

Abstract
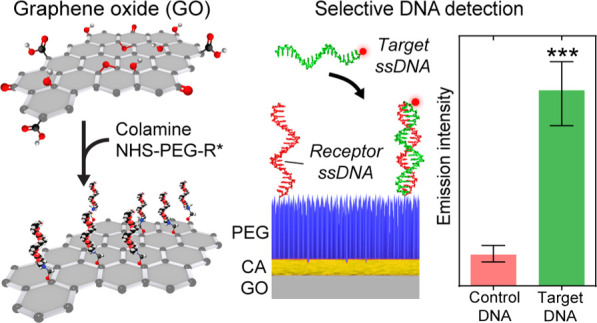
Graphene oxide (GO) has immense potential for widespread use in diverse in vitro and in vivo biomedical applications owing to its thermal and chemical resistance, excellent electrical properties and solubility, and high surface-to-volume ratio. However, development of GO-based biological nanocomposites and biosensors has been hampered by its poor intrinsic biocompatibility and difficult covalent biofunctionalization across its lattice. Many studies exploit the strategy of chemically modifying GO by noncovalent and reversible attachment of (bio)molecules or sole covalent biofunctionalization of residual moieties at the lattice edges, resulting in a low coating coverage and a largely bioincompatible composite. Here, we address these problems and present a facile yet powerful method for the covalent biofunctionalization of GO using colamine (CA) and the poly(ethylene glycol) cross-linker that results in a vast improvement in the biomolecular coating density and heterogeneity across the entire GO lattice. We further demonstrate that our biofunctionalized GO with CA as the cross-linker provides superior nonspecific biomolecule adhesion suppression with increased biomarker detection sensitivity in a DNA-biosensing assay compared to the (3-aminopropyl)triethoxysilane cross-linker. Our optimized biofunctionalization method will aid the development of GO-based in situ applications including biosensors, tissue nanocomposites, and drug carriers.
Keywords: graphene oxide, poly(ethylene glycol), colamine, APTES, DNA biosensor, surface chemistry, bioconjugation
Introduction
Since its discovery in 2004, graphene in both pristine and oxidized forms has been proposed for a wide variety of applications, ranging from nanocomposite material development to biomedical applications.1−4 This two-dimensional (2D) material’s unique combination of characteristics, such as a high surface-to-volume ratio, excellent electrical properties, chemical resistance, thermal stability, and solubility in aqueous solutions, makes graphene oxide (GO) of particular interest for biomedical applications including label-free biosensing and, more recently, as a drug delivery carrier.1,3,5−7 For example, feasibility studies have shown that GO can be used as a drug carrier for the treatment of cancer, multiple sclerosis, and Alzheimer’s disease; in gene therapy; as an early disease-detecting biosensor for Parkinson’s disease, Alzheimer’s disease, and cancer; in glucose immunosensing; as a grafting composite for tissue engineering; and in cell imaging among other applications.1,3,5,8−13
From a physicochemical point of view, these applications share the same critical surface composition requirements. To render a material functional for biomedical applications, an organochemical coating has to be added, which provides the means to covalently immobilize biosensing receptor biomolecules, such as antibodies, nucleic acids, and others. Ideally, the coating should provide homogeneous surface coverage with a high (bio)molecule density, stable chemical attachment, and high biocompatibility, while suppressing nonspecific biomolecule and cell adhesion.14−16 These characteristics have been shown to directly impact the target biomolecule selectivity and sensitivity (in the case of biosensors) and trigger immune response in different organisms.6,14−19
The challenges of biocompatibility and suppression of nonspecific biomolecule adhesion have been recently addressed using poly(ethylene glycol) (PEG) polymer coatings.12,20,21 Since GO is a nondegradable material, its solubility and in vivo biocompatibility can be significantly improved by applying physically and chemically inert PEG polymer coatings that cap the GO surface functionalities and decrease its hazard.22 Recent studies have evidenced that PEG-functionalized GO has significantly decreased toxicity to diverse human cell lines and living organisms by more than twofold compared to its pristine form at concentrations as high as 100 μg/mL.10,22−25 However, achieving chemically stable, dense, and homogeneous PEG functionalization of the entire GO lattice remains difficult.21 Currently, immobilization of PEG and biomolecules to GO is primarily realized using weak noncovalent and reversible binding strategies to attach (bio)molecules via electrostatic forces (e.g., π-stacking and van der Waals forces). Only a few approaches have been developed for covalent attachment of chemical moieties to residual oxygen-containing functional groups of GO.12,20 Candidate moieties for possible covalent attachment locations on GO include randomly distributed epoxides and relatively abundant hydroxyl groups within the GO hexagonal lattice as well as less-abundant carboxylic groups and ketones located at the edges of GO substrates.1,5,26 Covalent (bio)conjugation can be achieved either via chemical reduction using hydrazine, but also removes most of the other oxygen-containing functional groups, or via peptide binding through exposure to N-(3-dimethyl-aminopropyl-N′-ethyl carbodiimide) (EDC) or N,N′-dicyclohexyl carbodiimide, combined with N-hydroxy-succinimide (NHS) reactive groups.9,27 However, this strategy carries a notable drawback: particularly for GO flakes with larger dimensions, the use of edge carboxylic groups cannot provide a high-density coating on the GO lattice, which leads to both insufficient biocompatibility and decreased biosensing sensitivity.17,18
To improve the GO coating stability and coverage, other recent developments have focused on the covalent attachment of alkoxysilane cross-linkers to surface hydroxyl groups scattered across the GO plane. 3-Aminopropyl-triethoxysilane (APTES) has been a common choice, even though APTES modification can promote the formation of inhomogeneous and multilayered coatings, which render this approach largely inadequate for applications involving nanostructures.21,28−30 As a promising alternative, the colamine (CA) (β-hydroxyethylamine) cross-linker, originating from single-molecule atomic force spectroscopy, has recently shown superior functionalization performance for nanoscale semiconductor structures, for which it can provide dense and homogeneous monolayer coatings.21,31−33 Comparable with the covalent attachment of APTES to surface hydroxyl groups, CA forms covalent ether links with hydroxyl groups via a condensation reaction.31,32,34,35 In the case of GO, CA covalently ligates to both epoxides and abundant hydroxyl groups across the GO material plane, resulting in a significant increase in overall coating coverage.
In this work, we demonstrate the applicability of CA as a GO-compatible surface cross-linker superior to APTES. CA provides a highly uniform monolayer coating for subsequent dense covalent PEG immobilization to render GO biocompatible. In a step-wise and bottom-up fashion (Scheme 1), we characterize on a qualitative and quantitative level the surface-coating efficiency of CA and PEG as well as their performance as suppressors of nonspecific biomolecule adhesion compared to APTES–PEG. As a proof of concept, we covalently functionalize PEGylated CA and APTES-coated GO substrates with a DNA receptor and detect specific ssDNA “probe” sequences. Compared to APTES functionalization, the CA–PEG coating method presented here provides improved sensitivity due to the superior suppression of nonspecific adhesion of incompatible ssDNA sequences.
Scheme 1. Bottom-Up Fabrication of Biocompatible GO Sheets Functionalized with Biologically Active Molecules.

From top left: after (1) exfoliation and oxidation of graphite to GO, (2) CA was covalently coupled to both epoxide and hydroxyl groups on the GO surface plane. A secondary biocompatible layer consisting of NHS–PEG–R* was (3) covalently attached to primary amino groups provided by the CA coating. In our study, we (4) covalently attached NH2-conjugated 40-nt ssDNA to a carboxylic PEG-moiety (R*). Diverse binding strategies using different functional groups (R*) on PEG and target molecules (R#) are possible using this methodology (i.e., peptide binding, sulfhydryl cross-linking, and click chemistry). (Dashed square box) Examples of biomedically relevant (bio)molecules and drugs that can be immobilized on functional PEG-coated GO.
Experimental Section
Preparation of GO Sheets
GO was prepared from natural crystalline colloidal graphite using a modified Hummers and Offeman method.36 Pure graphite flakes (Ma-399.5 RG) were obtained from NGS Trading and Consulting GmbH with an average size of 45 μm.1 For exfoliation, 69 mL of concentrated H2SO4 was added to a mixture containing 3 g of graphite flakes and 1.5 g of NaNO3 and cooled to 0 °C. Afterward, 9 g of KMnO4 was gently titrated into the mixture, while ensuring that the reaction temperature remained below 20 °C. The mixture was then heated to 35 °C while stirring for a further 30 min. 138 mL of water was added slowly to produce a large exotherm, up to 98 °C, and the temperature was maintained for 15 min. Afterward, the sample mixture was cooled using an ice bath for 10 min. 420 mL of water and 3 mL of H2O2 were added, producing another exotherm. After air cooling, the mixture was filtered with a polyester fiber filter (Millipore, a 0.22 μm pore size, Merck Millipore, Germany). The filtrate was centrifuged at 4000 rpm for 4 h, and the supernatant was discarded. The remaining material was washed several times with 200 mL of ddH2O, 200 mL of 30% (v/v) HCl, and 200 mL of ethanol. After each washing step, the mixture was filtered and centrifuged as described above. The resulting material was coagulated with 200 mL of ether and filtered using a polytetrafluoroethylene membrane (Merck Millipore, Germany) with a pore size of 0.45 μm. The resulting solid was vacuum-dried at 14 psi for 12 h at room temperature.
Scanning Electron Microscopy of Graphene Oxide
GO flake preparations were imaged using a desktop scanning electron microscope (FEI NovaNanoSEM, ThermoFischer) on prime Si + SiO2 (dry) wafers (Microchemicals GmbH, Germany). Scanning electron microscopy (SEM) imaging was performed using a voltage of 5–15 kV and a 5 mm working distance.
X-Ray Photoelectron Spectroscopy of Amino-Functionalized GO
High-resolution (HR) X-ray photoelectron spectroscopy (XPS) spectra were recorded with a ThermoFisher K-Alpha system using Al Kα radiation with a photon energy of 1486.7 eV. The samples were loaded into the spectrometer without further purification and immobilized on a copper tape (Plano GmbH, G3397). For carbon and nitrogen spectra, 40 high-resolution XPS spectra were recorded using a spot size of 400 μm, a 50 eV pass energy, and a 0.1 eV step size. Charge neutralization was applied to minimize the effect of surface charges. All peaks were calibrated, referencing the carbon 1s peak to 285 eV. Additional high-resolution spectra were used to determine the binding states by deconvoluting the peaks using Thermo Avantage software (v5.952). After smart-type background subtraction, the peaks were fitted using Gaussian–Lorentzian product functions. The atomic ratios were quantitatively determined by correcting the peak areas with the TPP-2M sensitivity factor and compared with their corresponding peaks to verify their goodness of fit.
Raman Spectrometry
Graphite and GO samples were deposited on Si and SiO2 wafers by drop casting. Confocal Raman spectroscopy (inVia Reflex, Renishaw, UK) was performed using a 785 nm diode laser at 10% intensity with a signal integration time of 10 s, coupled to a low-noise CCD detector (Centrus, Renishaw, UK). Sample positioning was executed manually, and 5–10 graphite and GO substrates were measured for each experimental condition. Raman spectral analyses were performed with RStudio.
Atomic Force Microscopy
Atomic Force Microscopy (AFM) topography images (AFM Dimension FastScan, Brucker) were obtained in air in tapping mode using silicon nitride cantilevers (FASTSCAN-A, Bruker) with a nominal spring constant of ∼18 N/m and a resonance frequency of ∼800 kHz. The dimensions and height of the GO substrates (five measurements per substrate) were extracted and analyzed with Gwyddion.37
GO Surface Functionalization
The GO surface functionalization procedure with CA in this study is a modified version of a method described previously for SPM techniques.33,38 Here, GO substrates were sonicated in anhydrous dimethyl sulfoxide (DMSO, Sigma, USA) or dimethyl fumarate (DMF, Sigma-Aldrich) for 30 min and then incubated for 12 h in DMSO or DMF containing 5 M 2-aminoethanol hydrochloride (CA, Sigma-Aldrich, USA). Afterward, 100 μL of the GO solution was deposited onto a cleaned borosilicate cover glass support; then washed once with either DMSO or DMF, respectively, twice with ethanol, and once with Milli-Q H2O; and then dried in a nitrogen flow.
For silanization with APTES, GO substrates were sonicated in anhydrous DMSO for 30 min and washed with Milli-Q H2O three times. The GO solution was deposited on clean borosilicate glass supports and washed with ethanol and once again with Milli-Q H2O. The silanization with APTES was performed by adding 10 μL of 5% (v/v) APTES in toluene to the deposited GO flakes and incubated for 15 min at 75 °C. Afterward, the samples were rinsed thoroughly with methanol and Milli-Q H2O and then dried in a nitrogen flow.
The dried deposited GO sheets, with either CA or APTES functionalization, were PEGylated by depositing 2 mM heterobifunctional NHS–PEG–COOH (MW 3400, LaysanBio, USA) in anhydrous chloroform containing 0.5% (v/v) triethylamine for 1 h at room temperature in a closed, humid box. After PEGylation, the samples were washed five times with Milli-Q H2O and dried in a nitrogen flow. For covalent ssDNA attachment, NH2-labeled 40-nt ssDNA oligonucleotides (5′-NH2-CCACTCGTGACGCATTCACCTCAGCAGCACTCCT CCTCGG-3′, Purimex, Germany) were conjugated to the PEG-coated substrates via peptide binding at a concentration of 10 pmol/μL ssDNA in 100 mM 2-(N-morpholino)ethanesulfonic acid (MES, Sigma, USA, pH 4.7) buffer containing 50 mM 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC, Sigma, USA) for 1 h at room temperature. After covalent ssDNA immobilization, the support was washed twice with Milli-Q H2O, incubated for 10 min in a 10 mM KCl (potassium chloride, Sigma, USA) solution to eliminate nonspecifically adhered oligonucleotides, and finally incubated for 5 min in Milli-Q H2O, followed by drying in a nitrogen flow. The hybridization of the Atto647N-labeled, complementary 40-nt ssDNA strand (5′-Atto647N-CCGAGGAGGAGTG CTGCTGAGGTGAATGCGTCACGAGTGG-3′, Purimex, Germany) was carried out for 30 min at room temperature in TRIS–HCl buffer (20 mM TRIS, 100 mM NaCl, pH 7.5). Afterward, the samples were washed in 2× saline-sodium citrate (SSC, pH 7) buffer for 5 min and in 0.01× SSC buffer for another 5 min to remove nonspecifically adsorbed ssDNA.
Fluorescence Microscopy
The amount of GO-immobilized ssDNA was evaluated using an epifluorescence microscope (Nikon TE2000U, USA) with a Peltier-cooled back-illuminated EMCCD camera (IXON3, 1024 × 1024 pixels, Andor, Ireland) for sensitive fluorescence detection in combination with a 100× oil-immersion objective (CFI APO TIRF, NA. 1.45, Nikon, USA). Fluorophore excitation was achieved using a 150 W mercury-vapor lamp and a filter set (F41-008 AHF, Tübingen, Germany) specific to the excitation and emission wavelengths of Att647N (640, 690/20 nm, respectively). For each sample, the fluorescence intensity (in photon counts/s) was measured by taking the average over 10 areas consisting of 10 × 10 pixels. Each experiment was repeated three times to test the reproducibility and for statistical analysis.
Quantitative Evaluation of Nonspecific DNA Adhesion Suppression
Epifluorescence microscopy was also used to evaluate the amount of nonspecific adhesion of DNA to the different surface compositions. For these experiments, 24 × 24 mm borosilicate cover glasses (#1, Menzel GmbH, Germany) were cleaned by incubating them in a 5% (v/v) aqueous Hellmanex II-solution (Hellma GmbH, Germany) and sonicating for 20 min at 40 °C. Afterward, the cover glasses were washed thoroughly with Milli-Q H2O and sonicated again for 20 min in Milli-Q H2O. The cover glasses were dried in a nitrogen flow, and the generation of surface hydroxyl groups was carried out by applying oxygen plasma (SE80, Barrel Asher Plasma Technology, USA) for 15 min (50 sccm O2, 200 W, 100 mTorr). Either APTES (Sigma-Aldrich, USA) or CA (β-hydroxyethylamine, Sigma-Aldrich, USA) was then attached covalently to the oxidized cover glasses. The silanization with APTES was performed by adding 10 μL of 5% (v/v) APTES in toluene between two cleaned cover glasses and incubating for 15 min at 75 °C. The silanized glass supports were then rinsed thoroughly with methanol and Milli-Q H2O and then dried in a nitrogen flow. CA coating and PEGylation were performed as described for GO functionalization.
Nonspecific adhesion of ssDNA to cover glasses coated with APTES, CA, and the PEG cross-linker was tested using the fluorescently labeled ssDNA described above. To do so, 1 μM Atto647N-labeled 40-nt ssDNA in TRIS/HCl buffer (20 mM TRIS, 100 mM NaCl, pH 7.5) was added to the differently coated cover glasses and incubated for 1 h at room temperature in a dark, humid box. Afterward, the surfaces were washed three times with TRIS/HCl buffer for 5 min each, rinsed briefly with Milli-Q H2O, and dried gently in a nitrogen flow.
Quartz Crystal Microbalance with Dissipation Monitoring Data Acquisition and Sample Preparation
Surface functionalization with APTES and CA was monitored by quartz crystal microbalance with dissipation monitoring (QCM-D), where changes in resonance frequency Δf and dissipation ΔD correspond to variations in adsorbed mass and viscoelastic properties of the film, respectively.39 QCM-D measurements were performed with a QSense Analyzer system (Biolin Scientific, 578 Västra Frölunda, Sweden) and AT-cut quartz crystal chips coated with a ∼50 nm SiO2 film (Biolin Scientific). Δf and ΔD were monitored and recorded with sub-second temporal resolution at the fundamental tone (i = 1) and five overtones (i = 3, 5, 7, 9, 13), corresponding to resonance frequencies fi ≈ 5, 15, 25, 34, 45, 55, 65 MHz, respectively. Average (±SD) normalized frequency shifts, Δf = Δfi/i, and dissipation shifts, ΔD = ΔDi, for i = {5, 7, 9} are presented in Figures 1 and S1.
Figure 1.

CA provides a homogeneous single-layer coating of primary amines for subsequent PEGylation. (A) APTES and CA were covalently bound to silicon oxide surface hydroxyl groups. (B,C) Surface deposition of (B) APTES and (C) CA over time monitored via QCM-D. Δf and ΔD were measured with sub-second temporal resolution. Average (±SD) normalized frequency shifts, Δf = Δfi/i, and dissipation shifts, ΔD = ΔDi, for the overtones i = {5, 7, 9} are presented here and in Figure S1. (D) Areal molar density (gray) and estimated molecular layers (blue) of APTES and CA remaining bound after washing. (E) Example false-color wide-field fluorescence images for qualitative comparison of APTES (left) and CA (right) surface coating homogeneity via covalently coupled NHS-Atto647N fluorophores. (F) Comparison of the relative fluorescence of nonspecifically adhered 40-nt Atto-647N-labeled ssDNA on APTES, CA, and APTES and CA with covalently bound NHS–PEG–COOH and CA with covalently bound NHS–PEG–COOH (see Scheme 1) (N = 10 each), normalized to the value obtained for APTES. Statistical analysis consisted of an unpaired, two-tailed t-test (p: *** ≤ 0.001; ** ≤ 0.01). See also Figures S1 and S2.
Similar to the borosilicate cover glasses, SiO2-coated quartz crystals were treated with oxygen plasma (Diener Electric GmbH & Co., KG, Germany) for 5 min (25 sccm O2, 50 W) to generate surface silanol groups for covalent APTES and CA attachment, followed by washing with ddH2O and drying in a nitrogen stream. The system was operated at a constant flow rate of 10 μL/min using a peristaltic pump (MS-2/6, Ismatec GmbH, Germany) at room temperature (23 °C). Prior to the functionalization experiments, solvents (DMSO for CA and toluene for APTES) were injected into the flow cells until both Δf and ΔD reached a stationary value.
Results and Discussion
The aim of the study is to provide a versatile multistep functionalization method that fulfills the requirements of GO being implemented in diverse biomedical applications, such as chemical stability, biocompatibility, high (bio)molecule coverage, and high nonspecific biomolecule adhesion suppression performance.
The first step in functionalizing GO with high chemical stability is the essential covalent attachment of a surface cross-linker. Therefore, we compared the covalent coupling efficiency of the commonly used cross-linker APTES with our newly introduced CA to surface silanol groups on SiO2 supports (Figure 1A) as well as their ability to form surface mono- or multilayers as a function of reaction time. Here, we note that while GO is our target material, a first comparison of cross-linker coating efficiencies via fluorescence microscopy and QCM-D provides a more quantitatively accurate approach as GO preparations can vary in chemical composition and degree of structural defects.40
Following this reasoning, we first performed surface functionalization of SiO2-coated QCM-D crystals with APTES and CA, with both over 1 h (Figure 1B,C) and 12 h (Figure S1A,B), while monitoring mass deposition on the surface in real time. QCM-D measures changes in resonance frequency Δf and dissipation ΔD of the sensor crystal resulting from mass interactions upon the modification of the surface by cross-linkers.39 To a first approximation, the observed decrease in Δf for APTES (Figures 1B and S1A) and CA (Figures 1C and S1B) indicated an increase in the amount of adsorbed mass density over time, while the increase in ΔD verified the formation of a soft, viscoelastic surface coating. Strikingly, while Δf for CA deposition reached saturation within 20 min (Figures 1C and S1B), the shift in Δf for APTES decreased more slowly but steadily throughout a ∼12 h reaction time (Figures 1B and S1A). Subsequent washing with the solvent was performed to remove physisorbed cross-linkers and reveal the mass of cross-linkers that remained covalently bound (Figure 1D). For both reaction times, the areal mass density of APTES was ∼30-fold higher than that of CA (Figure 1D: gray bars). Considering that APTES can polymerize into large molecular chains, we reasoned that under the imperfectly anhydrogenous experimental conditions, the increase in APTES mass density has been caused by chain polymerization.
To estimate the number of surface layers formed by CA and APTES, while taking into account the van der Waals volumes of both cross-linkers (Figure 1D: blue bars), we found that APTES formed multiple layers over time (∼20 after 1 h, ∼130 after 12 h).41 This observation is in agreement with previous observations that have shown APTES forming hyperbranched polymers under hydrogenous conditions.29,30,42 In contrast, CA formed a monolayer after 12 h. The near-zero dissipation after the washing procedure supports the notion of a very thin-film deposition.
To verify the QCM-D results and assess the homogeneity of the APTES and CA coatings, we employed fluorescence microscopy with single-molecule detection sensitivity. We visualized the surface-grafted cross-linkers by covalently coupling NHS-ATTO647N fluorophores to the primary amino moieties provided by APTES and CA (Figure 1E).33 The APTES coating resulted in a highly inhomogeneous surface, resembling the previously observed island topography that resulted from thick APTES coatings.29,43 Prior studies identified that the polysiloxane structures within these islands are composed of a mixture of loosely bonded and 3D cross-linked APTES networks, as could also be observed in our fluorescence images.44 In contrast, the CA coating was homogeneously distributed across the surface with a consistent density. These results indicate that the use of CA is more advantageous than the use of APTES for the functionalization of nanoscale structures, where dense and homogeneous monolayers are vital.
Using the same methodology, we next coated SiO2 supports with APTES and CA in the presence and absence of conjugated NHS–PEG–COOH to probe the ability of each coating to suppress nonspecific ssDNA adhesion (Figure 1F). We made use of heterobifunctional NHS–PEG–COOH to allow the peptide-binding reaction between the PEG NHS group and the cross-linker primary amino group to occur without the addition of catalyzers. This approach prevents PEG coupling to the surface with both ends and ensures that the PEG COOH group remains freely accessible for subsequent covalent biomolecule attachment. After PEGylation, fluorophore-labeled 40-nt ssDNA was allowed to nonspecifically adhere to the different coatings, and quantitative fluorescence microscopy was used to measure the degree of nonspecific adhesion. Since GO exhibits strong autofluorescence in the green wavelengths (Figure S2), we chose Atto647N to label the ssDNA used in the adhesion and biosensing experiments.45 Compared to the commonly used APTES coating, CA coating exhibited a statistically significant decrease of ∼40% in nonspecific ssDNA adhesion. The decrease in adhesion can be explained by the denser and more homogeneous surface coverage by CA, while APTES coverage may have been more defective, exposing bare areas of SiO2 where electrostatic binding of DNA could occur more strongly.21 The addition of biocompatible PEG to APTES and CA improved the efficiency in suppressing nonspecific ssDNA adhesion by approximately ≥90% compared to APTES alone. This vast improvement likely resulted from both the chemically and physically inert character of PEG and the increased distance between the surface and biomolecule that PEG provides. Under the physiological conditions used here, the effective Debye length range in which strong electrostatic interactions occur is ∼1 nm, which can be overcome by the Flory radius of PEG (MW 3400) of ∼5 nm.46 In direct comparison, however, the CA coating with conjugated PEG was further able to significantly improve the suppression of nonspecific ssDNA adhesion with respect to APTES–PEG. This result further supports the notion that a dense and homogeneous monolayer composition for a surface cross-linker represents an important property for the design of coatings with high nonspecific biomolecule adhesion-suppressing performance.
Since a coating with CA conjugated with PEG largely fulfills the physicochemical requirements for coating inertness resulting in increased biocompatibility needed for in situ biomedical applications, we next focused on developing a procedure for coating GO with CA and PEG. As pictured in Scheme 1, the bottom-up workflow of our protocol for functionalizing GO can be divided into three major steps. First, graphite is chemically exfoliated and oxidized using a modified Hummers and Offeman method to produce GO (Figure S3A,B).36 Next, the hydroxyl and epoxide groups on the GO lattice are covalently functionalized with CA in an anhydrous solvent. Finally, the heterobifunctional NHS–PEG–COOH cross-linker is covalently coupled to the CA primary amine groups on the GO surface. We note here that the carboxyl functional group can be tailored to any other chemical bioconjugation strategy.
Following this workflow, we first prepared exfoliated GO and assessed dimension, layer thickness, and degree of oxidation via SEM, AFM, and Raman spectroscopy, respectively. AFM topography measurements (Figure 2A) revealed that the GO flakes mostly consisted of monolayers and few multilayers (Figure 2B,C), with an average GO layer thickness of 7 ± 4 (SD) nm, equivalent to 3–4 GO monolayers. The average surface area of the exfoliated GO flakes (Figure 2D: H2O) was 320 ± 60 μm2 (SEM), and they displayed a high surface-area-to-thickness ratio of ∼44,000. The surface-area-to-thickness ratio, a more appropriate parameter for thin-film and 2D materials than surface-to-volume ratio, is an important quality factor for achieving a high molecule/drug density at small carrier sizes and increased stability in nanocomposites.14 Raman spectroscopy (Figure S3C) revealed the characteristic spectral changes previously reported that are associated with successful oxidation of the graphite precursor. The GO flakes showed a slight shift of the G peak (graphite: 1580 ± 0.48 cm–1; GO: 1598 ± 0.57 cm–1), attributable to in-plane vibrations of sp2-bonded carbon atoms, as well as the appearance of a second-order vibrational D peak (GO: 2935 ± 0.52 cm–1), caused by defects in the hexagonal carbon lattice originating from existing oxidative functional groups.40,47 Due to the increased amount of disorder in oxidized GO in comparison to graphite, both peaks also became wider as has previously been observed for graphite.48
Figure 2.
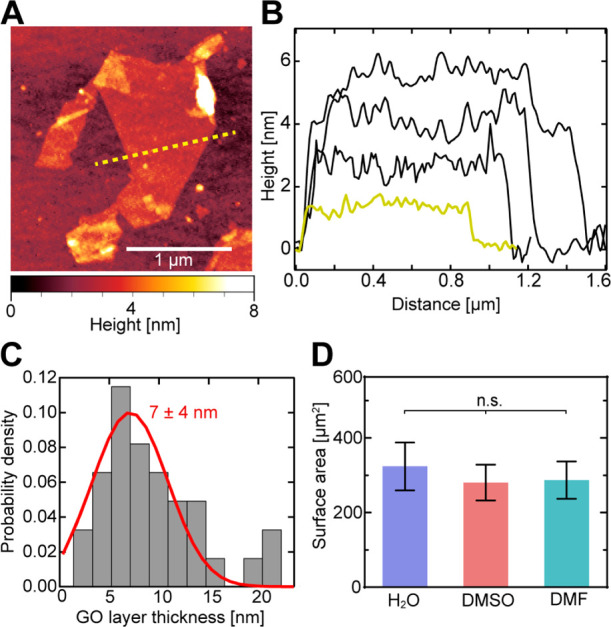
Dimensions and layer thickness of exfoliated GO flakes. (A) Example AFM topography of a single-layer GO flake with a minimum thickness of 1.4 ± 0.2 nm (the yellow dashed line indicates an example cross-section). (B) Example thickness profiles of four GO flakes [the yellow line corresponds to the example cross-section in (A)], showing GO flakes with thicknesses of 1–4 monolayers. (C) Measured GO flake thickness distribution. GO samples exhibit an average thickness of 7 ± 4 nm (SD; red line depicts Gaussian fit; N = 33), corresponding to 3–4 monolayers. (D) Average total surface area (±SD; N = 100) of GO flakes stored in H2O (for 7 days), DMSO, and DMF (for 24 h each). Statistical analysis consisted of one-way ANOVA with the Tukey post-hoc test (n.s. = nonsignificant). See also Figure S3.
Since we established that the functionalization of GO with CA should occur in an aprotic polar solvent over 12 h (Figure 1) to achieve a dense monolayer, we compared the ability of the two most commonly used solvents, DMSO and dimethyl formamide (DMF), to maintain the monodispersity of GO flakes, which is another important parameter for efficient functionalization in solution. Compared to H2O, a common storage solvent that maintains GO flake monodispersity, neither solvent showed a significant difference in flake dimensions, which would increase by agglomeration after 24 h (Figure 2D), indicating that either solvent would be suitable for maintaining monodispersity during the CA-coating process.
We next performed the covalent functionalization of GO with CA in both DMSO and DMF using the same approach as for SiO2 coating. The presence of CA and the ether formation of oxygen-containing functional groups (Figure 3A) upon successful GO functionalization were measured via HR XPS. Following functionalization, the presence of CA on the GO surface was detected by the emergence of a new peak for C–N bonds (286.17 eV) in the carbon HR-XPS spectrum (DMSO: Figure 3B; DMF: Figure S4A), together with a significant relative increase in N–C bond content (401.57 eV; Figure 3E) in the nitrogen HR-XPS spectrum (DMSO: Figure 3C; DMF: Figure S4B). The existing N–C and N–N (399.55 eV) bonds prior to GO functionalization most likely originated from residual contamination in the untreated GO sample and the fixation tape, respectively. Upon treatment with CA, the C–O–C peak (286.50 eV) notably increased in the oxygen HR-XPS spectrum (Figure 3E; DMSO: Figure 3D; DMF: Figure S4C), indicating successful ether formation between CA and GO surface hydroxyl and epoxide functional groups. The changes in N–C and C–O–C bond content upon CA coating (Figure 3E) were approximately the same in both solvents ([N–C]CA/[N–C]control ∼ 5; [C–O–C]CA/[C–O–C]control ∼ 2).
Figure 3.

XPS analysis of CA-functionalized GO. (A) Schematic of covalent binding of CA to surface hydroxyl and epoxide groups. (B) Carbon, (C) nitrogen, and (D) oxygen HR-XPS spectra before (top panels) and after (bottom panels) addition of CA in DMSO. (E) Relative atomic amounts of oxygen and nitrogen extracted from the multipeak fits to the HR-XPS spectra in (C,D) and Figure S3A-C. Statistical analysis was performed using a two-tailed, unpaired t-test (p: *** ≤ 0.001). See also Figure S4.
After successful covalent CA coating of GO flakes, we functionalized the substrates with biocompatible, heterobifunctional NHS–PEG–COOH using the same process as for SiO2 substrates. The ability of the CA–PEG coating on the GO substrates to allow biosensing of a specific biomolecule was tested next by preparing a DNA biosensor assay with our functionalized GO–CA–PEG substrates in comparison to GO–APTES–PEG samples.49−52 Commonly, such biosensor assays with electrically conductive transducers like GO are performed without labels by sensing changes in conductivity upon biomolecule binding to its specific receptor. However, coating coverage and local biomolecular interactions cannot be evaluated with such approaches. In contrast, fluorescence microscopy with single-molecule sensitivity can qualitatively and quantitatively evaluate multiple material characteristics, including PEG coverage, coupling of primary amino-labeled ssDNA to the heterobifunctional PEG, and the degree of nonspecific biomolecule adhesion.
To this end, nonfluorescent, primary amino-labeled 40-nt ssDNA (receptor) was covalently coupled to the GO–CA–PEG and GO–APTES–PEG coatings to the freely accessible carboxylic group of heterobifunctional PEG via peptide binding using EDC as a catalyzer.21 We then allowed complementary (target) Atto647N-labeled 40-nt ssDNA to hybridize with the receptor DNA on the PEGylated GO flakes during a 30 min incubation (Figure 4A) and assessed the coverage of receptor DNA immobilization on PEG after washing. False-color fluorescence microscopy of the functionalized GO–CA samples (Figure 4B) exemplified the successful PEGylation and receptor DNA immobilization with a dense coating across the entire GO lattice. Complementary photobleaching experiments (Figure 4B) of this sample revealed spatially uniform bleaching of target DNA-coupled fluorophores (Figure 4B, t = 6 min). Both the CA- and APTES-functionalized GO samples showed a comparable 2-fold decrease in overall intensity after 6 min of continuous fluorescence excitation (Figure 4C). This observation confirms that the fluorescence emission signal originated mainly from the fluorescently labeled target ssDNA and not from the autofluorescence of GO (Figure S2B). The slight irregularities in the coating layers commonly originate from defects in GO material integrity or the folding of differently sized GO sheets at the edges, as previously described.53
Figure 4.
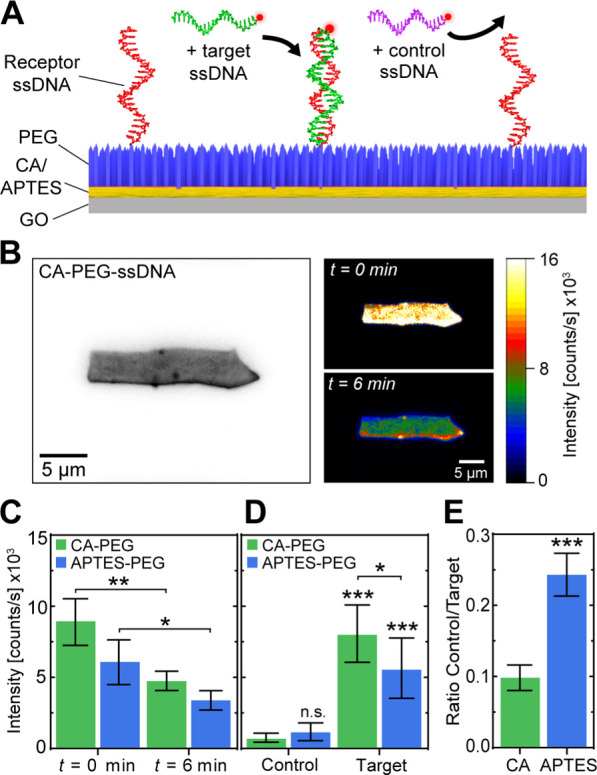
CA–PEG–functionalized GO detects target ssDNA in a biosensing assay more specific than APTES–PEG. (A) Schematic of the ssDNA detection assay on PEG–functionalized GO flakes. Noncomplementary 40-nt control ssDNA (purple) and complementary 40-nt target ssDNA (green) were fluorescently labeled with Atto647N for quantitative fluorescence detection. (B) Example images of (left) bright-field microscopy of a GO flake covalently functionalized with CA, PEG, and 40-nt receptor ssDNA and (right) fluorescence microscopy of the same GO flake after addition of complementary, Atto647N-labeled 40-nt target ssDNA after 0 min and 6 min of continuous fluorescence excitation. (C) Average fluorescence intensity (±SD) of GO samples (N = 3 each) coated with APTES–PEG–receptor ssDNA or CA–PEG–receptor ssDNA and with hybridized target ssDNA after 0 min and 6 min of photobleaching. (D) Average fluorescence intensity (±SD) of APTES–PEG–receptor ssDNA or CA–PEG–receptor ssDNA (N = 9 each) after the addition of 40-nt control ssDNA or complementary 40-nt target ssDNA. (E) Ratio of fluorescence intensities between control and target 40-nt ssDNA deposition resulting from (D). Statistical analyses were performed using a two-tailed, unpaired t-test (p: * ≤ 0.05; ** ≤ 0.01; *** ≤ 0.001; n.s. = nonsignificant).
Next, we replaced the complementary target ssDNA with noncomplementary control ssDNA to assess the nonspecific adhesion-suppressing efficacy of the functionalized CA–PEG and APTES–PEG coatings. Here, we added a high concentration (1 μM) of noncomplementary Atto647N-labeled 40-nt ssDNA to receptor ssDNA-functionalized GO–CA–PEG and GO–APTES–PEG flakes (Figure 4A) for an extended time, 1 h, to increase the probability of nonspecific ssDNA physisorption. After extensive washing with the physiological buffer, we observed a 10-fold lower fluorescence intensity compared to the hybridized target ssDNA on functionalized GO–CA–PEG samples (Figure 4D). In comparison, the functionalized GO–APTES–PEG samples exhibited only a 5-fold decrease in the fluorescence intensity. While these results demonstrate that the PEG coating was able to efficiently suppress nonspecific ssDNA adhesion in the case of both surface cross-linkers, nonspecific ssDNA adhesion was more than 2-fold lower in the case of GO–CA–PEG than for GO–APTES–PEG (Figure 4E). This observation indicates that the use of CA over APTES as a GO surface cross-linker results in a significantly increased biosensing sensitivity and specificity due to reduced unspecific adhesion of biomolecules.
Conclusions
Our results demonstrate dense covalent functionalization of the entire 2D GO lattice with biocompatible PEG and ssDNA using a multistep functionalization methodology. We found that CA was a superior surface cross-linker compared to traditional alkoxysilane (APTES) functionalization as it resulted in a denser and more homogeneous monolayer coating. The addition of covalently attached PEG to the CA coating strongly suppressed nonspecific biomolecule adhesion, which are prerequisites for its use in biomedical applications. In our method, heterobifunctional PEG cross-linkers further expand the method to other covalent coupling strategies by providing a second, variable functional group at the PEG terminus. Our proof-of-concept DNA-biosensing assay exhibited that our coating method increases the detection signal-to-noise ratio of the GO-based biosensors, facilitating the development of GO biosensor platforms for early disease detection.
This multistep functionalization methodology enhances the chemical robustness, coating coverage, and reliability of both GO-based biosensors and nanocomposites which may notably improve the applicability and performance of current and future designs. Importantly, the high PEG coverage achieved with this method may increase the biocompatibility of GO composites, therefore facilitating the use of GO in future in vivo biomedical applications, ranging from drug delivery and tissue printing to electrochemical measurements within cells and living organisms. The provided method using solely wet chemistry approaches further avoids the use of very specialized equipment, rendering this methodology accessible for a large range of laboratories.
Glossary
Abbreviations
- GO
graphene oxide
- PEG
poly(ethylene glycol)
- XPS
X-ray photoelectron spectroscopy
- CA
colamine
- APTES
(3-aminopropyl)triethoxysilane
- EDC
N-(3-dimethyl-aminopropyl-N′-ethyl carbodiimide)
- SEM
scanning electron microscopy
- DMSO
dimethyl sulfoxide
- DMF
dimethylformamide
- NHS
N-hydroxy-succinimide
- QCM(-D)
quartz crystal microbalance
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsanm.1c01522.
QCM measurements of APTES and CA coupling to SiO2 substrates over 12 h; autofluorescence of GO flakes; characterization of GO flakes with SEM and Raman spectroscopy; and XPS spectra for functionalized GO samples in the DMF solvent (PDF)
This work was supported by the Netherlands Organization for Scientific Research (NWO/OCW) as part of the Frontiers of Nanoscience program (2015SYN08) and the Brazilian funding agencies FAPESP (2010/18107-8, 2010/51748-7, 2012/51580-4, 2013/02300-1, and 2015/16611-4) and CNPq (479486/2012-3 and 429326/2018-1).
The authors declare no competing financial interest.
Supplementary Material
References
- Singh D. P.; Herrera C. E.; Singh B.; Singh S.; Singh R. K.; Kumar R. Graphene Oxide: An Efficient Material and Recent Approach for Biotechnological and Biomedical Applications. Mater. Sci. Eng., C 2018, 86, 173–197. 10.1016/j.msec.2018.01.004. [DOI] [PubMed] [Google Scholar]
- Novoselov K. S.; Geim A. K.; Morozov S. V.; Jiang D.; Zhang Y.; Dubonos S. V.; Grigorieva I. V.; Firsov A. A. Electric Field Effect in Atomically Thin Carbon Films. Science 2004, 306, 666–669. 10.1126/science.1102896. [DOI] [PubMed] [Google Scholar]
- Fang Y.; Wang E. Electrochemical Biosensors on Platforms of Graphene. Chem. Commun. 2013, 49, 9526. 10.1039/c3cc44735a. [DOI] [PubMed] [Google Scholar]
- Ferrand A.; Siaj M.; Claverie J. P. Graphene, the Swiss Army Knife of Nanomaterials Science. ACS Appl. Nano Mater. 2020, 3, 7305–7313. 10.1021/acsanm.0c02055. [DOI] [Google Scholar]
- Bollella P.; Fusco G.; Tortolini C.; Sanzò G.; Favero G.; Gorton L.; Antiochia R. Beyond Graphene: Electrochemical Sensors and Biosensors for Biomarkers Detection. Biosens. Bioelectron. 2017, 89, 152–166. 10.1016/j.bios.2016.03.068. [DOI] [PubMed] [Google Scholar]
- Pattnaik S.; Swain K.; Lin Z. Graphene and Graphene-Based Nanocomposites: Biomedical Applications and Biosafety. J. Mater. Chem. B 2016, 4, 7813–7831. 10.1039/C6TB02086K. [DOI] [PubMed] [Google Scholar]
- de Sousa M.; Visani de Luna L. A.; Fonseca L. C.; Giorgio S.; Alves O. L. Folic-Acid-Functionalized Graphene Oxide Nanocarrier: Synthetic Approaches, Characterization, Drug Delivery Study, and Antitumor Screening. ACS Appl. Nano Mater. 2018, 1, 922–932. 10.1021/acsanm.7b00324. [DOI] [Google Scholar]
- Zhang C.; Wang L.; Zhai T.; Wang X.; Dan Y.; Turng L.-S. The Surface Grafting of Graphene Oxide with Poly(Ethylene Glycol) as a Reinforcement for Poly(Lactic Acid) Nanocomposite Scaffolds for Potential Tissue Engineering Applications. J. Mech. Behav. Biomed. Mater. 2016, 53, 403–413. 10.1016/j.jmbbm.2015.08.043. [DOI] [PubMed] [Google Scholar]
- Masoudipour E.; Kashanian S.; Maleki N.; Karamyan A.; Omidfar K. A Novel Intracellular PH-Responsive Formulation for FTY720 Based on PEGylated Graphene Oxide Nano-Sheets. Drug Dev. Ind. Pharm. 2018, 44, 99–108. 10.1080/03639045.2017.1386194. [DOI] [PubMed] [Google Scholar]
- Yin F.; Hu K.; Chen Y.; Yu M.; Wang D.; Wang Q.; Yong K.-T.; Lu F.; Liang Y.; Li Z. SiRNA Delivery with PEGylated Graphene Oxide Nanosheets for Combined Photothermal and Genetherapy for Pancreatic Cancer. Theranostics 2017, 7, 1133–1148. 10.7150/thno.17841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza W.; Krupanidhi S. B. Engineering Defects in Graphene Oxide for Selective Ammonia and Enzyme-Free Glucose Sensing and Excellent Catalytic Performance for para-Nitrophenol Reduction. ACS Appl. Mater. Interfaces 2018, 10, 25285–25294. 10.1021/acsami.8b05162. [DOI] [PubMed] [Google Scholar]
- Díez-Pascual A. M.; Díez-Vicente A. L. Poly(Propylene Fumarate)/Polyethylene Glycol-Modified Graphene Oxide Nanocomposites for Tissue Engineering. ACS Appl. Mater. Interfaces 2016, 8, 17902–17914. 10.1021/acsami.6b05635. [DOI] [PubMed] [Google Scholar]
- Smith A. J.; Kelly N. L.; Figiel Ł.; Wan C.; Hanna J. V.; Farris S.; McNally T. Graphene Oxide Functionalized with 2-Ureido-4[1H]-pyrimidinone for Production of Nacre-Like Films. ACS Appl. Nano Mater. 2020, 3, 7161–7171. 10.1021/acsanm.0c01488. [DOI] [Google Scholar]
- Peña-Bahamonde J.; Nguyen H. N.; Fanourakis S. K.; Rodrigues D. F. Recent Advances in Graphene-Based Biosensor Technology with Applications in Life Sciences. J. Nanobiotechnol. 2018, 16, 75. 10.1186/s12951-018-0400-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicu L.; Leïchlé T. Biosensors and tools for surface functionalization from the macro- to the nanoscale: The way forward. J. Appl. Phys. 2008, 104, 111101. 10.1063/1.2973147. [DOI] [Google Scholar]
- Park J.-H.; Von Maltzahn G.; Zhang L.; Derfus A. M.; Simberg D.; Harris T. J.; Ruoslahti E.; Bhatia S. N.; Sailor M. J. Systematic Surface Engineering of Magnetic Nanoworms for in Vivo Tumor Targeting. Small 2009, 5, 694–700. 10.1002/smll.200801789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek I.; Skoda M.; Jarosz A.; Szukiewicz D. The Molecular Influence of Graphene and Graphene Oxide on the Immune System Under In Vitro and In Vivo Conditions. Arch. Immunol. Ther. Exp. 2016, 64, 195–215. 10.1007/s00005-015-0369-3. [DOI] [PubMed] [Google Scholar]
- Luo N.; Weber J. K.; Wang S.; Luan B.; Yue H.; Xi X.; Du J.; Yang Z.; Wei W.; Zhou R.; Ma G. PEGylated Graphene Oxide Elicits Strong Immunological Responses despite Surface Passivation. Nat. Commun. 2017, 8, 14537. 10.1038/ncomms14537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y.; Wang W.; Li Z.; Wang Y.; Zhang L.; Huang X.; Cai P. Comparative Proteomic Analysis Reveals Cytotoxicity Induced by Graphene Oxide Exposure in A549 Cells. J. Appl. Toxicol. 2021, 41, 1103–1114. 10.1002/jat.4096. [DOI] [PubMed] [Google Scholar]
- Han J.-L.; Xia X.; Tao Y.; Yun H.; Hou Y.-N.; Zhao C.-W.; Luo Q.; Cheng H.-Y.; Wang A.-J. Shielding Membrane Surface Carboxyl Groups by Covalent-Binding Graphene Oxide to Improve Anti-Fouling Property and the Simultaneous Promotion of Flux. Water Res. 2016, 102, 619–628. 10.1016/j.watres.2016.06.032. [DOI] [PubMed] [Google Scholar]
- Janissen R.; Sahoo P. K.; Santos C. A.; da Silva A. M.; von Zuben A. A. G.; Souto D. E. P.; Costa A. D. T.; Celedon P.; Zanchin N. I. T.; Almeida D. B.; Oliveira D. S.; Kubota L. T.; Cesar C. L.; de Souza A. P.; Cotta M. A. InP Nanowire Biosensor with Tailored Biofunctionalization: Ultrasensitive and Highly Selective Disease Biomarker Detection. Nano Lett. 2017, 17, 5938–5949. 10.1021/acs.nanolett.7b01803. [DOI] [PubMed] [Google Scholar]
- Seabra A. B.; Paula A. J.; de Lima R.; Alves O. L.; Durán N. Nanotoxicity of Graphene and Graphene Oxide. Chem. Res. Toxicol. 2014, 27, 159–168. 10.1021/tx400385x. [DOI] [PubMed] [Google Scholar]
- Pei X.; Zhu Z.; Gan Z.; Chen J.; Zhang X.; Cheng X.; Wan Q.; Wang J. PEGylated Nano-Graphene Oxide as a Nanocarrier for Delivering Mixed Anticancer Drugs to Improve Anticancer Activity. Sci. Rep. 2020, 10, 2717. 10.1038/s41598-020-59624-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saifullah B.; Buskaran K.; Shaikh R.; Barahuie F.; Fakurazi S.; Mohd Moklas M.; Hussein M. Graphene Oxide-PEG-Protocatechuic Acid Nanocomposite Formulation with Improved Anticancer Properties. Nanomaterials 2018, 8, 820. 10.3390/nano8100820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidram E.; Sulistio A.; Cho H.-J.; Amini A.; Harris T.; Zarrabi A.; Qiao G.; Stewart A.; Dunstan D. E. Targeted Graphene Oxide Networks: Cytotoxicity and Synergy with Anticancer Agents. ACS Appl. Mater. Interfaces 2018, 10, 43523–43532. 10.1021/acsami.8b17531. [DOI] [PubMed] [Google Scholar]
- Pal S.; Chakraborty M.; Sarkar N. Graphene Oxide Functionalized with 5-Aminophenanthroline for Selective Detection of Adenine through Fluorescence “Turn-Off–On” Response. ACS Appl. Nano Mater. 2020, 3, 3532–3539. 10.1021/acsanm.0c00212. [DOI] [Google Scholar]
- Xiong K.; Fan Q.; Wu T.; Shi H.; Chen L.; Yan M. Enhanced Bovine Serum Albumin Absorption on the N-Hydroxysuccinimide Activated Graphene Oxide and Its Corresponding Cell Affinity. Mater. Sci. Eng., C 2017, 81, 386–392. 10.1016/j.msec.2017.08.044. [DOI] [PubMed] [Google Scholar]
- Konnerth R.; Cervetti C.; Narita A.; Feng X.; Müllen K.; Hoyer A.; Burghard M.; Kern K.; Dressel M.; Bogani L. Tuning the Deposition of Molecular Graphene Nanoribbons by Surface Functionalization. Nanoscale 2015, 7, 12807–12811. 10.1039/C4NR07378A. [DOI] [PubMed] [Google Scholar]
- Metwalli E.; Haines D.; Becker O.; Conzone S.; Pantano C. G. Surface Characterizations of Mono-, Di-, and Tri-Aminosilane Treated Glass Substrates. J. Colloid Interface Sci. 2006, 298, 825–831. 10.1016/j.jcis.2006.03.045. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Sautter K.; Larsen A. M.; Findley D. A.; Davis R. C.; Samha H.; Linford M. R. Chemical Vapor Deposition of Three Aminosilanes on Silicon Dioxide: Surface Characterization, Stability, Effects of Silane Concentration, and Cyanine Dye Adsorption. Langmuir 2010, 26, 14648–14654. 10.1021/la.102447y. [DOI] [PubMed] [Google Scholar]
- Naz T.; Afzal A.; Siddiqi H. M.; Akhtar J.; Habib A.; Banski M.; Podhorodecki A. 2-Aminoethanol-Mediated Wet Chemical Synthesis of ZnO Nanostructures. Appl. Nanosci. 2015, 5, 425–433. 10.1007/s13204-014-0334-1. [DOI] [Google Scholar]
- Ebner A.; Hinterdorfer P.; Gruber H. J. Comparison of Different Aminofunctionalization Strategies for Attachment of Single Antibodies to AFM Cantilevers. Ultramicroscopy 2007, 107, 922–927. 10.1016/j.ultramic.2007.02.035. [DOI] [PubMed] [Google Scholar]
- Janissen R.; Oberbarnscheidt L.; Oesterhelt F. Optimized Straight Forward Procedure for Covalent Surface Immobilization of Different Biomolecules for Single Molecule Applications. Colloids Surf., B 2009, 71, 200–207. 10.1016/j.colsurfb.2009.02.011. [DOI] [PubMed] [Google Scholar]
- Stewart C.; Akhavan B.; Wise S. G.; Bilek M. M. M. A Review of Biomimetic Surface Functionalization for Bone-Integrating Orthopedic Implants: Mechanisms, Current Approaches, and Future Directions. Prog. Mater. Sci. 2019, 106, 100588. 10.1016/j.pmatsci.2019.100588. [DOI] [Google Scholar]
- Corobea M. S.; Albu M. G.; Ion R.; Cimpean A.; Miculescu F.; Antoniac I. V.; Raditoiu V.; Sirbu I.; Stoenescu M.; Voicu S. I.; Ghica M. V. Modification of Titanium Surface with Collagen and Doxycycline as a New Approach in Dental Implants. J. Adhes. Sci. Technol. 2015, 29, 2537–2550. 10.1080/01694243.2015.1073661. [DOI] [Google Scholar]
- Hummers W. S. Jr; Offeman R. E. Preparation of Graphitic Oxide. J. Am. Chem. Soc. 1958, 80, 1339. 10.1021/ja01539a017. [DOI] [Google Scholar]
- Nečas D.; Klapetek P. Gwyddion: An Open-Source Software for SPM Data Analysis. Open Phys. 2012, 10, 181. 10.2478/s11534-011-0096-2. [DOI] [Google Scholar]
- Janissen R.; Oesterhelt F. Directed Deposition of Single Molecules on Surfaces. J. Nanosci. Nanotechnol. 2010, 10, 5328–5332. 10.1166/jnn.2010.2382. [DOI] [PubMed] [Google Scholar]
- Reviakine I.; Johannsmann D.; Richter R. P. Hearing What You Cannot See and Visualizing What You Hear: Interpreting Quartz Crystal Microbalance Data from Solvated Interfaces. Anal. Chem. 2011, 83, 8838–8848. 10.1021/ac201778h. [DOI] [PubMed] [Google Scholar]
- Zheng B.; Gu G. X. Prediction of Graphene Oxide Functionalization Using Gradient Boosting: Implications for Material Chemical Composition Identification. ACS Appl. Nano Mater. 2021, 4, 3167–3174. 10.1021/acsanm.1c00384. [DOI] [Google Scholar]
- Zhao Y. H.; Abraham M. H.; Zissimos A. M. Fast Calculation of van Der Waals Volume as a Sum of Atomic and Bond Contributions and Its Application to Drug Compounds. J. Org. Chem. 2003, 68, 7368–7373. 10.1021/jo034808o. [DOI] [PubMed] [Google Scholar]
- Bhushan B.; Kwak K. J.; Gupta S.; Lee S. C. Nanoscale Adhesion, Friction and Wear Studies of Biomolecules on Silane Polymer-Coated Silica and Alumina-Based Surfaces. J. R. Soc., Interface 2009, 6, 719–733. 10.1098/rsif.2008.0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg E. T.; Bertilsson L.; Liedberg B.; Uvdal K.; Erlandsson R.; Elwing H.; Lundström I. Structure of 3-Aminopropyl Triethoxy Silane on Silicon Oxide. J. Colloid Interface Sci. 1991, 147, 103–118. 10.1016/0021-9797(91)90139-Y. [DOI] [Google Scholar]
- Wang D.; Jones F. R. Surface analytical study of the interaction between ?-amino propyl triethoxysilane and E-glass surface. J. Mater. Sci. 1993, 28, 2481–2488. 10.1007/BF01151683. [DOI] [Google Scholar]
- Shang J.; Ma L.; Li J.; Ai W.; Yu T.; Gurzadyan G. G. The Origin of Fluorescence from Graphene Oxide. Sci. Rep. 2012, 2, 792. 10.1038/srep00792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern E.; Wagner R.; Sigworth F. J.; Breaker R.; Fahmy T. M.; Reed M. A. Importance of the Debye Screening Length on Nanowire Field Effect Transistor Sensors. Nano Lett. 2007, 7, 3405–3409. 10.1021/nl071792z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski S.; Wierzbicki M.; Sawosz E.; Jung A.; Gielerak G.; Biernat J.; Jaremek H.; Łojkowski W.; Woźniak B.; Wojnarowicz J.; Stobinski L.; Malolepszy A.; Mazurkiewicz-Pawlicka M.; Lojkowski M.; Kurantowicz N.; Chwalibog A. Graphene Oxide-Based Nanocomposites Decorated with Silver Nanoparticles as an Antibacterial Agent. Nanoscale Res. Lett. 2018, 13, 116. 10.1186/s11671-018-2533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. J.; Lee S.-M.; Oh Y.-S.; Yang Y.-H.; Lim Y. S.; Yoon D. H.; Lee C.; Kim J.-Y.; Ruoff R. S. Unoxidized Graphene/Alumina Nanocomposite: Fracture- and Wear-Resistance Effects of Graphene on Alumina Matrix. Sci. Rep. 2015, 4, 5176. 10.1038/srep05176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Mendiola T.; Gutiérrez-Sánchez C.; Gibaja C.; Torres I.; Busó-Rogero C.; Pariente F.; Solera J.; Razavifar Z.; Palacios J. J.; Zamora F.; Lorenzo E. Functionalization of a Few-Layer Antimonene with Oligonucleotides for DNA Sensing. ACS Appl. Nano Mater. 2020, 3, 3625–3633. 10.1021/acsanm.0c00335. [DOI] [Google Scholar]
- Daǧcı Kıranşan K.; Topç̧u E. Conducting Polymer-Reduced Graphene Oxide Sponge Electrode for Electrochemical Detection Based on DNA Hybridization. ACS Appl. Nano Mater. 2020, 3, 5449–5462. 10.1021/acsanm.0c00782. [DOI] [Google Scholar]
- Ye Y.; Xie J.; Ye Y.; Cao X.; Zheng H.; Xu X.; Zhang Q. A Label-Free Electrochemical DNA Biosensor Based on Thionine Functionalized Reduced Graphene Oxide. Carbon 2018, 129, 730–737. 10.1016/j.carbon.2017.12.060. [DOI] [Google Scholar]
- Moreau A. L. D.; Janissen R.; Santos C. A.; Peroni L. A.; Stach-Machado D. R.; de Souza A. A.; de Souza A. P.; Cotta M. A. Highly-Sensitive and Label-Free Indium Phosphide Biosensor for Early Phytopathogen Diagnosis. Biosens. Bioelectron. 2012, 36, 62–68. 10.1016/j.bios.2012.03.038. [DOI] [PubMed] [Google Scholar]
- Dreyer D. R.; Park S.; Bielawski C. W.; Ruoff R. S. The Chemistry of Graphene Oxide. Chem. Soc. Rev. 2010, 39, 228–240. 10.1039/B917103G. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.