophilia B dog models with more than 8 years of data of an rAAV2‐FIX gene therapy.107 So far, no evidence of genotoxicity has emerged from long‐term follow‐up of human clinical trials.37
A recent report of the development of hepatocellular carcinoma (HCC) in a person with hemophilia B 1 year after receiving etranacogene dezaparvovec (AMT‐061) concluded that the HCC was not likely related to the study treatment.108
5.2. Limited patient eligibility for gene therapy
There is the potential that strict eligibility criteria for most clinical trials may lead to restricted indications for gene therapy, such as in healthy adult males. Primary exclusion criteria in most studies have generally included preexisting AAV Nabs, pediatric or elderly individuals, history of HIV or hepatitis B or hepatitis C virus infection, cardiovascular disease, tuberculosis or fungal infection, inflammatory diseases or use of immunomodulatory agents, malignancy, history of factor inhibitors, and other bleeding disorders.3
6. SUMMARY
Gene therapy represents a potential functional cure for people with hemophilia and is being developed to provide safe, durable, and effective factor expression. In vivo investigational rAAV gene therapy has been demonstrated to ameliorate the bleeding phenotype in adults, but long‐term safety and effectiveness remain to be established. Current research seeks to improve vector and other gene therapy attributes to achieve treatment success with simpler and more cost‐effective protocols and to expand access to individuals who are not currently candidates due to comorbidities, medical history, age, or other factors.
RELATIONSHIP DISCLOSURE
LL reports personal fees from Spark Therapeutics, and personal and other fees from LogicBio Therapeutics, outside the submitted work. In addition, LL has a patent new AAV capsid proteins for nucleic acid transfer patent, with royalties paid to LogicBio Therapeutics and Spark Therapeutics; a methods and AAV vectors for in vivo transduction patent pending; and an adeno‐associated virus capsid polypeptides and vectors pending. JMS reports nonfinancial support from Spark Therapeutics; and personal fees from Bayer, CSL Behring, Genentech, Sanofi, Spark Therapeutics, Takeda, Novo Nordisk, outside the submitted work. JFW was an employee and stockholder of Spark Therapeutics before the development of this manuscript. LAV was an employee and stockholder of Spark Therapeutics before the development of this manuscript. The funders had no role in the writing of the manuscript.
AUTHOR CONTRIBUTIONS
All the authors contributed equally to the development of this manuscript.
ACKNOWLEDGEMENTS
Writing and editorial assistance was provided to the authors by Charlotte Caine, PhD, of Axiom Healthcare Strategies and was funded by Spark Therapeutics.
Lisowski L, Staber JM, Wright JF, Valentino LA. The intersection of vector biology, gene therapy, and hemophilia. Res Pract Thromb Haemost. 2021;5:e12586. 10.1002/rth2.12586
Handling Editor: Dr Johnny Mahlangu
Funding information
Writing and editorial assistance was funded by Spark Therapeutics
Contributor Information
Janice M. Staber, @janice_staber.
Leonard A. Valentino, Email: lvalentino@hemophilia.org, @LenValentino1.
REFERENCES
- 1.Miesbach W, O'Mahony B, Key NS, Makris M. How to discuss gene therapy for haemophilia? A patient and physician perspective. Haemophilia. 2019;25(4):545‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.High KA, Roncarolo MG. Gene therapy. N Engl J Med. 2019;381(5):455‐464. [DOI] [PubMed] [Google Scholar]
- 3.Doshi BS, Arruda VR. Gene therapy for hemophilia: what does the future hold? Ther Adv Hematol. 2018;9(9):273‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cabanes‐Creus M, Westhaus A, Navarro RG, et al. Attenuation of heparan sulfate proteoglycan binding enhances in vivo transduction of human primary hepatocytes with AAV2. Mol Ther Methods Clin Dev. 2020;17:1139‐1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inagaki K, Piao C, Kotchey NM, Wu X, Nakai H. Frequency and spectrum of genomic integration of recombinant adeno‐associated virus serotype 8 vector in neonatal mouse liver. J Virol. 2008;82(19):9513‐9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nguyen GN, Everett JK, Kafle S, et al. A long‐term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat Biotechnol. 2021;39(1):47‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu J, Huang X, Yang Y. Innate immune response to adenoviral vectors is mediated by both toll‐like receptor‐dependent and ‐independent pathways. J Virol. 2007;81(7):3170‐3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuranda K, Jean‐Alphonse P, Leborgne C, et al. Exposure to wild‐type AAV drives distinct capsid immunity profiles in humans. J Clin Invest. 2018;128(12):5267‐5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zaiss AK, Cotter MJ, White LR, et al. Complement is an essential component of the immune response to adeno‐associated virus vectors. J Virol. 2008;82(6):2727‐2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Denard J, Marolleau B, Jenny C, et al. C‐reactive protein (CRP) is essential for efficient systemic transduction of recombinant adeno‐associated virus vector 1 (rAAV‐1) and rAAV‐6 in mice. J Virol. 2013;87(19):10784‐10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muhuri M, Maeda Y, Ma H, et al. Overcoming innate immune barriers that impede AAV gene therapy vectors. J Clin Invest. 2021;131(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia. Haemophilia. 2020;26(Suppl 6):1‐158. [DOI] [PubMed] [Google Scholar]
- 13.Valentino LA, Khair K. Prophylaxis for hemophilia A without inhibitors: treatment options and considerations. Expert Rev Hematol. 2020;13(7):731‐743. [DOI] [PubMed] [Google Scholar]
- 14.Hemlibra (emicizumab‐kxwh) injection, for subcutaneous use [prescribing information]. F. Hoffman‐La Roche AG; 2017. [Google Scholar]
- 15.Pierce GF. Uncertainty in an era of transformative therapy for haemophilia: addressing the unknowns. Haemophilia. 2020. [DOI] [PubMed] [Google Scholar]
- 16.Milone MC, O'Doherty U. Clinical use of lentiviral vectors. Leukemia. 2018;32(7):1529‐1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doering CB, Denning G, Shields JE, et al. Preclinical development of a hematopoietic stem and progenitor cell bioengineered factor VIII lentiviral vector gene therapy for hemophilia A. Hum Gene Ther. 2018;29(10):1183‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcucci KT, Jadlowsky JK, Hwang WT, et al. Retroviral and lentiviral safety analysis of gene‐modified T cell products and infused HIV and oncology patients. Mol Ther. 2018;26(1):269‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gandara C, Affleck V, Stoll EA. Manufacture of third‐generation lentivirus for preclinical use, with process development considerations for translation to good manufacturing practice. Hum Gene Ther Methods. 2018;29(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cartier N, Hacein‐Bey‐Abina S, Bartholomae CC, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X‐linked adrenoleukodystrophy. Science. 2009;326(5954):818‐823. [DOI] [PubMed] [Google Scholar]
- 21.Thompson AA, Walters MC, Kwiatkowski J, et al. Gene therapy in patients with transfusion‐dependent beta‐thalassemia. N Engl J Med. 2018;378(16):1479‐1493. [DOI] [PubMed] [Google Scholar]
- 22.European Medicines Agency. Zynteglo (betibeglogene autotemcel). bluebird bio; 2020. [Google Scholar]
- 23.Chang AH, Stephan MT, Lisowski L, Sadelain M. Erythroid‐specific human factor IX delivery from in vivo selected hematopoietic stem cells following nonmyeloablative conditioning in hemophilia B mice. Mol Ther. 2008;16(10):1745‐1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mamcarz E, Zhou S, Lockey T, et al. Lentiviral gene therapy with low dose busulfan for infants with X‐SCID results in the development of a functional normal immune system: interim results of an ongoing phase I/II clinical study. Blood. 2019;134(suppl_1):2058. [Google Scholar]
- 25.Shi Q. Platelet‐targeted gene therapy for hemophilia. Mol Ther Methods Clin Dev. 2018;9:100‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Staber JM, Pollpeter MJ, Anderson CG, et al. Long‐term correction of hemophilia A mice following lentiviral mediated delivery of an optimized canine factor VIII gene. Gene Ther. 2017;24(11):742‐748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milani M, Annoni A, Moalli F, et al. Phagocytosis‐shielded lentiviral vectors improve liver gene therapy in nonhuman primates. Sci Transl Med. 2019;11(493):eaav7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cantore A, Ranzani M, Bartholomae CC, et al. Liver‐directed lentiviral gene therapy in a dog model of hemophilia B. Sci Transl Med. 2015;7(277):277ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID‐X1 patients. J Clin Invest. 2008;118(9):3143‐3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nidetz NF, McGee MC, Tse LV, et al. Adeno‐associated viral vector‐mediated immune responses: understanding barriers to gene delivery. Pharmacol Ther. 2020;207:107453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shirley JL, de Jong YP, Terhorst C, Herzog RW. Immune responses to viral gene therapy vectors. Mol Ther. 2020;28(3):709‐722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaudet D, Methot J, Dery S, et al. Efficacy and long‐term safety of alipogene tiparvovec (AAV1‐LPLS447X) gene therapy for lipoprotein lipase deficiency: an open‐label trial. Gene Ther. 2013;20(4):361‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luxturna (Voretigene Neparvovec‐rzyl) Intraocular Suspension for Subretinal Injection. Spark Therapeutics, Inc; 2017. [Google Scholar]
- 34.Zolgensma (Onasemnogene Abeparvovec‐xioi) Suspension for intravenous infusion. AveXis, Inc; 2019. [Google Scholar]
- 35.European Medicines Agency. Zolgensma. 2020.
- 36.Howard DB, Harvey BK. Assaying the stability and inactivation of AAV serotype 1 vectors. Hum Gene Ther Methods. 2017;28(1):39‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verdera HC, Kuranda K, Mingozzi F. AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol Ther. 2020;28(3):723‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monahan PE, Négrier C, Tarantino M, Valentino LA, Mingozzi F. Emerging immunogenicity and genotoxicity considerations of adeno‐associated virus vector gene therapy for hemophilia. J Clin Med. 2021;10(11):2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finn JD, Ozelo MC, Sabatino DE, et al. Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood. 2010;116(26):5842‐5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boutin S, Monteilhet V, Veron P, et al. Prevalence of serum IgG and neutralizing factors against adeno‐associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21(6):704‐712. [DOI] [PubMed] [Google Scholar]
- 41.Mingozzi F, High KA. Overcoming the host immune response to adeno‐associated virus gene delivery vectors: the race between clearance, tolerance, neutralization, and escape. Annu Rev Virol. 2017;4(1):511‐534. [DOI] [PubMed] [Google Scholar]
- 42.Recht M. Clinical outcomes in patients with and without pre‐existing neutralizing antibodies to the vector: 6 month data from the phase 3 HOPE‐B gene therapy trial of etranacogene dezaparvovec. Mol Ther. 2021;29(4, Supplement 1):1‐427.33308439 [Google Scholar]
- 43.Duan D, Systemic AAV. Micro‐dystrophin gene therapy for duchenne muscular dystrophy. Mol Ther. 2018;26(10):2337‐2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solid Biosciences Provides SGT‐001 Program Update 2020 [Available from: https://www.solidbio.com/about/media/press‐releases/solid‐biosciences‐provides‐sgt‐001‐program‐update].
- 45.Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122(1):23‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiang Z, Kurupati RK, Li Y, et al. The effect of CpG sequences on capsid‐specific CD8(+) T cell responses to AAV vector gene transfer. Mol Ther. 2020;28(3):771‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Konkle BA, Walsh CE, Escobar MA, et al. BAX 335 hemophilia B gene therapy clinical trial results: potential impact of CpG sequences on gene expression. Blood. 2021;137(6):763‐774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sonntag F, Schmidt K, Kleinschmidt JA. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc Natl Acad Sci U S A. 2010;107(22):10220‐10225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McLaughlin SK, Collis P, Hermonat PL, Muzyczka N. Adeno‐associated virus general transduction vectors: analysis of proviral structures. J Virol. 1988;62(6):1963‐1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Logan GJ, Alexander IE. Adeno‐associated virus vectors: immunobiology and potential use for immune modulation. Curr Gene Ther. 2012;12(4):333‐343. [DOI] [PubMed] [Google Scholar]
- 51.Asokan A, Schaffer DV, Samulski RJ. The AAV vector toolkit: poised at the clinical crossroads. Mol Ther. 2012;20(4):699‐708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lisowski L, Dane AP, Chu K, et al. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014;506(7488):382‐386. 10.1038/nature12875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holehonnur R, Luong JA, Chaturvedi D, et al. Adeno‐associated viral serotypes produce differing titers and differentially transduce neurons within the rat basal and lateral amygdala. BMC Neurosci. 2014;15:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zinn E, Vandenberghe LH. Adeno‐associated virus: fit to serve. Curr Opin Virol. 2014;8:90‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Urabe M, Ding C, Kotin RM. Insect cells as a factory to produce adeno‐associated virus type 2 vectors. Hum Gene Ther. 2002;13(16):1935‐1943. [DOI] [PubMed] [Google Scholar]
- 56.Russell DW, Hirata RK. Human gene targeting by viral vectors. Nat Genet. 1998;18(4):325‐330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grimm D, Kay MA. From virus evolution to vector revolution: use of naturally occurring serotypes of adeno‐associated virus (AAV) as novel vectors for human gene therapy. Curr Gene Ther. 2003;3(4):281‐304. [DOI] [PubMed] [Google Scholar]
- 58.Logan GJ, Dane AP, Hallwirth CV, et al. Identification of liver‐specific enhancer‐promoter activity in the 3′ untranslated region of the wild‐type AAV2 genome. Nat Genet. 2017;49(8):1267‐1273. [DOI] [PubMed] [Google Scholar]
- 59.Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno‐associated viruses. J Infect Dis. 2009;199(3):381‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perocheau DP, Cunningham S, Lee J, et al. Age‐related seroprevalence of antibodies against AAV‐LK03 in a UK population cohort. Hum Gene Ther. 2019;30(1):79‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aydemir F, Salganik M, Resztak J, et al. Mutants at the 2‐fold interface of adeno‐associated virus type 2 (AAV2) structural proteins suggest a role in viral transcription for AAV capsids. J Virol. 2016;90(16):7196‐7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Manno CS, Pierce GF, Arruda VR, et al. Successful transduction of liver in hemophilia by AAV‐factor IX and limitations imposed by the host immune response. Nat Med. 2006;12(3):342‐347. [DOI] [PubMed] [Google Scholar]
- 63.Mount JD, Herzog RW, Tillson DM, et al. Sustained phenotypic correction of hemophilia B dogs with a factor IX null mutation by liver‐directed gene therapy. Blood. 2002;99(8):2670‐2676. [DOI] [PubMed] [Google Scholar]
- 64.Chandler RJ, Sands MS, Venditti CP. Recombinant adeno‐associated viral integration and genotoxicity: insights from animal models. Hum Gene Ther. 2017;28(4):314‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Colella P, Ronzitti G, Mingozzi F. Emerging issues in AAV‐mediated in vivo gene therapy. Mol Ther Methods Clin Dev. 2018;8:87‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Z, Lisowski L, Finegold MJ, Nakai H, Kay MA, Grompe M. AAV vectors containing rDNA homology display increased chromosomal integration and transgene persistence. Mol Ther. 2012;20(10):1902‐1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sohlenius‐Sternbeck AK. Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements. Toxicol In Vitro. 2006;20(8):1582‐1586. [DOI] [PubMed] [Google Scholar]
- 68.Ronzitti G, Gross D‐A, Mingozzi F. Human immune responses to adeno‐associated virus (AAV) vectors. Front Immunol. 2020;11(670). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pipe S, Leebeek FWG, Ferreira V, Sawyer EK, Pasi J. Clinical considerations for capsid choice in the development of liver‐targeted AAV‐based gene transfer. Mol Ther Methods Clin Dev. 2019;15:170‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paulk NK, Pekrun K, Zhu E, et al. Bioengineered AAV capsids with combined high human liver transduction in vivo and unique humoral seroreactivity. Mol Ther. 2018;26(1):289‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pei X, Shao W, Xing A, et al. Development of AAV variants with human hepatocyte tropism and neutralizing antibody escape capacity. Mol Ther Methods Clin Dev. 2020;18:259‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maheshri N, Koerber JT, Kaspar BK, Schaffer DV. Directed evolution of adeno‐associated virus yields enhanced gene delivery vectors. Nat Biotechnol. 2006;24(2):198‐204. [DOI] [PubMed] [Google Scholar]
- 73.Grimm D, Lee JS, Wang L, et al. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno‐associated viruses. J Virol. 2008;82(12):5887‐5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cabanes‐Creus M, Ginn SL, Amaya AK, et al. Codon‐optimization of wild‐type adeno‐associated virus capsid sequences enhances DNA family shuffling while conserving functionality. Mol Ther Methods Clin Dev. 2019;12:71‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Azuma H, Paulk N, Ranade A, et al. Robust expansion of human hepatocytes in Fah‐/‐/Rag2‐/‐/Il2rg‐/‐ mice. Nat Biotechnol. 2007;25(8):903‐910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nathwani AC, Tuddenham EG, Rangarajan S, et al. Adenovirus‐associated virus vector‐mediated gene transfer in hemophilia B. N Engl J Med. 2011;365(25):2357‐2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Davidoff AM, Gray JT, Ng CY, et al. Comparison of the ability of adeno‐associated viral vectors pseudotyped with serotype 2, 5, and 8 capsid proteins to mediate efficient transduction of the liver in murine and nonhuman primate models. Mol Ther. 2005;11(6):875‐888. [DOI] [PubMed] [Google Scholar]
- 78.Cabanes‐Creus M, Hallwirth CV, Westhaus A, et al. Restoring the natural tropism of AAV2 vectors for human liver. Sci Transl Med. 2020;12(560):eaba3312. [DOI] [PubMed] [Google Scholar]
- 79.Shestopal SA, Hao JJ, Karnaukhova E, et al. Expression and characterization of a codon‐optimized blood coagulation factor VIII. J Thromb Haemost. 2017;15(4):709‐720. [DOI] [PubMed] [Google Scholar]
- 80.Gernoux G, Wilson JM, Mueller C. Regulatory and exhausted T cell responses to AAV capsid. Hum Gene Ther. 2017;28(4):338‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kattenhorn LM, Tipper CH, Stoica L, et al. Adeno‐associated virus gene therapy for liver disease. Hum Gene Ther. 2016;27(12):947‐961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cogger VC, O'Reilly JN, Warren A, Le Couteur DG. A standardized method for the analysis of liver sinusoidal endothelial cells and their fenestrations by scanning electron microscopy. J Vis Exp. 2015;98:e52698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jacobs F, Gordts SC, Muthuramu I, De Geest B. The liver as a target organ for gene therapy: state of the art, challenges, and future perspectives. Pharmaceuticals (Basel). 2012;5(12):1372‐1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Johnson TN, Tucker GT, Tanner MS, Rostami‐Hodjegan A. Changes in liver volume from birth to adulthood: a meta‐analysis. Liver Transpl. 2005;11(12):1481‐1493. [DOI] [PubMed] [Google Scholar]
- 85.Manno CS, Chew AJ, Hutchison S, et al. AAV‐mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood. 2003;101(8):2963‐2972. [DOI] [PubMed] [Google Scholar]
- 86.Jiang H, Pierce GF, Ozelo MC, et al. Evidence of multiyear factor IX expression by AAV‐mediated gene transfer to skeletal muscle in an individual with severe hemophilia B. Mol Ther. 2006;14(3):452‐455. [DOI] [PubMed] [Google Scholar]
- 87.Nathwani AC, Reiss UM, Tuddenham EG, et al. Long‐term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371(21):1994‐2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nathwani AC, Reiss U, Tuddenham E, et al. Adeno‐associated mediated gene transfer for hemophilia B: 8 year follow up and impact of removing “empty viral particles” on safety and efficacy of gene transfer. Blood. 2018;132(suppl 1):491. [Google Scholar]
- 89.Simioni P, Tormene D, Tognin G, et al. X‐linked thrombophilia with a mutant factor IX (factor IX Padua). N Engl J Med. 2009;361(17):1671‐1675. [DOI] [PubMed] [Google Scholar]
- 90.Monahan PE, Sun J, Gui T, et al. Employing a gain‐of‐function factor IX variant R338L to advance the efficacy and safety of hemophilia B human gene therapy: preclinical evaluation supporting an ongoing adeno‐associated virus clinical trial. Hum Gene Ther. 2015;26(2):69‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.George LA, Sullivan SK, Rasko JEJ, et al. Efficacy and safety in 15 hemophilia B patients treated with the AAV gene therapy vector fidanacogene elaparvovec and followed for at least 1 year. Blood. 2019;134(suppl 1):3347. [Google Scholar]
- 92.Pipe SW, Recht M, Key NS, et al. First data from the phase 3 HOPE‐B gene therapy trial: efficacy and safety of etranacogene dezaparvovec (AAV5‐Padua hFIX variant; AMT‐061) in adults with severe or moderate‐severe hemophilia B treated irrespective of pre‐existing anti‐capsid neutralizing antibodies. Blood. 2020;136(suppl 2):LBA‐6. [Google Scholar]
- 93.Pipe S, Rajasekhar A, Everington T, et al. 101HEMB01 is a phase 1/2 open‐label, single ascending dose‐finding trial of DTX101 (AAVrh10FIX) in patients with moderate/severe hemophilia B that demonstrated meaningful but transient expression of human factor IX (hFIX). Blood. 2017;130:3331. [Google Scholar]
- 94.Chowdary P. editor. Follow‐up on a novel adeno‐associated virus (AAV) gene therapy (FLT180a) achieving normal FIX activity levels in severe haemophilia B (HB) patients (B‐AMAZE study). Annual Congress of European Association for Haemophilia and Allied Disorders (EAHAD). 2021; virtual: Haemophilia.
- 95.Nathwani AC. Data update from the B‐AMAZE phase 1/2 study–verbrinacogene setparvovec (FLT180a); virtual: Freeline corporate presentation. 2020. Available from: https://www.freeline.life/media/1349/presentation‐freeline‐company‐update‐14‐dec‐2020.pdf.
- 96.Rangarajan S, Walsh L, Lester W, et al. AAV5‐factor VIII gene transfer in severe hemophilia A. N Engl J Med. 2017;377(26):2519‐2530. [DOI] [PubMed] [Google Scholar]
- 97.Pasi KJ, Rangarajan S, Mitchell N, Lester W, Laffan M & Madan B et al. First‐in‐human evidence of durable therapeutic efficacy and safety of AAV gene therapy over three‐years with valoctocogene roxaparvovec for severe hemophilia A (BMN 270‐271 study). 64th Annual Meeting of Society of Thrombosis and Haemostasis Research. 2020;OC05‐2‐AB.
- 98.Pasi J. editor. First‐in‐human four‐year follow‐up study of durable therapeutic efficacy and safety: AAV gene therapy with valoctocogene roxaparvovec for severe hemophilia A. World Federation of Hemophilia Virtual Summit. 2020; virtual.
- 99.George LEE, Ragni M, Sullivan S, et al. Phase I/II trial of SPK‐8011: stable and durable FVIII expression for >2 years with significant ABR improvements in initial dose cohorts following AAV‐mediated FVIII gene transfer for hemophilia A. Res Pract Thromb Haemost. 2020;4. [Google Scholar]
- 100.Ferrante F. editor. First‐in‐human AAVhu37‐based gene therapy study in severe haemophilia A ‐ BAY 2599023 has stable and sustained expression of FVIII. Annual Congress of European Association for Haemophilia and Allied Disorders (EAHAD); 2021; virtual: Haemophilia.
- 101.Leavitt AD, Konkle BA, Stine K, et al. Updated follow‐up of the Alta Study, a phase 1/2 study of giroctocogene fitelparvovec (SB‐525) gene therapy in adults with severe hemophilia A. Blood. 2020;136(suppl 1):12. [Google Scholar]
- 102.George LA, Ragni MV, Rasko JEJ, et al. Long‐term follow‐up of the first in human intravascular delivery of AAV for gene transfer: AAV2‐hFIX16 for severe hemophilia B. Mol Ther. 2020;28(9):2073‐2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Salas D, Kwikkers KL, Zabaleta N, et al. Immunoadsorption enables successful rAAV5‐mediated repeated hepatic gene delivery in nonhuman primates. Blood Adv. 2019;3(17):2632‐2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Leborgne C, Barbon E, Alexander JM, et al. IgG‐cleaving endopeptidase enables in vivo gene therapy in the presence of anti‐AAV neutralizing antibodies. Nat Med. 2020;26(7):1096‐1101. [DOI] [PubMed] [Google Scholar]
- 105.Elmore ZC, Oh DK, Simon KE, Fanous MM, Asokan A. Rescuing AAV gene transfer from neutralizing antibodies with an IgG‐degrading enzyme. JCI. Insight. 2020;5(19):e139881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Karimipoor M. Study break: recent advances in hemophilia gene therapy. Iran Biomed J. 2019;23(1):7‐8. [PMC free article] [PubMed] [Google Scholar]
- 107.Niemeyer GP, Herzog RW, Mount J, et al. Long‐term correction of inhibitor‐prone hemophilia B dogs treated with liver‐directed AAV2‐mediated factor IX gene therapy. Blood. 2009;113(4):797‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.uniQure announces findings from reported case of hepatocellular carcinoma (HCC) in hemophilia B gene therapy program [press release]. 2021.
- 109.Brown BD, Venneri MA, Zingale A, Sergi Sergi L, Naldini L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat Med. 2006;12(5):585‐591. [DOI] [PubMed] [Google Scholar]
- 110.Miesbach W, Meijer K, Coppens M, et al. Gene therapy with adeno‐associated virus vector 5‐human factor IX in adults with hemophilia B. Blood. 2018;131(9):1022‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sankar AD, Weyand AC, Pipe SW. The evolution of recombinant factor replacement for hemophilia. Transfus Apher Sci. 2019;58(5):596‐600. [DOI] [PubMed] [Google Scholar]
- 112.Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet. 2016;388(10040):187‐197. [DOI] [PubMed] [Google Scholar]
- 113.Di Minno MN, Ambrosino P, Franchini M, Coppola A, Di Minno G. Arthropathy in patients with moderate hemophilia: a systematic review of the literature. Semin Thromb Hemost. 2013;39(7):723‐731. [DOI] [PubMed] [Google Scholar]
- 114.Den Uijl IE, Mauser Bunschoten EP, Roosendaal G, et al. Clinical severity of haemophilia A: does the classification of the 1950s still stand? Haemophilia. 2011;17(6):849‐853. [DOI] [PubMed] [Google Scholar]
- 115.Soucie JM, Monahan PE, Kulkarni R, Konkle BA, Mazepa MA, Network USHTC. The frequency of joint hemorrhages and procedures in nonsevere hemophilia A vs B. Blood Adv. 2018;2(16):2136‐2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.GUIDANCE DOCUMENT: Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs). In: Research CfBEa, editor. 2020.
- 117.George LA, Sullivan SK, Giermasz A, et al. Hemophilia B gene therapy with a high‐specific‐activity factor IX variant. N Engl J Med. 2017;377(23):2215‐2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Leebeek FWG, Meijer K, Coppens M, et al. AMT‐060 gene therapy in adults with severe or moderate‐severe hemophilia B confirm stable FIX expression and durable reductions in bleeding and factor IX consumption for up to 5 years. Blood. 2020;136(suppl 1):26. [Google Scholar]
- 119.Pipe S. editor. First data from the phase 3 HOPE‐B gene therapy trial: efficacy and safety of etranacogene dezaparvovec (AAV5‐Padua hFIX variant; AMT‐061) in adults with severe or moderate‐severe hemophilia B treated irrespective of pre‐existing anti‐capsid neutralizing antibodies. Annual Congress of the American Society of Hematology (ASH). 2020;136(Suppl 2):LBA‐6. [Google Scholar]
- 120.Von Drygalski A, Giermasz A, Castaman G, et al. Etranacogene dezaparvovec (AMT‐061 phase 2b): normal/near normal FIX activity and bleed cessation in hemophilia B. Blood Adv. 2019;3(21):3241‐3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pasi KJ, Rangarajan S, Mitchell N, et al. Multiyear follow‐up of AAV5‐hFVIII‐SQ gene therapy for hemophilia A. N Engl J Med. 2020;382(1):29‐40. [DOI] [PubMed] [Google Scholar]
- 122.BioMarin announces positive phase 3 gene therapy trial results in adults with severe hemophilia A; study met all primary and secondary efficacy endpoints in one‐year data set [press release]. 2021.
- 123.Sullivan SK, editor. SPK‐8016: Preliminary results from a phase 1/2 clinical trial of gene therapy for hemophilia A. Annual Congress of European Association for Haemophilia and Allied Disorders (EAHAD); 2021; Virtual: Haemophilia.
- 124.Pipe SW, Ferrante F, Reis M, et al. First‐in‐human gene therapy study of AAVhu37 capsid vector technology in severe hemophilia A ‐ BAY 2599023 has broad patient eligibility and stable and sustained long‐term expression of FVIII. Blood. 2020;136(Suppl 1):44‐45. [Google Scholar]
- 125.Chapin J, editor. Results from a phase 1/2 safety and dose escaaltion study of TAK‐754, an AAV8 vector with a codon‐optimized B‐domain‐deleted factor VIII transgene in severe hemophilia A. Annual Congress of European Association for Haemophilia and Allied Disorders (EAHAD); 2021 3‐5 February, 2021; Virtual: Haemophilia.