

Abstract
Background
Antiseizure drugs (ASDs) are the primary therapy for epilepsy, with more than 20 drugs introduced into clinical practice to date. These drugs are typically grouped by their mechanisms of action and therapeutic spectrum. This article aims to educate non‐neurologists and medical students about the new frontiers in the pharmacology of ASDs and presents the current state of the literature on the efficacy and tolerability of these agents.
Methods
Randomized controlled trials, observational studies, and evidence‐based meta‐analyses of ASD efficacy and tolerability as initial monotherapy for epileptic seizures and syndromes were identified in PubMed, EMBASE, the Cochrane Library, and Elsevier Clinical Pharmacology.
Results
The choice of ASD varies primarily according to the seizure type. Practical guidelines for ASD selection in patients with new‐onset and drug‐resistant epilepsy were recently published. The guidelines have shown that the newer‐generation drugs, which have unique mechanistic and pharmacokinetic properties, are better tolerated but have similar efficacy compared with the older drugs. Several ASDs are effective as first‐line monotherapy in focal seizures, including lamotrigine, carbamazepine, phenytoin, levetiracetam, and zonisamide. Valproate remains the first‐line drug for many patients with generalized and unclassified epilepsies. However, valproate should be avoided, if possible, in women of childbearing potential because of teratogenicity. Toxicity profile precludes several drugs from use as first‐line treatment, for example, vigabatrin, felbamate, and rufinamide.
Conclusions
Antiseizure drugs have different pharmacologic profiles that should be considered when selecting and prescribing these agents for epilepsy. These include pharmacokinetic properties, propensity for drug‐drug interactions, and adverse effects.
Keywords: antiseizure drug, epilepsy, seizure types
Antiseizure drugs (ASDs) have many different pharmacologic profiles that are relevant when selecting and prescribing these medications in patients with epilepsy. Some second‐generation ASDs have shown advantages in drug tolerability, drug‐drug interactions, and teratogenicity. Disappointingly, however, none of these medications appear to be more efficacious than first‐generation ASDs.
![]()
1. INTRODUCTION
Epilepsy is a chronic disorder of brain function characterized by recurrent seizures.1 At any one time, about 1% of the population requires medication to treat epilepsy.2 The antiseizure drug (ASD) is the mainstay of treatment. The choice of ASDs varies with different seizure types and epileptic syndromes.3 The appropriately chosen ASD provides adequate seizure control in 60%–70% of patients.4 There is no general agreement on when treatment should start after a “first seizure,” but randomized controlled trials (RCTs) in adult and pediatric patients have shown that early treatment reduces the recurrence for at least two years from the first seizure.5 The ASDs are often administered orally for a long period of time to prevent seizure recurrence. In designing a therapeutic approach, the use of a single drug (monotherapy) is preferred because of fewer adverse effects. However, 30% of the patients will continue to have uncontrolled seizures and multiple drugs are often used simultaneously.6 Monotherapy trials of two ASDs that are appropriate first‐line treatment for the patient's seizure type usually be initiated before combinations are tried. Patients who do not achieve seizure control following adequate trials with two or more appropriate drugs are considered drug‐resistant.6 The International League Against Epilepsy (ILAE), in 2010, has proposed the definition of drug‐resistant epilepsy (often used interchangeably with “'medically refractory”' and “'intractable”) as “failure of adequate trials of two tolerated, appropriately chosen and used antiseizure drug schedules (whether as monotherapies or in combination) to achieve sustained seizure freedom.”7
The older‐generation ASDs were introduced into clinical practice more than four decades ago. Research has shown that 30%–40% of people treated with an older ASD as monotherapy (including carbamazepine and valproate) experience adverse effects that contribute to treatment failure.8 This spurred a continuing search for new ASDs with novel molecular targets that could provide optimal care for patients with epilepsy.9, 10 The past three decades have seen the licensing of about 20 newer‐ (second‐ and third‐) generation ASDs with unique mechanisms of action and pharmacokinetics following a period of relative paucity.11 This advent has expanded the epilepsy therapeutic armamentarium and allowed the drugs to match the individual patient's characteristics. The American Academy of Neurology (AAN) subcommittee reports in 2004 observed that newer ASDs were not different in controlling seizures but have better tolerability, particularly fewer neurotoxic adverse effects.12, 13 In 2017, the ILAE published a new classification for seizure types and epilepsy syndrome to improve our understanding of epilepsy and include missing seizure types.14, 15, 16, 17 This classification replaced the previous versions published in 198118 and 1989,19 and extended in 2010,20 and has been of paramount importance to accurately classify the patient's seizure type(s). In 2018, the AAN and American Epilepsy Society (AES) subcommittee published updated guidelines for ASD selection in adult and pediatric patients with new‐onset and treatment‐resistant epilepsy.21, 22 However, the guidelines primarily relied on studies that compare the first‐ and second‐generation ASDs and found limited data for third‐generation drugs. Given the rapid advance in the development of ASDs in recent years and the continuous updates in definitions, classifications and treatment guidelines for seizure types, and epilepsy syndromes, this article aims to provide non‐neurologists and medical students with a complete overview of the new frontiers in the neuropharmacology of antiseizure drugs. The ASDs classification, pharmacokinetics, mechanisms of action, clinical use, and adverse effects are reviewed herein based on a comprehensive assessment of the current literature and recently published US and UK treatment guidelines.
1.1. Definition and epidemiology of epilepsy
Epilepsy is ”a neurological disorder that is characterized by an enduring predisposition to generate epileptic seizures and the associated cognitive, psychological, and social consequences”.1 Epilepsy is the third most common neurologic disorder; almost 10% of people will experience a seizure during their lives.2 The prevalence of epilepsy is 6.4 cases per 1,000 persons, and the annual incidence is 67.8/100,000 person‐years.23
1.2. Classification of seizure types and epilepsy syndromes
The ILAE classification framework, which was revised in 2017, is the key tool for the diagnosis of individuals presenting with seizures.14, 15, 16, 17 According to that classification, epileptic seizures are classified into “focal,” “generalized,” and “unknown” seizures, while epilepsy is classified into “focal,” “generalized,” “combined generalized and focal,” and “unknown” epilepsy. Focal‐onset seizures can be further described as focal aware (previously called simple partial), impaired awareness (complex partial), or focal to bilateral (secondarily generalized) tonic‐clonic seizures. Generalized seizures are classified into generalized tonic‐clonic (formerly idiopathic) seizures, motor (myoclonic and other motor) seizures, and nonmotor (absence) seizures. Etiologies of seizures and epilepsy syndromes have been reintroduced in the 2017 ILAE classification of seizure types and epilepsy syndromes, to include “genetic,” “structural,” “metabolic,” “infectious,” “immune,” and “unknown.” For a detailed description of seizure types epilepsy syndromes, see the ILAE Web site.24
1.3. Drug treatment of epilepsy
Epilepsy can persist for years and often for the patient's lifetime. ASDs are the primary therapy for epilepsy and are symptomatic treatments that control seizures.3 The choice of ASDs varies with different seizure types and epileptic syndromes.3 ASDs have many different pharmacologic profiles that are relevant when selecting and prescribing these agents in patients with epilepsy. This includes pharmacokinetic properties, propensity for drug‐drug interactions, and adverse‐effect profiles and toxicities. All older‐generation ASDs have acute dose‐related effects, primarily neurological effects.25, 26 This led to treatment failure in 30%–40% of people with epilepsy receiving an older drug as monotherapy.8 Hence, there has been a great deal of research into formulating better ASDs, primarily spurred by the fact that the older‐generation ASDs do not provide optimal safety, tolerability or seizure control for many patients with epilepsy. Over the past three decades, 20 newer‐generation ASDs have been approved for clinical use,11 and it was hoped that these drugs would have better efficacy in controlling seizures than older‐generation drugs. However, the AAN subcommittee reports in 2004 and 2018 observed that the newer‐generation ASDs were “not different” in controlling seizures, but better tolerated than the older drugs.12, 13, 21, 22
1.4. Classifications of ASDs
ASDs are classified as older (first‐) generation or newer (second‐ and third‐) generation agents. The older‐generation ASDs introduced into clinical practice more than four decades ago include phenobarbital, phenytoin, primidone, ethosuximide, valproate, carbamazepine, clonazepam, and clobazam. The “second‐generation” ASDs, which have been approved for the treatment of epilepsy since the late 1980s, include, in chronological order, vigabatrin, oxcarbazepine, lamotrigine, gabapentin, felbamate, topiramate, tiagabine, levetiracetam, and zonisamide. The third‐generation ASDs include, pregabalin, fosphenytoin, lacosamide, rufinamide, eslicarbazepine, retigabine (also known as ezogabine), perampanel, brivaracetam, cannabidiol, stiripentol, cenobamate, and fenfluramine.11, 12, 27, 28 The newer ASDs differ substantially in their mechanisms of action, spectra of activity, pharmacokinetics, and adverse effects profiles. Figure 1 presents the classification and year of introduction of ASDs available now. Current information on the other drugs in the pipeline can be found on the Epilepsy Foundation Web site.29
FIGURE 1.
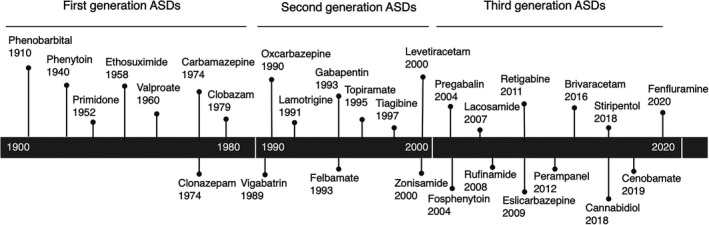
Classification and year of introduction of antiseizure drugs
1.5. Pharmacokinetics and drug interactions of ASDs
ASDs are commonly used for long periods of time, and consideration of their pharmacokinetic properties is essential for avoiding toxicity and drug interactions.30 An ideal ASD should demonstrate complete absorption, linear kinetics, a long elimination half‐life, and allowing once or twice‐daily dosing. Other favorable properties include low protein binding, lack of active metabolites, and clearance by kidneys. Generally, all ASDs are well absorbed after oral administration, have good bioavailability, and readily cross the blood‐brain barrier. Many ASDs are medium to long‐acting drugs (have half‐lives of >12 hours) and can be administered twice or three times a day. Phenobarbital, phenytoin, zonisamide, eslicarbazepine, and perampanel can often be administered once daily. Extended‐release preparations of drugs that have short half‐lives (eg, carbamazepine, valproate, levetiracetam, and lamotrigine) may decrease the incidence of adverse effects and allow once‐daily dosing. For optimum therapy, therapeutic drug concentration should be monitored in individual patients for some drugs. Examples include carbamazepine, phenytoin, and valproate. Drug levels can be helpful (1) to guide dose adjustments, (2) when breakthrough seizures occur, (3) when an interacting medication is added, (4) during pregnancy, (5) to assess compliance, and (6) to determine whether adverse effects are related to drug levels. Clinically relevant pharmacokinetic profiles of ASDs are summarized in Table 1.
TABLE 1.
Pharmacokinetic profiles of antiseizure drugs
| Antiseizure drug | Bioavailability % | Peak concentration (hr) | Plasma protein binding (%) | Elimination half‐life (hr) | Route of elimination | Therapeutic serum concentration (mcg/mL) |
|---|---|---|---|---|---|---|
| Brivaracetam | ~ 95 | 1 | ≤ 20 | 7‐10 | ++ | 0.2‐2 |
| Carbamazepine | 75‐85 | 4‐5 | 70‐80 | 10‐17 | ++++ | 4‐11 |
| Cannabidiol | 10‐20 | 2.5‐5 | >94 | 56‐61 | ++++ | NE |
| Cenobamate | 88 | 1‐4 | 60 | 50‐60 | +++ | NE |
| Clobazam | 90‐100 | 1‐3 | 80‐90 | 36‐42 | ++++ | 0.03‐3 |
| Clonazepam | >80 | 1‐4 | 80‐90 | 24‐48 | +++ | 10‐70a |
| Eslicarbazepine | >90 | 1‐4 | <40 | 13‐20 | ++++ | 5‐35 |
| Ethosuximide | 95‐100 | 3‐7 | 0 | 30‐60 | ++ | 40‐100 |
| Felbamate | >90 | 3‐5 | 22‐36 | 16‐22 | ++ | 30‐60 |
| Gabapentin | 50 | 2‐3 | 0 | 5‐9 | ‐ | 3‐21 |
| Lacosamide | 100 | 1‐2 | <30 | 12‐14 | + | 3‐10 |
| Lamotrigine | ~ 90 | 1‐3 | 55 | 8‐35 | +++ | 3‐13 |
| Levetiracetam | ~ 95 | 1‐2 | <10 | 6‐8 | ‐ | 5‐41 |
| Oxcarbazepine | 100 | 4‐5 | 75 | 10‐17 | ++++ | 3‐36 |
| Perampanel | 100 | 0.5‐3 | 95‐96 | 70‐110 | +++ | 0.1‐1 |
| Phenobarbital | >90 | 0.5‐4 | 55 | 90 | ++ | 12‐30 |
| Phenytoin | 85‐90 | 5‐7 | 90 | 24 | +++b | 10‐20 |
| Pregabalin | ~90 | 1‐2 | 0 | 4.5‐7 | ‐ | 2‐6 |
| Primidone | >90 | 2‐6 | 10 | 8‐15 | ++ | 8‐12 |
| Rufinamide | >90 | 4‐6 | 35 | 6‐10 | ++ | 4.5‐31 |
| Stiripentol | Variable | 2‐3 | 99 | 4.5‐13 | + | 4‐22 |
| Tiagabine | ~90 | 0.5‐2 | 96 | 2‐9 | +++ | 0.02‐0.2 |
| Topiramate | ~80 | 2‐4 | 15 | 20‐30 | + | 2‐10 |
| Valproate | >90 | 2‐4 | 90 | 15 | ++++ | 50‐100 |
| Vigabatrin | 100 | 1 | 0 | 5‐8 | ‐ | 20−160a |
| Zonisamide | >90 | 2‐6 | 40‐60 | 50‐68 | ++ | 10‐38 |
NE, not established
++++Extensive hepatic metabolism and active metabolite(s)
+++Extensive hepatic metabolism but no active metabolite(s)
++Hepatic metabolism (with or without active metabolites) and renal excretion.
+Variable (or moderate) hepatic metabolism (with or without active metabolites)
‐Renal excretion (unchanged). No hepatic metabolism
ng/mL
Saturable
In general, the ASD should be started at a low dose, with increments over several weeks to establish an effective and tolerable regimen. Some agents do not require titration, such as gabapentin and levetiracetam. Clinicians should be aware of certain factors that affect dosing. These factors include nonlinear relationships between dose and drug exposure (eg, phenytoin) and the influence of hepatic or renal impairment on clearance. Metabolism of phenytoin is nonlinear; elimination kinetics shift from first order at a low dose to zero‐order at moderate to high dose. As a subsequent, any small increases in the dose may increase phenytoin half‐life and serum concentration, and a patient may quickly develop toxicity. A common clinical error is to increase the dosage directly from 300 mg/d to 400 mg/d; toxicity frequently occurs at a variable time thereafter. Hepatic biotransformation of valproate may lead to hepatotoxicity. Hence, the liver function test is recommended in individual patients.
Most ASDs are metabolized by hepatic enzymes. Carbamazepine, oxcarbazepine, eslicarbazepine, phenobarbital, phenytoin, and primidone are inducers of hepatic cytochrome P450 enzyme and may decrease the effects of other drugs administered concomitantly (eg, valproate). Valproate and clobazam are inhibitors of hepatic enzymes and most likely to elevate the plasma concentration of other drugs administered concomitantly (eg, carbamazepine, ethosuximide, phenytoin, phenobarbital, and lamotrigine). Drug‐drug interactions with ASDs are complex since the drugs are often used in combination. These interactions may lead to either inadequate seizure control or drug toxicity. Of the newer drugs, levetiracetam, gabapentin, pregabalin, and vigabatrin are unique. These drugs are eliminated unchanged by the kidney and have no drug‐drug interactions. Lamotrigine, perampanel, tiagabine, topiramate, and zonisamide undergo hepatic drug metabolism and have potential drug interactions. Oxcarbazepine, felbamate, and topiramate selectively induce the hepatic metabolism of the oral contraceptive pill, failing birth control.
1.6. Mechanisms of actions of ASDs
A propensity for seizure generation occurs when there is an imbalance favoring excitation of neurons over inhibition. ASDs actions can generally be viewed in the context of inhibition of excitation or strengthening of inhibition, or both. Inhibition of excitation can be produced by effects on intrinsic excitability mechanisms in excitatory neurons (eg, inhibition of sodium and calcium channel), or on excitatory synaptic transmission (eg, glutamate AMPA and NMDA receptors and synaptic vesicle protein 2A). Enhancement of inhibition is produced by the increased availability of γ‐aminobutyric acid (GABA), increased activation of GABAA receptors, the mediators of inhibition in cortical areas relevant to seizures, and modulation of voltage‐gated potassium channel of the Kv7. For some drugs, the precise mechanism of action is not known (eg, valproate, zonisamide, and rufinamide), and some have multiple targets (eg, topiramate and felbamate).31 The ASD mechanisms of action are summarized in Table 2 and discussed in detail in the remainder sections.
TABLE 2.
Mechanistic categorization of current antiseizure drugs based on foremost targets at therapeutic concentrations
| Target and mechanisma | Antiseizure drug |
|---|---|
| Inhibition of voltage‐gated sodium channels | Phenytoin, fosphenytoin, carbamazepine, cenobamate, lamotrigine, oxcarbazepine, eslicarbazepine, lacosamide, and possibly topiramate, zonisamide, rufinamide, and phenobarbital |
| Inhibition of α2δ subunit of voltage‐gated calcium channels | Gabapentin, pregabalin |
| Inhibition of T type voltage‐gated calcium channels | Ethosuximide |
| Activation of GABAA receptor | Phenobarbital, benzodiazepines, and possibly topiramate, felbamate, retigabine, and stiripentol. |
| Inhibition of GABA transporter (selective) | Tiagabine |
| Inhibition of GABA transaminase enzyme | Vigabatrin |
| Modulation of synaptic vesicle protein 2A | Levetiracetam, brivaracetam |
| Various actions on multiple targets | Valproate, felbamate, topiramate, zonisamide, and cannabidiol |
| Opening KCNQ2‐5 (Kv7.2‐Kv7.5) voltage‐gated potassium channels | Retigabine (ezogabine)b |
| Inhibition of NMDA‐type glutamate receptors | Felbamate, topiramate and phenobarbital |
| Inhibition of AMPA‐type glutamate receptors | Perampanel |
Fenfluramine's mechanism of action for the treatment of seizures associated with Dravet syndrome is unknown.
Production of the drug retigabine has been discontinued by the manufacturer, and it is no longer available.
1.7. Voltage‐gated sodium channels blockade
At therapeutic concentrations, carbamazepine, cenobamate, lacosamide, lamotrigine, phenytoin, and zonisamide block voltage‐gated sodium channel in neuronal membranes. These agents act mainly on action potential firing and do not directly alter excitatory or inhibitory synaptic responses. However, the effect on action potentials translates into reduced transmitter output at synapses. Topiramate may also act, in part, by this mechanism. Phenobarbital and valproate may act on the sodium channel at high doses. This action is rate‐dependent (ie, the block increases with increased frequency of neuronal discharge).31
1.8. GABA‐related targets
Benzodiazepines interact with specific receptors (GABAA receptors) and increase the frequency of GABA‐mediated chloride ion channel opening (facilitate inhibitory effects of GABA). Phenobarbital and other barbiturates increase the duration of GABA‐mediated chloride ion channel opening. Vigabatrin, an analog of GABA, irreversibly inactivates GABA transaminase, the enzyme responsible for the termination of action of GABA. Valproate also can inhibit GABA transaminase at high doses. Tiagabine selectively inhibits the GAT‐1 GABA transporter, causing prolongation of GABA‐mediated inhibitory synaptic responses. Gabapentin and pregabalin are structural analogs of GABA but do not act through effects on any mechanism related to GABA‐mediated neurotransmission. Instead, the drugs bind to the α2δ subunit of the voltage‐gated calcium channel and decrease glutamate release at excitatory synapses. Other drugs that may facilitate the inhibitory actions of GABA include felbamate and topiramate.31
1.9. Calcium channel blockade
Ethosuximide inhibits low voltage‐gated T type calcium channels in thalamocortical neurons. Valproate may exert similar action. Gabapentin and pregabalin bind to the α2δ subunit of voltage‐gated calcium channel and may decrease glutamate release at excitatory synapses (the precise mechanism is not known).31
1.10. Glutamate synapses and other mechanisms
Levetiracetam binds to the synaptic vesicle protein 2A (SV2A) receptors on glutamate‐containing transmitter vesicles, where it is believed to inhibit excitatory neurotransmitters release (vesicular exocytosis) and thereby reduce synaptic excitability. Retigabine opens KCNQ2‐5 (Kv7.2‐Kv7.5) voltage‐gated potassium channel in presynaptic nerve terminals and thereby inhibits glutamate release. In addition to its action on calcium and sodium channel, valproate may enhance voltage‐gated potassium channel permeability, causing hyperpolarization. Perampanel is a potent noncompetitive antagonist of the AMPA receptors, a subtype of the ionotropic glutamate receptors that are the primary mediators of synaptic excitation in the central nervous system. Phenobarbital may block glutamate AMPA receptors and its action on voltage‐activated calcium channel and GABA‐chloride ion channel. Felbamate blocks glutamate NMDA receptors, with selectivity for those containing the GluN2B subunit. The drug also produces a barbiturate‐like potentiation of GABAA receptor responses. The possible sites for the action of topiramate include voltage‐gated sodium channel, GABAA receptor subtypes, and glutamate AMPA receptors.31
1.11. Principles of ASD selection for specific seizure type(s)
Treatment should be considered in patients reporting more than one unprovoked seizure or after a single seizure if the risk of recurrence is high.1 The initial ASD should be individualized on the basis of seizure type and/or epilepsy syndrome and slowly titrated up to a target dosage. Other important selection criteria include patient characteristics, drug efficacy, adverse‐effect profile, potential drug‐drug interactions, and cost.32 Combination therapy should be considered after the failure of two monotherapies.33 The therapeutic spectrum of ASDs can be categorized into (1) broad‐spectrum drugs used for both focal and generalized seizures, including, in alphabetical order, brivaracetam, clobazam, felbamate, lamotrigine, levetiracetam, perampanel, rufinamide, topiramate, valproate, and zonisamide; (2) narrow‐spectrum drugs used primarily for focal and focal to bilateral (secondarily generalized) tonic‐clonic seizures including carbamazepine, cenobamate, eslicarbazepine, gabapentin, lacosamide, oxcarbazepine, phenobarbital, phenytoin, pregabalin, primidone, stiripentol, tiagabine, and vigabatrin; and (3) a narrow‐spectrum drug used primarily for generalized absence seizures which is ethosuximide.
Table 3 presents the efficacy of ASDs against common seizure types and epilepsy syndrome. The data supplemented in the table are based primarily on the results of RCTs comparing the newer versus older drugs, including the SANAD studies34, 35, 36, 37 and others,38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62 and recently published meta‐analyses.63, 64, 65, 66, 67, 68, 69 Table 4 and the remainder sections of this article present guidance for ASD selection for the specific seizure type according to the 2017 ILAE classification of seizure types and epilepsy syndromes.14, 15, 16, 17 The guidance is based mainly on the current state of the literature on the efficacy and tolerability of ASDs and the recently published practical guidelines. The most popular treatment guidelines applied herein are the guidelines published by AAN & AES in 2004, 2015, and 2018,12, 13, 21, 22, 70 the ILAE in 2006 and 2013,71, 72 the UK National Institute for Health and Care Excellence in 2012,73, 74 and the Scottish Intercollegiate Guidelines Network in 2018.75, 76
TABLE 3.
Efficacy of antiseizure drugs against common seizure types and epilepsy syndromes
| Antiseizure drug/ seizure type | Focal seizures | GTCS | Absence | Myoclonic | Lennox–Gastaut syndrome | Infantile spasm | Dravet's syndrome |
|---|---|---|---|---|---|---|---|
| Brivaracetam | + | + | + | ||||
| Cannabidiol | + | +a | + | ||||
| Carbamazepine | + | + | – | – | – | ||
| Cenobamate | + | ||||||
| Clobazam | + | + | + | +e | |||
| Clonazepam | + | + | + | ||||
| Eslicarbazepine | + | – | – | ||||
| Ethosuximide | + | ||||||
| Felbamate | +b | + | |||||
| Fenfluramine | + | ||||||
| Gabapentin | + | ?+ | – | – | – | ||
| Lacosamide | + | ?+ | |||||
| Lamotrigine | + | + | + | ?+c | + | ||
| Levetiracetam | + | + | ?+ | + | |||
| Oxcarbazepine | + | + | – | – | – | ||
| Perampanel | + | + | |||||
| Phenobarbital | + | + | – | ?+ | |||
| Phenytoin | + | + | – | – | – | ||
| Pregabalin | + | – | |||||
| Primidone | + | + | – | ||||
| Retigabineg | + | ||||||
| Rufinamide | + | + | |||||
| Stiripentol | +f | ||||||
| Tiagabine | + | – | – | ||||
| Topiramate | + | + | + | + | + | +e | |
| Valproate | + | + | + | +d | + | + | +e |
| Vigabatrin | + | ?+ | – | – | +a | ||
| Zonisamide | + | + | ?+ | + | + |
GTCS; generalized tonic‐clonic seizure
Note that although there is evidence to support the use of these drugs for these seizure types, the drugs may not be indicated for this use by the US Food and Drug Administration.
+Effective;?+ possibly effective; – worsen seizure.
Especially when associated with tuberous sclerosis complex.
Can cause aplastic anemia and severe hepatitis, used only for patients who respond poorly to other agents.
Possibly effective but may worsen myoclonic seizures in some cases.
Preferred in patients with concomitant GTCS or myoclonic seizures (myoclonic absence seizure).
None of these is very effective in Dravet's syndrome.
In combination with clobazam and valproate.
Has been discontinued by the manufacturer, and it is no longer available.
TABLE 4.
Recommendations for add‐on and monotherapy in adults and pediatric patients >4 years of age with new‐onset epilepsy based on an assessment of current literature and published guidelines
| Seizure type |
First‐line (Monotherapy or add‐on) |
Second‐line (Monotherapy or add‐on) |
Third‐line (Add‐on) |
|---|---|---|---|
| Focal‐onset seizures, including focal to bilateral tonic‐clonic seizure | Lamotriginea |
Carbamazepine Levetiracetam Zonisamide Phenytoin Valproate Topiramateb Oxcarbazepineb Gabapentinb Phenobarbitalc Brivaracetamd Eslicarbazepined Lacosamided Perampaneld |
Cenobamatee Clobazam Retigabine Felbamate Rufinamide Pregabalin Tiagabine Vigabatrin |
| Generalized tonic‐clonic seizures (GTCS) | Valproatef |
Carbamazepine Phenytoin Lamotrigine Topiramate Levetiracetam Brivaracetam Perampanel Zonisamide Clobazam Phenobarbital |
|
| Myoclonic seizure | Valproate |
Lamotrigineg Topiramate Levetiracetam Brivaracetam Clonazepam Zonisamide |
|
| Absence seizures |
Ethosuximide Valproate |
Lamotrigine Clonazepam Levetiracetam |
|
| Unclassified seizuresh | Valproate |
Lamotrigine Levetiracetam Topiramate Zonisamide |
Evidence is insufficient to consider gabapentin, oxcarbazepine, or topiramate instead of lamotrigine in patients with new‐onset focal epilepsy. Gabapentin may be considered first‐line monotherapy in patients aged ≥60 years.
Often regarded as second‐line treatment in adults because of sedation and behavioral problems.
Received FDA approval for extrapolation of efficacy as monotherapy across individuals with focal seizures (Kanner AM, et al Neurology 2018; 91:82‐90).
Evidence is insufficient to consider the use of clobazam, felbamate, tiagabine, vigabatrin, or third‐generation antiseizure drugs as monotherapies in treating new‐onset focal epilepsy.
Valproate should be avoided, if possible, in women of childbearing potential.
May worsen myoclonic seizures in some cases.
Evidence is insufficient to support efficacy of newer antiseizure drugs in unclassified generalized tonic‐clonic seizures.
Lamotrigine was superior to levetiracetam and zonisamide for time to 12‐month remission and should remain a first‐line treatment for new‐onset focal epilepsy.
1.12. Drugs effective for focal‐onset seizures
Focal‐onset seizures account for 60% of all epilepsies. The RCTs and meta‐analyses have demonstrated comparable efficacy of several ASDs in controlling new‐onset focal seizures.22, 34, 37, 38, 39, 40, 41, 42, 43, 44, 46, 58, 62, 63, 64, 72, 77, 78, 79, 80, 81, 82, 83 The drug of the first choice for “focal” and “focal to bilateral” (secondarily generalized) tonic‐clonic seizures is lamotrigine.22, 34, 37 Phenytoin is the drug of choice for the urgent treatment of new‐onset or recurrent focal epilepsy. The other most widely used first‐line drugs for focal seizures include levetiracetam and zonisamide.22, 34 Carbamazepine is considered a second‐line treatment for new‐onset focal epilepsy owing to its unfavorable efficacy‐to‐tolerability profile.22, 34 Oxcarbazepine, topiramate, and valproate can be used, but they may not be as effective as lamotrigine and carbamazepine.22 Phenobarbital (and its derivative primidone) is often regarded as a second‐line treatment in adults because of sedation and behavioral problems.22 However, phenobarbital is a primary drug for neonatal seizures.
Given the unique mechanistic and pharmacokinetic profiles of the third‐generation ASDs, the United States Food and Drug Administration (FDA) recently issued a strategy that allows extrapolation of these drugs across populations as add‐on or monotherapy.22 Accordingly, brivaracetam, eslicarbazepine, lacosamide, and perampanel received FDA approval as initial monotherapies for new‐onset focal epilepsy.22 Individual monotherapy studies showed similar efficacy of second‐generation (levetiracetam and zonisamide) and third‐generation (eslicarbazepine and lacosamide) ASDs compared with controlled‐release carbamazepine for the treatment of new‐onset focal epilepsy.63 Vigabatrin use appears to be less efficacious than carbamazepine use and may not be offered; furthermore, tolerability profile precludes vigabatrin use as first‐line therapy.22 The drugs used as add‐on (adjunctive) therapy in focal seizures include gabapentin, pregabalin, felbamate, rufinamide, cenobamate, clobazam, retigabine, and tiagabine. However, the AAN/AES guidelines recommend gabapentin and lamotrigine, as first‐line monotherapy in patients aged ≥60 years with new‐onset focal epilepsy.22 A recent systematic review and network meta‐analysis showed that lacosamide, lamotrigine, and levetiracetam had the highest probability of ranking best for achieving seizure freedom in the elderly with new‐onset epilepsy.64 The FDA determined that the efficacy of ASDs for focal‐onset seizures in adults can be extrapolated downward to children four years of age and above.22
For drug‐resistant focal epilepsy in adults, eslicarbazepine can be used as monotherapy while the immediate‐release pregabalin, perampanel, lacosamide, eslicarbazepine, extended‐release topiramate, rufinamide, clobazam, felbamate, and vigabatrin should be considered as add‐on therapy.21 However, vigabatrin, felbamate, and rufinamide are not first‐line agents because of the retinopathy risk with vigabatrin, the modest benefit with rufinamide, and the hepatotoxicity and hematotoxicity risk with felbamate. In pediatric patients, levetiracetam, oxcarbazepine, and zonisamide should be considered as add‐on therapy.21
1.13. Drugs effective for generalized tonic‐clonic seizures (GTCS)
There has been a limited number of ASDs that can be used as first‐line agents for GTCS. Valproate remains the first‐line drug for many patients with generalized and unclassified epilepsies,22, 35, 36, 72 while phenobarbital (or primidone) is a primary drug in infants (for neonatal seizures) and an alternative in adults. However, valproate should not be prescribed for women of childbearing potential because of its dose‐dependent teratogenic profile; unless other ASDs cannot control the seizures, in which case the dose should be kept as low as possible.84 Indeed, some patients with genetic generalized epilepsy can control their seizures only with valproate. The other most widely used monotherapies for GTCS include lamotrigine, levetiracetam, brivaracetam, topiramate, clobazam, and zonisamide.22 However, there appears to be no evidence to support any second‐generation or third‐generation ASDs to be as efficacious as valproate monotherapy for generalized and unclassified epilepsies.35, 36 The combination of lamotrigine and valproate is believed to be particularly efficacious.85 Phenytoin and carbamazepine are effective but may worsen certain seizure types in generalized epilepsies, including absence epilepsy, juvenile myoclonic epilepsy, and Dravet's syndrome.86 Vigabatrin, tiagabine, oxcarbazepine, and possibly gabapentin are other drugs that may worsen these seizure types. Seizures of most patients with generalized epilepsy are easily controlled with appropriate medication. However, immediate‐release and extended‐release lamotrigine use should be considered add‐on therapy to decrease seizure frequency in drug‐resistant GTCS in adults.21 Levetiracetam should also be effective in both drug‐resistant GTCS and drug‐resistant juvenile myoclonic seizures. Other drugs used as add‐on therapy in GTCS include perampanel and lacosamide.
1.14. Drugs effective for focal seizures and certain generalized onset seizure types
A variety of drugs are primarily used to treat focal seizures; these drugs have also been effective in certain generalized onset seizure types. These drugs are lamotrigine, levetiracetam, brivaracetam, perampanel, phenobarbital, primidone, and felbamate.
1.15. Drugs effective for myoclonic seizures
Valproate is widely used for myoclonic seizures, such as in juvenile myoclonic epilepsy.87 Lamotrigine is possibly effective but may worsen myoclonic seizures in some cases. Other drugs effective in treating this seizure type are levetiracetam, brivaracetam, zonisamide, topiramate, and clonazepam.
1.16. Drugs effective for generalized absence seizures
Ethosuximide is the first‐line drug. It is often used in uncomplicated absence seizures if patients can tolerate its gastrointestinal side effects.22, 45 Despite the long half‐life (~ 40 hours), ethosuximide is generally administered in two or even three divided doses to minimize the adverse gastrointestinal effects. Valproate is preferred in patients with concomitant GTCS or myoclonic seizures (myoclonic absence seizure). Clonazepam is effective as an alternative drug but has the disadvantages of causing sedation and tolerance. Lamotrigine, levetiracetam, and zonisamide are also used in the absence seizures but not as effective as ethosuximide or valproate. Unless there are compelling reasons based on adverse events profile, ethosuximide or valproate use should be considered before lamotrigine in treating absence seizures.45 Lamotrigine should be considered in women of childbearing age because of its better tolerability and fewer fetal risks compared with valproate.
1.17. Drugs effective for other epilepsy syndromes
In combination with lamotrigine and a benzodiazepine (such as clobazam), valproate is the most widely used treatment for atonic seizures (eg, in the Lennox‐Gastaut syndrome). Other drugs used for the atonic seizures in Lennox‐Gastaut syndrome include topiramate, felbamate, and rufinamide.85 Because felbamate can cause aplastic anemia and severe hepatitis, it is used as add‐on therapy in patients who respond poorly to other agents. For infantile spasms (West's syndrome), valproate, topiramate, zonisamide, or a benzodiazepine (such as clonazepam or nitrazepam) is effective.85 Vigabatrin and everolimus are used if associated with tuberous sclerosis complex (a rare genetic disease that causes non‐cancerous (benign) tumors to grow in the brain and other organs). However, infantile spasms are primarily treated with intramuscular adrenocorticotropic hormone (ACTH) or oral corticosteroids such as prednisone or hydrocortisone (unknown mechanism). For Dravet's syndrome (the severe myoclonic epilepsy of infancy), valproate, topiramate, and clobazam can be used, although none of these is very effective.88 Stiripentol, a modulator of GABAA receptors, is often used in conjunction with clobazam or valproate.88 Cannabidiol was approved in 2018 to treat Dravet's syndromes66 in addition to Lennox‐Gastaut syndrome68 and infantile spasms associated with tuberous sclerosis complex. Recent meta‐analyses showed that adjunctive cannabidiol was associated with a greater reduction in convulsive seizure frequency than placebo in patients with Lennox‐Gastaut syndrome68 and children with Dravet syndrome.66 Fenfluramine is another new drug approved in 2020 for Dravet's syndrome. The drug is available only through a restricted drug distribution program, under a risk evaluation and mitigation strategy (REMS) because of the risk of valvular heart disease and pulmonary arterial hypertension. Recent studies, however, showed that fenfluramine provided a significantly greater reduction in convulsive seizure frequency compared with placebo and was generally well tolerated, with no observed valvular heart disease or pulmonary arterial hypertension.65, 89
1.18. Drugs effective for status epilepticus
Status epilepticus (SE) is a life‐threatening emergency that requires immediate treatment.90 In clinical practice, it is now generally accepted that a seizure lasting >5 minutes for GTCS and 10 minutes for focal seizures with or without impairment of consciousness should be treated as status epilepticus. Figure 2 presents the SE treatment algorithm. According to the evidence‐based guideline for the treatment of convulsive SE in children and adults by the AES in 2016,91 the first‐line treatment for SE is a benzodiazepine (either intravenous lorazepam or intravenous diazepam or intramuscular midazolam). If the preferred options are not available, rectal diazepam, intranasal midazolam, buccal midazolam, or intravenous phenobarbital is acceptable alternatives.
FIGURE 2.

Status Epilepticus treatment algorithm*
*Adopted from Glauser T, et al Epilepsy Curr. 2016; 16:48‐6. Refer to publication for complete recommendations.
ABCD, airway, breathing, circulation, disability (Neurologic); B, buccal; ECG, electrocardiogram; ED, emergency department; EEG, electroencephalogram; IM, intramuscular; IN, intranasal; IV, intravenous; PE, phenytoin sodium equivalents; SE, status epilepticus.
If the seizure continues, then second‐line treatment is administered.69 Intravenous fosphenytoin is preferred over phenytoin as the latter may cause cardiotoxicity. With phenytoin intravenous administration, there is also a risk of the potentially serious “purple glove syndrome,” in which a purplish‐black discoloration accompanied by edema and pain occurs distal to the site of injection. Fosphenytoin has a lower incidence of purple glove syndrome. The second‐line treatment also includes intravenous valproate or intravenous levetiracetam. Intravenous phenobarbital can be alternative (if not given already) but has a long half‐life causing persistent adverse effects, including severe sedation, respiratory depression, and hypotension. A recent network meta‐analysis compared efficacy and tolerability of intravenous valproate, phenytoin, diazepam, phenobarbital, lacosamide, and levetiracetam in adults with benzodiazepine‐resistant convulsive SE and suggested that phenobarbital had the greatest probabilities of being best in the achievement of SE control and seizure freedom, whereas valproate and lacosamide ranked best for the safety outcomes (respiratory depression and hypotension).69
If the second therapy fails to stop the seizures, another second‐line agent is often tried. Treatment‐resistant SE occurs when seizures continue or recur at least 30 minutes after treatment with first‐ and second‐line agents and should be treated with anesthetic doses of pentobarbital, propofol, midazolam, or thiopental. Dosing and frequency are available in the proposed algorithm for convulsive SE.91 Acute repetitive seizures (seizure clusters), in which there is complete recovery between seizures, are treated with benzodiazepines. Diazepam rectal gel is the only approved treatment for the out‐of‐hospital treatment of acute repetitive seizures.92
1.19. Adverse effects of ASDs
The adverse effects of selected ASDs are listed in Table 5. The first‐generation ASDs have acute dose‐related effects, primarily neurological effects such as sedation, dizziness, unsteadiness, blurred vision, diplopia, and tremor, in addition to neurocognitive and psychiatric symptoms.25, 26, 32 These effects are found across the different ASDs26, 62 and often mild and reversible. However, some drugs are better tolerated than others; for example, lamotrigine and levetiracetam are better tolerated than carbamazepine in elderly patients.93 Psychiatric adverse effects include depression, anxiety, irritability, impaired concentration, mood changes, hyperactivity, and, in rare cases, psychosis. Although the newer ASDs are touted as better tolerated than older drugs,12 psychiatric adverse effects are common with levetiracetam, topiramate, zonisamide, vigabatrin, and perampanel. Lamotrigine, carbamazepine, valproate, gabapentin, and pregabalin, in contrast, have mood‐stabilizing effects in some patients and less frequently cause behavioral or psychiatric effects.94
TABLE 5.
Common and serious adverse effects of selected antiseizure drugs
| Antiseizure drug | Systemic adverse effects | Neurologic adverse effects | Rare idiosyncratic reactions |
|---|---|---|---|
| Brivaracetam | Nausea, vomiting, constipation, fatigue | Headache, somnolence, dizziness, abnormal coordination, nystagmus, mood changes | |
| Carbamazepine | Nausea, vomiting, diarrhea, a plastic anemia, leukopenia, hyponatremia (common reason for discontinuation), hepatotoxicity, rash, pruritus | Ataxia, dizziness, blurred vision, diplopia, headache | Erythematous maculopapular rash (Steven‐Johnson syndrome and toxic epidermal necrolysis), teratogenicity |
| Cenobamate | Nausea, vomiting, fatigue, hyperkalemia, QT shortening | Somnolence, dizziness, headache, balance disorder, diplopia | Drug reaction with eosinophilia and systemic symptoms (DRESS)/multiorgan hypersensitivity (at high doses) |
| Eslicarbazepine | Nausea, vomiting, diarrhea, hyponatremia, rash | Dizziness, drowsiness, headache, somnolence, diplopia, ataxia, blurred vision, tremor | |
| Ethosuximide | Nausea, vomiting | Sleep disturbance, drowsiness, hyperactivity | |
| Felbamate | Nausea, vomiting, anorexia, weight loss | Insomnia, dizziness, headache, ataxia | Aplastic anemia, severe hepatitis/hepatic failure |
| Gabapentin | Infrequent | Somnolence, dizziness, ataxia, headache, tremor, and fatigue | |
| Lacosamide | Nausea, vomiting, increased cardiac conduction (PR interval) | Dizziness, ataxia, diplopia, headache | |
| Lamotrigine | Nausea, rash, cardiac arrhythmias | Dizziness, tremor, diplopia | Steven‐Johnson syndrome |
| Levetiracetam | Fatigue, infection, anemia, leukopenia | Somnolence, dizziness, agitation, anxiety, irritability, depression, psychosis | |
| Oxcarbazepine | Nausea, rash, hyponatremia (more common) | Somnolence, headache, dizziness, vertigo, ataxia, diplopia | |
| Perampanel | Weight gain, fatigue, nausea | Dizziness, somnolence, irritability, gait disturbance, falls (with high dose), aggression, mood alteration | |
| Phenobarbital | Nausea, rash | Somnolence, ataxia, dizziness, confusion, cognitive dysfunction, tolerance, dependence | |
| Phenytoin | Gingival hyperplasia, hirsutism, megaloblastic anemia, peripheral neuropathy, osteoporosis, rash | Nystagmus (early sign of phenytoin administration), diplopia, ataxia, somnolence | |
| Pregabalin | Weight gain, peripheral edema, dry mouth | Somnolence, dizziness, ataxia, headache, and tremor | |
| Rufinamide | Nausea, vomiting, leukopenia, cardiac conduction (QT interval shortening) | Somnolence, fatigue, dizziness, ataxia, headache, diplopia | |
| Tiagabine | Abdominal pain, nausea, lack of energy | Dizziness, difficulty concentrating, somnolence, nervousness, tremor, language problems | |
| Topiramate | Anorexia, weight loss, paresthesia, fatigue | Nervousness, psychomotor slowing, language problems, depression, anxiety, mood problems, tremor | Acute glaucoma (may require prompt drug withdrawal). |
| Valproate | Gastrointestinal irritation, weight gain, hair loss, easy bruising | Ataxia, somnolence, tremor | Hepatotoxicity, teratogenicity, and thrombocytopenia |
| Vigabatrin | Fatigue | Somnolence, headache, dizziness, agitation, confusion, psychosis. | Irreversible bilateral concentric visual field defect |
| Zonisamide | Weight loss, nausea, anorexia | Somnolence, dizziness, confusion, headache, psychosis | Potentially serious skin rashes |
Most women with epilepsy who become pregnant require continued ASD therapy for seizure control. If possible, valproate, carbamazepine, phenytoin, phenobarbital, and topiramate should be avoided in women of childbearing potentials. Recent analysis from several international pregnancy registers suggests that in utero exposure to valproate during the first trimester is associated with a threefold increased risk of congenital malformations, commonly neural tube defects (spina bifida) and cardiovascular, orofacial, and digital abnormalities.95 The use of valproate during the first trimester is also associated with cognitive impairments. Carbamazepine may cause neural tube defects and craniofacial anomalies. Fetal hydantoin syndrome is related to the use of phenytoin. Treatment with topiramate during the first trimester of pregnancy is associated with a 10‐fold increase in oral clefts risk. Phenobarbital can cause congenital malformations, most often cardiac defects. No ASD is known to be entirely safe for the developing fetus. However, lamotrigine and levetiracetam have the lowest risks of major congenital malformations and may be safer, particularly for cognition compared with valproate.85
Valproate causes hepatotoxicity in children less than two years of age. Carbamazepine and lamotrigine may cause life‐threatening Steven‐Johnson syndrome and toxic epidermal necrolysis, which is strongly associated with the HLA‐B*1502 allele. Asians, who have a 10‐fold increased risk of the drug‐induced Stevens‐Johnson syndrome compared with other ethnic groups, should be tested before starting the drug. The use of zonisamide is also associated with severe skin reactions. Aplastic anemia and acute hepatic failure have limited the use of felbamate to severe and drug‐resistant epilepsy. Overdose toxicity with benzodiazepines and barbiturates may cause respiratory depression. Management is primarily supportive (airway management, mechanical ventilation) and flumazenil in benzodiazepine overdose. Withdrawal from ASDs should be accomplished gradually (over a 1‐ to 3‐month period or longer) to avoid the occurrence of severe seizures or status epilepticus. Physical dependence occurs with barbiturates and benzodiazepines, and there is a well‐recognized risk of rebound seizures with abrupt withdrawal. However, withdrawal is less likely to be a problem with ethosuximide. Withdrawal is believed to be successful in patients with generalized epilepsies who exhibit a single seizure type, whereas the longer duration of epilepsy, an abnormal neurologic examination, an abnormal EEG, and certain epilepsy syndromes, including juvenile myoclonic epilepsy, are associated with increased risk of recurrence.85
Clinical studies suggest a possible association of lamotrigine, levetiracetam, and topiramate with suicidality. In 2008, the FDA issued an alert that ASDs, as a class, may be associated with an increased risk of suicidality based on an analysis of data from placebo‐controlled add‐on clinical trials of ASDs in patients with drug‐resistant epilepsy, although this is still highly controversial.96, 97 Long‐term treatment with ASDs is associated with a twofold to threefold increased risk of osteoporosis and bone fractures.98 In addition, increased body weight and fat are common in patients using valproate, carbamazepine, gabapentin, pregabalin, vigabatrin, and perampanel and can lead to serious health consequences associated with obesity and increased cardiovascular disease risk.99 In March 2021, the FDA issued an alert that lamotrigine may be associated with an increased risk of cardiac arrhythmias in people with underlying cardiac disease.100 However, this risk is not apparent in healthy individuals.101
1.20. The new frontiers in epilepsy pharmacology
Since 1989, nearly 20 second‐generation and third‐generation antiseizure drugs (ASDs) with different mechanisms of action have reached the market, resulting in a greatly increased range of treatment options for patients and prescribers.102, 103 Some second‐generation ASDs have shown advantages in drug tolerability, drug‐drug interactions, and teratogenicity and thus offer valuable individualized options in treating epilepsy. Disappointingly, however, none of these medications appear to be more efficacious than first‐generation ASDs, and none have substantially reduced the proportion of patients with drug‐resistant epilepsy, highlighting the need for novel strategies in epilepsy drug development. Given the favorable pharmacokinetic characteristics and adverse‐effect profiles for the third‐generation medications, additional evidence to further define their efficacy is crucial to the future treatment of epilepsy. The high incidence of drug‐resistance and unwanted adverse effects caused by taking long‐term ASDs raise substantial concern. In particular, there is an urgent need for developing novel drugs that act beyond the membrane ion channels and neural transmissions. Deciphering the genetics that underpins the mechanisms that generate seizures is one of the promising areas in the field.104 Many epilepsy genes have been identified, including genes that increase the risk of different types of epilepsy, such as generalized and focal epilepsy and developmental and epileptic encephalopathies. These genes have a role in ion channel and synaptic dysfunction in addition to transcriptional regulation. Most mutations occur in the protein‐coding exons, the sodium and potassium channel, and glutamate receptors.32 Ganaxolone, sirolimus, everolimus, cannabidiol, fenfluramine, stiripentol, and memantine are new selective disease‐modifying therapies recently approved for severe forms of genetic epilepsies.32 Data to support gene‐therapy treatments need to be established in future research, and there are often other disorders that these therapies can improve, including acquired epilepsies.104 Furthermore, with a deeper understanding of the cellular and molecular mechanisms of epileptogenesis, non‐classical novel antiseizure agents, such as neurosteroid and allopregnanolone, with their effect on both GABAergic and glutaminergic systems, may shed light on finding a new approach to treat epilepsy, which is not only controlling seizures but also retarding epileptogenesis.105 Novel pharmacological approaches built on gene therapies and epileptogenesis may overcome many of the adverse effects of currently available treatment options.
2. CONCLUSIONS
Antiseizure drugs (ASDs) have many different pharmacologic profiles that are relevant when selecting and prescribing these medications in patients with epilepsy. These include pharmacokinetic properties, propensity for drug‐drug interactions, and adverse‐effect profile and toxicities. This article reviewed the current state of the literature about the clinical pharmacology of ASDs, based primarily on the most recent evidence for the efficacy and tolerability of the currently available drugs and the recent US and UK treatment guidelines. It is anticipated that Table 4 in this review (recommendations for ASD selection) will require updating as future studies yield more detailed results. It is hoped that this review remains a succinct and practical guide to assist non‐neurologists, particularly the primary healthcare practitioners, in patient care decisions. The medical students may also wish to read this review to improve their understanding of epilepsy and its pharmacological treatment. Finally, the content of this review is not intended to include all legitimate criteria for choosing to use or exclude a specific drug, nor recommended as a substitute for current scientific and clinical information. Physicians are encouraged to carefully review the literature in addition to the full guidelines to understand all recommendations associated with patient care and be confident that the information contained in this work is accurate, particularly for new or infrequently used drugs. Physicians are also encouraged to obtain information about drug dosing and frequency from the product information sheet included in each drug package. Complete information on US Food and Drug Administration (FDA) labeling for each drug can be accessed using the FDA searchable database (FDALabel).
CONFLICT OF INTEREST
The author declares no conflict of interest.
AUTHOR CONTRIBUTIONS
The author has made substantial contributions to the conception and design of the work, and acquisition, analysis, and interpretation of data for the work; drafting the work and revising it critically for important intellectual content; final approval of the version to be published; and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Hakami T. Neuropharmacology of Antiseizure Drugs. Neuropsychopharmacol Rep. 2021;41:336–351. 10.1002/npr2.12196
Funding information
The author received no financial support for the research, authorship, and/or publication of this article
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article.
REFERENCES
- 1.Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014;55:475‐82. [DOI] [PubMed] [Google Scholar]
- 2.Hauser WA, Beghi E. First seizure definitions and worldwide incidence and mortality. Epilepsia. 2008;49(Suppl 1):8‐12. [DOI] [PubMed] [Google Scholar]
- 3.Pitkanen A. Therapeutic approaches to epileptogenesis–hope on the horizon. Epilepsia. 2010;51(Suppl 3):2‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314‐9. [DOI] [PubMed] [Google Scholar]
- 5.Marson A, Jacoby A, Johnson A, Kim L, Gamble C, Chadwick D, et al. Immediate versus deferred antiepileptic drug treatment for early epilepsy and single seizures: a randomised controlled trial. Lancet. 2005;365:2007‐13. [DOI] [PubMed] [Google Scholar]
- 6.Kwan P, Brodie MJ. Drug treatment of epilepsy: when does it fail and how to optimize its use? CNS Spectr. 2004;9:110‐9. [DOI] [PubMed] [Google Scholar]
- 7.Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Allen Hauser W, Mathern G, et al. Definition of drug resistant epilepsy: consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010;51:1069‐77. [DOI] [PubMed] [Google Scholar]
- 8.Mattson RH, Cramer JA, Collins JF, Smith DB, Delgado‐Escueta AV, Browne TR, et al. Comparison of carbamazepine, phenobarbital, phenytoin, and primidone in partial and secondarily generalized tonic‐clonic seizures. N Engl J Med. 1985;313:145‐51. [DOI] [PubMed] [Google Scholar]
- 9.French JA, White HS, Klitgaard H, Holmes GL, Privitera MD, Cole AJ, et al. Development of new treatment approaches for epilepsy: unmet needs and opportunities. Epilepsia. 2013;54(Suppl 4):3‐12. [DOI] [PubMed] [Google Scholar]
- 10.Wilcox KS, Dixon‐Salazar T, Sills GJ, Ben‐Menachem E, Steve White H, Porter RJ, et al. Issues related to development of new antiseizure treatments. Epilepsia. 2013;54(Suppl 4):24‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stephen LJ, Brodie MJ. Pharmacotherapy of epilepsy: newly approved and developmental agents. CNS Drugs. 2011;25:89‐107. [DOI] [PubMed] [Google Scholar]
- 12.French JA, Kanner AM, Bautista J, Abou‐Khalil B, Browne T, Harden CL, et al. Efficacy and tolerability of the new antiepileptic drugs I: treatment of new onset epilepsy: report of the Therapeutics and Technology Assessment Subcommittee and Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2004;62:1252‐60. [DOI] [PubMed] [Google Scholar]
- 13.French JA, Kanner AM, Bautista J, Abou‐Khalil B, Browne T, Harden CL, et al. Efficacy and tolerability of the new antiepileptic drugs II: treatment of refractory epilepsy: report of the Therapeutics and Technology Assessment Subcommittee and Quality Standards Subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2004;62:1261‐73. [DOI] [PubMed] [Google Scholar]
- 14.Fisher RS. An overview of the 2017 ILAE operational classification of seizure types. Epilepsy Behav. 2017;70:271‐3. [DOI] [PubMed] [Google Scholar]
- 15.Fisher RS, Cross JH, D'Souza C, et al. Instruction manual for the ILAE 2017 operational classification of seizure types. Epilepsia. 2017;58:531‐42. [DOI] [PubMed] [Google Scholar]
- 16.Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:522‐30. [DOI] [PubMed] [Google Scholar]
- 17.Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia. 2017;58:512‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Proposal for revised clinical and electroencephalographic classification of epileptic seizures. From the commission on classification and terminology of the international league against epilepsy. Epilepsia. 1981;22:489‐501. [DOI] [PubMed] [Google Scholar]
- 19.Proposal for revised classification of epilepsies and epileptic syndromes. Commission on classification and terminology of the international league against epilepsy. Epilepsia. 1989;30:389‐99. [DOI] [PubMed] [Google Scholar]
- 20.Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676‐85. [DOI] [PubMed] [Google Scholar]
- 21.Kanner AM, Ashman E, Gloss D, Harden C, Bourgeois B, Bautista JF, et al. Practice guideline update summary: efficacy and tolerability of the new antiepileptic drugs II: treatment‐resistant epilepsy: report of the guideline development, dissemination, and implementation subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2018;91:82‐90. [DOI] [PubMed] [Google Scholar]
- 22.Kanner AM, Ashman E, Gloss D, Harden C, Bourgeois B, Bautista JF, et al. Practice guideline update summary: efficacy and tolerability of the new antiepileptic drugs I: treatment of new‐onset epilepsy: report of the guideline development, dissemination, and implementation subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2018;91:74‐81. [DOI] [PubMed] [Google Scholar]
- 23.Fiest KM, Sauro KM, Wiebe S, Patten SB, Kwon C‐S, Dykeman J, et al. Prevalence and incidence of epilepsy: a systematic review and meta‐analysis of international studies. Neurology. 2017;88:296‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Epilepsy Foundation . Epilepsy Pipeline Tracker, https://www.epilepsy.com/pipeline‐listing‐page (accessed April 25, 2021)
- 25.Baker GA, Jacoby A, Buck D, Stalgis C, Monnet D, et al. Quality of life of people with epilepsy: a European study. Epilepsia. 1997;38:353‐62. [DOI] [PubMed] [Google Scholar]
- 26.Loscher W, Schmidt D. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia. 2011;52:657‐78. [DOI] [PubMed] [Google Scholar]
- 27.Marson AG, Kadir ZA, Hutton JL, Chadwick DW, et al. The new antiepileptic drugs: a systematic review of their efficacy and tolerability. Epilepsia. 1997;38:859‐80. [DOI] [PubMed] [Google Scholar]
- 28.Vajda FJ. Pharmacotherapy of epilepsy: new armamentarium, new issues. J Clin Neurosci Off J Neurosurg Soci Australa. 2007;14:813‐23. [DOI] [PubMed] [Google Scholar]
- 29.International League Against Epilepsy . Epilepsy Syndromes, https://www.epilepsydiagnosis.org/syndrome/epilepsy‐syndrome‐groupoverview.html (accessed April 25, 2021)
- 30.Zaccara G, Perucca E. Interactions between antiepileptic drugs, and between antiepileptic drugs and other drugs. Epileptic Disord. 2014;16:409‐31. [DOI] [PubMed] [Google Scholar]
- 31.Sills GJ, Rogawski MA. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology. 2020;168:107966. [DOI] [PubMed] [Google Scholar]
- 32.Devinsky O, Vezzani A, O'Brien TJ, Jette N, Scheffer IE, de Curtis M, et al. Epilepsy. Nat Rev Dis Primers. 2018;4:18024. [DOI] [PubMed] [Google Scholar]
- 33.Karceski S, Morrell M, Carpenter D. The expert consensus guideline series treatment of epilepsy. Epilepsy Behav. 2001;2:A1‐A50. [DOI] [PubMed] [Google Scholar]
- 34.Marson AG, Al‐Kharusi AM, Alwaidh M, Appleton R, Baker GA, Chadwick DW, et al. The SANAD study of effectiveness of carbamazepine, gabapentin, lamotrigine, oxcarbazepine, or topiramate for treatment of partial epilepsy: an unblinded randomised controlled trial. Lancet. 2007;369:1000‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marson AG, Al‐Kharusi AM, Alwaidh M, Appleton R, Baker GA, Chadwick DW, et al. The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalised and unclassifiable epilepsy: an unblinded randomised controlled trial. Lancet. 2007;369:1016‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marson A, Burnside G, Appleton R, Smith D, Leach JP, Sills G, et al. The SANAD II study of the effectiveness and cost‐effectiveness of valproate versus levetiracetam for newly diagnosed generalised and unclassifiable epilepsy: an open‐label, non‐inferiority, multicentre, phase 4, randomised controlled trial. Lancet. 2021;397:1375‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marson A, Burnside G, Appleton R, Smith D, Leach JP, Sills G, et al. The SANAD II study of the effectiveness and cost‐effectiveness of levetiracetam, zonisamide, or lamotrigine for newly diagnosed focal epilepsy: an open‐label, non‐inferiority, multicentre, phase 4, randomised controlled trial. Lancet. 2021;397:1363‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Privitera MD, Brodie MJ, Mattson RH, Chadwick DW, Neto W, Wang S, et al. Topiramate, carbamazepine and valproate monotherapy: double‐blind comparison in newly diagnosed epilepsy. Acta Neurol Scand. 2003;107:165‐75. [DOI] [PubMed] [Google Scholar]
- 39.Rowan AJ, Ramsay RE, Collins JF, Pryor F, Boardman KD, Uthman BM, et al. New onset geriatric epilepsy: a randomized study of gabapentin, lamotrigine, and carbamazepine. Neurology. 2005;64:1868‐73. [DOI] [PubMed] [Google Scholar]
- 40.Brodie MJ, Perucca E, Ryvlin P, Ben‐Menachem E, Meencke H‐J, et al. Comparison of levetiracetam and controlled‐release carbamazepine in newly diagnosed epilepsy. Neurology. 2007;68:402‐8. [DOI] [PubMed] [Google Scholar]
- 41.Brodie MJ, Overstall PW, Multicentre GL. Multicentre, double‐blind, randomised comparison between lamotrigine and carbamazepine in elderly patients with newly diagnosed epilepsy. The UK Lamotrigine Elderly Study Group. Epilepsy Res. 1999;37:81‐7. [DOI] [PubMed] [Google Scholar]
- 42.Baulac M, Brodie MJ, Patten A, Segieth J, Giorgi L, et al. Efficacy and tolerability of zonisamide versus controlled‐release carbamazepine for newly diagnosed partial epilepsy: a phase 3, randomised, double‐blind, non‐inferiority trial. Lancet Neurol. 2012;11:579‐88. [DOI] [PubMed] [Google Scholar]
- 43.Chadwick D. Safety and efficacy of vigabatrin and carbamazepine in newly diagnosed epilepsy: a multicentre randomised double‐blind study. Vigabatrin European Monotherapy Study Group. Lancet. 1999;354:13‐9. [DOI] [PubMed] [Google Scholar]
- 44.Kwan P, Brodie MJ, Kälviäinen R, Yurkewicz L, Weaver J, Knapp LE, et al. Efficacy and safety of pregabalin versus lamotrigine in patients with newly diagnosed partial seizures: a phase 3, double‐blind, randomised, parallel‐group trial. Lancet Neurol. 2011;10:881‐90. [DOI] [PubMed] [Google Scholar]
- 45.Glauser TA, Cnaan A, Shinnar S, et al. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy: initial monotherapy outcomes at 12 months. Epilepsia. 2013;54:141‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saetre E, Perucca E, Isojärvi J, Gjerstad L, et al. An international multicenter randomized double‐blind controlled trial of lamotrigine and sustained‐release carbamazepine in the treatment of newly diagnosed epilepsy in the elderly. Epilepsia. 2007;48:1292‐302. [DOI] [PubMed] [Google Scholar]
- 47.French JA, Krauss GL, Wechsler RT, Wang X‐F, DiVentura B, Brandt C, et al. Perampanel for tonic‐clonic seizures in idiopathic generalized epilepsy A randomized trial. Neurology. 2015;85:950‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.French J, Brandt C, Friedman D, Biton V, Knapp L, Pitman V, et al. Adjunctive use of controlled‐release pregabalin in adults with treatment‐resistant partial seizures: a double‐blind, randomized, placebo‐controlled trial. Epilepsia. 2014;55:1220‐8. [DOI] [PubMed] [Google Scholar]
- 49.Brodie MJ, Rosenfeld WE, Vazquez B, Sachdeo R, Perdomo C, Mann A, et al. Rufinamide for the adjunctive treatment of partial seizures in adults and adolescents: a randomized placebo‐controlled trial. Epilepsia. 2009;50:1899‐909. [DOI] [PubMed] [Google Scholar]
- 50.Brodie MJ, Lerche H, Gil‐Nagel A, Elger C, Hall S, Shin P, et al. Efficacy and safety of adjunctive ezogabine (retigabine) in refractory partial epilepsy. Neurology. 2010;75:1817‐24. [DOI] [PubMed] [Google Scholar]
- 51.French JA, Mosier M, Walker S, Sommerville K, Sussman N, et al. A double‐blind, placebo‐controlled study of vigabatrin three g/day in patients with uncontrolled complex partial seizures. Vigabatrin Protocol 024 Investigative Cohort. Neurology. 1996;46:54‐61. [DOI] [PubMed] [Google Scholar]
- 52.Elger C, Halász P, Maia J, Almeida L, Soares‐da‐Silva P, et al. Efficacy and safety of eslicarbazepine acetate as adjunctive treatment in adults with refractory partial‐onset seizures: a randomized, double‐blind, placebo‐controlled, parallel‐group phase III study. Epilepsia. 2009;50:454‐63. [DOI] [PubMed] [Google Scholar]
- 53.French JA, Baroldi P, Brittain ST, Johnson JK, et al. Efficacy and safety of extended‐release oxcarbazepine (Oxtellar XR) as adjunctive therapy in patients with refractory partial‐onset seizures: a randomized controlled trial. Acta Neurol Scand. 2014;129:143‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.French JA, Costantini C, Brodsky A, von Rosenstiel P, et al. Adjunctive brivaracetam for refractory partial‐onset seizures: a randomized, controlled trial. Neurology. 2010;75:519‐25. [DOI] [PubMed] [Google Scholar]
- 55.Baulac M, Rosenow F, Toledo M, Terada K, Li T, De Backer M, et al. Efficacy, safety, and tolerability of lacosamide monotherapy versus controlled‐release carbamazepine in patients with newly diagnosed epilepsy: a phase 3, randomised, double‐blind, non‐inferiority trial. Lancet Neurol. 2017;16:43‐54. [DOI] [PubMed] [Google Scholar]
- 56.French JA, Abou‐Khalil BW, Leroy RF, Yacubian EMT, Shin P, Hall S, et al. Randomized, double‐blind, placebo‐controlled trial of ezogabine (retigabine) in partial epilepsy. Neurology. 2011;76:1555‐63. [DOI] [PubMed] [Google Scholar]
- 57.Dam M, Ekberg R, Loyning Y, et al. A double‐blind study comparing oxcarbazepine and carbamazepine in patients with newly diagnosed, previously untreated epilepsy. Epilepsy Res. 1989;3:70‐6. [DOI] [PubMed] [Google Scholar]
- 58.Kalviainen R, Aikia M, Saukkonen AM, et al. Vigabatrin vs carbamazepine monotherapy in patients with newly diagnosed epilepsy. A randomized, controlled study. Arch Neurol. 1995;52:989‐96. [DOI] [PubMed] [Google Scholar]
- 59.Ramsay E, Faught E, Krumholz A, Naritoku D, Privitera M, Schwarzman L, et al. Efficacy, tolerability, and safety of rapid initiation of topiramate versus phenytoin in patients with new‐onset epilepsy: a randomized double‐blind clinical trial. Epilepsia. 2010;51:1970‐7. [DOI] [PubMed] [Google Scholar]
- 60.Trinka E, Marson AG, Van Paesschen W, Kälviäinen R, Marovac J, Duncan B, et al. KOMET: an unblinded, randomised, two parallel‐group, stratified trial comparing the effectiveness of levetiracetam with controlled‐release carbamazepine and extended‐release sodium valproate as monotherapy in patients with newly diagnosed epilepsy. J Neurol Neurosurg Psychiatry. 2013;84:1138‐47. [DOI] [PubMed] [Google Scholar]
- 61.Trinka E, Ben‐Menachem E, Kowacs PA, Elger C, Keller B, Löffler K, et al. Efficacy and safety of eslicarbazepine acetate versus controlled‐release carbamazepine monotherapy in newly diagnosed epilepsy: a phase III double‐blind, randomized, parallel‐group, multicenter study. Epilepsia. 2018;59:479‐91. [DOI] [PubMed] [Google Scholar]
- 62.Hakami T, Todaro M, Petrovski S, MacGregor L, Velakoulis D, Tan M, et al. Substitution monotherapy with levetiracetam vs older antiepileptic drugs: a randomized comparative trial. Arch Neurol. 2012;69:1563‐71. [DOI] [PubMed] [Google Scholar]
- 63.Lattanzi S, Zaccara G, Giovannelli F, Grillo E, Nardone R, Silvestrini M, et al. Antiepileptic monotherapy in newly diagnosed focal epilepsy. A network meta‐analysis. Acta Neurol Scand. 2019;139:33‐41. [DOI] [PubMed] [Google Scholar]
- 64.Lattanzi S, Trinka E, Del Giovane C, Nardone R, Silvestrini M, Brigo F, et al. Antiepileptic drug monotherapy for epilepsy in the elderly: A systematic review and network meta‐analysis. Epilepsia. 2019;60:2245‐54. [DOI] [PubMed] [Google Scholar]
- 65.Sharawat IK, Panda PK, Kasinathan A, Panda P, Dawman L, Joshi K, et al. Efficacy and tolerability of fenfluramine in patients with Dravet syndrome: A systematic review and meta‐analysis. Seizure. 2021;85:119‐26. [DOI] [PubMed] [Google Scholar]
- 66.Lattanzi S, Brigo F, Trinka E, Zaccara G, Striano P, Del Giovane C, et al. Adjunctive cannabidiol in patients with dravet syndrome: a systematic review and meta‐analysis of efficacy and safety. CNS Drugs. 2020;34:229‐41. [DOI] [PubMed] [Google Scholar]
- 67.Veroniki AA, Cogo E, Rios P, Straus SE, Finkelstein Y, Kealey R, et al. Comparative safety of anti‐epileptic drugs during pregnancy: a systematic review and network meta‐analysis of congenital malformations and prenatal outcomes. BMC Med. 2017;15:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lattanzi S, Brigo F, Cagnetti C, Trinka E, Silvestrini M, et al. Efficacy and safety of adjunctive cannabidiol in patients with Lennox‐Gastaut syndrome: a systematic review and meta‐analysis. CNS Drugs. 2018;32:905‐16. [DOI] [PubMed] [Google Scholar]
- 69.Brigo F, Del Giovane C, Nardone R, Trinka E, Lattanzi S, et al. Intravenous antiepileptic drugs in adults with benzodiazepine‐resistant convulsive status epilepticus: a systematic review and network meta‐analysis. Epilepsy Behav. 2019;101:106466. [DOI] [PubMed] [Google Scholar]
- 70.Krumholz A, Shinnar S, French J, et al. Evidence‐based guideline: management of an unprovoked first seizure in adults: report of the guideline development subcommittee of the American Academy of Neurology and the American Epilepsy Society. Neurology. 2015;85:1526‐7. [DOI] [PubMed] [Google Scholar]
- 71.Glauser T, Ben‐Menachem E, Bourgeois B, Cnaan A, Chadwick D, Guerreiro C, et al. ILAE treatment guidelines: evidence‐based analysis of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 2006;47:1094‐120. [DOI] [PubMed] [Google Scholar]
- 72.Glauser T, Ben‐Menachem E, Bourgeois B, Cnaan A, Guerreiro C, Kälviäinen R, et al. Updated ILAE evidence review of antiepileptic drug efficacy and effectiveness as initial monotherapy for epileptic seizures and syndromes. Epilepsia. 2013;54:551‐63. [DOI] [PubMed] [Google Scholar]
- 73.Appleton RE, Freeman A, Cross JH. Diagnosis and management of the epilepsies in children: a summary of the partial update of the 2012 NICE epilepsy guideline. Arch Dis Child. 2012;97:1073‐6. [DOI] [PubMed] [Google Scholar]
- 74.Nunes VD, Sawyer L, Neilson J, Sarri G, Cross JH, et al. Diagnosis and management of the epilepsies in adults and children: summary of updated NICE guidance. BMJ. 2012;344:e281. [DOI] [PubMed] [Google Scholar]
- 75.Network SIG. SIGN 143‐Diagnosis and management of epilepsy in adults. 2018.
- 76.Network SIG. Diagnosis and Management of Epilepsy in adults. SIGN national clinical guideline 143. Edinburgh: SIGN; 2015. [Google Scholar]
- 77.Stephen LJ, Sills GJ, Leach JP, Butler E, Parker P, Hitiris N, et al. Sodium valproate versus lamotrigine: a randomised comparison of efficacy, tolerability and effects on circulating androgenic hormones in newly diagnosed epilepsy. Epilepsy Res. 2007;75:122‐9. [DOI] [PubMed] [Google Scholar]
- 78.Brodie MJ, Richens A, Yuen AW. Double‐blind comparison of lamotrigine and carbamazepine in newly diagnosed epilepsy. UK Lamotrigine/Carbamazepine Monotherapy Trial Group. Lancet. 1995;345:476‐9. [DOI] [PubMed] [Google Scholar]
- 79.Reunanen M, Dam M, Yuen AW. A randomised open multicentre comparative trial of lamotrigine and carbamazepine as monotherapy in patients with newly diagnosed or recurrent epilepsy. Epilepsy Res. 1996;23:149‐55. [DOI] [PubMed] [Google Scholar]
- 80.Nieto‐Barrera M, Brozmanova M, Capovilla G, Christe W, Pedersen B, Kane K, et al. A comparison of monotherapy with lamotrigine or carbamazepine in patients with newly diagnosed partial epilepsy. Epilepsy Res. 2001;46:145‐55. [DOI] [PubMed] [Google Scholar]
- 81.Tanganelli P, Regesta G. Vigabatrin vs. carbamazepine monotherapy in newly diagnosed focal epilepsy: a randomized response conditional cross‐over study. Epilepsy Res. 1996;25:257‐62. [DOI] [PubMed] [Google Scholar]
- 82.Aikia M, Jutila L, Salmenpera T, Mervaala E, Kalviainen R, et al. Comparison of the cognitive effects of tiagabine and carbamazepine as monotherapy in newly diagnosed adult patients with partial epilepsy: pooled analysis of two long‐term, randomized, follow‐up studies. Epilepsia. 2006;47:1121‐7. [DOI] [PubMed] [Google Scholar]
- 83.Christe W, Krämer G, Vigonius U, Pohlmann H, Steinhoff BJ, Brodie MJ, et al. A double‐blind controlled clinical trial: oxcarbazepine versus sodium valproate in adults with newly diagnosed epilepsy. Epilepsy Res. 1997;26:451‐60. [DOI] [PubMed] [Google Scholar]
- 84.Tomson T, Battino D, Perucca E. Teratogenicity of antiepileptic drugs. Curr Opin Neurol. 2019;32:246‐52. [DOI] [PubMed] [Google Scholar]
- 85.Roger JP, Michael AR. Antiseizure drugs. In: Katzung BG, editor. Basic & clinical pharmacology. McGraw‐Hill; 2018, p. 409‐39. [Google Scholar]
- 86.French JA. Seizure exacerbation by antiepileptic drugs. Epilepsy Curr. 2005;5:192‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Silvennoinen K, Lange N, Zagaglia S, Balestrini S, Androsova G, Wassenaar M, et al. Comparative effectiveness of antiepileptic drugs in juvenile myoclonic epilepsy. Epilepsia Open. 2019;4:420‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wallace A, Wirrell E, Kenney‐Jung DL. Pharmacotherapy for dravet syndrome. Paediatr Drugs. 2016;18:197‐208. [DOI] [PubMed] [Google Scholar]
- 89.Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double‐blind, placebo‐controlled trial. Lancet. 2019;394:2243‐54. [DOI] [PubMed] [Google Scholar]
- 90.Trinka E, Cock H, Hesdorffer D, Rossetti AO, Scheffer IE, Shinnar S, et al. A definition and classification of status epilepticus–report of the ILAE task force on classification of status epilepticus. Epilepsia. 2015;56:1515‐23. [DOI] [PubMed] [Google Scholar]
- 91.Glauser T, Shinnar S, Gloss D, Alldredge B, Arya R, Bainbridge J, et al. Evidence‐based guideline: treatment of convulsive status epilepticus in children and adults: report of the guideline committee of the American Epilepsy Society. Epilepsy Curr. 2016;16:48‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Haut SR. Seizure clusters: characteristics and treatment. Curr Opin Neurol. 2015;28:143‐50. [DOI] [PubMed] [Google Scholar]
- 93.Werhahn KJ, Trinka E, Dobesberger J, Unterberger I, Baum P, Deckert‐Schmitz M, et al. A randomized, double‐blind comparison of antiepileptic drug treatment in the elderly with new‐onset focal epilepsy. Epilepsia. 2015;56:450‐9. [DOI] [PubMed] [Google Scholar]
- 94.Ettinger AB. Psychotropic effects of antiepileptic drugs. Neurology. 2006;67:1916‐25. [DOI] [PubMed] [Google Scholar]
- 95.Tomson T, Battino D, Bonizzoni E, Craig J, Lindhout D, Sabers A, et al. Dose‐dependent risk of malformations with antiepileptic drugs: an analysis of data from the EURAP epilepsy and pregnancy registry. Lancet Neurol. 2011;10:609‐17. [DOI] [PubMed] [Google Scholar]
- 96.Patorno E, Bohn RL, Wahl PM, et al. Anticonvulsant medications and the risk of suicide, attempted suicide, or violent death. JAMA. 2010;303(14):1401‐9. [DOI] [PubMed] [Google Scholar]
- 97.Gibbons RD, Hur K, Brown CH, Mann JJ, et al. Relationship between antiepileptic drugs and suicide attempts in patients with bipolar disorder. Arch Gen Psychiatry. 2009;66:1354‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shiek Ahmad B, Hill KD, O'Brien TJ, Gorelik A, Habib N, Wark JD, et al. Falls and fractures in patients chronically treated with antiepileptic drugs. Neurology. 2012;79:145‐51. [DOI] [PubMed] [Google Scholar]
- 99.Chukwu J, Delanty N, Webb D, Cavalleri GL, et al. Weight change, genetics and antiepileptic drugs. Expert Rev Clin Pharmacol. 2014;7:43‐51. [DOI] [PubMed] [Google Scholar]
- 100.French JA, Perucca E, Sander JW, Bergfeldt L, Baulac M, Auerbach DS, et al. FDA safety warning on the cardiac effects of lamotrigine: an advisory from the Ad Hoc ILAE/AES task force. Epilepsy Curr. 2021: 1535759721996344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dixon R, Alexander S, Brickel N. Effect of lamotrigine on the PR interval in healthy subjects. Br J Clin Pharmacol. 2011;71:961‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Perucca E, Brodie MJ, Kwan P, Tomson T, et al. 30 years of second‐generation antiseizure medications: impact and future perspectives. Lancet Neurol. 2020;19:544‐56. [DOI] [PubMed] [Google Scholar]
- 103.Chen Z, Brodie MJ, Kwan P. What has been the impact of new drug treatments on epilepsy? Curr Opin Neurol. 2020;33:185‐90. [DOI] [PubMed] [Google Scholar]
- 104.Drew L. Gene therapy targets epilepsy. Nature. 2018;564:S10‐1. [DOI] [PubMed] [Google Scholar]
- 105.Rogawski MA, Zolkowska D. Anticonvulsant activity of steroids. Google Patents. 2021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article.