

Summary
Brain function relies almost solely on glucose as an energy substrate. The main model of brain metabolism proposes that glucose is taken up and converted into lactate by astrocytes to fuel the energy-demanding neuronal activity underlying plasticity and memory. Whether direct neuronal glucose uptake is required for memory formation remains elusive. We uncover, in Drosophila, a mechanism of glucose shuttling to neurons from cortex glia, an exclusively perisomatic glial subtype, upon formation of olfactory long-term memory (LTM). In vivo imaging reveals that, downstream of cholinergic activation of cortex glia, autocrine insulin signaling increases glucose concentration in glia. Glucose is then transferred from glia to the neuronal somata in the olfactory memory center to fuel the pentose phosphate pathway and allow LTM formation. In contrast, our results indicate that the increase in neuronal glucose metabolism, although crucial for LTM formation, is not routed to glycolysis.
Keywords: glia, long-term memory, glucose, energy metabolism, pentose phosphate pathway, insulin, Drosophila, mushroom body, nicotinic acetylcholine receptor
Graphical abstract
Highlights
-
•
Neuronal glucose metabolism is increased upon long-term memory formation
-
•
Glial cells shuttle glucose to neurons following insulin signaling activation
-
•
Glucose fuels the neuronal pentose phosphate pathway
Higher brain functions are associated with increased metabolic demand. Through in vivo imaging and behavioral approaches, de Tredern et al. reveal that the neuronal pentose phosphate pathway is crucial for long-term memory formation, supported by glucose transfer from glia to neurons.
Introduction
In humans, the brain consumes about 20% of whole-body energy (Attwell and Laughlin, 2001). It is currently assumed that most of the brain’s energy is consumed by neurons for molecular processes that support neurotransmission, such as action potential generation, ion concentration restoration, or vesicular recycling (Alle et al., 2009; Harris et al., 2012). Glucose is the major energy metabolite consumed by the brain under physiological conditions (Siesjö, 1978) that can be stored locally as glycogen (Magistretti and Allaman, 2015). Glycogen is not stored in the energy-demanding neurons but in neighboring glia in various species (Kis et al., 2015; Magistretti and Allaman, 2015). This compartmentation imposes the existence of coupling mechanisms between neurons and glia so that neuronal energy needs and supply are matched precisely. At the synapse, the astrocyte-neuron lactate shuttle (ANLS) is one proposed mechanism that couples glutamate uptake and recycling by astrocytes with transfer of glycolysis-derived lactate to neurons (Pellerin and Magistretti, 1994; Magistretti and Allaman, 2015). In this scenario, lactate would then be converted into pyruvate to fuel neuronal oxidative phosphorylation and produce ATP. This effectively explains how the energy supply can be adjusted to local synaptic activity. However, this paradigm is still debated; other studies suggest that glucose itself is used preferentially by neurons upon acute activation (Bak et al., 2009; Díaz-García et al., 2017; Ashrafi et al., 2017; Bak and Walls, 2018; Barros and Weber, 2018), whereas lactate uptake would occur mostly at rest (Yellen, 2018). In response to sensory stimulation, neurons metabolize glucose for energy production through glycolysis (Díaz-García et al., 2017). In contrast, it has also been shown that glycolysis is actively inhibited in neurons, with glucose preferentially fueling the pentose phosphate pathway (PPP) to protect them against oxidative stress (Herrero-Mendez et al., 2009). Thus, the fate of glucose in neurons is still unclear, although some studies have started to address the question directly by cell-type-specific in vivo visualization of activity-dependent glucose consumption (Díaz-García et al., 2017, 2019; Keller et al., 2021). In particular, how glucose is used by neurons upon memory formation remains elusive.
Long-term memory (LTM) is a paramount example of a cognitive process featuring a broad spectrum of energy-demanding events, such as activity-dependent synaptic plasticity, gene expression regulation and de novo protein synthesis. Several studies in rodents have shown that lactate supply to neurons from astrocytes is necessary for LTM formation but dispensable for short-term memory (Newman et al., 2011; Suzuki et al., 2011; Gao et al., 2016). After conditioning, glycogen-derived lactate is transported from astrocytes to neurons to fuel neuronal activity and local protein translation at the synapses (Descalzi et al., 2019). However, it is doubtful that the cost of LTM only stems from protein synthesis in a limited number of synapses. As an illustration, it has been reported that, in Drosophila, the high metabolic cost of LTM formation can imbalance the energy budget of the whole organism (Mery and Kawecki, 2005) so that adaptive mechanisms exist to shut down LTM formation under nutritional deficit (Plaçais and Preat, 2013). This suggests that other signaling events and regulations, uncoupled from synaptic activity, occur at the scale of the whole cell, representing additional sinks in energy fuels, whose nature remains to be identified. Notably, it is still unknown whether, in addition to lactate, neuronal glucose uptake is also increased to form LTM.
The genetic tractability and corresponding versatile analytic tools available in Drosophila offer the possibility to manipulate specific pathways in precisely defined cell types, making it a choice organism to address this question. The best-established memory paradigm in Drosophila is associative olfactory conditioning, resulting from pairing of an odorant with electric shocks (Tully et al., 1994). Although memory decays within a few hours after a single pairing (1× training), stable protein synthesis-dependent LTM can be formed by subjecting flies to repeated conditioning cycles separated by rest intervals (5× spaced training) (Tully et al., 1994; Yin et al., 1994).
Compared with the fine anatomical and functional dissection of neuronal circuits underlying Drosophila memory (Cognigni et al., 2018), our knowledge of the role of Drosophila glia in memory processes is scarce (Yamazaki et al., 2014; Matsuno et al., 2015). In particular, neuron-glia metabolic coupling during memory formation has not yet been studied in Drosophila. The Drosophila brain contains about 100,000 neurons and 15,000 glial cells (Kremer et al., 2017). All neuronal somata segregate in the periphery of the brain, in a region called the cortex. Neurons project a single neurite toward the center of the brain, in the neuropil, where it can ramify and elaborate synapses. Matching this organization, glial cells are classified into five categories (Awasaki et al., 2008). Two layers of perineurial and subperineural glia form a physical and chemical barrier similar to the mammalian blood-brain barrier, isolating the central nervous system from hemolymph-borne molecules. Immediately underneath lies the cortex region, where neuronal somata are enwrapped individually by processes from cortex glial cells, forming a honeycomb-shaped structure (Freeman, 2015). Within the neuropil, ensheathing glia delimit major brain structures, and astrocyte-like glial cells infiltrate the neuropil in relatively close apposition to the synapses.
Brain energetics in Drosophila have remained understudied so far, but recent studies have revealed strong similarities between flies and vertebrates in terms of metabolic coupling between neurons and glia. From the main circulating sugar trehalose, Drosophila glial cells produce and release glucose, lactate, and alanine (Volkenhoff et al., 2015), which are taken up by neurons and, hence, may serve as potential fuel. It has also been shown that all glial cell types, in addition to neurons, can take up glucose (Volkenhoff et al., 2018). Nonetheless, how production of energy substrates is dispatched and regulated within the different glial cell types and according to different brain functions is unknown. The core neuronal assembly underlying olfactory learning and memory is the mushroom body (MB), a bilateral structure of about 2,000 neurons in each brain hemisphere. MB-intrinsic neurons, called Kenyon cells (KCs), are cholinergic neurons (Barnstedt et al., 2016) with somata packed in the dorso-posterior part of the brain (Figure S1A).
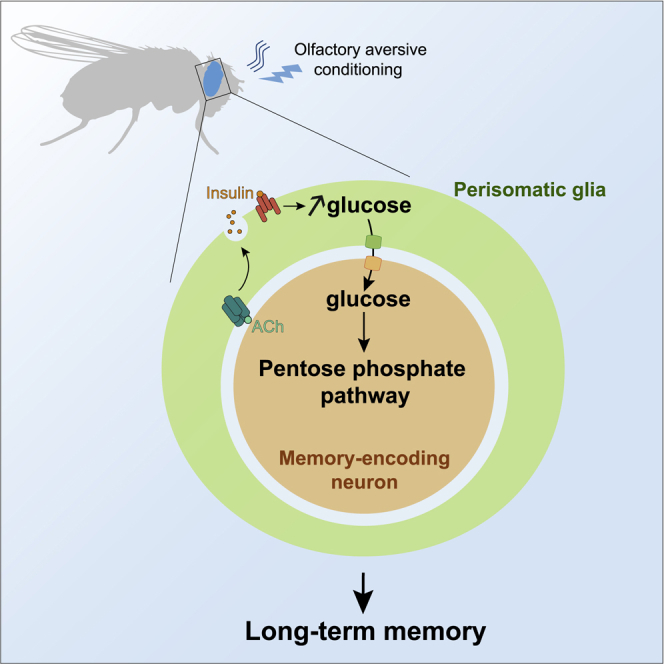
We have previously reported that, in Drosophila, LTM formation involves upregulation of energy metabolism because MB neurons increase their pyruvate consumption after spaced training (Plaçais et al., 2017). In this work, we examined the neuronal reliance on metabolic fuels during LTM formation by addressing glucose dynamics and the role of glia. We first established a specific need for glucose uptake and consumption by MB neurons for LTM. We identified cortex glia as the source of increased glucose supply for neuronal somata. We further showed that activation of cholinergic signaling in cortex glia triggers insulin-dependent autocrine regulation of intracellular glucose concentration. Finally, we revealed that the purpose of the LTM-specific neuronal glucose uptake is to fuel the PPP independent of neuronal pyruvate consumption. Our results demonstrate the critical role of prolonged neuronal glucose consumption, supported by a glucose shuttle between glial cells and neuronal somata during the early phase of memory consolidation.
Results
Glucose uptake is increased in MB neurons during the first hours of LTM formation
The Drosophila glucose transporter gene Glut1, a homolog of Glut1 and Glut3 in mammals (Escher and Rasmuson-Lestander, 1999), is strongly expressed in Drosophila neurons (Volkenhoff et al., 2018). To address the role of potential glucose uptake by MB neurons for LTM, we knocked down Glut1 expression selectively in adult MB neurons. For this, we expressed an RNAi directed against Glut1 under control of the MB neuron-selective driver VT30559-GAL4 (Plaçais et al., 2017) together with the thermosensitive GAL4 inhibitor GAL80ts expressed ubiquitously (tub-GAL80ts; McGuire et al., 2003) to restrict RNAi expression solely to adulthood (STAR Methods). We found that knocking down Glut1 in MB neurons impaired LTM after spaced training (Figure 1A). LTM was not impaired when RNAi expression was not induced (Figure S1B1), ruling out any developmental effect because of leaky RNAi expression in this experiment. Sensory acuity for the relevant stimuli (Figure S1B3) was normal in the flies of interest. To address whether this impairment was specific to LTM, we assessed memory following two other paradigms. The first paradigm was 3 h after a single-cycle training, which elicits a shorter-lasting memory than LTM. The second paradigm was 24 h after a massed training, which consists of multiple consecutive cycles and elicits formation of another long-lasting memory that is distinct from LTM and does not involve de novo protein synthesis (Tully et al., 1994; Isabel et al., 2004; Plaçais et al., 2012, 2017). Memory was not impaired under either of these conditions (Figures 1A and S1B2).
Figure 1.

Glucose uptake is increased in MB neurons during the first hours of LTM formation
(A) Knockdown of Glut1 in MB neurons at the adult stage disrupted LTM compared with the genotypic controls after 5× spaced training (n = 18, F2,53 = 6.79, p = 0.0024) but did not affect memory after 5× massed training (n = 11–14, F2,38 = 0.36, p = 0.70).
(B) Images of the FLII12Pglu-700μδ6 FRET glucose sensor expressed in MB neurons showing the CFP (cyan fluorescent protein, labeled in blue) and YFP (yellow fluorescent protein, labeled in yellow) channels for one brain (scale bar, 20 μm). The dashed lines indicate the regions of interest.
(C) Glucose concentration in the somata of MB neurons following application of validamycin A (4 mM, dashed line) decreased faster in flies after 5× spaced training compared with flies conditioned with a non-associative spaced unpaired training protocol (n = 19–20, t37 = 3.66, p = 0.0008).
(D) No difference in glucose decrease following validamycin application was observed between flies subjected to massed training or to a non-associative massed unpaired protocol (n = 12, t22 = 0.18, p = 0.86). Flies used in this experiment also carried an inducible cortex glia GAL4 driver but no effector (tub-GAL80ts/+; 54H02-GAL4/+) to match the conditions used for other experiments performed in parallel, displayed in Figures 2, 3, 4, and 5.
All data are presented as mean ± SEM. Asterisks illustrate the significance level of the t test, or of the least significant pairwise comparison following an ANOVA, with the following nomenclature: ∗∗p < 0.01; ∗∗∗p < 0.001; ns: not significant, p > 0.05. See also Figure S1.
Because our behavioral results strongly suggest an important role of neuronal glucose metabolism in LTM formation, we conducted functional imaging experiments to monitor glucose uptake and consumption by MB neurons (Figure 1B). We used a genetically encoded fluorescence resonance energy transfer (FRET) glucose sensor, FLII12Pglu-700μδ6 (Takanaga et al., 2008), whose functionality has already been validated in Drosophila brain neurons (Plaçais et al., 2017; Volkenhoff et al., 2018). To measure the net glucose consumption by MB neurons using the FRET glucose sensor, we devised an experimental strategy that consists of acutely blocking new glucose synthesis in the brain and measuring the resulting decrease in glucose concentration over time in neurons of interest. Glucose is supplied to the fly brain upon uptake and breakdown of trehalose, the circulating sugar in the fly body. Degradation of trehalose into glucose is catalyzed by the enzyme trehalase, highly expressed and functional in surface glia, the counterpart of the mammalian blood-brain barrier (Volkenhoff et al., 2015). In the brain, glucose is therefore available for neurons and glial cells. Validamycin A is a water-soluble specific inhibitor of trehalase that has been characterized previously as a potential pesticide in various insects, including Drosophila (Gruber et al., 2013). We monitored glucose concentration in MB somata during acute application of validamycin A, interrupting glucose supply to the brain. In control flies, treatment with validamycin A resulted in a decrease in intracellular glucose over time as expected (Figure 1C). We then performed similar experiments in flies within 2 h after olfactory conditioning. Strikingly, the observed decrease in glucose occurred earlier in flies that had been subjected to spaced training than in flies submitted to non-associative spaced unpaired conditioning that does not allow memory formation (Figure 1C). This effect persisted for 5 h following olfactory conditioning but faded after 12 h (Figure S1E). No such effect could be observed following massed training or single-cycle training (Figures 1D and S1D). These results suggest that the rate of glucose consumption in MB neurons is increased after spaced training.
However, it could be argued that, after spaced training, the rate of glucose consumption would be unchanged, whereas the baseline glucose concentration would be lower in MB neurons. Assuming that the glucose sensor would be initially saturated, this could also result in an earlier observed decrease. We reasoned that use of a lower-affinity glucose sensor would allow us, under the same protocol, to measure the initial glucose level with higher sensitivity, making it possible to rule out this alternative explanation. The single-fluorophore glucose sensor iGlucoSnFR (Keller et al., 2021) displays a 10 times lower affinity for glucose (Kd = 7.7 mM in vitro; Keller et al., 2021) compared with the FLII12Pglu-700μδ6 glucose sensor (Kd ≈ 0.7 mM in vitro; Takanaga et al., 2008). Using this low-affinity sensor, we verified that there was no difference in basal glucose level between flies subjected to spaced or non-associative unpaired training (Figure S1C). We also confirmed with this sensor that spaced training induced a higher rate of glucose decrease following validamycin application (Figure S1C). These results demonstrate that glucose uptake and consumption by MB neuronal somata is increased when LTM formation is initiated.
Glucose transfer from cortex glia to MB neurons is required for LTM
Having established that glucose uptake and consumption by MB neurons is necessary for LTM formation, we sought to determine the source of this glucose. Glial cells provide metabolic support to neurons. To identify which glial subtype could export glucose to feed MB neurons, we selectively knocked down the glucose transporter Glut1 in each of the glial cell types contacting neurons: astrocyte-like, ensheathing, and cortex glia (Figure S2D). LTM was normal when Glut1 RNAi was expressed in adult astrocyte-like glia via the Alrm-GAL4 driver (Figure S2E) or in ensheathing glia via the 56F03-GAL4 driver (Figure S2F). However, selective knockdown of Glut1 in adult cortex glia using the 54H02-GAL4 driver impaired LTM performance after spaced training (Figure 2A). Sensory controls were normal, and without RNAi induction, LTM was normal (Figures S2B1 and S2B2). Moreover, memory after massed training was not affected by Glut1 knockdown in cortex glia (Figures 2A). Expression of Glut1 RNAi exclusively in cortex glia significantly decreased Glut1 mRNA levels in whole heads by about 25% (Figure S2A), confirming that cortex glia express Glut1 in addition to neurons (Volkenhoff et al., 2018). We replicated these behavioral experiments using a second, non-overlapping Glut1 RNAi (Figure S2C). These results indicate that glucose transport across cortex glia is required specifically for LTM.
Figure 2.

Glucose is shuttled from cortex glia to MB neuron somata during LTM formation
(A) Knockdown of Glut1 in adult cortex glia disrupted LTM compared with the genotypic controls after 5× spaced training (n = 13/14, F2,39 = 5.40, p = 0.0088) but did not affect 24-h memory after 5× massed training (n = 12, F2,35 = 0.03, p = 0.97).
(B) The low-affinity glucose sensor iGlucoSnFR was expressed in cortex glia. An orange dashed lines delimit the cortex glia in the posterior part of the brain, where the somata of MB neurons are located. The blue dashed line delimits cortex glia in the anterior brain, a region that does not contain MB neurons. Scale bar, 30 μm.
(C) The basal glucose concentration increased in MB-neuron-surrounding cortex glia (orange) after 5× spaced training compared with after a non-associative spaced unpaired training protocol (two-tailed unpaired t test, n = 16, t30 = 2.72, p = 0.011). The basal glucose concentration in a control anterior region containing no MB neurons (blue) was not changed after 5× spaced training compared with after a non-associative spaced unpaired training protocols (two-tailed unpaired t test, n = 13/14, t25 = 0.011, p = 0.99).
(D) Spaced training failed to elicit a faster decrease in glucose in MB neurons upon knockdown of Glut1 in cortex glia at the adult stage (two-tailed unpaired t test, n = 12, t22 = 0.93, p = 0.36). Data were collected in parallel with the data presented in Figure 1C.
All data are presented as mean ± SEM. Asterisks illustrate the significance level of the t test or of the least significant pairwise comparison following an ANOVA, with the following nomenclature: ∗p < 0.05; ns: not significant, p > 0.05. See also Figure S2.
Because MB neuron somata increased their glucose uptake (Figure 1), we hypothesized that spaced training would involve a flux of glucose from cortex glia to MB neuron somata, which might rely on a rise in glucose availability in cortex glia. Indeed, we observed that the glucose levels in cortex glia, measured with the low-affinity glucose sensor (Figure 2B), were higher in flies subjected to spaced training compared with non-associative training (Figure 2C). Strikingly, this increased glucose level was observed specifically in the posterior cortex glia region surrounding MB neuron somata, whereas the glucose level in cortex glia of an anterior brain domain was unchanged (Figure 2C). These results reveal that spaced training induces a local increase in glucose concentration in cortex glia in the MB region. Because the basal glucose concentration was unchanged in MB neurons (Figure S1C), these results are consistent with the existence of a glia-to-neuron glucose gradient after spaced conditioning, which could drive glucose transport from cortex glia to MB neurons, where it is then metabolized to sustain LTM.
Finally, to test whether Glut1 in cortex glia mediates glucose export to fuel MB neuron somata, we conducted glucose imaging experiments in MB neurons using the FLII12Pglu-700μδ6 glucose FRET sensor with Glut1 knockdown in adult cortex glia. As expected if cortex glia were the source of glucose for neurons, spaced training failed to induce upregulation of glucose metabolism under that condition (Figure 2D). Overall, these results establish the existence of a glucose flux between cortex glia and MB neuron somata sustaining LTM formation.
A cholinergic signal activates cortex glia during LTM formation
The existence of a glucose shuttle between cortex glia and MB neurons hints at a dialog between the two cell types. Therefore, we first investigated the nature of the signal that cortex glia might receive from neurons upon LTM formation. Acetylcholine is the main excitatory neurotransmitter in the fly brain (Yasuyama and Salvaterra, 1999). In particular, MB neurons activate their downstream output neurons through cholinergic synapses (Barnstedt et al., 2016). We thus investigated whether an acetylcholine receptor is required in cortex glia for LTM. Because the nicotinic receptor subunit nAChRα7 is expressed and functional in mammalian glia (Shen and Yakel, 2012; Shytle et al., 2004; Vélez-Fort et al., 2009; Papouin et al., 2017), we tested whether expression of its homologous gene in cortex glia was necessary for LTM. Knocking down nAChRα7 in adult cortex glia strongly impaired LTM performance after spaced training (Figure 3A). LTM was not impaired when RNAi expression was not induced (Figure S3A1). The same genetic manipulation did not affect memory after massed or single-cycle training (Figures 3A and S3A2) or sensory acuity in naive flies (Figure S3A3). We replicated these results using a second, non-overlapping nAChRα7 RNAi (Figure S3B). These data suggest that a cholinergic signal acting on nAChRα7 receptors activates cortex glia, specifically during LTM formation.
Figure 3.

nAChRα7 activation increases glucose concentration in cortex glia
(A) nAChRα7 knockdown in adult cortex glia disrupted LTM compared with the genotypic controls after 5× spaced training (n = 9, F2,26 = 7.46, p = 0.003) but did not affect memory measured 24 h after 5× massed training (n = 8, F2,23 = 0.25, p = 0.78).
(B) The fluorescent calcium reporter GCaMP6f was expressed in cortex glia. A representative 2-photon microscopy image in one focal plane is shown, with the corresponding regions of interest in the dorsal part of the cortex glia in which calcium dynamics were analyzed (dashed lines). Scale bar, 40 μm.
(C) In vivo imaging of calcium dynamics in cortex glia was monitored following nicotine application to the brain. Left panel: average traces of calcium dynamics in response to 30 s of nicotine perfusion at 50 μM (red line) (n = 27–29) in flies of the indicated genotypes. Right panel: quantification of the mean response during the 10–60 s after the end of nicotine perfusion (indicated by the black line on the time traces) for both genotypes. The calcium response following nicotine perfusion was decreased significantly because of nAChRα7 knockdown in cortex glia (n = 27–29, t54 = 2.12, p = 0.039).
(D) A representative 2-photon microscopy image of the FLII12Pglu-700μδ6 FRET glucose sensor expressed in cortex glia (dashed lines). Scale bar, 40 μm.
(E) After 30 s of 50 μM nicotine stimulation (red line), the glucose concentration was sustainably increased in cortex glia. Decreasing the expression levels of nAChRα7 in adult cortex glia lowered the nicotine-induced glucose elevation, as measured during the 200-s quantification window (black line) (n = 10, t18 = 3.65, p = 0.002).
(F) Spaced training failed to elicit a faster decrease in glucose concentration in MB neurons upon knockdown of nAChRα7 in cortex glia at the adult stage (n = 12, t22 = 0.76, p = 0.46).
Data were collected in parallel with the data presented in Figure 1C. All data are presented as mean ± SEM. Asterisks illustrate the significance level of the t test or of the least significant pairwise comparison following an ANOVA, with the following nomenclature: ∗p < 0.05; ∗∗p < 0.01; ns: not significant, p > 0.05. See also Figure S3.
The nAChRα7 subunit exhibits high calcium permeability in mammals (Liu et al., 2015). To assess whether Drosophila nAChRα7 activation also results in calcium entry, we measured the dynamics of intracellular calcium levels in cortex glia using a GCaMP6f fluorescent reporter in vivo in response to nicotine, which is a specific agonist of nicotinic acetylcholine receptors (Figure 3B). Following a 30-s nicotine perfusion at 50 μM, we measured a sustained increase in calcium concentration in cortex glia (Figures 3C and S3C). Importantly, nAChRα7 knockdown significantly reduced this calcium elevation (Figure 3C). This calcium increase was reduced more strongly when nAChRα7 RNAi was expressed along with Dicer-2, a ribonuclease enhancing RNAi efficiency (Figure S3C). This provides functional evidence that nAChRα7 can indeed mediate calcium signaling in cortex glia upon its activation.
We hypothesized that this cholinergic activation triggers an increase in cortex glia glucose concentration because spaced training elicited an increase in basal glucose level in cortex glia (Figure 2C). To test this hypothesis, we performed in vivo imaging experiments in flies expressing the glucose sensor FLII12Pglu-700μδ6 in cortex glia (Figure 3D). In these flies, the same nicotine treatment as used previously for calcium imaging (Figure 3C) induced a sustained increase in glucose concentration in cortex glia (Figure 3E). Remarkably, this effect was sensitive to knockdown of nAChRα7 in adult cortex glia (Figure 3E). These results reveal that cholinergic activation of cortex glia triggers a rise in glucose concentration.
To test whether nAChRα7 activation in cortex glia is necessary for shuttling of glucose to MB neuron somata, we performed glucose imaging experiments in MB neurons using nAChRα7 knockdown in adult cortex glia. Under that condition, spaced training failed to increase neuronal glucose consumption (Figure 3F).
Finally, we addressed whether the nicotine-induced rise in glucose concentration could be mediated by Glut1. We did not observe any change in the nicotine-induced increase in glucose concentration in cortex glia when Glut1 was knocked down in adult cortex glia (Figure S3D). This result indicates that Glut1 does not mediate nicotine-induced glucose uptake in cortex glia; instead, Glut1 in cortex glia is responsible for glucose export from cortex glia to MB neurons because it is required for glucose uptake and consumption in MB neurons (Figure 2D). These experiments demonstrate that cortex glia activation through nAChRα7 signaling results in an increase in glial glucose levels and glucose export via Glut1 to MB neurons to sustain LTM.
Insulin receptor signaling in adult cortex glia is necessary for LTM
Insulin signaling is involved in regulation of carbohydrate metabolism in mammals (Wilcox, 2005) and insects (Mattila and Hietakangas, 2017). Furthermore, in the brain, the insulin receptor has been shown to mediate glucose uptake in mammalian astrocytes (García-Cáceres et al., 2016). In Drosophila, insulin signaling is acutely required for LTM but dispensable for immediate memory (Chambers et al., 2015). Consequently, we wanted to find out whether insulin signaling could be necessary for LTM formation by regulating the glucose concentration increase in cortex glia. We examined the Drosophila insulin receptor InR expression pattern in the adult brain with immunohistochemistry using a specific antibody (Puig and Tjian, 2005). InR was highly expressed in cortex glia (Figure 4A), a finding confirmed by published single-cell transcriptomic results (Davie et al., 2018; Figure S4A). Therefore, we wanted to find out whether InR expression in cortex glia is necessary for LTM. Indeed, InR knockdown in adult cortex glia caused an LTM defect (Figure 4C). The sensory acuity controls were normal (Figure S4B2), and LTM was not impaired without induction of RNAi expression (Figure S4B1). Moreover, InR knockdown in adult cortex glia did not affect memory after massed training (Figure 4C). We confirmed these results using a second, non-overlapping InR RNAi (Figure S4C). Thus, InR is required in cortex glia specifically during LTM formation.
Figure 4.

InR signaling pathway regulates LTM formation
(A) Brains from 54H02-GAL4 > UAS-mCD8::GFP flies were immunostained with anti-GFP (green) and anti-InR (red) antibodies. The bottom panel shows a magnification of the region above the MB calyx (white rectangle). InR strongly colocalized with the cortex glial membrane. Scale bar, 40 μm.
(B) Scheme of the canonical insulin receptor (InR) signaling pathway. Upon insulin binding to InR, the insulin receptor signal (IRS) activates the PI3K enzyme. The PI3K92E catalyzes formation of the membrane lipid PiP3 from PiP2. PiP3 recruits PDK1 kinase, activating Akt1.
(C) InR knockdown in cortex glia, induced at adulthood only, disrupted LTM compared with genotypic controls (n = 24,F2,71 = 4.95, p = 0.0098) but did not affect 24-h memory after 5× massed training (n = 16, F2,47 = 0.94, p = 0.40).
(D) PI3K92E knockdown in adult cortex glia disrupted LTM compared with the genotypic controls (n = 19, F2,56 = 4.88, p = 0.01) but did not affect 24-h memory after 5× massed training (n = 10, F2,29 = 0.13, p = 0.88).
(E) Akt1 knockdown in adult cortex glia disrupted LTM compared with genotypic controls (n = 17, F2,50 = 8.37, p = 0.0008) but did not affect 24-h memory after 5× massed training (n = 9–12, F2,32 = 1.17, p = 0.32).
(F) Spaced training failed to elicit a faster decrease in glucose concentration in MB neurons upon knockdown of InR in cortex glia at the adult stage (n = 10/11, t19 = 0.42, p = 0.68).
Data were collected in parallel with the data presented in Figure 1C. All data are presented as mean ± SEM. Asterisks illustrate the significance level of the t test, or of the least significant pairwise comparison following an ANOVA, with the following nomenclature: ∗p < 0.05; ns: not significant, p > 0.05. See also Figure S4.
To verify that the canonical InR signaling is recruited for LTM, we targeted two interactors downstream of InR: the catalytic subunit of the Drosophila phosphatidylinositol 3-kinase (PI3K) subunit 92E and Akt1 (Figure 4B). As expected, knockdown of PI3K92E or Akt1 in adult cortex glia impaired LTM performance (Figures 4D and 4E). PI3K92E or Akt1 knockdown did not affect memory after massed training (Figures 4D and 4E). The sensory acuity controls were normal (Figures S4D2 and S4E2), and LTM was not impaired without induction of RNAi expression (Figures S4D1 and S4E1).
Finally, to address whether cortex glia InR is necessary for the increased glucose uptake and consumption in MB neuron somata, we conducted glucose imaging experiments in MB neurons using InR knockdown in adult cortex glia. Spaced training failed to induce upregulation of glucose metabolism under that condition (Figure 4F). Overall, these results reveal that insulin signaling in cortex glia is essential for LTM formation and participates in glucose shuttling to MB neurons.
Autocrine insulinergic signaling in cortex glia underlies LTM formation
Because cholinergic activation through nAChRα7 and InR is involved in glucose shuttling to neurons, we hypothesized that InR in cortex glia might be activated in an autocrine manner following cholinergic activation. This would imply that insulin-like peptides are released through exocytosis by cortex glia after spaced training. In accordance with this scheme, cholinergic activation elicited a calcium increase in cortex glia, which has been reported to trigger exocytosis in astrocytes in mammals (Hamilton and Attwell, 2010) as well as in flies (Ma et al., 2016). We first attempted to disrupt vesicular release in cortex glia through expression of tetanus toxin (TNT; Sweeney et al., 1995). This yielded specific impairment of LTM (Figure S5B). According to our data, increased glucose consumption in MB neurons occurs during the first 2 h after spaced training (Figure 1C). We therefore tested whether vesicular release from cortex glia occurred within the early frame of spaced training. Expression of the dominant-negative temperature-sensitive Shibirets (Shits) transgenic allele (Kitamoto, 2001) allows blocking vesicular recycling in the cells where it is expressed, including glial cells (Sousa-Nunes et al., 2011; Melom and Littleton, 2013; Ma et al., 2016; Artiushin et al., 2018), with acute temporal control provided by merely switching flies to a restrictive temperature (33°C). We first expressed Shits in cortex glia and blocked vesicular release during 6 h after spaced training by exposing flies to the restrictive temperature. Under this procedure, flies in which cortex glia were silenced displayed impaired LTM performance compared with genotypic controls (Figure 5A). Silencing cortex glia for 6 h after massed training left 24-h memory performance intact (Figure 5A). LTM performance was normal in flies of interest that had not been subjected to high-temperature treatment (Figure S5A). We then narrowed down the silencing period to the first 2 h after spaced training, which was sufficient to impair LTM (Figure 5A). These results define a narrow time frame during which vesicular release in cortex glia occurs for LTM. Pursuing our hypothesis, we then investigated whether an insulin-like peptide (Ilp) in cortex glia is necessary for LTM formation. Eight different genes encode Ilps in the fly genome (Colombani et al., 2012; Grönke et al., 2010). We found that expressing RNAi against Ilp4 in adult cortex glia impaired LTM (Figure 5B), whereas knockdown of other Ilps did not affect LTM performance (Figure S5C). In an experiment without induction of Ilp4 RNAi, LTM was normal (Figure S5D1), as were olfactory and shock acuity after RNAi induction (Figure S5D3). Moreover, Ilp4 knockdown did not affect memory after 5× massed training and 1× training (Figures 5B and S5D2). We confirmed these results with a second specific, non-overlapping RNAi directed against Ilp4 (Figure S5E). We examined Ilp4 distribution in the adult brain using a specific antibody (Grönke et al., 2010). Indeed, Ilp4 distribution colocalized with cortex glia (Figure 5C), supporting our behavioral data and showing that Ilp4 is mainly expressed in cortex glia. Our results thus demonstrate that Ilp4 is required in adult cortex glia specifically for LTM.
Figure 5.

Ilp4 expression in cortex glia is necessary for LTM
(A) The scheme above memory graphs indicates the time course of temperature treatments. Blocking exocytosis in cortex glia using Shits for 6 h (n = 19; F2,56 = 8.65, p = 0.0006) or only 2 h (n = 14, F2,41 = 3.93, p = 0.028) after conditioning yielded a LTM defect. Blocking cortex glia for 6 h after 5× massed training had no effect on 24-h memory performance (n = 25, F2,74 = 2.24, p = 0.11).
(B) Ilp4 knockdown in adult cortex glia disrupted LTM, tested 24 h after 5× spaced training (n = 18, F2,53 = 4.24, p = 0.020) but did not affect memory tested 24 h after 5× massed training (n = 16, F2,47 = 0.38, p = 0.68).
(C) Immunostaining against GFP (green) and Ilp4 (red) performed on brains from 54H02-GAL4 > UAS-mCD8::GFP flies revealed localization of Ilp4 in cortex glia. Scale bar, 10 μm.
(D) Co-expression of Ilp4 RNAi and constitutively active Akt1 (myr-Akt1) restores normal memory as opposed to Ilp4 RNAi expression alone (n = 14–18, F4,78 = 6.36, p = 0.0002).
(E) After 30 s of 50 μM nicotine stimulation (red line), the glucose concentration was sustainably increased in cortex glia. Decreasing the expression levels of Ilp4 and InR in adult cortex glia lowered the nicotine-induced glucose elevation (n = 15/16, F2,46 = 4.40, p = 0.02).
(F) Spaced training failed to elicit a faster decrease in glucose concentration in MB neurons upon knockdown of Ilp4 in cortex glia at the adult stage (n = 11, t20 = 0.11, p = 0.91).
Data were collected in parallel with the data presented in Figure 1C. All data are presented as mean ± SEM. Asterisks illustrate the significance level of the t test, or of the least significant pairwise comparison following an ANOVA, with the following nomenclature: ∗p < 0.05; ∗∗p < 0.01; ns: not significant, p > 0.05. See also Figure S5.
If the function of Ilp4 is to autocrinally activate InR, then activating the InR pathway should rescue the LTM defect induced by Ilp4 loss of function. We expressed myr-Akt1, a constitutively active form of Akt1 (Zheng et al., 2007), together with Ilp4 RNAi in adult cortex glia. Although expression of Ilp4 RNAi alone in cortex glia induced the LTM defect, its co-expression with myr-Akt1 restored normal LTM performance (Figure 5D). These results further indicate that autocrine Ilp4-InR signaling in cortex glia is necessary to initiate LTM formation after spaced training.
Our hypothesis postulates that autocrine Ilp4-InR signaling connects cholinergic activation to glucose regulation in cortex glia. We performed in vivo glucose imaging experiments upon nicotine stimulation while interfering with insulin signaling. As expected, the nicotine-induced increase in glucose concentration in cortex glia was reduced when InR or Ilp4 was knocked down in adult cortex glia (Figure 5E).
Finally, because InR in cortex glia is necessary for the increased glucose consumption in MB neurons (Figure 4F), we verified that expression of its ligand Ilp4 was also required for this purpose. We therefore conducted glucose imaging experiments in MB neurons using Ilp4 knockdown in adult cortex glia. Spaced training failed to induce upregulation of glucose metabolism under that condition (Figure 5F). These results reveal a sequential chain of signaling events in cortex glia that link cholinergic activation to a glucose increase through Ilp4-InR signaling, all feeding the increased glucose demand of MB neurons to sustain LTM.
MB neurons take up glucose to fuel the PPP
In the previous section, we demonstrated the need for increased glucose uptake and consumption by MB neurons for LTM. We next addressed the fate of this glucose in MB neurons. Upon entry, glucose is phosphorylated rapidly and directed to (1) glycolysis to generate pyruvate and ATP, providing energy, or (2) the PPP to generate ribulose-5-phosphate and NADPH (the reduced form of Nicotinamide Adenine Dinucleotide Phosphate, which is synthesized through the reduction of NADP+), providing building blocks for nucleotide biosynthesis and reducing power, respectively (Figure S6A). Ribulose-5-phosphate can also be re-routed to glycolytic intermediates and oxidized to pyruvate. Using the pyruvate FRET sensor Pyronic (San Martín et al., 2014), we previously published a protocol enabling measurement of mitochondrial pyruvate consumption by measuring pyruvate accumulation following application of sodium azide, a blocker of mitochondrial complex IV (Plaçais et al., 2017). In this study, we demonstrated that, upon spaced training, pyruvate flux in MB neuron vertical lobes (an anatomical structure encompassing MB neuron axons; Figure S1A) was increased (Plaçais et al., 2017). Because pyruvate is the end product of glycolysis, we applied the same protocol under a Glut1 knockdown condition to assess whether glucose uptake by MB neurons serves to fuel the increased demand for pyruvate. After a direct application of sodium azide (NaN3) at the surface of the brain being imaged, we observed faster accumulation of pyruvate in spaced-trained flies compared with flies that had non-associative training (Figure 6A1), as reported previously (Plaçais et al., 2017). Flies expressing a Glut1 RNAi in the MB also showed a difference of the same magnitude between the two training protocols (Figure 6A2), revealing that the enhanced glucose and pyruvate uptake by MB neurons after spaced training are independent. Moreover, expressing an RNAi against phosphofructokinase (PFK), a rate-limiting glycolytic enzyme, in adult MB neurons had no effect on LTM performance (Figure 6B). These data suggest that the additional glucose provided to MB neurons after spaced training is not consumed by glycolysis.
Figure 6.

Glucose fuels the MB neuron PPP for LTM formation
(A) Pyruvate imaging in MB vertical lobes.
(A1) As reported previously, spaced training elicited faster pyruvate accumulation following sodium azide (NaN3) application (5 mM) compared with non-associative unpaired training (n = 10/11, t19 = 2.17, p = 0.043).
(A2) With Glut1 knockdown, spaced training still elicited an increase in pyruvate accumulation compared with non-associative training (n = 11, t20 = 2.28, p = 0.034).
(B) PFK knockdown in adult MB neurons did not affect LTM (n = 12, F2,35 = 0.10, p = 0.90).
(C) Zw, 6PGL, or Pgd knockdown in adult cortex glia induced LTM defects (n = 17/18, F6,120 = 7.40, p < 0.0001) but did not affect 24-h memory after 5× massed training (n = 12, F6,81 = 0.45, p = 0.84).
(D) Glucose imaging in MB neuron somata.
(D1) Variations in glucose concentration in the somata of MB neurons were monitored following application of validamycin A (4 mM) at the time point marked by a dashed line. Glucose concentration in the somata of MB neurons decreased faster in flies after 5× spaced training compared with flies conditioned with a non-associative spaced unpaired training protocol (n = 10/11, t19 = 3.32, p = 0.0036).
(D2) Spaced training failed to induce a faster decrease in glucose concentration in MB neuron somata when 6PGL was knocked down (n = 11/12, t21 = 0.77, p = 0.45).
(D3) Spaced training failed to induce a faster decrease in glucose concentration in MB neuron somata when Pgd was knocked down (n = 11/12, t21 = 0.043, p = 0.97).
All data are presented as mean ± SEM. Asterisks illustrate the significance level of the t test, or of the least significant pairwise comparison following an ANOVA, with the following nomenclature: ∗p < 0.05; ∗∗p < 0.01; ns: not significant, p > 0.05. See also Figure S6.
We then addressed whether glucose is incorporated in the PPP. The first three reactions of the PPP form the so-called oxidative branch. They are catalyzed by a glucose-6-phosphate dehydrogenase called Zwischenferment (Zw) in Drosophila, 6-phosphogluconolactonase (6PGL; encoded by the CG17333 gene), and 6-phosphogluconate dehydrogenase (Pgd). Knockdown of Zw, 6PGL, or Pgd in adult MB neurons induced an LTM defect (Figure 6C). LTM was not impaired when RNAi was not induced (Figure S6B1), and sensory acuity controls after induction were normal (Figure S6B2). Memory performance was normal after massed training when expressing Zw, 6PGL, or Pgd RNAi (Figure 6C), showing that PPP is involved specifically in LTM formation.
Finally, we performed glucose imaging experiments in MB neuron somata while expressing RNAi against 6PGL or Pgd in adult MB neurons. As expected, spaced training triggered an increase in glucose consumption rate in flies expressing no RNAi (Figure 6D1), but it failed to induce upregulation of glucose consumption after 6PGL (Figure 6D2) or Pgd (Figure 6D3) knockdown. These results confirm that the glucose shuttled from cortex glia to MB neurons in the first hours following spaced training is consumed by the PPP in KCs, allowing LTM formation (Figure 7).
Figure 7.
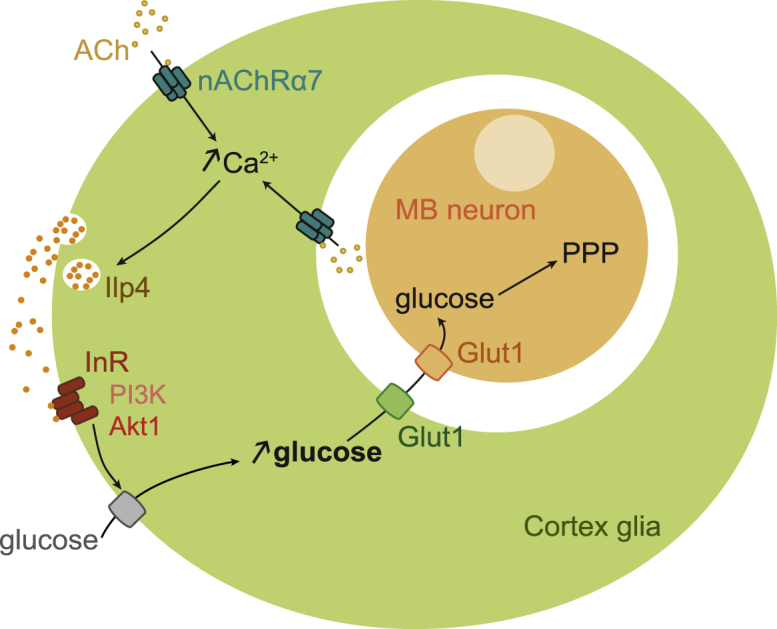
Model of cortex glia-MB neuron glucose shuttling during LTM consolidation
During LTM formation, acetylcholine (ACh) activates nAChRa7 in cortex glia. Whether ACh comes from cholinergic MB neurons or from other neurons was not addressed in this study and remains to be determined. The resulting increase in intracellular calcium concentration in cortex glia triggers Ilp4 secretion and autocrine action on InR and its canonical interactors PI3K and Akt1. Insulin signaling results in an increase in glucose concentration through an undetermined glucose transporter. Subsequently, glucose is shuttled toward the somata of MB neurons via Glut1 in both cell types. Glucose is then used to feed the PPP.
Discussion
In vertebrates, glial cells that are in contact with neuronal somata include protoplasmic astrocytes, which are intimately linked to synapses and neuronal cell bodies (Halassa et al., 2007; Allen and Barres, 2009), satellite glial cells in sensory ganglia (Hanani, 2005), and perineuronal oligodendrocytes (Takasaki et al., 2010). However, none of these cell types is associated exclusively with neuronal somata, making it difficult to address the specificities of neuron-glia coupling at the cell body level in vertebrates. Because of its clear separation between neuropil glia and the glia that enwraps neuronal somata, the Drosophila brain is well suited for uncovering specific interactions between glia and neuronal somata that could be relevant in other species.
Fueling the PPP of MBs for LTM
We demonstrate here that, during LTM formation, MB neuronal somata increase their glucose metabolism via Glut1-dependent glucose import. In accordance with our data, strong Glut1-dependent glucose uptake by neurons has been reported in Drosophila (Volkenhoff et al., 2018). We further report that PPP-dependent glucose consumption is critical specifically for neuronal plasticity underlying LTM formation. This is consistent with a study reporting that synaptic plasticity-related genes are upregulated in human brain regions that perform high levels of nonoxidative glucose metabolization (Goyal et al., 2014). Human subjects performing a cognitive task show increased levels of nonoxidative glucose consumption that persists for at least 40 min after the end of the experiment (Madsen et al., 1995). These observations, in concordance with our data, support participation of nonoxidative glucose consumption, potentially via the PPP, in long-lasting neuronal processes such as plasticity.
Models of neuron-glia metabolic compartmentation in mammals postulate that the PPP shunt is favored in neurons for antioxidant purposes, bypassing glycolytic energy production (Herrero-Mendez et al., 2009). Whether active inhibition of glycolysis favors glucose routing to the PPP in our model remains to be determined. Alternatively, the PPP could be stimulated by the redox state of MB neurons because the activity of glucose-6-phosphate dehydrogenase, a PPP rate-limiting enzyme, is increased when the NADPH/NADP+ ratio is low (Holten et al., 1976). A major function of the PPP is to produce NADPH, which is necessary to maintain reducing power in neurons and readily alleviate oxidative stress induced by reactive oxygen species (ROS) and reactive nitrogen species (RNS) that provoke cell damage (Bolaños and Almeida, 2010). ROS and RNS productions increase with neuronal O2 consumption. We established that LTM involves upregulation of mitochondrial pyruvate consumption and pyruvate dehydrogenase activity (Plaçais et al., 2017). Thus, mobilization of neuronal PPP could be a consequence of increased energy metabolism in MB neurons as a means to preserve neuronal integrity. ROS are also important regulators of physiological functions. It has been reported that astrocytic mitochondrial ROS are necessary for proper neuronal function and particularly short-term memory (Vicente-Gutierrez et al., 2019). ROS, in particular hydrogen peroxide, positively regulate hippocampal long-term potentiation and consequent LTM formation (Thiels et al., 2000). This intriguing duality of ROS effects on neuronal state suggests the existence of regulation processes that can balance its positive and negative effects. In this context, the PPP can participate in such regulation because it can modulate antioxidant levels and, thus, ROS concentration within neurons. Further investigation is needed to determine the role of the PPP for LTM.
Independent glucose and pyruvate regulation in MB neurons for LTM
We previously reported that the activity of the pyruvate dehydrogenase complex in MB neurons is also necessary for LTM (Plaçais et al., 2017). A corresponding increase in pyruvate metabolism has been measured in the axonal branches of MB neurons (Plaçais et al., 2017). However, our data from the present report also reveal that impairing neuronal glycolysis does not prevent LTM formation and that disrupting neuronal glucose import through Glut1 knockdown does not prevent neurons from increasing pyruvate consumption upon LTM formation. These data suggest that neuronal import of other pyruvate-producing energy metabolites may occur. Indeed, Drosophila glia can release lactate and amino acids, both metabolizable by neurons (Volkenhoff et al., 2015). Our model is therefore consistent with the notion that neurons can consume glucose through the PPP while still importing glucose-derived substrates from glia, as shown for other species in the context of LTM formation (Newman et al., 2011; Suzuki et al., 2011).
Cortex glia sense neuronal activity
Recent research in Drosophila has highlighted the crucial roles interactions between glia and neuronal somata play in the maintenance of neuronal excitability to prevent seizures (Kunduri et al., 2018; Melom and Littleton, 2013; Weiss et al., 2019). Our work additionally shows that perisomatic cortex glia are sensitive to acetylcholine and modulate their metabolism in response to cholinergic activation, which might influence neuronal excitability. Mammalian astrocytes are also sensitive to cholinergic activation in response to neuronal activity, suggesting a conserved neuron-glia dialog mechanism across species (Araque et al., 2002; Takata et al., 2011). The origin of the acetylcholine that activates cortex glia for LTM remains to be determined. Because acetylcholine is quickly broken down by acetylcholinesterase when it is released at the synapse, it is unlikely that it could diffuse away from the neuropil region and reach the cortex. Instead, it is probable that local release of acetylcholine occurs directly in the cortex, close to cortex glial cells.
We speculate that acetylcholine could be released by active neuronal somata, such as MB neurons, which could locally activate their enwrapping cortex glial cells. Indeed, we report that LTM formation (and therefore MB neuron activation) induces an increase in glucose concentration in cortex glia, suggesting a dialog between the two cell types. Calcium-dependent somatic release of neurotransmitters has already been described in various species, such as mice, chickens, and leeches (Corsetti et al., 2012; Del-Bel and De-Miguel, 2018; Trueta et al., 2003), as well as in cultured Drosophila neurons (Yao et al., 2000). Interestingly, the calcium concentration is increased in MB neuron somata upon their physiological activation by odorants (Lüdke et al., 2018). Moreover, calcium signaling in the MB neuron nucleus (located in the soma) is required for LTM but dispensable for other memory phases (Weislogel et al., 2013). Therefore, a somatic calcium increase might occur in MB neurons as a result of repeated, spaced odorant exposure during LTM conditioning. However, it remains to be demonstrated whether somatic release of acetylcholine could be responsible for cortex glia activation.
Autocrine insulin activation of cortex glia
In mammals, peripheral glucose is directly available in the blood, but it is known that insulin enhances glucose uptake in particular through translocation of Glut4 to the plasma membrane. Interestingly, Glut4 and insulin signaling are important regulators of memory processes in the hippocampus (McNay et al., 2010; Pearson-Leary and McNay, 2016; Pearson-Leary et al., 2018). Remarkably, we found that inducible knockdown of InR in cortex glia dampened glucose concentration increases in cortex glia and impaired LTM. One previous report has revealed that InR mediates glucose intake in astrocytes (García-Cáceres et al., 2016). Therefore, it seems that insulin signaling in glial cells is a conserved pathway for regulating brain glucose availability. Indeed, Drosophila harbors functional InR signaling in fat cells, allowing glucose uptake and storage in a manner similar to what occurs in mammalian adipose tissue in response to insulin (Zhang et al., 2011; Crivat et al., 2013; van Dam et al., 2020). Although the glucose transporter Glut4 is known to modulate insulin-dependent glucose uptake in mammalian adipose tissue and skeletal muscle, a Drosophila insulin-sensitive glucose transporter has not yet been identified (Crivat et al., 2013). Thus, the mechanism by which InR triggers glucose increases in cortex glia remains to be determined.
Remarkably, we found that InR activation is autocrine because Ilp4 knockdown in adult cortex glia impairs LTM. Although Ilp4 is one of the two most conserved insulin-like peptides in the fly genome among Drosophila species, its functions have remained uncharacterized until now (Grönke et al., 2010). Here we show that, in the brain, Ilp4 is mainly expressed in cortex glia and is instrumental during LTM formation. A large body of literature suggests that reduced brain insulin signaling, reduced glucose metabolism, and cognitive impairments are causally linked, although a clear mechanistic picture of how the three phenomena are interconnected remains to be identified (Chen et al., 2011; Arnold et al., 2018; Frazier et al., 2019; Soto et al., 2019). In this context, the mechanism we unveiled in Drosophila could be instructive for future investigations in mammals because it promotes glial cells (rather than neurons) as prominent targets of insulin signaling and as a relevant source of insulin for memory processes.
Limitations of the study
Although we described a neuron-glia dialog occurring at the level of neuronal somata, we did not demonstrate direct activation of cortex glia through release of neurotransmitters from MB neuron cell bodies.
Mechanisms underlying glia-neuron metabolic coupling seem to be conserved between Drosophila and mammalian models. Still, the Drosophila brain is not vascularized, which may account for some differences in nutrient delivery to the brain between species. We will await studies from other laboratories to confirm our results.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GFP (mouse) | Sigma-Aldrich | Cat. G6539; RRID: AB_259941 |
| Anti-Ilp4 (rabbit) | Grönke et al., 2010 | N/A |
| Anti-InR (rabbit) | Puig and Tjian, 2005 | N/A |
| Alexa 488 anti-mouse IgG (goat) | Invitrogen | Cat. A-11029, RRID: AB_2534088 |
| Alexa 594 anti-rabbit IgG (goat) | Invitrogen | Cat. A-11037, RRID: AB_2534095 |
| Chemicals, peptides, and recombinant proteins | ||
| 4-methylcyclohexanol (98%) | Sigma-Aldrich | Cat. 153095 |
| 3-octanol (99%) | Sigma-Aldrich | Cat. 218405 |
| (-)-nicotine (> 99%) | Sigma-Aldrich | Cat. N3876 |
| Validamycin A | Sigma-Aldrich | Cat. 32347 |
| NaCl | Sigma-Aldrich | Cat. S9625 |
| KCl | Sigma-Aldrich | Cat. P3911 |
| D-Glucose | Sigma-Aldrich | Cat. G8270 |
| D-Trehalose dihydrate | Sigma-Aldrich | Cat. T9531 |
| Sucrose | Sigma-Aldrich | Cat. S9378 |
| CaCl2 | Sigma-Aldrich | Cat. C3881 |
| MgCl2 | Sigma-Aldrich | Cat. M9272 |
| HEPES-hemisodium salt | Sigma-Aldrich | Cat. H7637 |
| Sodium Azide | Sigma-Aldrich | Cat. S2002 |
| Paraffin oil | VWR | Cat. 24679 |
| Prolong Mounting Medium | Lifetechnology | Cat. P36930 |
| Experimental models: Organisms/strains | ||
| VT30559-GAL4 (III) | VDRC | 206077 |
| tubulin-GAL80ts (II) | BDSC | 7019 |
| tubulin-GAL80ts;VT30559-GAL4 | Plaçais et al., 2017 | N/A |
| UAS-Glut1 RNAi KK108683 | VDRC | 101365 |
| UAS-Glut1 RNAi GD4552 | VDRC | 13326 |
| 13F02-LexA (II) | BDSC | 52460 |
| LexAop-FLII12Pglu-700μδ6 | This report | N/A |
| LexAop-iGlucoSnFR | BDSC | 82993 |
| UAS-FLII12Pglu-700μδ6 | Plaçais et al., 2017 | N/A |
| UAS-FLII12Pglu-700μδ6, VT30559-GAL4 | This report | N/A |
| 54H02-GAL4 (III) | M. Freeman (Coutinho-Budd et al., 2017) | N/A |
| tubulin-GAL80ts; 54H02-GAL4 | This report | N/A |
| Alrm-GAL4 (III) | M. Freeman (Doherty et al., 2009) | N/A |
| 56F03-GAL4 (III) | BDSC | 39157 |
| tubulin-GAL80ts; Alrm-GAL4 | This report | N/A |
| tubulin-GAL80ts; 56F03-GAL4 | This report | N/A |
| 54H02-LexA (III) | This report | N/A |
| tubulin-GAL80ts, 13F02-LexA; 54H02-GAL4 | This report | N/A |
| LexAop-FLII12Pglu-700μδ6; UAS-Glut1 RNAi GD4552 | This report | N/A |
| UAS-nAChRα7 RNAi TRiP JF02570 | BDSC | 27251 |
| UAS-nAChRα7 RNAi KK108471 | VDRC | 100756 |
| UAS-GCaMP6f | BDSC | 42747 |
| UAS-GCaMP6f; UAS-nAChRα7 RNAi JF02570 | This report | N/A |
| tubulin-GAL80ts; 54H02-GAL4, UAS-FLII12Pglu-700μδ6 | This report | N/A |
| LexAop-FLII12Pglu-700μδ6; UAS-nAChRα7 RNAi JF02570 | This report | N/A |
| tubulin-GAL80ts; 54H02-GAL4, UAS-Dcr-2 | This report | N/A |
| UAS-mCD8::GFP | Plaçais et al., 2012 | N/A |
| UAS-InR RNAi TRiP HMS03166 | BDSC | 51518 |
| UAS-InR RNAi GD104 | VDRC | 992 |
| UAS-PI3K92E RNAi JF02770 | BDSC | 27690 |
| UAS-Akt1 RNAi HMS00007 | BDSC | 33615 |
| LexAop-FLII12Pglu-700μδ6; UAS-InR RNAi TRiP HMS03166 | This report | N/A |
| UAS-Ilp4 RNAi TRiP HMS02660 | BDSC | 43288 |
| UAS-Ilp4 RNAi TRiP HMS00547 | BDSC | 33682 |
| UAS-shits | Plaçais et al., 2012 | N/A |
| UAS-TNT | BDSC | 28838 |
| UAS-myr-Akt1 | BDSC | 80935 |
| UAS-Ilp4 RNAi TRiP HMS02660; UAS-myr-Akt1 | This report | N/A |
| LexAop-FLII12Pglu-700μδ6; UAS-Ilp4 RNAi TRiP HMS02660 | This report | N/A |
| UAS-Ilp1 RNAi TRiP HMS00648 | BDSC | 32861 |
| UAS-Ilp2 RNAi TRiP HMS00476 | BDSC | 32475 |
| UAS-Ilp3 RNAi TRiP HMS00546 | BDSC | 33681 |
| UAS-Ilp5 RNAi TRiP HMS00548 | BDSC | 33683 |
| UAS-Ilp6 RNAi TRiP HMS00549 | BDSC | 33684 |
| UAS-Ilp7 RNAi TRiP HMS00649 | BDSC | 32862 |
| UAS-Ilp8 RNAi GD1670 | VDRC | 9420 |
| UAS-PFK RNAi HMS01324 | BDSC | 34336 |
| tubulin-GAL80ts; VT30559-GAL4, UAS-Pyronic | Plaçais et al., 2017 | N/A |
| UAS-Zw RNAi KK108898 | VDRC | 101507 |
| UAS-6PGL RNAi HMS02626 | BDSC | 42933 |
| UAS-Pgd RNAi HMC05959 | BDSC | 65078 |
| tubulin-GAL80ts, 13F02-LexA; VT30559-GAL4 | This report | N/A |
| LexAop-FLII12Pglu-700μδ6; UAS-6PGL RNAi HMS02626 | This report | N/A |
| LexAop-FLII12Pglu-700μδ6; UAS-Pgd RNAi HMC05959 | This report | N/A |
| Oligonucleotides | ||
| Primer Glut1 forward: GGCGGAATGTTCATCTTCTC | This report | N/A |
| Primer Glut1 reverse: GGACATCCAGTCGATCATTTC | This report | N/A |
| Recombinant DNA | ||
| 2250 SIN-cPPT-PGK-FLIIP- glu700-WHV plasmid | G. Bonvento | N/A |
| pJFRC19 plasmid | Pfeiffer et al., 2010 | Addgene #26224 |
| Software and algorithms | ||
| GraphPad Prism 7 | GraphPad Software, San Diego, CA, 2007 | https://www.graphpad.com:443/ |
| MATLAB V2013 | MathWorks, Natick, MA | https://fr.mathworks.com/ |
| ImageJ | ImageJ | https://imagej.nih.gov/ij/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Thomas Preat (thomas.preat@espci.fr).
Materials availability
Materials generated in this study will be made available upon reasonable request to the lead contact.
Experimental model and subject details
Flies were raised on standard medium at 18, 23 or 25°C (depending on the experiments, see method details) and 60% humidity in a 12-h light/dark cycle. The study was performed on 1-4-day-old adult flies. For behavior experiments, both male and female were used. Otherwise, female flies were used because of their larger size. All flies obtained from libraries or received after the injection of transgenes (except flies from the TRiP RNAi collection) were outcrossed for 5 generations to a reference strain carrying the w1118 mutation in an otherwise Canton Special (Canton S) genetic background. Because TRiP RNAi transgenes are labeled by a y+ marker, these lines were outcrossed to a y1w67c23 strain in an otherwise Canton S background.
Method details
Behavior Experiments
For behavior experiments, flies were raised on standard medium at 18°C and 60% humidity in a 12-h light/dark cycle. We used the TARGET system (McGuire et al., 2003) to inducibly express RNAi constructs exclusively in adult flies, and not during development. To achieve the induction of RNAi expression, adult flies were kept at 30.5°C for 2-3 days before conditioning. Otherwise, experimental flies (0-3 days old) were transferred to fresh bottles containing standard medium on the day before conditioning.
The behavior experiments, including the sample sizes, were conducted similarly to other studies from our laboratory (Pascual and Préat, 2001). Groups of 20-50 flies were subjected to one of the following olfactory conditioning protocols: a single cycle, five consecutive associative training cycles (5 x massed training), or five associative cycles spaced by 15-min inter-trial intervals (5 x spaced training). Non-associative control protocols (unpaired protocols) were also employed for imaging experiments. Conditioning was performed using previously described barrel-type machines that allow parallel training of up to 6 groups. Throughout the conditioning protocol, each barrel was plugged into a constant air flow at 2 L·min-1. For a single cycle of associative training, flies were first exposed to an odorant (the CS+) for 1 min while 12 pulses of 5 s long 60 V electric shocks were delivered; flies were then exposed 45 s later to a second odorant without shocks (the CS-) for 1 min. The odorants 3-octanol and 4-methylcyclohexanol, diluted in paraffin oil at a final concentration of 2.79·10−1 g·L-1, were alternately used as conditioned stimuli. During unpaired conditionings, the odour and shock stimuli were delivered separately in time, with shocks occurring 3 min before the first odorant.
Flies were kept on standard medium between conditioning and the memory test, in an incubator set at 18°C. For experiments involving blockade with Shits, flies were transferred to preheated bottles in a 33°C room, immediately after the end of the last cycle of the training protocol. After spending the appropriate time period at 33°C (2 or 6 h), flies were returned to an incubator set at the permissive temperature of 18°C. The time course of the temperature shifts employed in an experiment are displayed above the memory performance graph in each figure.
The memory test was performed in a T-maze apparatus, typically 24 h after massed or spaced training and 3 h after single-cycle training. Each arm of the T-maze was connected to a bottle containing 3-octanol and 4-methylcyclohexanol, diluted in paraffin oil at a final concentration identical to the one used for conditioning. Flies were given 1 minute to choose between either arm of the T-maze. A score was calculated as the number of flies avoiding the conditioned odour minus the number of flies preferring the conditioned odour, divided by the total number of flies. A single performance index value is the average of two scores obtained from two groups of genotypically identical flies conditioned in two reciprocal experiments, using either odorant (3-octanol and 4-methylcyclohexanol) as the CS+. The indicated ‘n’ is the number of independent performance index values for each genotype.
The shock response tests were performed at 25°C by placing flies in two connected compartments; electric shocks were provided in only one of the compartments. Flies were given 1 min to move freely in these compartments, after which they were trapped, collected and counted. The compartment where the electric shocks were delivered was alternated between two consecutive groups. Shock avoidance was calculated as for the memory test.
Because the delivery of electric shocks can modify olfactory acuity, our olfactory avoidance tests were performed on flies that had first been presented another odour paired with electric shocks. Innate odour avoidance was measured in a T-maze similar to those used for memory tests, in which one arm of the T-maze was connected to a bottle with odour diluted in paraffin oil and the other arm was connected to a bottle with paraffin oil only. Naive flies were given the choice between the two arms during 1 min. The odour-interlaced side was alternated for successive tested groups. Odour concentrations used in this assay were the same as for memory assays. At these concentrations, both odorants are innately repulsive.
Immunohistochemistry Experiments
Before dissection, whole adult female flies (2-4 days old) were fixed in 4% formaldehyde in PBST (PBS containing 1% Triton X-100) at 4°C overnight. Brains were dissected in PBST solution and rinsed three times for 20 min in PBST, blocked with 2% bovine serum albumin in PBST for 2 h at room temperature, and then incubated with primary antibodies at 1:400 (mouse anti-GFP, Sigma-Aldrich), 1:1000 (rabbit anti-InR) and 1:200 (rabbit anti-Ilp4) in the blocking buffer at 4°C overnight. The following day, brains were rinsed 3 times for 20 min in PBST, and incubated with secondary antibodies at 1:400 (Invitrogen, goat anti-mouse Alexa 488 Cat. 11029 and goat anti-rabbit Alexa 594 Cat. 11037) in PBST-BSA 2% solution for 3 h at room temperature. Brains were further rinsed 3 times for 20 min in PBST, and were mounted in ProLong Mounting Medium (Lifetechnology) for imaging. Images were acquired with a Nikon A1R confocal microscope. Confocal images were imported into ImageJ for analyses.
In vivo Imaging
Calcium imaging experiments were performed by expressing GCaMP6f (UAS-GCaMP6f in attP40) in cortex glia using the 54H02-GAL4 driver. Glucose imaging experiments were performed on flies expressing the glucose sensor in MB neurons via the VT30559-GAL4 driver or 13F02-LexA driver, or in cortex glia using the 54H02-GAL4 or 54H02-LexA driver, in combination with either UAS-FLII12Pglu-700μδ6 (random insertion on chromosome III (Plaçais et al., 2017)), LexAop-FLII12Pglu-700μδ6 (in attP18, generated for this study) or LexAop-iGlucoSnFR (Keller et al., 2021). Pyruvate imaging experiments were performed on flies expressing the pyruvate sensor in MB neurons via the VT30559-GAL4 driver in combination with UAS-Pyronic (Plaçais et al., 2017). RNAis were expressed in MB neurons using the inducible tub-GAL80ts; VT30559-GAL4 driver, or in cortex glia using the inducible tub-GAL80ts; 54H02-GAL4 driver.
For imaging experiments, flies were raised at either 23°C (if the experiment involved the use of GAL80ts) or 25°C to increase the expression level of genetically-encoded sensors. To achieve the induction of RNAi expression, adult flies were kept at 30.5°C for 2-3 days before conditioning.
As in all previous imaging work from our laboratory, all in vivo imaging was performed on female flies, which are preferred since their bigger size facilitates surgery.
For experiments on conditioned flies, data were collected indiscriminately from 30 min to 2 h after training, unless indicated otherwise. A single fly was picked and prepared for imaging as previously described (Plaçais et al., 2012). Briefly, the head capsule was opened and the brain exposed by gently removing the superior tracheae. The head capsule was bathed in artificial haemolymph solution for the duration of the preparation. The composition of this solution was: NaCl 130 mM (Sigma S9625), KCl 5 mM (Sigma P3911), MgCl2 2 mM (Sigma M9272), CaCl2 2 mM (Sigma C3881), D-Trehalose 5 mM (Sigma T9531), Sucrose 30 mM (Sigma S9378), and HEPES-hemisodium salt 5 mM (Sigma H7637). At the end of surgery, any remaining solution was absorbed and a fresh 90 μL droplet of this solution was applied on top of the brain. Note that calcium imaging was performed using a slightly distinct recipe found in our former studies (NaCl 130 mM (Sigma S9625), KCl 5 mM (Sigma P3911), MgCl2 2 mM (Sigma M9272), CaCl2 2 mM (Sigma C3881), Sucrose 36 mM (Sigma S9378), and HEPES-hemisodium salt 5 mM (Sigma H7637)). Two-photon imaging was performed using a Leica TCS-SP5 upright microscope equipped with a 25x, 0.95 NA, water-immersion objective, and two distinct sets of emission filters depending on the sensor used. Two-photon excitation was achieved using a Mai Tai DeepSee laser tuned to either 820 nm (FLII12Pglu-700μδ6 glucose sensor), 825 nm (Pyronic pyruvate sensor), 905 nm (iGlucoSnFR glucose sensor), or 910 nm (GCaMP6f). The frame rate was 1 or 2 images per second, depending on the experiment.
For nicotine experiments, nicotine was freshly diluted from a commercial liquid (Sigma N3876) into the saline used for imaging on each experimental day. A perfusion setup at a flux of 2.5 mL·min-1 enabled the time-restricted application of 50 μM nicotine on top of the brain. Baseline recording was performed during 1 min, after which the saline supply was switched to drug supply. The solution reached the in vivo preparation within 30 s. The stimulation was maintained for 30 s, before switching back to the saline perfusion for an additional 5 min.
For the glucose consumption experiments, validamycin A (Sigma 32347), a trehalase-selective inhibitor, was directly diluted into artificial haemolymph solution at a final concentration of 40 mM, aliquoted, and stored at −20°C. A freshly thawed aliquot was used for every fly. After 1 min of baseline acquisition, 10 μL of the solution was added to the 90 μL saline droplet on top of the brain, bringing validamycin A to a final concentration of 4 mM. The signal was then acquired for another 12 min. Image analysis was performed using a custom-written MATLAB script. Regions of interest (ROI) were delimited by hand around the labeled regions of interest (cortex glia or all MB neuron somata). For glucose imaging using the FLII12Pglu-700μδ6 glucose sensor, the average intensity of the YFP and CFP channels over each ROI were calculated over time after background subtraction. The FRET ratio (YFP/CFP) of the FLII12Pglu-700μδ6 glucose sensor was computed to obtain a signal positively correlated with the glucose concentration. This ratio was normalized by a baseline value calculated over the 30 s preceding drug injection. The area over the curve (AOC) was computed in order to obtain values positively correlated with glucose consumption. AOC was calculated as the integral between 200 s and 900 s.
The low-affinity glucose sensor iGlucoSnFR was used to estimate the baseline glucose concentration. For this, the trace of fluorescence decrease following validamycin A application was normalized to the value of the stable floor plateau that ended all of the recordings, corresponding to the fully unbound state of the sensor. The decrease was recorded for 900 s after the drug application and the floor value was calculated as the average over the last 150 s of the recording.
For calcium imaging, the GCaMP6f signal was calculated over time after background subtraction and normalized by a baseline value calculated over the 30 s preceding drug injection.
Pyruvate imaging experiments were performed according to a previously well-characterized protocol (Plaçais et al., 2017). After 1 min of baseline acquisition, 10 μl of a 50 mM sodium azide solution (prepared in the same artificial haemolymph solution) were injected into the 90-μl droplet bathing the fly’s brain, bringing sodium azide to a final concentration of 5 mM. ROI were delimited by hand around each visible vertical lobe, and the average intensity of both mTFP and Venus channels over each ROI were calculated over time after background subtraction. The Pyronic sensor was designed so that FRET from mTFP to Venus decreases when pyruvate concentration increases. To obtain a signal that positively correlates with pyruvate concentration, the inverse FRET ratio was computed as mTFP intensity divided by Venus intensity. This ratio was normalized by a baseline value calculated over the 30 s preceding drug injection. The slope was calculated between 10 and 70% of the plateau.
The indicated ‘n’ is the number of animals that were assayed in each condition.
Generation of Transgenic Flies
The 2250 SIN-cPPT-PGK-FLIIP-glu700-WHV plasmid was digested by BamHI and XbaI. The resulting 2,395 bp fragment was purified by electrophoresis and cloned into the pJFRC19 plasmid (13XLexAop2-IVS-myr::GFP) previously digested by BglII and XbaI to remove the myr::GFP sequence. The resulting construct was verified by restriction. Transgenic fly strains were obtained by site-specific embryonic injection of the resulting vector in the attP18 landing site (chromosome 1), which was outsourced to Rainbow Transgenic Flies, Inc (CA, USA).
Quantification and statistical analysis
All data are presented as mean ± SEM. For behavior experiments, 2 groups of about 30 flies were reciprocally conditioned, using respectively octanol or methylcyclohexanol as the CS+. The memory score was calculated from the performance of two groups as described above, which represents an experimental replicate. For imaging experiments, one replicate corresponds to one fly brain. Comparisons of the data series between two conditions were achieved by a two-tailed unpaired t test (Figures 1C, 1D, 2C, 2D, 3C–3F, 4F, 5F, 6A, 6D, S1C–S1E, S3C, and S3D). Comparisons between more than two distinct groups were made using a one-way ANOVA test, followed by Newman-Keuls pairwise comparisons between the experimental groups and their controls (Figures 1A, 2A, 3A, 4C–4E, 5A, 5B, 5D 5E, 6B, 6C, S1B, S2A–S2C, S2E, S2F, S3A, S3B, S4A–S4E, S5A–S5E, and S6B). ANOVA results are presented as the value of the Fisher distribution F(x,y) obtained from the data, where x is the number of degrees of freedom between groups and y is the total number of degrees of freedom for the distribution. Statistical tests were performed using the GraphPad Prism 7 software. In the figures, asterisks illustrate the significance level of the t test, or of the least significant pairwise post-hoc comparison following an ANOVA, with the following nomenclature: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ns: not significant, p > 0.05.
Acknowledgments
We thank Susan Broughton and Linda Partridge for providing the anti-Ilp4 antibody. We thank Oscar Puig and Jaakko Mattila for providing the anti-InR antibody. We also thank Bruno Weber for critical comments on the manuscript. This work was funded by the European Research Council (EnergyMemo Advanced Grant 741550). E.d.T. was funded by a doctoral fellowship from the French Ministry of Research and a fourth-year PhD grant from the Fondation pour la Recherche Médicale.
Author contributions
Conceptualization, P.-Y.P., T.P., Y.R., and E.d.T.; investigation, E.d.T., P.-Y.P., Y.R., J.M., and L.P.; writing – original draft, E.d.T., P.-Y.P., and Y.R.; writing – review & editing, T.P.; supervision, P.-Y.P. and T.P.; funding Acquisition, T.P.
Declaration of interests
The authors declare no competing interests.
Published: August 24, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109620.
Contributor Information
Pierre-Yves Plaçais, Email: pierre-yves.placais@espci.fr.
Thomas Preat, Email: thomas.preat@espci.fr.
Supporting citations
The following references appear in the Supplemental information: Chell and Brand (2010); Naganos et al. (2012).
Supplemental information
Data and code availability
Data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Alle H., Roth A., Geiger J.R.P. Energy-efficient action potentials in hippocampal mossy fibers. Science. 2009;325:1405–1408. doi: 10.1126/science.1174331. [DOI] [PubMed] [Google Scholar]
- Allen N.J., Barres B.A. Neuroscience: Glia - more than just brain glue. Nature. 2009;457:675–677. doi: 10.1038/457675a. [DOI] [PubMed] [Google Scholar]
- Araque A., Martín E.D., Perea G., Arellano J.I., Buño W. Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J. Neurosci. 2002;22:2443–2450. doi: 10.1523/JNEUROSCI.22-07-02443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold S.E., Arvanitakis Z., Macauley-Rambach S.L., Koenig A.M., Wang H.-Y., Ahima R.S., Craft S., Gandy S., Buettner C., Stoeckel L.E. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat. Rev. Neurol. 2018;14:168–181. doi: 10.1038/nrneurol.2017.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artiushin G., Zhang S.L., Tricoire H., Sehgal A. Endocytosis at the Drosophila blood-brain barrier as a function for sleep. eLife. 2018;7:e43326. doi: 10.7554/eLife.43326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G., Wu Z., Farrell R.J., Ryan T.A. GLUT4 Mobilization Supports Energetic Demands of Active Synapses. Neuron. 2017;93:606–615.e3. doi: 10.1016/j.neuron.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D., Laughlin S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 2001;21:1133–1145. doi: 10.1097/00004647-200110000-00001. [DOI] [PubMed] [Google Scholar]
- Awasaki T., Lai S.-L., Ito K., Lee T. Organization and postembryonic development of glial cells in the adult central brain of Drosophila. J. Neurosci. 2008;28:13742–13753. doi: 10.1523/JNEUROSCI.4844-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak L.K., Walls A.B. CrossTalk opposing view: lack of evidence supporting an astrocyte-to-neuron lactate shuttle coupling neuronal activity to glucose utilisation in the brain. J. Physiol. 2018;596:351–353. doi: 10.1113/JP274945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak L.K., Walls A.B., Schousboe A., Ring A., Sonnewald U., Waagepetersen H.S. Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J. Neurochem. 2009;109(Suppl 1):87–93. doi: 10.1111/j.1471-4159.2009.05943.x. [DOI] [PubMed] [Google Scholar]
- Barnstedt O., Owald D., Felsenberg J., Brain R., Moszynski J.-P., Talbot C.B., Perrat P.N., Waddell S. Memory-Relevant Mushroom Body Output Synapses Are Cholinergic. Neuron. 2016;89:1237–1247. doi: 10.1016/j.neuron.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros L.F., Weber B. CrossTalk proposal: an important astrocyte-to-neuron lactate shuttle couples neuronal activity to glucose utilisation in the brain. J. Physiol. 2018;596:347–350. doi: 10.1113/JP274944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolaños J.P., Almeida A. The pentose-phosphate pathway in neuronal survival against nitrosative stress. IUBMB Life. 2010;62:14–18. doi: 10.1002/iub.280. [DOI] [PubMed] [Google Scholar]
- Chambers D.B., Androschuk A., Rosenfelt C., Langer S., Harding M., Bolduc F.V. Insulin signaling is acutely required for long-term memory in Drosophila. Front. Neural Circuits. 2015;9:8. doi: 10.3389/fncir.2015.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chell J.M., Brand A.H. Nutrition-responsive glia control exit of neural stem cells from quiescence. Cell. 2010;143:1161–1173. doi: 10.1016/j.cell.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.Y., Stern S.A., Garcia-Osta A., Saunier-Rebori B., Pollonini G., Bambah-Mukku D., Blitzer R.D., Alberini C.M. A critical role for IGF-II in memory consolidation and enhancement. Nature. 2011;469:491–497. doi: 10.1038/nature09667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cognigni P., Felsenberg J., Waddell S. Do the right thing: neural network mechanisms of memory formation, expression and update in Drosophila. Curr. Opin. Neurobiol. 2018;49:51–58. doi: 10.1016/j.conb.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombani J., Andersen D.S., Léopold P. Secreted peptide Dilp8 coordinates Drosophila tissue growth with developmental timing. Science. 2012;336:582–585. doi: 10.1126/science.1216689. [DOI] [PubMed] [Google Scholar]
- Corsetti V., Mozzetta C., Biagioni S., Augusti Tocco G., Tata A.M. The mechanisms and possible sites of acetylcholine release during chick primary sensory neuron differentiation. Life Sci. 2012;91:783–788. doi: 10.1016/j.lfs.2012.08.026. [DOI] [PubMed] [Google Scholar]
- Coutinho-Budd J.C., Sheehan A.E., Freeman M.R. The secreted neurotrophin Spätzle 3 promotes glial morphogenesis and supports neuronal survival and function. Genes Dev. 2017;31:2023–2038. doi: 10.1101/gad.305888.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crivat G., Lizunov V.A., Li C.R., Stenkula K.G., Zimmerberg J., Cushman S.W., Pick L. Insulin stimulates translocation of human GLUT4 to the membrane in fat bodies of transgenic Drosophila melanogaster. PLoS ONE. 2013;8:e77953. doi: 10.1371/journal.pone.0077953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie K., Janssens J., Koldere D., De Waegeneer M., Pech U., Kreft Ł., Aibar S., Makhzami S., Christiaens V., Bravo González-Blas C. A Single-Cell Transcriptome Atlas of the Aging Drosophila Brain. Cell. 2018;174:982–998.e20. doi: 10.1016/j.cell.2018.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del-Bel E., De-Miguel F.F. Extrasynaptic Neurotransmission Mediated by Exocytosis and Diffusive Release of Transmitter Substances. Front. Synaptic Neurosci. 2018;10:13. doi: 10.3389/fnsyn.2018.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descalzi G., Gao V., Steinman M.Q., Suzuki A., Alberini C.M. Lactate from astrocytes fuels learning-induced mRNA translation in excitatory and inhibitory neurons. Commun. Biol. 2019;2:247. doi: 10.1038/s42003-019-0495-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-García C.M., Mongeon R., Lahmann C., Koveal D., Zucker H., Yellen G. Neuronal Stimulation Triggers Neuronal Glycolysis and Not Lactate Uptake. Cell Metab. 2017;26:361–374.e4. doi: 10.1016/j.cmet.2017.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-García C.M., Lahmann C., Martínez-François J.R., Li B., Koveal D., Nathwani N., Rahman M., Keller J.P., Marvin J.S., Looger L.L., Yellen G. Quantitative in vivo imaging of neuronal glucose concentrations with a genetically encoded fluorescence lifetime sensor. J. Neurosci. Res. 2019;97:946–960. doi: 10.1002/jnr.24433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty J., Logan M.A., Taşdemir O.E., Freeman M.R. Ensheathing glia function as phagocytes in the adult Drosophila brain. J. Neurosci. 2009;29:4768–4781. doi: 10.1523/JNEUROSCI.5951-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escher S.A., Rasmuson-Lestander A. The Drosophila glucose transporter gene: cDNA sequence, phylogenetic comparisons, analysis of functional sites and secondary structures. Hereditas. 1999;130:95–103. doi: 10.1111/j.1601-5223.1999.00095.x. [DOI] [PubMed] [Google Scholar]
- Frazier H.N., Ghoweri A.O., Anderson K.L., Lin R.-L., Porter N.M., Thibault O. Broadening the definition of brain insulin resistance in aging and Alzheimer’s disease. Exp. Neurol. 2019;313:79–87. doi: 10.1016/j.expneurol.2018.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman M.R. Drosophila Central Nervous System Glia. Cold Spring Harb. Perspect. Biol. 2015;7:020552. doi: 10.1101/cshperspect.a020552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao V., Suzuki A., Magistretti P.J., Lengacher S., Pollonini G., Steinman M.Q., Alberini C.M. Astrocytic β2-adrenergic receptors mediate hippocampal long-term memory consolidation. Proc. Natl. Acad. Sci. USA. 2016;113:8526–8531. doi: 10.1073/pnas.1605063113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Cáceres C., Quarta C., Varela L., Gao Y., Gruber T., Legutko B., Jastroch M., Johansson P., Ninkovic J., Yi C.-X. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell. 2016;166:867–880. doi: 10.1016/j.cell.2016.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal M.S., Hawrylycz M., Miller J.A., Snyder A.Z., Raichle M.E. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 2014;19:49–57. doi: 10.1016/j.cmet.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grönke S., Clarke D.-F., Broughton S., Andrews T.D., Partridge L. Molecular evolution and functional characterization of Drosophila insulin-like peptides. PLoS Genet. 2010;6:e1000857. doi: 10.1371/journal.pgen.1000857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber F., Knapek S., Fujita M., Matsuo K., Bräcker L., Shinzato N., Siwanowicz I., Tanimura T., Tanimoto H. Suppression of conditioned odor approach by feeding is independent of taste and nutritional value in Drosophila. Curr. Biol. 2013;23:507–514. doi: 10.1016/j.cub.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Halassa M.M., Fellin T., Takano H., Dong J.-H., Haydon P.G. Synaptic islands defined by the territory of a single astrocyte. J. Neurosci. 2007;27:6473–6477. doi: 10.1523/JNEUROSCI.1419-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton N.B., Attwell D. Do astrocytes really exocytose neurotransmitters? Nat. Rev. Neurosci. 2010;11:227–238. doi: 10.1038/nrn2803. [DOI] [PubMed] [Google Scholar]
- Hanani M. Satellite glial cells in sensory ganglia: from form to function. Brain Res. Brain Res. Rev. 2005;48:457–476. doi: 10.1016/j.brainresrev.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Harris J.J., Jolivet R., Attwell D. Synaptic energy use and supply. Neuron. 2012;75:762–777. doi: 10.1016/j.neuron.2012.08.019. [DOI] [PubMed] [Google Scholar]
- Herrero-Mendez A., Almeida A., Fernández E., Maestre C., Moncada S., Bolaños J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009;11:747–752. doi: 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- Holten D., Procsal D., Chang H.-L. Regulation of pentose phosphate pathway dehydrogenases by NADP+/NADPH ratios. Biochem. Biophys. Res. Commun. 1976;68:436–441. doi: 10.1016/0006-291x(76)91164-5. [DOI] [PubMed] [Google Scholar]
- Isabel G., Pascual A., Preat T. Exclusive consolidated memory phases in Drosophila. Science. 2004;304:1024–1027. doi: 10.1126/science.1094932. [DOI] [PubMed] [Google Scholar]
- Keller J.P., Marvin J.S., Lacin H., Lemon W.C., Shea J., Kim S., Lee R.T., Koyama M., Keller P.J., Looger L.L. In vivo glucose imaging in multiple model organisms with an engineered single-wavelength sensor. Cell Rep. 2021;35:109284. doi: 10.1016/j.celrep.2021.109284. [DOI] [PubMed] [Google Scholar]
- Kis V., Barti B., Lippai M., Sass M. Specialized Cortex Glial Cells Accumulate Lipid Droplets in Drosophila melanogaster. PLoS ONE. 2015;10:e0131250. doi: 10.1371/journal.pone.0131250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamoto T. Conditional modification of behavior in Drosophila by targeted expression of a temperature-sensitive shibire allele in defined neurons. J. Neurobiol. 2001;47:81–92. doi: 10.1002/neu.1018. [DOI] [PubMed] [Google Scholar]
- Kremer M.C., Jung C., Batelli S., Rubin G.M., Gaul U. The glia of the adult Drosophila nervous system. Glia. 2017;65:606–638. doi: 10.1002/glia.23115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunduri G., Turner-Evans D., Konya Y., Izumi Y., Nagashima K., Lockett S., Holthuis J., Bamba T., Acharya U., Acharya J.K. Defective cortex glia plasma membrane structure underlies light-induced epilepsy in cpes mutants. Proc. Natl. Acad. Sci. USA. 2018;115:E8919–E8928. doi: 10.1073/pnas.1808463115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zeng X., Hui Y., Zhu C., Wu J., Taylor D.H., Ji J., Fan W., Huang Z., Hu J. Activation of α7 nicotinic acetylcholine receptors protects astrocytes against oxidative stress-induced apoptosis: implications for Parkinson’s disease. Neuropharmacology. 2015;91:87–96. doi: 10.1016/j.neuropharm.2014.11.028. [DOI] [PubMed] [Google Scholar]
- Lüdke A., Raiser G., Nehrkorn J., Herz A.V.M., Galizia C.G., Szyszka P. Calcium in Kenyon Cell Somata as a Substrate for an Olfactory Sensory Memory in Drosophila. Front. Cell. Neurosci. 2018;12:128. doi: 10.3389/fncel.2018.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z., Stork T., Bergles D.E., Freeman M.R. Neuromodulators signal through astrocytes to alter neural circuit activity and behaviour. Nature. 2016;539:428–432. doi: 10.1038/nature20145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen P.L., Hasselbalch S.G., Hagemann L.P., Olsen K.S., Bülow J., Holm S., Wildschiødtz G., Paulson O.B., Lassen N.A. Persistent resetting of the cerebral oxygen/glucose uptake ratio by brain activation: evidence obtained with the Kety-Schmidt technique. J. Cereb. Blood Flow Metab. 1995;15:485–491. doi: 10.1038/jcbfm.1995.60. [DOI] [PubMed] [Google Scholar]
- Magistretti P.J., Allaman I. A cellular perspective on brain energy metabolism and functional imaging. Neuron. 2015;86:883–901. doi: 10.1016/j.neuron.2015.03.035. [DOI] [PubMed] [Google Scholar]
- Matsuno M., Horiuchi J., Yuasa Y., Ofusa K., Miyashita T., Masuda T., Saitoe M. Long-term memory formation in Drosophila requires training-dependent glial transcription. J. Neurosci. 2015;35:5557–5565. doi: 10.1523/JNEUROSCI.3865-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila J., Hietakangas V. Regulation of Carbohydrate Energy Metabolism in Drosophila melanogaster. Genetics. 2017;207:1231–1253. doi: 10.1534/genetics.117.199885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire S.E., Le P.T., Osborn A.J., Matsumoto K., Davis R.L. Spatiotemporal rescue of memory dysfunction in Drosophila. Science. 2003;302:1765–1768. doi: 10.1126/science.1089035. [DOI] [PubMed] [Google Scholar]
- McNay E.C., Ong C.T., McCrimmon R.J., Cresswell J., Bogan J.S., Sherwin R.S. Hippocampal memory processes are modulated by insulin and high-fat-induced insulin resistance. Neurobiol. Learn. Mem. 2010;93:546–553. doi: 10.1016/j.nlm.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melom J.E., Littleton J.T. Mutation of a NCKX eliminates glial microdomain calcium oscillations and enhances seizure susceptibility. J. Neurosci. 2013;33:1169–1178. doi: 10.1523/JNEUROSCI.3920-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mery F., Kawecki T.J. A cost of long-term memory in Drosophila. Science. 2005;308:1148. doi: 10.1126/science.1111331. [DOI] [PubMed] [Google Scholar]
- Naganos S., Horiuchi J., Saitoe M. Mutations in the Drosophila insulin receptor substrate, CHICO, impair olfactory associative learning. Neurosci. Res. 2012;73:49–55. doi: 10.1016/j.neures.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Newman L.A., Korol D.L., Gold P.E. Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS ONE. 2011;6:e28427. doi: 10.1371/journal.pone.0028427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papouin T., Dunphy J.M., Tolman M., Dineley K.T., Haydon P.G. Septal Cholinergic Neuromodulation Tunes the Astrocyte-Dependent Gating of Hippocampal NMDA Receptors to Wakefulness. Neuron. 2017;94:840–854.e7. doi: 10.1016/j.neuron.2017.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual A., Préat T. Localization of long-term memory within the Drosophila mushroom body. Science. 2001;294:1115–1117. doi: 10.1126/science.1064200. [DOI] [PubMed] [Google Scholar]
- Pearson-Leary J., McNay E.C. Novel Roles for the Insulin-Regulated Glucose Transporter-4 in Hippocampally Dependent Memory. J. Neurosci. 2016;36:11851–11864. doi: 10.1523/JNEUROSCI.1700-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson-Leary J., Jahagirdar V., Sage J., McNay E.C. Insulin modulates hippocampally-mediated spatial working memory via glucose transporter-4. Behav. Brain Res. 2018;338:32–39. doi: 10.1016/j.bbr.2017.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L., Magistretti P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer B.D., Ngo T.-T.B., Hibbard K.L., Murphy C., Jenett A., Truman J.W., Rubin G.M. Refinement of tools for targeted gene expression in Drosophila. Genetics. 2010;186:735–755. doi: 10.1534/genetics.110.119917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaçais P.-Y., Preat T. To favor survival under food shortage, the brain disables costly memory. Science. 2013;339:440–442. doi: 10.1126/science.1226018. [DOI] [PubMed] [Google Scholar]
- Plaçais P.-Y., Trannoy S., Isabel G., Aso Y., Siwanowicz I., Belliart-Guérin G., Vernier P., Birman S., Tanimoto H., Preat T. Slow oscillations in two pairs of dopaminergic neurons gate long-term memory formation in Drosophila. Nat. Neurosci. 2012;15:592–599. doi: 10.1038/nn.3055. [DOI] [PubMed] [Google Scholar]
- Plaçais P.-Y., de Tredern É., Scheunemann L., Trannoy S., Goguel V., Han K.-A., Isabel G., Preat T. Upregulated energy metabolism in the Drosophila mushroom body is the trigger for long-term memory. Nat. Commun. 2017;8:15510. doi: 10.1038/ncomms15510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O., Tjian R. Transcriptional feedback control of insulin receptor by dFOXO/FOXO1. Genes Dev. 2005;19:2435–2446. doi: 10.1101/gad.1340505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Martín A., Ceballo S., Baeza-Lehnert F., Lerchundi R., Valdebenito R., Contreras-Baeza Y., Alegría K., Barros L.F. Imaging mitochondrial flux in single cells with a FRET sensor for pyruvate. PLoS ONE. 2014;9:e85780. doi: 10.1371/journal.pone.0085780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J.X., Yakel J.L. Functional α7 nicotinic ACh receptors on astrocytes in rat hippocampal CA1 slices. J. Mol. Neurosci. 2012;48:14–21. doi: 10.1007/s12031-012-9719-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shytle R.D., Mori T., Townsend K., Vendrame M., Sun N., Zeng J., Ehrhart J., Silver A.A., Sanberg P.R., Tan J. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J. Neurochem. 2004;89:337–343. doi: 10.1046/j.1471-4159.2004.02347.x. [DOI] [PubMed] [Google Scholar]
- Siesjö B.K. Brain energy metabolism and catecholaminergic activity in hypoxia, hypercapnia and ischemia. J. Neural Transm. Suppl. 1978:17–22. [PubMed] [Google Scholar]
- Soto M., Cai W., Konishi M., Kahn C.R. Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc. Natl. Acad. Sci. USA. 2019;116:6379–6384. doi: 10.1073/pnas.1817391116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa-Nunes R., Yee L.L., Gould A.P. Fat cells reactivate quiescent neuroblasts via TOR and glial insulin relays in Drosophila. Nature. 2011;471:508–512. doi: 10.1038/nature09867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A., Stern S.A., Bozdagi O., Huntley G.W., Walker R.H., Magistretti P.J., Alberini C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144:810–823. doi: 10.1016/j.cell.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney S.T., Broadie K., Keane J., Niemann H., O’Kane C.J. Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron. 1995;14:341–351. doi: 10.1016/0896-6273(95)90290-2. [DOI] [PubMed] [Google Scholar]
- Takanaga H., Chaudhuri B., Frommer W.B. GLUT1 and GLUT9 as major contributors to glucose influx in HepG2 cells identified by a high sensitivity intramolecular FRET glucose sensor. Biochim. Biophys. Acta. 2008;1778:1091–1099. doi: 10.1016/j.bbamem.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasaki C., Yamasaki M., Uchigashima M., Konno K., Yanagawa Y., Watanabe M. Cytochemical and cytological properties of perineuronal oligodendrocytes in the mouse cortex. Eur. J. Neurosci. 2010;32:1326–1336. doi: 10.1111/j.1460-9568.2010.07377.x. [DOI] [PubMed] [Google Scholar]
- Takata N., Mishima T., Hisatsune C., Nagai T., Ebisui E., Mikoshiba K., Hirase H. Astrocyte calcium signaling transforms cholinergic modulation to cortical plasticity in vivo. J. Neurosci. 2011;31:18155–18165. doi: 10.1523/JNEUROSCI.5289-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiels E., Urban N.N., Gonzalez-Burgos G.R., Kanterewicz B.I., Barrionuevo G., Chu C.T., Oury T.D., Klann E. Impairment of long-term potentiation and associative memory in mice that overexpress extracellular superoxide dismutase. J. Neurosci. 2000;20:7631–7639. doi: 10.1523/JNEUROSCI.20-20-07631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trueta C., Méndez B., De-Miguel F.F. Somatic exocytosis of serotonin mediated by L-type calcium channels in cultured leech neurones. J. Physiol. 2003;547:405–416. doi: 10.1113/jphysiol.2002.030684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully T., Preat T., Boynton S.C., Del Vecchio M. Genetic dissection of consolidated memory in Drosophila. Cell. 1994;79:35–47. doi: 10.1016/0092-8674(94)90398-0. [DOI] [PubMed] [Google Scholar]
- van Dam E., van Leeuwen L.A.G., Dos Santos E., James J., Best L., Lennicke C., Vincent A.J., Marinos G., Foley A., Buricova M. Sugar-Induced Obesity and Insulin Resistance Are Uncoupled from Shortened Survival in Drosophila. Cell Metab. 2020;31:710–725.e7. doi: 10.1016/j.cmet.2020.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vélez-Fort M., Audinat E., Angulo M.C. Functional alpha 7-containing nicotinic receptors of NG2-expressing cells in the hippocampus. Glia. 2009;57:1104–1114. doi: 10.1002/glia.20834. [DOI] [PubMed] [Google Scholar]
- Vicente-Gutierrez C., Bonora N., Bobo-Jimenez V., Jimenez-Blasco D., Lopez-Fabuel I., Fernandez E., Josephine C., Bonvento G., Enriquez J.A., Almeida A., Bolaños J.P. Astrocytic mitochondrial ROS modulate brain metabolism and mouse behaviour. Nat. Metab. 2019;1:201–211. doi: 10.1038/s42255-018-0031-6. [DOI] [PubMed] [Google Scholar]
- Volkenhoff A., Weiler A., Letzel M., Stehling M., Klämbt C., Schirmeier S. Glial Glycolysis Is Essential for Neuronal Survival in Drosophila. Cell Metab. 2015;22:437–447. doi: 10.1016/j.cmet.2015.07.006. [DOI] [PubMed] [Google Scholar]
- Volkenhoff A., Hirrlinger J., Kappel J.M., Klämbt C., Schirmeier S. Live imaging using a FRET glucose sensor reveals glucose delivery to all cell types in the Drosophila brain. J. Insect Physiol. 2018;106:55–64. doi: 10.1016/j.jinsphys.2017.07.010. [DOI] [PubMed] [Google Scholar]
- Weislogel J.-M., Bengtson C.P., Müller M.K., Hörtzsch J.N., Bujard M., Schuster C.M., Bading H. Requirement for nuclear calcium signaling in Drosophila long-term memory. Sci. Signal. 2013;6:ra33. doi: 10.1126/scisignal.2003598. [DOI] [PubMed] [Google Scholar]
- Weiss S., Melom J.E., Ormerod K.G., Zhang Y.V., Littleton J.T. Glial Ca2+signaling links endocytosis to K+ buffering around neuronal somas to regulate excitability. eLife. 2019;8:e44186. doi: 10.7554/eLife.44186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005;26:19–39. [PMC free article] [PubMed] [Google Scholar]
- Yamazaki D., Horiuchi J., Ueno K., Ueno T., Saeki S., Matsuno M., Naganos S., Miyashita T., Hirano Y., Nishikawa H. Glial dysfunction causes age-related memory impairment in Drosophila. Neuron. 2014;84:753–763. doi: 10.1016/j.neuron.2014.09.039. [DOI] [PubMed] [Google Scholar]
- Yao W.-D., Rusch J., Poo Mm., Wu C.-F. Spontaneous acetylcholine secretion from developing growth cones of Drosophila central neurons in culture: effects of cAMP-pathway mutations. J. Neurosci. 2000;20:2626–2637. doi: 10.1523/JNEUROSCI.20-07-02626.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuyama K., Salvaterra P.M. Localization of choline acetyltransferase-expressing neurons in Drosophila nervous system. Microsc. Res. Tech. 1999;45:65–79. doi: 10.1002/(SICI)1097-0029(19990415)45:2<65::AID-JEMT2>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Yellen G. Fueling thought: Management of glycolysis and oxidative phosphorylation in neuronal metabolism. J. Cell Biol. 2018;217:2235–2246. doi: 10.1083/jcb.201803152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J.C., Wallach J.S., Del Vecchio M., Wilder E.L., Zhou H., Quinn W.G., Tully T. Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell. 1994;79:49–58. doi: 10.1016/0092-8674(94)90399-9. [DOI] [PubMed] [Google Scholar]
- Zhang W., Thompson B.J., Hietakangas V., Cohen S.M. MAPK/ERK signaling regulates insulin sensitivity to control glucose metabolism in Drosophila. PLoS Genet. 2011;7:e1002429. doi: 10.1371/journal.pgen.1002429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X., Yang Z., Yue Z., Alvarez J.D., Sehgal A. FOXO and insulin signaling regulate sensitivity of the circadian clock to oxidative stress. Proc. Natl. Acad. Sci. USA. 2007;104:15899–15904. doi: 10.1073/pnas.0701599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.