

Abstract
Slow delayed rectifier (IKs) channels consist of KCNQ1 and KCNE1. IKs functions as a ‘repolarization reserve’ in the heart by providing extra current for ventricular action potential shortening during β-adrenergic stimulation. There have been debates as to how KCNQ1 and KCNE1 traffic in cells, where they associate to form IKs channels, and the distribution pattern of IKs channels relative to β-adrenergic signaling complex. We used experimental strategies not previously applied to KCNQ1, KCNE1 or IKs, to provide new insights into these issues. ‘Retention-using-selected-hook’ experiments showed that newly translated KCNE1 constitutively trafficked through the conventional secretory path to the cell surface. KCNQ1 largely stayed in the endoplasmic reticulum, although dynamic KCNQ1 vesicles were observed in the submembrane region. Disulfide-bonded KCNQ1/KCNE1 constructs reported preferential association after they had reached cell surface. In-situ proximity ligation assay (PLA) detected IKs channels in surface sarcolemma but not t-tubules of ventricular myocytes, similar to the reported location of adenylate cyclase 9/yotiao. Fluorescent protein (FP)-tagged KCNQ1 and KCNE1, in conjunction with antibodies targeting their extracellular epitopes, detected distinct cell surface and cytoplasmic pools of both proteins in myocytes. We conclude that in cardiomyocytes KCNQ1 and KCNE1 traffic by different routes to surface sarcolemma where they assemble into IKs channels. This mode of delayed channel assembly helps IKs fulfill its function of repolarization reserve. Proteomic experiments revealed a novel KCNQ1 interactor, microtubule plus-end binding protein 1 (EB1). EB1 dimer (active form) bound KCNQ1 and increased its surface level. An LQT1 mutation, Y111C, reduced KCNQ1 binding to EB1 dimer.
Keywords: cardiac electrophysiology, repolarization reserve, protein trafficking, proteomics
INTRODUCTION
The slow delayed rectifier (IKs) channel functions as a ‘repolarization reserve’ in the heart (Sarkar & Sobie, 2016). During exercise or under emotional stress, β-adrenergic tone is high and heart rates are fast. More repolarizing currents are needed to shorten ventricular action potentials, to allow sufficient diastole for ventricular filling. IKs responds to β-adrenergic stimulation by becoming larger (amplitude response) and activating faster (gating response) (Marx et al., 2002). As such, IKs provides the extra current needed for action potential shorting during high β-adrenergic tone (Jost et al., 2005; Banyasz et al., 2014).
IKs channel has two major components: a voltage-gated KCNQ1 channel and regulatory KCNE1 subunits (Sanguinetti et al., 1996; Xu et al., 2013). Loss-of-function mutations in KCNQ1 and KCNE1 have been linked to long QT syndrome types 1 and 5, while gain-of-function mutations in KCNQ1 have been linked to short QT syndrome type 2 (Splawski et al., 2000; Borggrefe et al., 2005). Excessive IKs has been observed in atrial myocytes from patients of chronic atrial fibrillation (Caballero et al., 2010). These clinical data indicate the importance of a dynamic control of IKs current amplitude to support its role of ‘repolarization reserve’, yet preventing excessive IKs when not needed. Although the issues of how KCNQ1 and KCNE1 traffic and interact in cells have been studied for many years, there remain debates among investigators and discrepancies among reports. These debates or discrepancies are described below, in the context of four major questions we address in this study. We used experimental approaches not previously applied to the studies of KCNQ1, KCNE1 or IKs, thus providing new insights into the issues under debates.
First, where are IKs channels located in adult ventricular myocytes?
To achieve a timely response to β-adrenergic stimulation, cell surface IKs channels need to be close to adenylate cyclase 9 (AC9) and yotiao (also known as A-kinase anchoring protein 9). This is because IKs response to β-adrenergic stimulation relies on cAMP production by AC9 (Li et al., 2019), and recruitment of protein kinase A (PKA) and protein phosphatase 1A (PP1A) by yotiao to its vicinity (Marx et al., 2002). It has been shown that in adult mouse ventricular myocytes, AC9 and yotiao both cluster to the surface sarcolemma (SS) but not t-tubules (TT) (Li et al., 2019). Yet it has been proposed that KCNQ1, and likely the IKs channels, are highly enriched in t-tubules instead of surface sarcolemma (Rasmussen et al., 2003; Balse et al., 2012) (Oliveras et al., 2020). Therefore, there appears to be a discrepancy between AC9/yotiao and IKs channels in terms of their subcellular distribution pattern. A common caveat in previous studies is the use of immunofluorescence of KCNQ1 and KCNE1 and their colocalization to infer IKs location (Jiang et al., 2017; Oliveras et al., 2020). In this study, we used a much more stringent approach, in situ proximity ligation assay (PLA) (Soderberg et al., 2008), to detect KCNQ1/KCNE1 assembly in adult ventricular myocytes.
Second, are there separate (cell surface and cytoplasmic) pools of KCNQ1 and KCNE1 in adult ventricular myocytes? If yes, where are they?
Previously, we used biochemical experiments to propose that in guinea pig hearts native KCNQ1 is mainly in a cytoplasmic compartment (junctional sarcoplasmic reticulum, jSR), while native KCNE1 is mainly in the sarcolemma (SL) (Jiang et al., 2017). Such segregation of KCNQ1 and KCNE1 into different subcellular compartments appears contradicting the conventional view that KCNQ1 and KCNE1 should be well colocalized because they are obligate partners in cardiac myocytes. Here we used antibodies binding extracellular epitopes on KCNQ1 and KCNE1 to distinguish cell surface KCNQ1 and KCNE1 from their cytoplasmic counterparts.
Third, how do KCNQ1 and KCNE1 traffic from endoplasmic reticulum (ER, where they are translated) to the cell surface, and how do they interact with each other during the journey?
Previously, we proposed that KCNQ1 and KCNE1 traffic by separate routes from ER to the sarcolemma in adult ventricular myocytes (Jiang et al., 2017). This concept appears at adds with the view that KCNE1 requires KCNQ1 to reach cell surface (Chandrasekhar et al., 2006), or KCNE1 regulates KCNQ1 trafficking to cell surface (Krumerman et al., 2004). One major challenge in mapping proteins’ trafficking routes is the synchronization of their movements in cells. In this study, we used a novel approach ‘retention using selected hook’ or RUSH (Boncompain et al., 2012) to synchronize the timing of KCNQ1 and KCNE1 exit from the ER. This allowed us to track their trafficking paths and timing of reaching different cellular compartments.
Fourth, where are KCNQ1 and KCNE1 assembled into IKs channels?
Different scenarios have been proposed: KCNQ1 and KCNE1 assemble early while they are in the ER (Bas et al., 2011), in a post-ER but pre-Golgi vesicular compartment (Vanoye et al., 2010), at the Golgi (David et al., 2013), or at the ER/plasma membrane junctions (Oliveras et al., 2020). In this study, we used a reporter of functional KCNQ1/KCNE1 assembly to quantitatively compare the degree of their assembly under the control conditions vs when they were forced to stay inside cells.
Finally, we used affinity purification followed by liquid chromatography/tandem mass spectrometry (LC/MS-MS) to search for novel KCNQ1 and KCNE1 interactors in cells. The proteomic data support our experimental findings: KCNQ1 and KCNE1 traffic by separate routes from ER to the surface membrane, and they cluster to the same subdomains in surface membrane to assemble into IKs channels. We also verified a novel KCNQ1 interactor identified by proteomics, microtubule plus-end binding protein 1 (EB1). We showed that EB1 dimer binds and facilitates KCNQ1 to reach the surface membrane, and an LQT1-associated mutation (Y111C) reduces KCNQ1 binding to EB1 dimer.
MATERIALS AND METHODS
Molecular constructs
The following plasmids were from AddGene: Str-Ii_VSVG-SBP-eGFP (from Franck Perez, Addgene plasmid # 65300) (Boncompain et al., 2012), DsRed2-ER-5 (from Michael Davidson, Addgene plasmid # 55836), pECFP-SEC31A (from David Stephens, Addgene plasmid # 66612), pMXs-IP spGFP-ERGIC53 (from Noboru Mizushima, Addgene plasmid # 38270), and EB1-tandem dimer tdTomato (from Erik Dent, Addgene plasmid # 50825).
KCNQ1-GFP (Q1-GFP), with eGFP fused to the C-terminus of the 676 aa KCNQ1 isoform 1, was a gift from Andrew Tinker (Mashanov et al., 2010). An LQT1-associated mutation, Y111C (tyrosine at position 111 mutated to cysteine), was created in the Q1-GFP background. KCNE1-dsR (E1-dsR) has been described previously (Jiang et al., 2017). HA-KCNE1-dsR (HA-E1-dsR) was created by inserting an HA epitope (YPYDVPDYA) between aa 20 and 21 in the extracellular domain of E1-dsR. KCNQ1-Q147C (Q1-Q147C) had glutamine at position 147 mutated to cysteine in a cysteine-removed 548 aa KCNQ1 isoform 0 background. KCNE1-G40C (E1-G40C) had glycine at position 40 mutated to cysteine. Q1-Q147C and E1-G40C have been described previously (Wang et al., 2011). All in-house mutations were created using Q5 Site-directed mutagenesis kit, and confirmed by direct DNA sequencing.
The ER-hooked_HA-E1-dsR and ER-hooked_Q1-GFP were created using Str-Ii_VSVG-SBP-EGFP as the starting material by GenScript. Str-Ii_VSVG-SBP-EGFP has an ER-membrane embedded protein (Ii, an isoform of the human invariant chain of the major histocompatibility complex with an N-terminal arginine-based motif retaining it in the ER), a core streptavidin peptide (Str) fused to its N-terminus in the cytosolic side of the ER membrane, an internal ribosome entry (IRE) sequence, followed by wild-type vesicular stomatitis virus protein G (VSVG) fused with streptavidin binding peptide (SBP) and eGFP. Q1-GFP was fused with SBP and a 7-Gly peptide, (G)7, in between to allow free SBP binding to Str. HA-E1-dsR was fused with SBP and a 7-Gly peptide in between. For both, Asc I and Pac I enzyme sites were created at 5’ and 3’ ends, respectively. The VSVG-SBP-EGFP was removed from plasmid 65300 using Asc I and Pac I cuts, followed by directed subcloning of the Q1-GFP-(G)7-SBP or HA-E1-dsR-(G)7-SBP to create the ER-hooked Q1-GFP and ER-hooked HA-E1-dsR constructs under CMV promoter for mammalian cell expression. Both constructs were confirmed by DNA sequencing.
Adenovirus production
HA-E1-dsR and Q1-GFP were subcloned into Adeno_shuttle vector and, after confirming gene function by patch clamp, immunoblotting, and imaging experiments, they were sent to Vector Biolabs for adenoviral production into adenoviral-type 5, ΔE1/E3. The viral titers were 4.5×1010 and 5×1010 PFU/ml, respectively, where PFU stands for ‘plaque formation units’.
Cardiac myocyte isolation, culture and adenovirus transduction
The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. The animal protocol (AM10294) has been reviewed and approved annually by the Institutional Animal Care and Use Committee of Virginia Commonwealth University.
Myocytes were isolated from guinea pig or Sprague Dawley rats, 2–4 month old, male. The procedures of myocyte isolation, culture, and adenovirus incubation have been described in our publication (Jiang et al., 2017). Briefly, the aorta was cannulated and mounted on a Langendorff apparatus. The heart was perfused with the following solutions sequentially (all at 36°C, oxygenated): normal Tyrode’s (mM: NaCl 146, MgCl2 0.5, HEPES 5, dextrose 5.5, KCl 4, CaCl2 2, pH 7.3) for 5 min, nominally Ca-free Tyrode’s with 1% fat-free bovine serum albumin (BSA) for 5 min, and the same Ca-free, BSA-containing solution with collagenase (type II, Worthington, 1 mg/ml), for 20–30 min. The atria were removed, and the ventricular tissue chunks were gently shaken in Kraftbruhe (KB) medium to release single myocytes. Myocytes were allowed to recover in KB for 1 hr at room temperature (RT), and switched to nominally Ca free Tyrode’s supplemented with BDM (5 mM), L-carnitine (2 mM), taurine (5 mM), glutamate (2 mM) and BSA (1%) for 1 hr at RT. Myocytes were plated on mouse laminin-coated coverslips or dishes and incubated in medium 199 supplemented with creatine (5 mM), L-carnitine (5 mM), taurine (5 mM), BSA (0.2%), FCS (5%), cytochalasin D (0.2 uM), and penicillin/streptomycin, in 36°C CO2 moist incubator. After culture for 2 hr, adenovirus(es) was(were) added to the culture medium at 107 PFU/ml each final concentration. Incubation with adenovirus(es) continued for specified amounts of time (noted in text or figure legends), before experiments.
COS-7 and HEK293 cell culture and transfection
The procedures have been described in our previous publication (Jiang et al., 2017). Briefly, cells were maintained in DMEM supplemented with 10% FCS, non-essential amino acids, penicillin and streptomycin in 36°C CO2 moist incubator. Cells were plated on matrigel-coated coverslips or dishes, and when reaching ~70% confluency, transfected with cDNAs facilitated by lipofectamine 2000 for 5–6 hr in base DMEM. After removing cDNAs, cells were cultured in complete DMEM for 24–36 hr before experiments.
Patch clamp experiments
Currents of rat and guinea pig ventricular myocytes were recorded using the whole-cell patch clamp mode. Rat myocytes were superfused with normal Tyrode’s solution (composition given above, with CaCl2 2 mM) containing 1 uM nisoldipine at RT. Guinea pig myocytes were superfused with Na- and Ca-free Tyrode’s solution (NaCl and CaCl2 replaced by equimolar choline-Cl and MgCl2) at 33°C. Patch pipette solution contained (in mM): K-aspartate 120, KCl 20, EGTA 10, ATP (K) 10, MgCl2 1, HEPES 10, pH 7.3. Command voltage and current recording were controlled by Clampex of pCalmp 10 via Axopatch 200B/1440A interface and low-pass filter (cut off 10 kHz). Currents were analyzed with Clampfit. Numerical data were exported to Excel or SigmaPlot for processing and graph production.
In situ proximity ligation assay (PLA) experiments (Soderberg et al., 2008)
Myocytes transduced with Q1-GFP and HA-E1-dsR and cultured for 24–36 hr were fixed (4% paraformaldehyde in 1xPBS, at RT, 10 min), permeabilized (0.1% triton X-100 in 1xPBS, at RT, for 10 min), and incubated with GFP mouse Ab and dsR rabbit Ab overnight at 4°C. After removing the Abs, the PLA procedure was performed using DUO92-14 kit (Sigma Aldrich) following manufacturer’s instructions. Far red fluorophore was incorporated to mark sites of proximity between GFP mouse and dsR rabbit Abs.
Retention using selected hook (RUSH) experiments (Boncompain et al., 2012)
COS-7 cells transfected with ER-hooked constructs (described above under ‘Molecular constructs’) were cultured under the control conditions for 36 hr. Then biotin was added to the medium (final concentration 100 uM) and incubation continued at 36°C in CO2 moist incubator for specified amounts of time (noted in text or figure legends). Biotin competed off Str-Ii binding to SBP on the Q1-GFP, HA-E1-dsR or VSVG-GFP, releasing them from the ER hook. After biotin incubation, cells were fixed for imaging experiments. GFP and dsR were labeled with GFP goat Ab/Alexa488 donkey anti-goat and dsR rabbit Ab/Alexa568 donkey anti-rabbit, respectively.
Immunoblot experiments
Unless otherwise noted, immunoblot experiments were run under reducing conditions. Protein samples were mixed with 2x or 5x sample buffer containing reducing agent (5% mercaptoethanol), and incubated at 50°C for 15 min before loading. Proteins were fractionated by sodium dodecylsulfate/polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to PVDF membranes. The membranes were blocked by incubation in 1x TBST with 10% nonfat dry milk (RT, 1 hr) and then incubated with primary Abs diluted in 1xTBST with 5% BSA (4°C overnight, or 2 hr at RT). Membranes were then incubated with horse radish peroxidase (HRP)-conjugated secondary Ab and immunoreactive bands were visualized with enhanced chemiluminescence kit using an imager FluorChem M. Band intensities were quantified using AlphaView SA, and numerical data were exported to Excel for processing.
Quantification of cell surface KCNQ1 by biotinylation
To biotinylate cell surface proteins, live COS-7 cells expressing Q1-Q147C/E1-G40C. Q1-GFP or Q1-GFP Y111C were incubated with membrane-impermeable, amine-reactive biotin (EZ-Link Sulfo-NHS-SS-Biotin) at 0.25 mg/ml, for 30 min at 4°C. Biotinylation reaction was quenched by 100 mM glycine in 1xPBS. Cells were lysed in 1% Triton X-100 lysis buffer and 90% of the whole cell lysate (WCL) was incubated with neutravidin beads to bind biotinylated (cell surface) proteins. The other 10% WCL and the eluent from neutravidin beads (biotinylated fraction) were fractionated by SDS-PAGE, transferred to PVDF membranes, and probed with an Ab targeting KCNQ1. KCNQ1 band intensities in the WCL lane (W) and in the biotinylated fraction lane (b) were quantified by densitometry, and the results were used to calculate % KCNQ1 on the cell surface: [b/(W*9+b)]*100%.
Quantification of disulfide bonded Q1-Q147C/E1-G40C
COS-7 cells transfected with Q1-Q147C/E1-G40C were cultured for 24 hr at 16°C to allow protein translation without ER exit. The medium was switched to 36°C and culture continued under the control conditions, or in the presence of brefeldin A (BFA, 25 ug/ml) for 24 hr before experiments. Cells were lysed. WCL was divided into two equal aliquots. One incubated with dithiothreitol (DTT, 50 mM, RT, 30 min) to break disulfide bonds and the other not (+DTT and -DTT). WCLs were mixed with 2x sample buffer without mercaptoethanol, and fractionated on SDS-PAGE. After proteins were transferred to PVDF membrane, the membrane was probed with an Ab targeting KCNQ1.
Microscopy experiments and image data analysis
Imaging experiments were done using Zeiss 710 (confocal microscopy, diffraction-limited), Zeiss 880 (Airyscan microscopy, xy resolution improved ~ 1.7 fold), Nikon structured illumination microscopy (SIM, xy resolution improved ~ 2 fold), or Olympus total internal reflection fluorescence (TIRF) microscope. COS-7 and myocytes were imaged using x63 and x40 oil immersion objectives, respectively. Fluorophores were sequentially excited by laser lines: 405 nm (Hoechst dye), 488 nm (GFP or Alexa488), 561 nm (dsRed or Alexa568), and 633 nm (Alexa647). The emitted lights were collected with appropriate band-pass or long-pass filters to avoid ‘bleed through’.
To image fixed COS-7 cells or myocytes, cells were plated on matrigel- or laminin-coated no. 1.5 coverslips, and transfected or transduced as specified in text or figure legends. For experiments testing cell surface proteins, live COS-7 cells or fixed but un-permeabilized myocytes were incubated with antibodies targeting epitopes in the extracellular domains of KCNQ1 and KCNE1, followed by Alexa-conjugated secondary Ab targeting the surface-bound primary Ab. COS-7 cells were then fixed. COS-7 cells or myocytes were permeabilized (0.1% Triton X-100 in 1xPBS, RT, 10 min), and incubated with Abs targeting intracellular epitopes. Coverslips were mounted on glass slides with ProLong Diamond anti-fade mountant (Molecular probes).
To image live COS-7 cells, cells were plated on matrigel-coated no. 1.5 glass-bottom microwell dishes (MatTek), transfected as described in text. Nuclei were stained with Hoechst dye. Cells were imaged in phenol red-free DMEM with 5% FCS at 37°C.
Immunoprecipitation experiments
HEK293 cells transfected with Q1-GFP and HA-E1-dsR or untransfected HEK293 cells (as negative control), or rat ventricular myocytes transduced with Q1-GFP and HA-E1-dsR were used for immunoprecipitation experiments. Whole cell lysate (WCL) in 1% Triton X-100 lysis buffer was precleared with protein A/G coated magnetic beads. The beads were collected as negative control ([-] IP) for rat ventricular myocytes. Precleared WCL was divided into equal aliquots, and each aliquot was incubated with fresh protein A/G coated magnetic beads with immunoprecipitating Abs (specified in text or figure legends) overnight at 4°C. The beads were collected as positive [+] IP. [-] and [+] IP beads were washed, and bound proteins were eluted by 2x sample buffer with 5% mercaptoethanol, at 50°C for 30 min. [-] and [+] IP were fractionated side-by-side with WCL (as direct input) by SDS-PAGE, followed by transfer to PVDF membranes. The membranes were probed with Abs noted in text or figure legends.
Proteomic experiments and data analysis
Nine immunoprecipitate samples ([+]: 2 GFP rabbit, 2 GFP goat, 2 dsR rabbit Ab from WCL of HEK293 cells transfected with Q1-GFP and HA-E1-dsR, and [-]: 1 each of GFP rabbit, GFP goat, dsR rabbit Ab from WCL of untransfected HEK293 cells) were quality checked and subject to protein identification using liquid chromatography/tandem mass spectrometry (LC/MS-MS, by Poochon Scientific). The samples were fractionated by 4–12% SDS-PAGE, followed by in-gel trypsin digestion. Tryptic peptide mixtures were cleaned up with C18 Zip-tip, and analyzed by LC/MS-MS using a Thermo Scientific Q-Exactive hybrid Quadrupole-Orbitrap Mass Spectrometer and a Thermo Dionex UltiMate 3000 RSLCnano System. Peptide mixture was loaded onto a peptide trap cartridge at a flow rate of 5 ul/min. The trapped peptides were eluted onto a reverse-phase PicoFrit column (New Objective, Woburn, MA) using a linear gradient of acetonitrile (3–36%) in 0.1% formic acid. The elution time was 60 min, at a flow rate of 0.3 ul/min. Eluted peptides were ionized and sprayed into the mass spectrometer, using a Nanospray Flex Ion Source ES071 (Thermo) under the following settings: spray voltage 1.8 kV, capillary temperature 250°C. Raw data files were searched against the Uniprot human protein database using the Proteome Discoverer 1.4 software based on the SEQUEST algorithm. Carbamidomethylation (+57.021 Da) of cysteines was a fixed modification, and oxidation of methionine and deamidation of glutamine and asparagine (+0.98402 Da) were set as dynamic modifications. The minimum peptide length was specified to be 5 amino acids. The precursor mass tolerance was set to 15 ppm, whereas the fragment mass tolerance was set to 0.05 Da. The maximum false peptide discovery rate was specified as 0.01. The resulting Proteome Discoverer Report contains all assembled proteins with peptide sequences and peptide spectrum match counts (#PSM).
The protein lists in combined GFP rabbit and goat Ab immunoprecipitate samples (4) and in DsRed rabbit Ab immunoprecipitate samples (2) were analyzed in the following manner:
The #PSM of all [+] samples were combined (ΣPSM of [+]). Those with ΣPSM of [+] ≥ 2 were retained.
Of the above, those absent in [-] ((ΣPSM of [-] = 0) and those having ratio of ΣPSM ([+]/[−]) ≥ 2 were retained. There were a total of 109 proteins in GFP Ab IP (Table S1), and 109 proteins in dsRed Ab IP (Table S2). Both tables are available as on-line supporting information.
The two protein lists were compared and divided into three lists: shared, KCNQ1-GFP unique and HA-KCNE1-dsR unique. These proteins were manually curated based on molecular function and/or cellular compartment.
Antibodies and reagents
Primary Abs used were: KCNE1 mouse Ab (H00003753-M01, AbNova), HA mouse Ab (MMS-101P, Covance), dsRed rabbit Ab (catalog # 632496, Clontech), KCNQ1 rabbit Ab targeting the extracellular domain of KCNQ1 (APC-168, Alomone), KCNQ1 goat Ab (C20, Santa Cruz biotechnology), GFP goat Ab (ab5450, AbCam), GFP rabbit Ab (ab290, AbCam), GFP mouse Ab (MA5-15256, Thermo Fisher), and EB1 rat Ab (ab53358, AbCam). Alexa-conjugated secondary Abs and WGA were purchased from Molecular Probes. HRP-conjugated secondary Abs were from Thermo Fisher.
Statistical analysis
All group data are reported by dot plots superimposed on bar graph of Mean±SD. Comparison between two groups was by two-tailed t-test assuming unequal variance (Excel). Comparison among more than two groups was by one-way ANOVA (SigmaStat v. 4.0). If p value was < 0.01, this was followed by Tukey’s pairwise tests. A p value < 0.05 is considered significant.
RESULTS
Fluorescent protein (FP)- and epitope-tagged KCNQ1 and KCNE1 expressed in adult rat ventricular myocytes behave like their native counterparts
We used Q1-GFP and HA-E1-dsRed as KCNQ1 and KCNE1 surrogates. These FP-tagged constructs offered two major advantages: allowing us to observe their distribution without antibodies, and serving as positive control (vs myocytes or cells without gene transfer) in the validation of experimental strategies. We used adult rat ventricular myocytes as the expression system. They have low background KCNQ1 and KCNE1 expression (Wu et al., 2006; Jiang et al., 2017; Oliveras et al., 2020), and their sarcolemma has distinct subdomains (t-tubule ‘TT’, surface sarcolemma ‘SS’, and intercalated disc ‘ICD’) important for data interpretation.
Fig. 1A, left, shows that when expressed in rat ventricular myocytes Q1-GFP was in prominent transverse striations, while HA-E1-dsR was prominent on the lateral surface (XY plane views). The two overlapped sporadically on or close to the lateral surface (XZ cross-sectional views) and in the intercalated disc region at cell ends. These patterns are similar to their native counterparts in ventricular myocytes from young adult rats reported previously (Jiang et al., 2017). They are also similar to the distribution patterns of native KCNQ1 and KCNE1 in guinea pig ventricular myocytes, the most commonly used model for studying cardiac IKs (Fig. 1B, left). Patch clamp recording revealed robust IKs currents in rat ventricular myocytes transduced with Q1-GFP and HA-E1-dsR, but not control myocytes (Fig. 1A, right). The slow rates of activation and deactivation and the positive voltage range of channel activation are similar to the native IKs recorded from young adult rat ventricular myocytes (as HMR1556-sensitive currents) (Jiang et al., 2017). They are also similar to native IKs recorded from guinea pig ventricular myocytes (Fig. 1B, right). These observations support the validity of using Q1-GFP and HA-E1-dsR expressed in rat ventricular myocytes to investigate how native KCNQ1 and KCNE1 traffic and interact in the myocyte environment.
Fig. 1. Fluorescent protein- and epitope-tagged KCNQ1 and KCNE1 retain their distribution patterns in cardiac myocytes and normal IKs channel function.

(A) Properties of Q1-GFP and HA-E1-dsR expressed in rat ventricular myocytes. Left: Confocal images of Q1-GFP and HA-E1-dsR in a rat ventricular myocyte. Shown are XY plane view at the central z-plane and a selected XZ plane view (locations noted by the white dashed lines in XY plane view). Overlap between Q1-GFP and HA-E1-dsR in the XZ plane view is marked by asterisks in the ‘merge’ panel. Right: Currents recorded from rat ventricular myocytes transduced with Q1-GFP and HA-E1-dsR (upper) and time-control (myocyte isolated from the same heart and cultured for the same duration without adenoviruses, middle). The voltage clamp protocol is diagrammed in the inset. The lower panel shows Boltzmann fit to the voltage-dependence of channel activation in transduced myocytes, estimated by the relationship between the test pulse voltages (Vt) and the peak tail current amplitudes (Ipeak): degree of activation =(Imax/Ipeak)/(1+exp((V0.5-Vt)/k)), where Imax, V0.5 and k are the estimated maximal peak tail current amplitude of the cell, half-maximum activation voltage and slope factor, respectively. The parameter values are listed in the inset. Data were pooled from four myocytes of one heart. (B) Properties of native KCNQ1 and KCNE1 in guinea pig ventricular myocytes. Left: Confocal images of immunofluorescence of native KCNQ1 and KCNE1 in a guinea ventricular myocyte. KCNQ1 and KCNE1 were labeled with goat and mouse primary antibodies, followed by Alexa-488 donkey anti-goat and Alexa-568 donkey anti-mouse secondary Abs. The format of this panel is the same as that of left panel in (A). Right: Native IKs recorded from guinea pig ventricular myocytes. Representative current traces are shown on top, with a diagram of the voltage clamp protocol, and Boltzmann fit to the voltage dependence of channel activation is shown on bottom, with the estimated parameter values in the inset. Data were pooled from nine myocytes of three hearts.
IKs channels are mainly present in the surface sarcolemma but not t-tubules in adult ventricular myocytes
We used in situ proximity ligation assay (PLA) (Soderberg et al., 2008) to locate functional Q1-GFP and HA-E1-dsR assemblies in adult ventricular myocytes. Fig. 2A depicts the PLA procedure. PLA detects locations where PLA probes bind that are ≤ 40 nm apart. In a functional Q1-GFP/HA-E1-dsR assembly, the distance between GFP mouse Ab and dsR rabbit Ab is estimated to be ≤ 37 nm, based on: (1) The cryoEM structure of KCNQ1/KCNE3 assembly (6V01.pdb), (Sun and MacKinnon (2020), which is similar to the KCNQ1/KCNE1 assembly in having KCNE3 occupying the cleft spaces between KCNQ1 subunits (Xu et al., 2013). In this cryoEM structure, the C-termini of KCNQ1 and KCNE3 are ~ 7 nm apart. (2) The dimensions of eGFP (2Y0G.pdb) and monomer dsRed (2VAD.pdb) (long-axes ~ 5 nm). (3) The length of antibodies (~ 10 nm).
Fig. 2. Localize KCNQ1/KCNE1 assembly in cardiac myocytes by in situ proximity ligation assay (PLA).

Rat ventricular myocytes transduced with Q1-GFP and HA-E1-dsR were cultured for 24–36 hr before experiments. (A) Diagram of PLA procedure. Primary antibodies (1st Abs) were GFP mouse Ab and dsRed rabbit Ab, binding to the FP tags in the cytoplasmic compartment. At sites where the 1st Abs were ≤ 40 nm apart, binding of nucleotide-conjugated PLA probes to the 1st Abs allowed nucleotide ligation and amplification. During the latter reaction, far red fluorophore was incorporated to mark these sites. Not shown in the diagram: after PLA procedure, myocytes were incubated with Alexa488 goat anti-mouse and Alexa568 goat anti-rabbit Abs to label total Q1-GFP and HA-E1-dsR. (B) Representative images of Q1-GFP, HA-E1-dsR and PLA (pseudo-colored white) from a myocyte. To better appreciate the 3D distribution pattern of PLA signals in the myocyte, the PLA image is shown as z-projection of maxima. A total of 29 Z-slices was acquired, and 20 Z-slices (excluding the bottom 6 and the top 3 slices to avoid interference by cell surface PLA signals) were used to create the z-projection. (C) Quantification of Q1-GFP, HA-E1-dsR and PLA signals in cell periphery and cell interior. Cell periphery was defined as an area 2 um wide within the cellular contour, while cell interior was the area within cell periphery. Shown are data points from individual myocytes (symbols, 8 myocytes from 2 hearts) and mean±SD (bar graphs). The three groups of data were subject to one-way ANOVA (p=0.001), followed by Tukey pairwise tests. PLA signals have higher % in cell periphery, and lower % in cell interior, than Q1-GFP and HA-E1-dsR.
Fig. 3 shows that no PLA signals were detected in control myocytes (not incubated with adenoviruses) or when only Q1-GFP was expressed. PLA signals were detected when both Q1-GFP and HA-E1-dsR were expressed. The higher the expression level, the stronger the PLA signals (36 vs 24 hr culture of adenovirus-transduced myocytes). These data indicate that non-specific probe binding or non-specific nucleotide ligation was minimal.
Fig. 3. Check for non-specific signals from in situ proximity ligation assay (PLA) used to detect KCNQ1/KCNE1 assembly.

Rat ventricular myocytes were incubated for the noted numbers of hours without or with adenovirus transduction of Q1-GFP and HA-E1-dsR. The PLA procedure and incubation with Alexa conjugated secondary Abs were the same as those described for Fig. 2. Asterisks mark non-specific PLA signals in nuclei of myocytes after 36 hr culture.
To have a complete view of the distribution pattern of PLA signals and their relationship to Q1-GFP and HA-E1-dsR, we acquired z-stack images of the fluorescence signals in every myocyte studied (8 myocytes, from 2 independent experiment). To quantify their distribution on lateral surface vs in cell interior (including t-tubules and cytoplasmic compartments), we collapsed the 3D z-stacks into 2D images by z-stack projection of maximal signals. The top and bottom slices were not included to avoid interference from signals on the top and bottom cell surfaces. A representative example is depicted in Fig. 2B. The PLA signals were mainly localized to the lateral surface. On the other hand, there were abundant Q1-GFP and HA-E1-dsR signals in the cell interior. This distinction offers another support for the specificity of PLA signals: PLA detected Q1-GFP/HA-E1-dsR assemblies, but not unassembled Q1-GFP and HA-E1-dsR even when they were abundant and close to each other.
We quantified the percentage of signals in the cell periphery, defined as a space 2 um wide within the cellular contour. Cell interior was defined as cellular area within the periphery. Data summarized in Fig. 2C confirms the consistency of images presented in Fig. 2B: PLA signals were much more abundant in cell periphery, and much less in cell interior, than Q1-GFP or HA-E1-dsR. Clustering of PLA signals to the lateral surface, without transverse striation pattern in cell interior, indicates that IKs channels are mainly localized to the surface sarcolemma, instead of t-tubules. This distribution pattern is consistent with proximity between cell surface IKs channels and AC9 and yotiao (Li et al., 2019), which is important for a timely IKs response to β-adrenergic stimulation.
Cell surface KCNQ1 and KCNE1 are on the surface sarcolemma but not t-tubules, and there are distinct cytoplasmic pools of KCNQ1 and KCNE1 in adult ventricular myocytes
We used a Q1 rabbit Ab that targets aa 284–297 in the extracellular domain of KCNQ1 (termed ‘Q1 ext Ab’) to label cell surface Q1. To test its ability and specificity in detecting cell surface targets, we exposed live COS-7 cells transiently transfected with Q1-GFP to the Q1 ext Ab, followed by Alexa-conjugated secondary Ab. Fig. 4A depicts two adjacent COS-7 cells, one expressing Q1-GFP (cell 1) and the other not (cell 2). Immunofluorescence signals of Q1 ext Ab (Q1 ext Ab IF) was clearly detected as puncta in the periphery of cell 1. No Q1 ext Ab IF was detected in cell 2. We used HA mouse Ab to detect cell surface HA-E1-dsR. Fig. 4B shows that no immunofluorescence signal of HA mAb (HA Ab IF) was detectable in myocytes not transduced with HA-E1-dsR, confirming its specificity.
Fig. 4. Check the specificity of Q1 ext Ab and HA Ab in detecting cell surface Q1-GFP and HA-E1-dsR.

(A) In unpermeabilized COS-7 cells, Q1 ext Ab IF signals were detected in cell 1 expressing abundant Q1-GFP, but not in cell 2 not expressing Q1-GFP based on the GFP fluorescence (GFP FP). Cellular contours are marked by dotted white line (cell 1) or dashed yellow line (cell 2). (B) HA Ab immunofluorescence (HA Ab IF) was not detectable in control rat ventricular myocytes without or with cell membrane permeabilization. Myocyte images were shown by fluorescence of wheat germ agglutinin (WGA).
Fig. 5Aa shows that in myocytes expressing Q1-GFP, the total pool of this protein detected by GFP fluorescence signals (GFP FP) showed the typical pattern of transverse striations. There were also strong perinuclear GFP signals, representing high translation activity in the nuclear envelope after adenovirus-mediated gene transfer. Cell surface Q1-GFP was detected by incubating unpermeabilized myocytes to Q1 ext Ab, followed by Alexa-conjugated secondary Ab. Cell surface Q1-GFP was mainly in myocyte periphery. There were no perinuclear signals or striations in myocyte interior. The fluorescence profiles across the unpermeabilized myocyte in Fig. 5Ba show clear distinction between cell surface and total pools of Q1-GFP. We used the fluorescence profiles to quantify the percentage of signals in cell periphery. Data summary in Fig. 5Ca supports the consistency among myocytes: the % of Q1 ext Ab IF in cell periphery was much higher than that of GFP FP.
Fig. 5. Distinguish between cell surface and total KCNQ1 and KCNE1 in cardiac myocytes.

Adult rat ventricular myocytes transduced with Q1-GFP and HA-E1-dsR were fixed. Myocytes labeled as ‘Unpermeabilized’ were incubated with a Q1 rabbit Ab targeting aa 284 −297 in the extracellular domain of KCNQ1 (Q1 ext Ab), or HA mouse Ab (targeting HA epitope in the extracellular domain of HA-E1-dsR). This was followed by Alexa647-conjugated secondary Abs. Myocytes labeled as ‘Permeabilized’ were incubated with 0.1% Triton X-100 at room temperature for 10 min to permeabilize the cell membrane. This was followed by incubation with the same primary (Q1 ext Ab and HA Ab) and secondary Abs. (A) Representative confocal images. Immunofluorescence (IF) signals from Q1 ext Ab and HA Ab report cell surface proteins (Unpermeabilized) or total proteins (Permeabilized), while fluorescence signals from GFP and dsRed (GFP FP and dsRed FP) report total Q1-GFP and HA-E1-dsR. White dashed lines denote where the fluorescence signal profiles in (B) were determined. (B) Profiles of fluorescence signals across myocyte width. Black and magenta traces represent FP and IF signals, respectively. The fluorescence profiles and cell widths were normalized to values between 0 and 1.0. Gray shading denotes where cell periphery signals in (C) were quantified. (C) Quantification of FP and IF signals in cell periphery of unpermeabilized myocytes. The percentage (%) of areas beneath the profile in the 0–0.2 and 0.8–1 cell width ranges was quantified for Q1-GFP and Q1-ext Ab IF (18 myocytes from 5 hearts), and for HA-E1-dsR and HA Ab IF (9 myocytes from 3 hearts). Shown are data points (circles) from individual myocytes and mean±SD (bar graphs), with p values by t-test.
Exposing permeabilized myocytes to Q1 ext Ab revealed that the distribution patterns of Q1 ext Ab IF and GFP FP were qualitatively the same: strong transverse striations and perinuclear signals. This is supported by the fluorescence profiles in Fig. 5Ba. These observations confirm that cell surface Q1-GFP is on surface sarcolemma but not t-tubules. The prominent transverse striation pattern seen in total Q1-GFP (as well as immunofluorescence of native KCNQ1 in guinea pig ventricular myocyte, Fig. 1B) represents a cytoplasmic protein pool. The most likely candidate for the cytoplasmic location is the junctional SR, which is adjacent to the z-lines and cannot be distinguished from t-tubules at the light microscopy level. Cytoplasmic pool in jSR is consistent with our previous biochemical experiments on native KCNQ1 in guinea pig heart (Jiang et al., 2017).
Fig. 5Ab shows that in myocytes expressing HA-E1-dsR, the total protein pool detected by the dsRed fluorescence (dsRed FP) had the typical distribution pattern: prominent on lateral surface and in vesicles, and dim in striations. Cell surface HA-E1-dsR was labeled by exposing unpermeabilized myocytes to HA mAb followed by Alexa-conjugated secondary Ab. Cell surface HA-E1-dsR was exclusively on the surface sarcolemma. There were no striations or vesicles. The fluorescence profiles across the myocyte width support the clear distinction between dsRed FP and HA Ab IF in the unpermeabilized myocyte (Fig. 5Bb). Data summary in Fig. 5Cb supports the consistency among myocytes: the % of HA Ab IF in cell periphery was much higher than that of dsRed FP. Exposing permeabilized myocytes to the HA Ab showed that the distribution patterns of HA Ab IF and dsRed FP overlapped (Fig. 5Ab and 5Bb). Therefore, similar to Q1-GFP, cell surface HA-E1-dsR is on surface sarcolemma but not t-tubules. The dim transverse striation pattern seen in total HA-E1-dsR represents a cytoplasmic pool, possibly the jSR as is the case for Q1-GFP.
KCNQ1 and KCNE1 traffic through separate routes from endoplasmic reticulum to the plasma membrane
We used a strategy ‘retention using selective hook’, or RUSH (Boncompain et al., 2012), to synchronize ER exit of Q1-GFP and HA-E1-dsR. This allowed us to monitor their paths and time courses of forward trafficking. The molecular construct and experimental protocol of RUSH are diagrammed in Fig. 6A. COS-7 cells transfected with the RUSH constructs were cultured under the control conditions before experiments. They were incubated with biotin (100 uM, 36°C) for specified durations and fixed. Q1-GFP and HA-E1-dsR were detected by immunofluorescence (GFP mouse Ab, and dsRed rabbit Ab, followed by Alexa-conjugated secondary Abs). Fig. 6B shows that before biotin incubation, Q1-GFP and HA-E1-dsR were retained in the ER (marked by junctophilin-2, JPH2) (Jiang et al., 2016). After 30 min biotin incubation, HA-E1-dsR entered vesicles while Q1-GFP appeared to stay in the ER. This observation suggested that Q1-GFP and HA-E1-dsR traffic differently after ER exit.
Fig. 6. Track KCNE1 and KCNQ1 movements after their ER exit by the ‘retention using selected hook’ (RUSH) strategy.
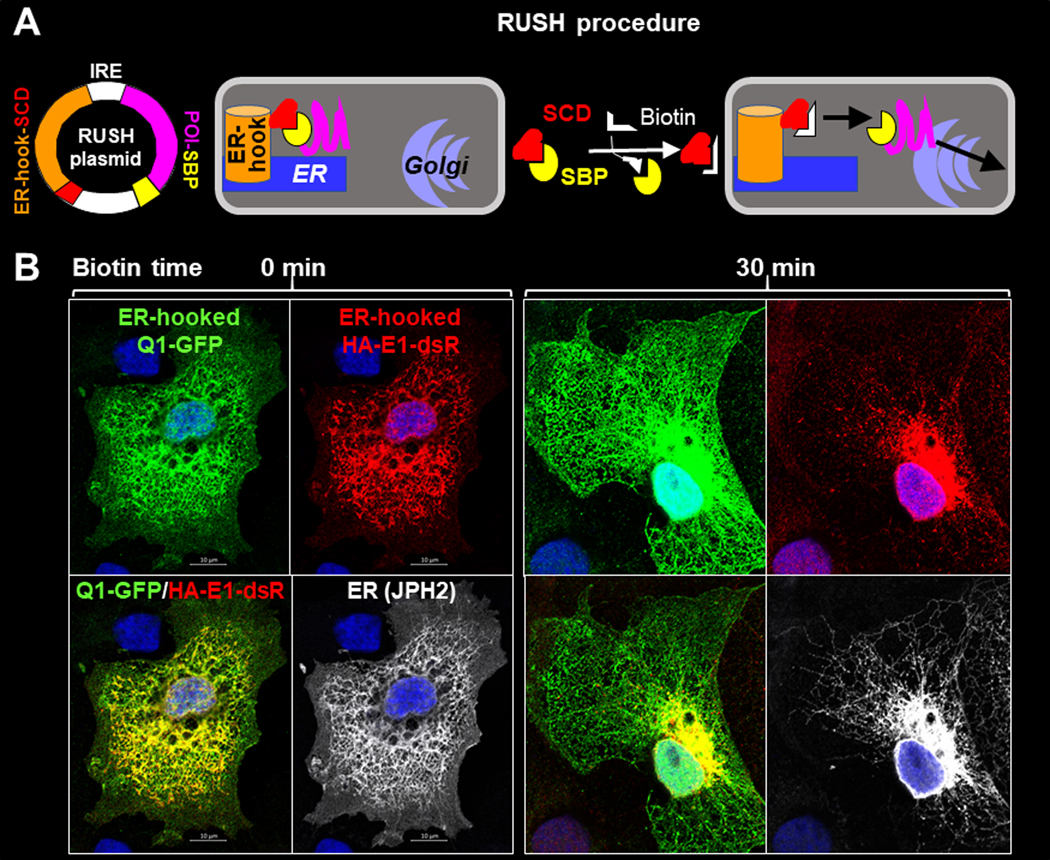
(A) Diagram of RUSH procedure. Protein-of-interest (POI) was fused with a streptavidin-binding-peptide (POI-SBP) and subcloned into a bi-cistronic plasmid (with internal ribosome entry ‘IRE’) that had an upstream ER-resident protein (an isoform of the human invariant chain of the major histocompatibility complex, Ii, retained in the ER membrane by a double arginine motif in its cytoplasmic domain (Boncompain et al., 2012)) fused with ‘streptavidin core domain’ (ER-hook-SCD). POI-SBP and ER-hook-SCD were translated as two independent proteins in the same cells. COS-7 cells were transfected with RUSH plasmid and cultured under the control conditions, during which the POI-SBP was retained at the ER by ER-hook-SCD. Biotin (membrane permeable) added to the culture medium would compete off POI-SBP from the ER-hook-SCD, allowing POI-SBP to exit ER and travel down its secretory path. (B) Fluorescence images of ER-hooked Q1-GFP, HA-E1-dsR and junctophilin-2 (JPH2, an ER marker) in COS-7 cells fixed before (0 min, left) or 30 min (right) after biotin application. Q1-GFP, HA-E1-dsR and JPH2 were detected by GFP mouse Ab/Alexa488 donkey anti-mouse, dsR rabbit Ab/Alexa568 donkey anti-rabbit, and JPH2 goat Ab/Alexa647 chicken anti-goat (pseudo-colored white).
To examine the nature of their trafficking paths, we expressed them separately. ER-hooked HA-E1-dsR was coexpressed with ER-hooked vesicular stromatitis virus protein G (VSVG, fused with GFP). VSVG served as a reporter for constitutive forward trafficking through the conventional secretory path (Lippincott-Schwartz et al., 2000). Fig. 7A shows that before biotin application, both HA-E1-dsR and VSVG-GFP were retained in the ER. After biotin application, both exited the ER in vesicles and clustered to the Golgi zone (30 – 60 min). By 120 min, both disappeared from the vesicular and Golgi compartments, and were present in the cell membrane.
Fig. 7. ER-hooked Q1-GFP and HA-E1-dsR traffic differently after released from the ER hook.

(A) Comparison of distribution patterns of ER-hooked HA-E1-dsR and ER-hooked VSVG (vesicular stomatitis virus protein G, a reporter of the constitutive anterograde trafficking pathway, tagged with GFP) (Lippincott-Schwartz et al., 2000). Shown are images of COS-7 cells coexpressing the two constructs fixed before and at specified time points after biotin application. *: Surface expression of HA-E1-dsR and VSVG-GFP. (B) Partial overlap among HA-E1-dsR vesicles after exiting the ER and mCFP tagged SEC31A (COPII component) and/or GFP tagged ERGIC-53 (marker of ER to Golgi intermediate component). (C) Comparison of distribution patterns of ER-hooked Q1-GFP and dsRed-ER (ER-resident protein fused with dsRed, as an ER marker). Shown are images of COS-7 cells coexpressing the two fixed before and at specified time points after biotin application. Images were immunofluorescence (dsR rabbit Ab/Alexa568 donkey anti-rabbit, GFP mouse Ab/Alexa488 donkey anti-mouse).
Fig. 7B depicts a COS-7 cell coexpressing HA-E1-dsR, mCFP-tagged SEC31A (component of COPII coat, a marker of ER exit sites) and GFP-tagged ERGIC-53 (a marker of ER to Golgi intermediate compartment). The cell had been incubated in brefeldin A (BFA) to prevent proteins’ ER exit (Klausner et al., 1992). After BFA washout, the restriction on ER exit was lifted. We observed HA-E1-dsR vesicles appearing outside ER bundles, partially overlapped with mCFP-SEC31A and GFP-ERGIC-53. Together data presented in Fig. 7A and 7B indicate that HA-E1-dsR trafficked through the conventional secretory path, from ER through Golgi to PM, in a constitutive manner.
ER-hooked Q1-GFP was coexpressed with dsRed-ER, an ER marker (Fig. 7C). Before and up to 120 min after biotin application, the Q1-GFP distribution pattern overlapped with that of dsRed-ER, indicating that most of the Q1-GFP stayed in the ER even after released from the ER-hook.
However, high-resolution imaging using structured illumination microscopy (SIM) revealed small submicron Q1-GFP vesicles outside the ER tubules after 45–60 min incubation with biotin (Fig. 8A). To investigate this further, we imaged movements and relationship of Q1-GFP (regular version) and dsRed-ER in live COS-7 cells at 37°C. The first series of experiments was done using Zeiss 880 in the Airyscan mode, which provided the sensitivity, spatial and time resolution required for detecting dynamic events of submicron objects. We focused on the peripheral region where the cell was ≤ 1 um thick (cross-section view, top of Fig. 8B). The time lapse images of Fig. 8B depict Q1-GFP-positive tubulo-vesicular structures that stretched out from the dsRed-ER-positive bundles, appearing close to the cell edge and disappearing between frames. Similar observations were obtained in a total of eight cells from two independent experiments. A plausible explanation for these events is Q1-GFP containing vehicles budding off ER in tubulo-vesicles, which then fused with the plasma membrane to deliver the cargo and disappeared. Q1-GFP on the cell surface could be seen as the ‘green hue’.
Fig. 8. Detect dynamic Q1-GFP vesicles outside the ER in the vicinity of plasma membrane.

Experiments were done in COS-7 cells. (A) Immunofluorescence images obtained with structured illumination microscopy (SIM) of ER-hooked Q1-GFP and dsRed-ER in the periphery of a COS-7 cell fixed after incubation with 100 uM biotin for 60 min. In the ‘merge’ panel, Q1-GFP was largely colocalized with dsRed-ER (yellow bundles). However, Q1-GFP vesicles of sub-micron sizes were detected outside the ER bundles, better seen in the enlarged view. (B) Confocal images of a live COS-7 cell expressing regular Q1-GFP and dsRed-ER, obtained with Zeiss 880 in the Airyscan mode at 37°C. Top: A cross-section view of the whole cell. The cell periphery was ~ 1 um thick. Bottom: Time-lapse images of Q1-GFP, dsRed-ER, and their merge in cell periphery. Arrows point to dynamic Q1-GFP-positive tubulo-vesicular structures that appeared in the vicinity of, but outside, ER in one frame and disappeared in the next. The hue in the green, but not the red, channel represents Q1-GFP on the cell surface. (C) Time lapse of simultaneous Q1-GFP (TIRF mode) and DsRed-ER (wide-field mode) images. Yellow arrows point to relatively stable Q1-GFP structure associated with ER. Green arrows point to a dynamic Q1-GFP vesicle, appearing in frame 2 outside the ER and disappearing in frame 3.
To seek more supporting evidence for a direct trafficking route of Q1-GFP from juxtamembrane ER to the surface, the second series of experiments was done using simultaneous TIRF/wide field imaging with an Olympus microscope. In this case, the camera chip was divided into 2 halves: one registered the green channel (TIRF, Q1-GFP) and the other the red channel (wide field, dsRed-ER). The two channels were separated by laser excitation lines and band-pass filters of the emission, and time difference between the two was negligible. Fig. 8C depicts Q1-GFP positive tubulo-vesicles just beneath the plasma membrane. Some were relative stationary and were associated with ER (yellow arrows). The green arrow points to a Q1-GFP tubulo-vesicle associated with ER (frame 1), a vesicle budding from the ER (frame 2), disappearing in Frame 3. A direct ER to PM transport of Q1-GFP by an unconventional secretory path (Nickel & Seedorf, 2008) is consistent with our observations that coexpressing JPH2 with KCNQ1 in COS-7 cells increased cell surface KCNQ1 protein level (Jiang et al., 2016), accompanied by an enlargement of ER-PM junctions (Jiang et al., 2019).
KCNQ1 and KCNE1 assemble preferentially on the cell surface
Experiments presented above showed that in adult ventricular myocytes IKs channels, as well as cell surface KCNQ1 and KCNE1, are localized on the surface sarcolemma. Importantly, although cytoplasmic KCNQ1 and KCNE1 partially overlapped in transverse striations along the z-lines, the IKs channels did not manifest any transverse striation pattern. This distinction indicates that cytoplasmic KCNQ1 and KCNE1 did not assemble into IKs channels despite their spatial proximity. Furthermore, RUSH experiments showed that KCNQ1 and KCNE1 trafficked by separate routes from ER to the cell surface. Do KCNQ1 and KCNE1 assemble into IKs channels after they have reached the cell surface?
To answer this question in a quantitative manner, we used a KCNQ1/KCNE1 pair that reported IKs assembly in immunoblot experiments. Previously we showed that cells expressing Q1-Q147C/E1-G40C had wild-type like IKs current under reducing conditions (DTT treatment, disrupting disulfide bonds). However, under non-reducing conditions (no DTT pretreatment), the two engineered Cys side chains could form a disulfide bond, locking the IKs channel in the open state (Wang et al., 2011). To allow disulfide bond formation, Cβ atoms of two Cys side chains need to come very close to each other, ≤ 0.46 nm apart (Careaga & Falke, 1992). This means that disulfide-bond between Q1-Q147C and E1-G40C (abbreviated as ‘Q1-S-S-E1’) occurred in the open state of a fully assembled IKs channel (Wang et al., 2011). The degree of Q1-S-S-E1 formation can be readily quantified by non-reducing SDS-PAGE: Q1-S-S-E1 migrated as an 80 kDa band, different from the Q1 and E1 bands (60 and 20 kDa, respectively). To test whether Q1-Q147C/E1-G40C assembly preferentially occurred in the cytoplasm vs on the cell surface, we used BFA to block protein exit to the cell surface. Fig. 9A depicts two scenarios. If Q1-Q147C/E1-G40C assembly preferentially occurred in the cytoplasm, BFA incubation would not reduce, or even increased, Q1-S-S-E1 formation. If Q1-Q147C/E1-G40C assembly preferentially occurred on the cell surface, BFA incubation would reduce Q1-S-S-E1 formation.
Fig. 9. Test preference of KCNQ1 and KCNE1 assembly inside cells or on cell surface.

(A) Experimental design. COS-7 cells transfected with Q1-Q147C and E1-G40C were cultured under the control conditions or in the presence of brefeldin A (BFA, 25 ug/ml, preventing proteins from reaching the cell surface). The two Cys side chains (147C on Q1 and 40C on E1) could come very close to each other to form a disulfide bond in the open state of a fully-assembled KCNQ1/KCNE1 channel. Therefore, the abundance of Q1-S-S-E1 reported the degree of KCNQ1/KCNE1 assembly. Two scenarios are depicted. Left: Q1 and E1 preferentially assemble inside cells. Forcing Q1-Q147C and E1-G40C to stay inside cells by BFA would not prevent, or even encourage, Q1-S-S-E1 formation. Right: Q1 and E1 preferentially assemble on the cell surface. BFA would prevent Q1-S-S-E1 formation. (B) Testing the effectiveness of BFA in preventing proteins from reaching the cell surface. Cell surface proteins were biotinylated with a membrane-impermeable, amine-reactive biotin derivative, and biotinylated fraction was purified from whole cell lysate using neutravidin bead pull-down. Top: A representative immunoblot image of W(CL) and b(iotinylated) fraction probed by Ab targeting Q1. Bottom: immunoblot of a cytoplasmic protein, actin, confirming no contamination by cytoplasmic proteins in biotinylated fraction. Lower panel of (D): densitometry quantification of % cell surface Q1. Under the control conditions, Q1-Q147C reached the same level of cell surface expression as Q1-WT. Incubation with BFA markedly reduced surface level of Q1-Q147C, indicating that BFA treatment effectively prevented Q1-Q147C (and E1-G40C, by inference) from reaching the cell surface. (C) Quantification of degree of Q1-S-S-E1 formation by non-reducing SDS-PAGE. Shown is a representative immunoblot image. WCL from cells expressing Q1-Q147C and E1-G40C cultured under the control conditions or in the presence of BFA (-BFA and +BFA, respectively), with or without reducing agent pretreatment (DTT, breaking disulfide bonds) were analyzed by immunoblot with Ab targeting Q1. The 80 kDa band represented disulfide-linked Q1-Q147C/E1-G40C, confirmed by DTT treatment which collapsed the 80 kDa band into the 60 kDa band. Upper panel of (D): quantification of KCNQ1/KCNE1 assembly (80 kDa divided by 60 kDa band intensity). Incubation with BFA significantly reduced KCNQ1/KCNE1 assembly, supporting the scenario shown on right in (A). In (D), symbols of different styles and colors represent data from individual experiments. For illustration purpose, the symbols are connected by lines of the same colors. T-test, ‘+BFA’ vs ‘-BFA’, p <0.05.
The effectiveness of BFA incubation in preventing proteins from reaching the cell surface was confirmed by quantifying cell surface proteins with biotinylation followed by neutravidin pull-down (representative immunoblot by reducing SDS-PAGE shown in Fig. 9B, and densitometry data summarized in the lower panel of Fig. 9D). We then quantified Q1-S-S-E1 formation in cells cultured under the control conditions vs in the presence of BFA. The Q1 immunoblot image in Fig. 9C shows that control WCL run on non-reducing SDS-PAGE had two bands at 80 and 60 kDa. Pretreatment of WCL with DTT before loading collapsed the 80 kDa band into the 60 kDa band, confirming the nature of the 80 kDa band as Q1-S-S-E1. WCL from BFA-treated cells had much lower 80 kDa band intensity than that of the control WCL. We used the ratio of 80 kDa to 60 kDa band intensities as a readout of Q1-S-S-E1 formation normalized by the Q1 expression level. Densitometry data are summarized in the upper panel of Fig. 9D: BFA incubation caused a 50% reduction in Q1-S-S-E1 formation. This result supports the scenario of preferential Q1-Q147C/E1-G40C assembly on the cell surface.
KCNQ1 and KCNE1 are closer to each other on the cell surface than in cytoplasm
To allow KCNQ1/KCNE1 assembly, the two must come close to each other on the cell surface. To test whether this is the case, we compared spatial proximity between Q1-GFP and HA-E1-dsR on the cell surface vs their cytoplasmic counterparts. Cell surface Q1-GFP and HA-E1-dsR were detected by incubating live COS-7 cells with Q1 ext rabbit Ab and HA mouse Ab, followed by Alexa647 goat anti-rabbit and Alexa405 goat anti-mouse Abs. Cytoplasmic Q1-GFP and HA-E1-dsR were detected by their fluorescent protein tags (Fig. 10A diagram). High density confocal images were acquired in z-stacks: xy pixel dimension 40 nm, z-steps 150 nm (a representative example shown in Fig. 10B). The raw images were deconvoluted, corrected for chromatic aberrations, and segmented. The segmented images (example shown in Fig. 10C) allowed us to discern distinct fluorescence foci, i.e. clusters of fluorescently labeled proteins, and calculated the nearest neighbor distances between them. Fig. 10D depicts the distribution of distances between nearest Q1-GFP neighbors to HA-E1-dsR on the cell surface and in cytoplasm. The median distances are 0.44 and 1.76 um, respectively, indicating that on average cell surface Q1-GFP and HA-E1-dsR were closer to each other than cytoplasmic counterparts.
Fig. 10. KCNQ1 and KCNE1 are closer to each other on cell surface than inside cells.

(A) Experimental design. Live COS-7 cells coexpressing Q1-GFP and HA-E1-dsR were incubated with Q1 ext rabbit Ab and HA mouse Ab, followed by Alexa647 goat anti-rabbit and Alexa405 goat anti-mouse. Therefore, far red and blue fluorescence signals reported cell surface Q1-GFP and HA-E1-dsR. Green and red fluorescence signals reported total Q1-GFP and HA-E1-dsR. (B) Representative raw images of far red and blue channels (cell surface Q1-GFP and HA-E1-dsR), and green/red channels (cytoplasmic counterparts). The raw images were deconvoluted, chromatic aberration corrected, and segmented to identify fluorescence foci. (C) Segmented images of the same cells, showing fluorescence foci. The coordinates of fluorescence foci were used to calculate the nearest neighbor distances. (D) Distribution of distances between nearest Q1-GFP neighbors to HA-E1-dsR on cell surface (top, far red foci to blue foci) and in cytoplasm (bottom, green foci to red foci) based on 22,648 and 47,119 nearest neighbor distances, respectively, from 10 cells. The median distances were 0.44 and 1.76 um, respectively.
The wider separation between Q1-GFP and HA-E1-dsR in the cytoplasmic compartment is consistent with their separate trafficking routes. How did they come close to each other when they had reached the cell surface? There are two possible mechanisms. First, they might preferentially cluster to the lipid raft domains of surface membrane, as has been shown for their native counterparts in guinea pig ventricular myocytes (Nakamura et al., 2007). Second, cell surface KCNQ1 and KCNE1 may concentrate in PIP2-enriched regions, due to electrostatic attraction between negatively charged PIP2 headgroups and the high positive charge densities in the juxtamembrane cytoplasmic domains of KCNQ1 and KCNE1 (Li et al., 2011; Zaydman & Cui, 2014).
Search for KCNQ1 and KCNE1 interactors using affinity purification and liquid chromatography/tandem mass spectrometry (LC/MS-MS)
During their separate journeys from ER to the plasma membrane, Q1-GFP and HA-E1-dsR may interact with different proteins (interactors). During their clustering to the same microdomains on cell surface and eventual assembly into functional IKs channels, Q1-GFP and HA-E1-dsR may interact with the same set of protein interactors. To search for their interactors in a global and unbiased manner, we immunoprecipitated Q1-GFP and HA-E1-dsR, and used LC/MS-MS to search for proteins co-purified as putative interactors. To take advantage of the much more complete human protein database than other species, we used human embryonic kidney (HEK293) cells as the expression system. HEK293 cells were cotransfected with Q1-GFP and HA-E1-dsR, or untransfected (the latter served as negative control, labeled as ‘-cDNA’ in Fig. 11A). Whole cell lysates (WCLs) were made under mild detergent conditions (1% Triton X-100) to preserve noncovalent protein-protein associations. Each WCL was divided into three equal aliquots, used for immunoprecipitation with dsR rabbit Ab, GFP rabbit Ab, and GFP goat Ab, respectively. To check the quality of the immunoprecipitates (IPs) and whether non-covalent protein-protein associations were preserved, we analyzed WCLs, negative and positive IPs ([-] and [+] IPs, from untransfected and transfected cells respectively) by immunoblots (Fig. 11A). The immunoblots show that dsR Ab immunoprecipitated HA-E1-dsR effectively. Furthermore, the presence of Q1-GFP in [+] IPs with dsR Ab, i.e. Q1-GFP/HA-E1-dsR coimmunoprecipitation, confirmed that non-covalent protein-protein association was preserved. Likewise, GFP rabbit Ab and GFP goat Ab were able to immunoprecipitate Q1-GFP, although differing in their preference for monomer vs dimer Q1-GFP. We also detected HA-E1-dsR in [+] IPs by both GFP Abs, confirming again the preservation of non-covalent protein-protein association. The ‘?’ in Fig. 11A denotes a band detected by GFP goat Ab in WCL from untransfected cells similar in size to Q1-GFP monomer. This was a consistent finding, although its nature is unclear.
Fig. 11. Check the quality of immunoprecipitates from HEK293 cells before LC/MS-MS, and validate a novel Q1-GFP interactor (microtubule plus-end binding protein 1, EB1) identified in proteomic experiments.

Two batches of HEK293 cells (Exp #1 and Exp #2) transfected with Q1-GFP and HA-E1-dsR, and one batch of untransfected HEK293 cells (-cDNA) were lysed in 1% Triton X-100 lysis buffer. Three equal fractions of WCL from each of the three were subject to immunoprecipitation with dsR rabbit Ab, GFP rabbit Ab and GFP goat Ab. (A) Immunoblot analysis of WCL and immunoprecipitates. IP lanes were loaded 5% of the total immunoprecipitates. After SDS-PAGE and protein transfer to PVDF membrane, the membrane was cut below the 75 kDa size marker (SM) band. The upper portion was probed with GFP goat Ab (WCL and IP with rabbit Abs) or GFP rabbit Ab (IP with GFP goat Ab). The lower portion was probed with HA mouse Ab. (B) Test native EB1 protein in immunoprecipitates with GFP rabbit Ab (left) and dsR rabbit Ab (right). WCL and three lanes for each IP reaction (Super = WCL after IP, [-] IP without Ab, [+] IP with Ab) were fractionated by SDS-PAGE. After proteins were transferred to PVDF membrane, the membrane was cut below 75 kDa size marker band. The upper portion was probed with GFP goat Ab. The lower portion was first probed with HA mouse Ab, and after stripping, reprobed with EB1 rat Ab. The green and red arrows point to expected Q1-GFP bands (100 kDa and 200 kDa, monomer and dimer) and HA-E1-dsR bands (42 kDa if unglycosylated, higher molecular weights if N- and/or O-glycosylated (Chandrasekhar et al., 2011)). Native EB1 migrated as monomer and dimer (30 and 60 kDa, respectively) (Chen et al., 2014).
The [+] and [-] IP samples were subject to LC/MS-MS analysis. Detailed description of the LC/MS-MS procedures is provided in the Methods section. Search of mass spectrometry data against the Uniprot human protein database produced nine protein lists, that were combined in the following manner before analysis: (1) protein lists from the four [+] IPs with GFP Abs were combined as [+] IP of Q1-GFP, (2) protein lists from the two [-] IPs with GFP Abs were combined as [-] IP of Q1-GFP, and (3) protein lists from the two [+] IPs with dsR Ab were combined as [+] IP of HA-E1-dsR. We used two filters to retain high-confidence proteins from these lists. First, we retained those proteins identified in the [+] IPs with at least two unique peptide-spectrum matches (ΣPSM ≥ 2). This filter eliminated potential artifacts due to opportunistic peptide matches. Second, from the above protein lists we retained those absent in the [-] IPs, and those detected in the [-] IPs but at a lower level than in the [+] IPs (ΣPSM ratio of [+] IP:[-] IP ≥ 2). In total, we found 109 proteins each in Q1-GFP and HA-E1-dsR immunoprecipitates. These proteins, along with their ΣPSM data, are listed in Tables S1 and S2 available as on-line Supporting information.
As a test of the validity of proteomics data, we focused on a novel putative KCNQ1 interactor: microtubule plus-end binding protein 1 (EB1). EB1 was found in Q1-GFP [+] IP, but not HA-E1-dsR [+] IP in LC/MS-MS experiments. Fig. 11B shows that an EB1 dimer band was highly abundant in the [+] IP by GFP rabbit Ab but very dim in the [+] IP by dsR rabbit Ab (ratio of band intensity 10:1). This is consistent with the proteomics data.
EB1 guides and facilitates KCNQ1 to reach cell surface
We further tested whether native EB1 preferentially bound Q1-GFP, but not HA-E1-dsR, in cardiac myocytes. Rat ventricular myocytes were transduced with Q1-GFP and HA-E1-dsR, cultured for 36 hr before lysis with 1% Triton X-100. The whole cell lysate was divided into three aliquots and subject to immunoprecipitation with GFP rabbit Ab, HA mouse Ab and EB1 rat Ab, respectively. WCL, negative control ([-] IP, WCL incubated with protein A/G beads without Abs), and [+] IP samples were fractionated by SDS-PAGE, and probed with Abs listed on the left side of Fig. 12A. We detected reciprocal coimmunoprecipitation between Q-GFP and native EB1 dimer. While the HA-E1-dsR was co-immunoprecipitated with Q1-GFP reciprocally, there was no EB1 band in the HA-E1-dsR [+] IP. The same results were obtained in two independent experiments, confirming that in cardiac myocytes EB1 interacted with Q1-GFP but not HA-E1-dsR. Furthermore, Q1-GFP preferentially binds EB1 dimer, but not monomer.
Fig. 12. Microtubule plus-end binding protein 1 (EB1) binds KCNQ1 in rat ventricular myocytes and is colocalized with myocyte surface KCNQ1.

Rat ventricular myocytes transduced with Q1-GFP and HA-E1-dsR were cultured for 36 hr and used for two types of experiments. (A) Coimmunoprecipitation: Three equal aliquots of myocyte whole cell lysate (WCL, in 1% Triton X-100) were incubated with immunoprecipitating (IP) antibodies: GFP rabbit Ab, HA mouse Ab, and EB1 rat Ab, in the presence of protein A/G beads. WCL, negative control ([-], WCL incubated with protein A/G beads without IP Ab) and positive immunoprecipitate ([+]) were fractionated by SDS-PAGE. Proteins were transferred to PVDF membrane, and the membrane was cut below the 75 kDa size marker. The upper portion was probed with GFP goat Ab and the lower portion was probed with dsR rabbit Abs (listed on the left). After ECL, the lower portion was stripped and reprobed with EB1 rat Ab. For illustration purpose, irrelevant bands were removed. EB1 monomer and dimer bands are noted as 1x and 2x. Q1-GFP and native EB1 dimer co-immunoprecipitated reciprocally (marked by $). HA-E1-dsR and Q1-GFP co-immunoprecipitated reciprocally (marked by * and #). However, there was no sign of co-immunoprecipitation between HA-E1-dsR and EB1. ‘IgG’: GFP rabbit Ab heavy chain detected by the secondary goat anti-rabbit Ab. (B) Immunofluorescence/confocal microscopy: Fixed myocytes without membrane permeabilization were incubated with Q1 ext rabbit Ab to label cell surface Q1. After membrane permeabilization, myocytes were incubated with GFP mouse Ab (binding total pool of Q1-GFP) and EB1 rat Ab (binding native EB1), followed by Alexa568-, Alexa488- and Alexa647-conjugated secondary Abs targeting rabbit, mouse and rat Abs, respectively. Shown are representative images from a myocyte at cell center and close to top surface, acquired with Zeiss 880 in the Airyscan mode.
Fig. 12B shows that in a rat ventricular myocyte transduced with Q1-GFP, cell surface Q1 (detected by Q1 ext Ab as described for Fig. 5) but not cytoplasmic Q1-GFP were highly colocalized with native EB1. In the cell center view, surface Q1 and cytoplasmic EB1 overlapped at the cell ends (intercalated disc, ICD, region). EB1 clustering to ICD is consistent with its established role of guiding connexin 43 to this area of myocytes (Shaw et al., 2007). In the cell surface view, both surface Q1 and subsarcolemmal EB1 were in distinct puncta overlapping with each other. Similar observations were obtained from 6 myocytes from two independent experiments examined in the same manner. These observations suggest that EB1 may guide Q1-GFP trafficking along microtubules toward the plus ends at myocyte periphery, thus helping deliver Q1-GFP to the cell surface.
To test whether this was the case, we compared the distribution pattern of Q1-GFP expressed alone or coexpressed with EB1-tdTomato in COS-7 cells (Fig. 13A). In cells transfected with both Q1-GFP and EB1-tdTomato, we could find cells expressing both (marked by ‘*’ in Fig. 13A) as well as cells expressing Q1-GFP without EB1-tdTomato (marked by ‘#’). The top panel of Fig. 13A contrasts these two groups of cells in the same field: in cells coexpressing both, Q1-GFP was prominent in the cell periphery, suggesting surface expression, whereas in cells expressing Q1-GFP alone, the protein was prominent in the ER but lacked cell periphery signals. The bottom panel of Fig. 13A confirms this difference by labeling cell surface Q1-GFP with Q1 ext Ab and Alexa647 anti-rabbit (live cell labeling, as described for Fig. 4A): the cell coexpressing Q1-GFP and EB1-tdTomato had more abundant Q1 ext Ab IF signals than an adjacent cell expressing Q1-GFP alone.
Fig. 13. EB1 promotes Q1-GFP to reach cell surface and while wild-type Q1-GFP preferentially binds EB1 dimer, an LQT1-associated, trafficking defective, mutation (Y111C) preferentially binds EB1 monomer.

(A) Fluorescence images of COS-7 cells co-transfected with Q1-GFP and EB1-tdTomato (tdTomato stands for ‘tandem dimer of Tomato proteins’, which behaves as a monomer). Top: In the same field, Q1-GFP in cells coexpressing both constructs had Q1-GFP in cell periphery (*) while those expressing Q1-GFP without EB1-tdTomato had Q1-GFP in the ER (#). Bottom: Q1-GFP in the cell coexpressing both constructs had more Q1-GFP on cell surface (detected by Q1 ext Ab IF under unpermeabilized conditions as described for Fig. 4A, *) than the cell expressing Q1-GFP alone (#). (B) Top: KCNQ1 N-terminal amino acid sequence (from aa 1 to 123, ending at the beginning of S1), highlighting two putative EB1-binding sites and position 111. Bottom: Fluorescence images of Q1-GFP (wild-type, ‘WT’, or Y111C), dsRed-ER, and their merge. Both WT and Y111C resided in the ER. (C) Immunoblot images of Q1-GFP WT and Y111C in whole cell lysate (W) and on cell surface (b, biotinylated fraction, as described for Fig. 9B). Immunoblot of actin confirmed lack of cytosolic proteins in biotinylated fraction. Y111C had much lower total and cell surface protein levels than WT. (D) Comparison of EB1 co-immunoprecipitation with Q1-GFP between WT and Y111C. The experimental procedure and presentation format are similar to those of Fig. 11B. Cells expressing Q1-GFP, WT or Y111C, were lysed in 1% Triton, and whole cell lysates were subject to immunoprecipitation with GFP rabbit Ab. WCL, super (WCL after immunoprecipitation), [-] IP (incubation with protein A/B beads without Ab), and [+] IP were analyzed by immunoblot with Q1 (C-20 goat) Ab and EB1 rat Ab. EB1 monomer and dimer bands are marked (1x and 2x). Experiments in (A), (B) and (C) were conducted in COS-7 expression. Experiment in (D) was conducted in HEK293 cells.
A trafficking defective LQT1 mutant prevents KCNQ1 binding to EB1 dimer
A search for EB1-binding motifs has identified a short amino acid sequence ‘SxIP’, conserved in many EB1-binding proteins (Hannappa et al., 2009). KCNQ1 harbors two putative EB1-binding motifs in its N-terminus (Fig. 13B, top), adjacent to an N-terminal trafficking motif of KCNQ1 (Dahimene et al., 2006). Three LQT1-associated mutations have been identified here: Y111C, L114P, and P117L (Dahimene et al., 2006). We studied how Y111C might affect KCNQ1 trafficking. When comparing the distribution patterns of Q1-GFP (wild-type, WT) and Q1-GFP-Y111C expressed in COS-7 cells (Fig. 13B, bottom) or HEK293 cells (data not shown), we could not differentiate between the two: both resided in the ER compartment labeled by dsRed-ER. However, biochemical experiments revealed that relative to WT, Y111C had much reduced total protein level (Fig. 13C, whole cell lysate lanes, immunoblot of Q1 vs actin), and was largely absent on the cell surface (biotinylated fraction lanes), confirming its trafficking defect and LQT1 association status. We then compared Q1-GFP/EB1 interaction between WT and Y111C using HEK293 expression. Fig. 13D shows that while Q1-GFP preferentially bound EB1 dimer (as shown above), Y111C preferentially bound EB1 monomer. The functional implication of this observation is presented in the Discussion.
DISCUSSION
New insights into IKs function in the heart
By applying novel approaches to the investigation of KCNQ1 and KCNE1 distribution in ventricular myocytes, our study provides the following new insights into how IKs serves its function as a repolarization reserve in the heart. First, using in situ proximity ligation assay to locate KCNQ1/KCNE1 assemblies, we showed that IKs channels are on the surface sarcolemma but not t-tubules. This location puts IKs channels in close proximity to the β-adrenergic signaling complex, adenylate cylase 9 and yotiao, previously shown to be localized on the surface sarcolemma of adult murine ventricular myocytes (Li et al., 2019). This spatial proximity is critical for a timely response of IKs to β-adrenergic stimulation: local cAMP produced by adenylate cyclase 9 rapidly activates PKA recruited by yotiao to the IKs channels (Marx et al., 2002; Li et al., 2012), which phosphorylates KCNQ1 to increase the IKs amplitude and accelerate its activation (Marx et al., 2002). Protein phosphatase 1A, also recruited by yotiao to the IKs channels (Marx et al., 2002), then dephosphorylates KCNQ1 to terminate the IKs response when β-adrenergic stimulation subsides. It is important to note that our data do not support the t-tubule localization of IKs channels proposed previously based on indirect evidence (Balse et al., 2012; Oliveras et al., 2020).
Second, by combining antibodies targeting extracellular epitopes on the KCNQ1 and KCNE1 constructs and the fluorescent protein tags, we showed that both KCNQ1 and KCNE1 have a cell surface pool and an intracellular pool. Cell surface KCNQ1 and KCNE1 are on the surface sarcolemma, similar to the IKs channels identified by PLA, but not t-tubules. Previously, we showed that the intracellular KCNQ1 pool is in the junctional SR (jSR), manifested as transverse striations along z-lines (Jiang et al., 2017). Intracellular KCNE1 is present in both vesicles and transverse striations. We propose that these vesicles represent transport vehicles during KCNE1 biogenesis as well as those in endocytosis/recycling. KCNE1 in transverse striations appears more prominent in rat ventricular myocytes (Wu et al., 2006; Oliveras et al., 2020) than in guinea pig or canine ventricular myocytes (Wang et al., 2013; Jiang et al., 2017). One possible explanation is cell surface KCNE1 redirected to jSR after association with KCNQ1 (Oliveras et al., 2020).
Third, using the novel RUSH approach and advanced imaging techniques, we showed that KCNE1 traveled constitutively through the conventional secretory pathway (ER, Golgi. and then plasma membrane), while KCNQ1 appeared to travel through an unconventional secretory pathway (from ER directly to plasma membrane) (Nickel & Seedorf, 2008). The observation that after release from the ER, KCNE1 rapidly reached the cell surface (< 120 min) in the absence of KCNQ1 does not support the previous claims that newly translated KCNE1 requires association with KCNQ1 to reach the cell surface (Chandrasekhar et al., 2006; Vanoye et al., 2010). In the context of IKs function in cardiac myocytes, unconventional trafficking of KCNQ1 directly from jSR to the sarcolemma expedites the formation of new IKs channels, critical for a timely response to β-adrenergic stimulation.
Fourth, using a quantifiable reporter of KCNQ1/KCNE1 assemblies, we showed that the IKs channels were preferentially formed on the cell surface, after both components have reached the plasma membrane. In the context of IKs function in cardiac myocytes, this is consistent with the concept of ‘segregation of KCNQ1 and KCNE1 until more IKs is needed’. Lone KCNE1 on the surface sarcolemma does not have IKs function. It is in a standby mode, waiting for KCNQ1 to exit the jSR compartment. KCNQ1-containing vesicles may travel along microtubules, guided by EB1 dimer, to the sarcolemma where KCNQ1 associates with KCNE1 to form IKs channels. Indeed, KCNQ1 mobilization along microtubules is required for the ‘amplitude response’ of IKs channels to β-adrenergic stimulation (Nicolas et al., 2008). This was revealed by the observations that microtubule depolymerization by colchicine prevents the amplitude response of IKs to β-adrenergic stimulation, despite intact gating response and intact PKA-mediated phosphorylation of both S27 in KCNQ1 and S43 in yotiao (Nicolas et al., 2008). Together, our data support a scenario where delayed KCNQ1 and KCNE1 assembly on the cell surface helps IKs fulfill its function as a repolarization reserve in the heart.
New insights into KCNQ1 and KCNE1 trafficking by affinity purification/proteomics
We divided the proteins found in Q1-GFP and HA-E1-dsR immunoprecipitates into three groups: uniquely in Q1-GFP immunoprecipitate (41), uniquely in HA-E1-dsR immunoprecipitate (41), and shared between the two (68, Venn diagram in Fig. 14A). We then manually curated these three groups of proteins, and selected those known to be involved in protein trafficking and distribution. These proteins are listed in Table 1, categorized by their cellular compartments or molecular functions (first column). Based on Table 1, in conjunction with information from our current and previous experimental findings (Wang et al., 2013; Jiang et al., 2017), we present a synthetic view of how KCNQ1 and KCNE1 traffic and assemble in cells (Fig. 14B).
Fig. 14. Search for KCNQ1 and KCNE1 interactors in their trafficking paths.

(A) Venn diagram of proteins found in KCNQ1-GFP immunoprecipitate (109 proteins) and in HA-KCNE1-dsR immunoprecipitate (109 proteins). Sixty eight are shared. (B) Cartoon of KCNQ1 and KCNE1 forward trafficking, assembly and endocytosis.
Table 1.
Manually curated list of proteins involved in membrane protein trafficking uniquely found in KCNQ1-GFP IP, HA-KCNE1-dsR IP, or shared between the two.
| Cellular compartment/molecular function | Uniquely in KCNQ1-GFP IP | Uniquely in HA-KCNE1-dsR IP | SHARED |
|---|---|---|---|
| ER proteins/Chaperones | Calnexin, Erlin-2, HSP70-related, HSP86, T-complex 1-ε | HSP75 T-complex 1-β, −γ | Calreticulin HSP70, HSP71, HSP90, T-complex 1-α, −η, −θ |
| ER-Golgi intermediate | - | ER-to-Golgi SNARE of 24 kDa | - |
| Traffic on microtubules | Kinesin-1, EB1 | Tubulin-β4B | Tubulin-α1B, β2A, β4, β5 |
| Traffic on actin filaments | - | - | Myosin-9, −10, actin α, actin β, actin β2-linke |
| 14–3-3 proteins | 14–3-3 α, β, γ | - | 14–3-3 ε, θ, ζ |
| Cytoskeleton | - | Vimentin | α-actinin-4, filamin-A |
| Ras-related proteins | Ras GPTase-activating-like protein, Rap-1b like protein | Rab10 | Rab5c |
| Lipid raft domains | - | - | Flotillin-1 |
| Cell-ECM adhesion | Vinculin, Talin-1 | - | - |
| Cell-cell adhesion | - | Plakophilin-1 | Desmocollin-1, Desmoglein-1 Desmoplakin |
The following are shared interactors between Q1-GFP and HA-E1-dsR, that provide clues as to how the two travel and distribute in cells:
Both Q1-GFP and HA-E1-dsR interacted with of ER proteins (calnexin, Erlin-2, and calreticulin) and chaperones (heat-shock proteins and isoforms of T-complex 1), reflecting their translation in the rough ER, and dependence on the chaperones for proper folding.
Both Q1-GFP and HA-E1-dsR interacted with α and β tubulins, consistent with cargo vesicles traveling on microtubule (MT) tracks.
Both Q1-GFP and HA-E1-dsR interacted with actin isoforms (α, β and β2-like) and conventional, non-muscle myosin-9 and myosin-10. These myosin motors move vesicles along actin filaments toward the barbed ends at the plasma membrane (Chantler et al., 2010), suggesting that KCNQ1- and KCNE1-containing cargo vesicles traffic on cortical actin filaments before fusion with the plasma membrane.
Both Q1-GFP and HA-E1-dsR interacted with actin-binding proteins: α-actinin-4 and filamin-A. HA-E1-dsR also interacted with intermediate filament, vimentin. These interactors may anchor KCNQ1 and KCNE1 on the cell surface by cortical cytoskeleton.
Both Q1-GFP and HA-E1-dsR interacted with flotillin-1, a lipid-raft resident protein. This is consistent with clustering of native KCNQ1 and KCNE1 to lipid rafts in guinea pig heart (Nakamura et al., 2007).
Both Q1-GFP and HA-E1-dsR interacted with Rab5c. HA-E1-dsR also interacted with Rab10. These small GTPases mediate endocytosis of plasma membrane proteins.
Both Q1-GFP and HA-E1-dsR interacted with 14-3-3 proteins. 14-3-3 proteins coordinate the organization of microtubules and actin filaments (Shikano et al., 2006). The functional implications of 14-3-3 interaction with KCNQ1 and KCNE1 require future investigation.
Both Q1-GFP and HA-E1-dsR interacted with components of desmosomal junction. These interactors may explain why surface KCNQ1 and KCNE1 cluster to the intercalated disc region of cardiac myocytes (Figs. 1A and 1C) (Balse et al., 2012).
Interactors unique to Q1-GFP or HA-E1-dsR also provide clues as to how the two travel separately in cells:
HA-E1-dsR uniquely bound ER-to-Golgi SNARE of 24 kDa (also called Sec22B). This, in conjunction with our observations that after exiting the ER, HA-E1-dsR entered a vesicular compartment overlapping with Sec31A (COPII component) and ERGIC-53 (ER-to-Golgi intermediate compartment), supports the view that HA-E1-dsR trafficked by the conventional secretory path from ER to Golgi and then cell surface (Lippincott-Schwartz et al., 2000). Importantly, there was no protein involved in ER to Golgi transport found in Q1-GFP immunoprecipitate. This is consistent with the notion that Q1-GFP trafficked from ER to plasma membrane bypassing the Golgi.
Q1-GFP, but not HA-E1-dsR, interacted with kinesin-1 and EB1. The motor protein, kinesin-1, pulls ER tubules out of the ER cisternae and move them along MT tracks toward cell periphery, creating the so-called ER sliding events (Friedman & Voeltz, 2011). We observed ER sliding events in live cell imaging of Q1-GFP, signaling Q1-GFP in ER tubules moving toward the cell periphery (Fig. 8B). Functional implications of Q1-GFP/EB1 interaction is presented below.
Q1-GFP, but not HA-E1-dsR, interacted with vinculin and talin-1. These two proteins, together with integrins, form a macromolecular complex at cell-extracellular matrix (ECM) junctions (Henderson et al., 2017). These interactors may explain why cell surface KCNQ1 clusters to surface sarcolemma (facing ECM) but not t-tubules in cardiac myocytes.
Q1-GFP interacted with Rap1b-like protein. This may be related to the interaction of Q1-GFP with vinculin and talin-1, because Rap1 is involved in integrin activation (Henderson et al., 2017).
Functional implications of preferential KCNQ1/EB1 dimer interaction
End binding protein 1 (EB1) is the master switch for binding of microtubule plus-end binding proteins (+TIPS) to microtubules’ growing ends toward the cell periphery (Hommappa et al., 2006). Structurally, EB1 can be divided into three domains: an N-terminal calponin-homology (CH) domain responsible for binding to microtubule plus ends, a C-terminal EB-homology (EBH) domain responsible for binding +TIPS, and a linker domain in between (Chen et al., 2014). Two studies of crystallography in conjunction with functional assays revealed that dimerization of EB1, through a coiled-coil mechanism at its C-terminus, can stabilize EB1 binding to both microtubule plus-ends and +TIPS (Hommappa et al., 2006; Chen et al., 2014), essentially defining EB1 dimer as the active form of the protein.
We showed that Q1-GFP preferentially binds EB1 dimer, instead of monomer, when expressed in both rat ventricular myocytes (Fig. 12) and HEK293 cells (Figs. 11 and 13). Our data support the notion that KCNQ1 binds the active form of EB1 and is guided by EB1 along microtubules to reach the cell surface. On the other hand, a trafficking defective mutant identified in LQT1 patients, Y111C (Dahimene et al., 2006), preferentially binds EB1 monomer but not dimer. Our data suggest a novel mechanism by which a mutation can impair trafficking to cell surface by preventing KCNQ1 interaction with the active form of EB1. Whether this mechanism is applicable to other trafficking defective KCNQ1 mutants requires further experimentation.
Technical limitations
With the RUSH approach, ideally we want to track Q1-GFP and HA-E1-dsR distribution in live cells after release from the ER hook. Unfortunately, the GFP and dsRed fluorescence signals from the RUSH constructs were very dim, requiring high laser power. This caused photobleaching and cytotoxicity, making time-lapse imaging of live cells of sufficient durations (60–120 min) unsuccessful. Repeated attempts to overcome this problem, by adjusting cDNA concentrations and culture time, were unsuccessful.
We believe there are two contributing factors. First, in our RUSH constructs the GFP and dsRed were fused at both N- and C-termini. Although similar configurations were reported in the original RUSH paper (Boncompain et al., 2012), it is not clear whether such configurations hindered chromophore formation in our constructs. Second, immunoblot of whole cell lysates showed that at the same cDNA concentration and after the same culture time, ER-hooked constructs produced about 10-fold less proteins than their non-hooked counterparts. This might be caused by ER stress suppressing protein translation.
Supplementary Material
KEY POINTS.
In adult ventricular myocytes, the IKs channels are distributed on the surface sarcolemma, not t-tubules.
In adult ventricular myocytes, KCNQ1 and KCNE1 have distinct cell surface and cytoplasmic pools.
KCNQ1 and KCNE1 traffic from the endoplasmic reticulum to plasma membrane by separate routes, and assemble into IKs channels on the cell surface.
Liquid chromatography/tandem mass spectrometry applied to affinity-purified KCNQ1 and KCNE1 interacting proteins reveals novel interactors involved in protein trafficking and assembly.
Microtubule plus-end binding protein 1 (EB1) binds KCNQ1 preferentially in its dimer form, and promotes KCNQ1 to reach the cell surface.
An LQT1-associated mutation, Y111C, reduces KCNQ1 binding to EB1 dimer.
ACKNOWLEDGEMENTS
Confocal microscopy was performed at the Virginia Commonwealth University - Department of Neurobiology & Anatomy Microscopy Facility, supported in part by NIH-NINDS Center Core Grant 5P30NS047463.
FUNDING
This study was supported by NIH/NHLBI (HL128610, HL96962, and HL94450).
Biography

Zach Wilson developed this work as a technician in Dr. Gea-Ny Tseng's lab in the Physiology and Biophysics Department at Virginia Commonwealth University, in Richmond, Virginia. The lab focuses on electrophysiology and ion channel regulation in myocytes. Previously, he worked as a technician in the Internal Medicine department of Virginia Commonwealth University, in a lab specializing in heart failure and reperfusion. Zach will be attending medical school starting in fall of 2021.
Footnotes
ADDITIONAL INFORMATION
Data Availability statement
All data supporting the results in this paper, except the proteomic data, are in the paper itself. The proteomic data are uploaded as supporting information (Table S1 and Table S2) and are available on-line. This is referenced in the proteomics experiments and data analysis of Methods section and the Results section.
CONFLICT OF INTEREST None.
REFERENCES
- Balse E, Steele DF, Abriel H, Coulombe A, Fedida D & Hatem SN. (2012). Dynamic of ion channel expression at the plasma membrane of cardiomyocytes. Physiological Review 92, 1317–1358. [DOI] [PubMed] [Google Scholar]
- Banyasz T, Jian Z, Horvath B, Khabbaz S, Izu LT & Chen-Izu Y. (2014). Beta-adrenergic stimulation reverses the IKr-IKs dominant pattern during cardiac action potential. Pflugers Arch 466, 2067–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bas T, Gao GY, Lvov A, Chandrasekhar KD, Gilmore R & Kobertz WR. (2011). Post-translational N-glycosylation of type I transmembrane KCNE1 peptides. Implications for membrane protein biogenesis and disease. J Biol Chem 286, 28150–28159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncompain G, Divoux S, Gareil N, de Forges H, Lescure A, Latreche L, Mercanti V, Jollivet F, Raposo G & Perez F. (2012). Synchronization of secretory protein traffic in populations of cells. Nature Methods 9, 493–498. [DOI] [PubMed] [Google Scholar]
- Borggrefe M, Wolpert C, Antzelevitch C, Veltmann C, Giustetto C, Gaita F & Schimpf R. (2005). Short QT syndrome genotype-phenotype correlatoins. J Electrocardiology 38, 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero R, de la Fuente MG, Gomez R, Barana A, Amoros I, Dolz-Gaiton P, Osuna L, Almendral J, Atienza F, Fernandez-Aviles F, Pita A, Rodriguez-Roda J, Pinto A, Tamargo J & Delpon E. (2010). In humans, chronic atrial fibrillation decreases the transient outward current and ultrarapid component of the delayed rectifier current differentially on each atria and increases the slow component of the delayed rectifier current in both. J Am Coll Cardiol 55, 2346–2354. [DOI] [PubMed] [Google Scholar]
- Careaga CL & Falke JJ. (1992). Thermal motions of surface α-helices in the D-galactose chemosensory receptor. Detection by disulfide trapping. Journal of Molecular Biology 226, 1219–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekhar KD, Bas T & Kobertz WR. (2006). KCNE1 subunits require co-assembly with K+ channels for efficient trafficking and cell surface expression. J Biol Chem 281, 40015–40023. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar KD, Lvov A, Terrenoire C, Gao GY, Kass RS & Kobertz WR. (2011). O-glycosylation of the cardiac IKs complex. J Physiol 589, 3721–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantler PD, Wylie SR, Wheeler-Jones CP & McGomnell IM. (2010). Conventional myosins-unconventional functions. Biophysical Review 2, 67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Luo Y, Li L, Ran J, Wang X, Gao S, Liu M, Li D, Shui W & Zhou J. (2014). Phosphoregulation of the dimerization and functions of end-binding protien 1. Protein Cell 5, 795–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahimene S, Alcolea S, Naud P, Jourdon P, Escande D, Brasseur R, Thomas A, Baro I & Merot J. (2006). The N-terminal juxtamembranous domain of KCNQ1 is critical for channel surface expression. Implications in the Romano-Ward LQT1 syndrome. Circ Res 99, 1076–1083. [DOI] [PubMed] [Google Scholar]
- David J-P, Andersen MN, Olesen S-P, Rasmussen HB & Schmitt N. (2013). Trafficking of the IKs-complex in MDCK cells: site of subunit assembly and determinants of polarized localization. Traffic 14, 399–411. [DOI] [PubMed] [Google Scholar]
- Friedman JR & Voeltz GK. (2011). The ER in 3D: a multifunctional dynamic membrane network. Trends in Cell Biology 21, 709–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannappa S, Gouveia SM, Weisbrich A, Damberger FF, Bhavesh NS, Jawhari H, Grigoriev I, van Rijssel FJA, Buey RM, Lawera A, Jelesarov I, Winkler FK, Wuthrich K, Akhmanova A & Steinmetz MO. (2009). An EB1-binding motif acts as a microtubule tip localization signal. Cell 138, 366–376. [DOI] [PubMed] [Google Scholar]
- Henderson CA, Gomez CG, Novak SM, Lei M-M & Gregorio CC. (2017). Overview of the muscle cytoskeleton. Comprehensive Physiology 7, 891–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hommappa S, Okhrimenko O, Jaussi R, Jawhari H, Jelesarov I, Winkler FK & Steinmetz MO. (2006). Key interaction modes of dynamic +TIP networks. Molecular Cell 23, 663–671. [DOI] [PubMed] [Google Scholar]
- Jiang M, Hu J, White FKH, Williamson J, Klymchenko AS, Murthy A, Workman SW & Tseng G-N. (2019). S-palmitoylation of junctophilin-2 is critical for its role in tethering the sarcoplasmic reticulum to the plasma membrane. J Biol Chem 294, 13487–13501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Wang Y-H & Tseng G-N. (2017). Adult ventricular myocytes segregate KCNQ1 and KCNE1 to keep the IKs amplitude in check until when larger IKs is needed. Circ Arrhythm Electrophysiol 10, e005084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Zhang M, Howren M, Wang YH, Tan A, Balijepalli RC, Huizar JF & Tseng G-N. (2016). JPH-2 interacts with Cai-handling proteins and ion channels in dyads: contribution to premature ventricular contraction-induced cardiomyopathy. Heart Rhythm 13, 743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost N, Virag L, Bitay M, Takacs J, Lengyel C, Biliczki P, Nagy Z, Bogats G, Lathrop DA, Papp JG & Varro A. (2005). Restricting excessive cardiac action potential and QT prolongation. A vital role for IKs in human ventricular muscle. Circulation 112, 1392–1399. [DOI] [PubMed] [Google Scholar]
- Klausner RD, Donaldson JG & Lippincott-Schwartz J. (1992). Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol 116, 1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumerman A, Gao X-H, Bian J-S, Melman YF, Kagan A & McDonald TV. (2004). An LQT mutant minK alters KvLQT1 trafficking. Am J Physiol 286, C1453–C1463. [DOI] [PubMed] [Google Scholar]
- Li Y, Chen L, Kass RS & Dessauer CW. (2012). The A-kinase anchoring protein Yotiao facilitates complex formation between adenylyl cyclase type 9 and the IKs potassium channel in heart. J Biol Chem 287, 29815–29824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hof T, Baldwin TA, Chen L, Kass RK & Dessauer CW. (2019). Regulation of IKs potassium current by isoproterenol in adult cardiomyocytes requires type 9 adenylate cyclase. Cells 8, 981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zaydman MA, Wu D, Shi J, Guan M, Virgin-Downey B & Cui J. (2011). KCNE1 enhances phosphatidylinositol 4,5-bisphosphate (PIP2) sensitivity of IKs to modulate channel activity. PNAS 108, 9095–9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Roberts TH & Hirschberg K. (2000). Secretory protein trafficking and organelle dynamics in living cells. Ann Rev Cell Dev Biol 16, 557–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, Marks AR & Kass RS. (2002). Requirement of a macrocmolecular signaling complex for β adrenergic receptor modulaiton of the KCNQ1-KCNE1 potassium channel. Science 295, 496–499. [DOI] [PubMed] [Google Scholar]
- Mashanov GI, Nobels M, Harmer SC, Molloy JE & Tinker A. (2010). Direct observation of individual KCNQ1 potassium channels reveals their distinctive diffusive behavior. J Biol Chem 285, 3664–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Kurokawa J, Bai C-X, Asada K, Xu J, Oren RV, Zhu ZI, Clancy CE, Isobe M & Furukawa T. (2007). Progesterone regulates cardiac repolarization through a nongenomic pathway. An in vitro patch-clamp and computational modeling study. Circulation 116, 2913–2922. [DOI] [PubMed] [Google Scholar]
- Nickel W & Seedorf M. (2008). Unconventional mechanisms of protein transport to the cell surface of eukaryotic cells. Annu Rev Cell Dev Biol 24, 287–308. [DOI] [PubMed] [Google Scholar]
- Nicolas CS, Park K-H, Harchi AE, Camonis J, Kass RS, Escande D, Merot J, Loussouarn G, Le Bouffant F & Baro I. (2008). IKs response to protein kinase A-dependent KCNQ1 phosphorylation requires direct interaction with microtubules. Cardiov Res 79, 427–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveras A, Serrano-Novillo C, Moreno C, de la Cruz A, Valenzuela C, Soeller C, Comes N & Felipe A. (2020). The unconventional biogenesis of Kv7.1-KCNE1 complexes. Science Advances 6, eaay4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen HB, Moller M, Knaus H-G, Jensen BS, Olesen S-P & Jorgensen NK. (2003). Subcellular localization of the delayed rectifier K+ channels KCNQ1 and ERG1 in the rat heart. Am J Physiol 286, H1300–H1309. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL & Keating MT. (1996). Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature 384, 80–83. [DOI] [PubMed] [Google Scholar]
- Sarkar AX & Sobie EA. (2016). Quantification of repolarization reserve to understand interpatient variability in the response to proarrhythmic drugs: A computational analysis. Heart Rhythm 8, 1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RM, Fay AJ, Puthenveedu MA, von Zastrow M, Jan Y-N & Jan LY. (2007). Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell 128, 547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikano S, Coblitz B, Wu M & Li M. (2006). 14–3-3 proteins: regulation of endoplasmic reticulum localization and surface expression of membrane proteins. Trends in Cell Biology 16, 370–375. [DOI] [PubMed] [Google Scholar]
- Soderberg O, Leuchowius K-J, Gullberg M, Jarvius M, Weibrecht I, Larsson L-G & Landegren U. (2008). Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation. Methods 45, 227–232. [DOI] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, Lehmann MH, Priori SG, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM & Keating MT. (2000). Spectrum of mutations in long-QT syndrome genes KvLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 102, 1178–1185. [DOI] [PubMed] [Google Scholar]
- Sun J & MacKinnon R. (2020). Structural basis of human KCNQ1 modulation and gating. Cell 180, 340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoye CG, Welch RC, Tian C, Sanders CR & George ALJ. (2010). KCNQ1/KCNE1 assembly, co-translation not required. Channels (Austin) 4, 108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y-H, Jiang M, Xu X-L, Hsu K-L, Zhang M & Tseng G-N. (2011). Gating-related molecular motions in the extracellular domain of the IKs channel: implications for IKs channelopathy. J Memb Biol 239, 137–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y-H, Zankov DP, Jiang M, Zhang M, Henderson SC & Tseng G-N. (2013). [Ca]i elevation and oxidative stress induce KCNQ1 translocation from cytosol to cell surface and increase IKs in cardiac myocytes. J Biol Chem 288, 35358–35371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D-M, Jiang M, Zhang M, Liu X-S, Korolkova YV & Tseng G-N. (2006). KCNE2 is colocalized with KCNQ1 and KCNE1 in cardiac myocytes and may function as a negative modulator of IKs current amplitude in the heart. Heart Rhythm 3, 1469–1480. [DOI] [PubMed] [Google Scholar]
- Xu Y, Wang YH, Meng X-Y, Zhang M, Jiang M, Zhang H-X, Cui M & Tseng G-N. (2013). Building KCNQ1/KCNE1 docking models and probing their interactions by molecular dynamics simulations. Biophysical Journal 105, 2461–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaydman MA & Cui J. (2014). PIP2 regulation of KCNQ channels: biophysical and molecular mechanisms for lipid modulation of voltage-dependent gating. Frontiers in Physiology 5, article 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.