

Abstract
Dynamics of development can be followed by confocal time-lapse microscopy of live transgenic zebrafish embryos expressing fluorescence in specific tissues or cells. A difficulty with imaging whole embryo development is that zebrafish embryos grow substantially in length. When mounted as regularly done in 0.3–1% low melt agarose, the agarose imposes growth restriction, leading to distortions in the soft embryo body. Yet, to perform confocal time-lapse microscopy, the embryo must be immobilized. This article describes a layered mounting method for zebrafish embryos that restrict the motility of the embryos while allowing for the unrestricted growth. The mounting is performed in layers of agarose at different concentrations. To demonstrate the usability of this method, whole embryo vascular, neuronal and muscle development was imaged in transgenic fish for 55 consecutive hours. This mounting method can be used for easy, low-cost imaging of whole zebrafish embryos using inverted microscopes without requirements of molds or special equipment.
Keywords: Developmental Biology, Issue 155, zebrafish, embryo, confocal microscopy, time-lapse, mounting, transgenic
Introduction
The zebrafish has long been a model organism for developmental biology, and microscopy is the major method to visualize embryonic development. The advantages of using zebrafish embryos for developmental studies include small size, optical clarity, rapid development, and high fecundity of the adult fish. The generation of transgenic zebrafish lines expressing fluorescence in certain tissues or cells have allowed for a direct visualization of tissue development in ways not possible with larger vertebrate animals. In combination with time-lapse microscopy, details and dynamics of the tissue development can be readily studied.
A difficulty with imaging zebrafish development is that the embryos grow substantially in length; the embryo extends its length four times within the first 3 days of life1. Also, the body of the early embryo is soft, and easily becomes distorted if growth is restricted. Yet, to perform confocal microscopy, the embryo must be immobilized. To keep embryos in a fixed position for confocal time-lapse imaging, they are regularly anesthetized and mounted in 0.3–1% low-melt agarose. This has the advantage of allowing for some growth during imaging for a certain period of time, while restricting movements of the embryo. Parts of the embryo can efficiently be imaged like this. However, when using this method for imaging of the whole embryo for extended time periods, distortions are observed because of restricted growth caused by the agarose. Thus, other mounting methods are required. Kaufmann and colleagues have described an alternative mounting of zebrafish embryos for light sheet microscopy, such as selective plane illumination microscopy (SPIM), by mounting the embryos in fluorinated ethylene propylene (FEP) tubes containing low concentrations of agarose or methylcellulose2. This technique produces a superb visualization of embryogenesis over time. Schmid et al. describe mounting of up to six embryos in agarose in FEB tubes for light-sheet microscopy3 providing visualization of several embryos in one imaging session. Molds have been used to create embryo arrays for efficient mounting of larger numbers of embryos4. Masselkink et al. have constructed 3D printed plastic molds that can be used to make silicon casts that zebrafish embryos at different stages can be placed in, enabling mounting in a constant position for imaging, including confocal imaging5. 3D printing has also been used to make molds for consistent positioning of zebrafish embryos in 96-well format6. Some molds are customized for certain stages and may not permit unrestricted growth for long time periods, whereas other molds are more flexible. Recently, Weijts et al. published the design and fabrication of a four-well dish for live imaging of zebrafish embryos7. In this dish, the tail and trunk of anesthetized fish embryos are placed manually under a clear silicone roof attached just above a cover glass to form a pocket. The embryo is then fixed in this position by the addition of 0.4% agarose. This mounting allows for the imaging of the about 2 mm long posterior part (trunk and tail) of the embryo, and as up to 12 embryos can be mounted per well, the method allows for the imaging of multiple samples. Similarly, Hirsinger and Steventon recently presented a method where the head of the fish is mounted in agarose, while the tail can freely grow, and this method also efficiently facilitates imaging of the trunk and tail region of the embryo8.
This article describes a layered mounting method for zebrafish embryos that restrict the movements of the embryos while allowing for unrestricted growth. The advantages of this mounting method are that it is a low-cost, fast and easy method to mount embryos of various stages for imaging using any inverted microscope. The mounting permits long-term imaging of the whole body (head, trunk and tail) during embryo development. To showcase the usability of this method, whole embryo vascular, neuronal and muscle development was imaged in transgenic fish. Two embryos per session, at two wavelengths in 3D were imaged by time-lapse microscopy for 55 consecutive hours to render movies of tissue development.
Protocol
The animal work presented here was approved by the Institutional Animal Care and Use Committees (IACUCs) of the University of Houston and Indiana University.
1. Fish husbandry
NOTE: Work with vertebrate models requires an IACUC approved protocol. It should be conducted according to relevant national and international guidelines.
Maintain adult zebrafish as described in previously published literature9.
In the afternoon, place adult zebrafish in breeding tanks. Breed males to females at a ratio of 1:2.
2. Preparation of solutions
Make a stock solution of 1% low melt agarose in embryo media (E3: 5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4, adjusted to pH 7.0). Aliquot the stock solution into 1.5 mL tubes and store them at 4 °C.
-
Make a stock solution of 4% (w/v) Tricaine (MS-222) in distilled water. Store at 4 °C in a dark bottle.
CAUTION: Tricaine is toxic and should be weighed and dissolved in a fume hood.
Make a stock solution of 20 mM N-phenylthiourea (PTU) in distilled water. Store at −20 °C.
3. Preparation of embryos
-
After mating, harvest embryos in E3 in a Petri dish and incubate them at 26.5 °C for about 28 h before mounting.
NOTE: This slows down the development of the embryos so that the embryos are approximately at 30 somite stage at the beginning of imaging.
Anesthetize embryos in 0.016–0.020% Tricaine in E3. To inhibit pigmentation, add PTU to a concentration of 200 μM.
Dechorionate the embryos using forceps under a dissecting microscope. Using two forceps, grip and gently pull the chorion apart to release the embryo.
4. Mounting in agarose
NOTE: The developed mounting method requires two different concentrations of low-melt agarose in E3 with 0.02% Tricaine and PTU as needed. The first agarose solution contains an optimal concentration of agarose at which the distortion and motility are at a minimum. The optimization is described in step 5 below.
-
Heat the agarose solutions for the two layers (concentration defined in step 5 below and 1%) to 65 °C. Let the agarose cool down to approximately 30 °C just before mounting so that the embryo is not harmed by the heat. For mounting, use 35 mm glass bottom dishes with a No. 0 cover glass bottom. The cover glass attached to the bottom of the dish creates a 10 mm shallow (approx. 1.2 mm deep) well, in which the embryo is to be placed.
NOTE: In this case, the concentration with the least motility and distortions were between 0.025 to 0.040% agarose.
Gently place a dechorionated embryo with one of its lateral sides toward the bottom of the dish using a glass pipette or micropipette. If using a micropipette, cut the outer part of the tip to increase the size of the opening to fit the embryo (Figure 1A). Carefully remove any remaining E3 with a micropipette.
Add the first agarose solution to the small well created by the cover glass attached to the bottom of the dish to cover the embryo (Layer 1) (Figure 1B). Ensure that the agarose covers the small well but will not overflow it.
Cover the small well with a cover glass (22 mm x 22 mm) (Figure 1C) to create a narrow agarose filled space with the embryo between the two cover glasses.
Place a layer of 1% agarose solution on top of the cover glass all over the bottom of the dish (Layer 2) (Figure 1D). As this layer solidifies, it holds the cover glass in place.
-
Fill the remaining portion of the dish with E3 containing 0.02% Tricaine to keep the system hydrated (Layer 3) (Figure 1E).
NOTE: In this setup, the cover glass and 1% agarose protect the bottom layer from getting diluted.
Figure 1 : Description of mounting method.

(A) Add the zebrafish embryo to the small well created by the glass bottom in the 35 mm dish. (B) Add agarose layer 1 to the small well to cover the embryo. (C) Carefully place a cover glass over the small well. (D) Add agarose layer 2 on the whole bottom of the 35 mm dish. (E) Add E3 to the dish. (F) Schematic drawing of a cross section of the mounting set up. (G) Microscope image (5x objective) of the zebrafish embryo in the final montage.
5. Optimization of agarose solution for layer 1
To identify the optimal concentration of agarose for Layer 1, use a multiscale grid search approach. Mount embryos in increasing concentrations of agarose ranging from 0.01% to 1% followed by time-lapse imaging of embryo growth restriction and motility in the field of view. Identify the concentrations where both the distortion and motility are at a minimum.
To optimize the concentration of agarose further, mount the embryos using a finer range of concentrations of agarose (e.g., between 0.025 and 0.040% agarose) depending on the concentration found to be best in step 5.1 (e.g., 0.025%, 0.028%, 0.031%, etc.).
NOTE: In our laboratory, the optimal agarose concentration was around 0.03%.
6. Time-lapse imaging
NOTE: This mounting method works for any inverted microscope with time-lapse functionality for fluorescence and bright field imaging.
Perform time-lapse imaging for the whole embryo or parts of it for up to 55 h. For imaging of several embryos, use a stage adaptor that holds several dishes with one embryo mounted per dish. Rotate the dishes one by one so that the embryos are approximately horizontally positioned as determined by eye. For optimal embryo growth and development, use a microscope stage with an incubator set at 28.5 °C.
-
Select a low magnification objective in the microscope software. Under the eye piece, locate and finely align the embryos horizontally by rotating the dishes using bright field illumination and record their locations in the software. Create a filename for automatic saving of the data.
NOTE: In this experiment, a plan apo λ 4× 0.2 NA objective and the XY tab within the ND acquisition window were used to record the locations of the embryos. Data were saved in .nd2 format. The objective was selected by clicking on its icons in the Ti Pad tab.
-
Set the pinhole size, scan speed, image size, and zoom. Next, select a higher magnification objective for capturing the images.
NOTE: In this experiment, a plan-apo 10× 0.45 NA objective was used for imaging of whole embryos, and a super-fluor 20× 0.75 NA objective was used for the higher magnification imaging of parts of the embryo. The pinhole was set to 1.2 AU (19.2 μm for the 10x lens), the scan speed was set for a pixel dwell time of 2.4 μs and the image size was set to 1024 × 1024 pixels with a scan zoom of one, giving a pixel size of 1.24 μm for the 10x lens and 0.62 μm for the 20x lens.
-
Select the channels for the fluorescence to be imaged. Adjust the image capture settings one channel at a time. For each fluorescence channel adjust the laser power and the detector high voltage, making sure to collect the best dynamic range possible while avoiding the saturation and limiting photobleaching.
NOTE: In this experiment, GFP was imaged with the 488 green channel, (488 nm laser and emission between 500 and 550 nm), and RFP with the 561 red channel (561 nm laser and emission between 570 and 600 nm), and the transmission image was collected using the 561 nm laser and the transmitted light detector (TD channel).
-
To image the whole embryo with the 10x objective, image several fields of view with overlap and stitch them together using the microscope software.
NOTE: As the embryos will grow substantially in size during the imaging, make sure that there is space in the field of view anterior and posterior to the embryo. Use the Scan Large Image tab of the ND acquisition window and select a 4 × 1 pattern with 10% overlap to capture four adjacent fields of view.
-
Configure the settings for capturing the Z-stacks using the microscope software.
NOTE: Because the embryo is mounted close to the bottom of the glass dish, its center will move away from the bottom as it grows. Use the Z tab of the ND acquisition window and select the Asymmetrical relative range. With this method, the current focus plane is used as the Reference plane and the rest of the planes are asymmetrically distributed above and below to include the whole embryo within the Z-stack volume, with enough space to account for the specimen’s growth. In all experiments, the interval was set to 11 μm and the total range to about 45 planes.
-
In order to maintain the focus of multiple embryos during long-term time-lapse imaging, use an automated focus. If imaging several embryos, adjust the individual offset levels for each embryo to focus at the Reference plane.
NOTE: In this experiment, a laser-based focus stabilization system was used.
Set up time-lapse parameters in the microscope software and image for a duration of 55 h at about 1 h intervals. At each cycle, capture two embryo datasets sequentially and save data automatically after each cycle.
Process images using an on-line or off-line version of the microscope software. If more than one embryo was imaged, create independent files for each embryo by splitting the dataset based on the location of imaging. Use maximum intensity projections or a similar tool to convert 3D time data sets into 2D time data sets. Create movie files by using the Save as menu option, selecting .avi as the file format. Select the No compression option with 200 ms intervals for a playback speed of 5 frames /s.
NOTE: The original dataset of two embryos in this experiment was split using the Split locations function in the software (File | Import/Export | Split Multipoints). 3D time datasets were converted to 2D time data sets using the Maximum intensity projection function (Image | ND Processing| Create Maximum Intensity Projection Image in Z).
Representative Results
Development of the mounting method
The main aim of this work was to develop a low-cost mounting technique for time-lapse imaging of zebrafish development for extended periods of time. The layered mounting method was developed to allow for full growth of the fragile zebrafish embryo body, while restricting its movements. If the agarose concentration of layer 1 is too high, the embryos will become distorted and curved (Figure 2). Embryos grown at 0.1% and 0.5% agarose have shortened tails, distorted fins, and curved heads. On the contrary, if the agarose concentration is too low, the embryos will move out of the field of view during time-lapse microscopy, even though they are anesthetized, as the growing tail swings out from the embryo body and causes it to move. In our hands, the optimal agarose concentration varied between 0.028% and 0.034% agarose between different batches of agarose. Mounting below 0.025% agarose did not provide enough resistance for the embryo to stay in the field of view.
Figure 2: Embryo growth restriction and developmental delay in different concentrations of agarose.

Embryos were mounted in different agarose concentrations and their size and development imaged at 48 hpf. Images captured by a microscope equipped with a digital microscope color camera (2.5x objective) and the accompanying microscope software.
Extended time-lapse imaging of vascular, neuronal and muscle development
After optimizing the mounting method described above, time lapse confocal microscopy images were captured over a span of 55 h. We imaged live transgenic zebrafish expressing GFP or RFP in different tissues. An advantage of using transgenic fish with endogenous fluorescence for time lapse imaging is that the fluorescence molecules, such as GFP and RFP, are produced continuously in the live embryo, and, thus, it does not easily photo bleach. First, embryos of a cross of Tg(kdr1: EGFP)mitfab692/b69210 and Ubi-zebrabow11 were imaged. In these embryos, RFP is expressed in all cells of the embryo, which allows for visualization of the general embryo structure. GFP is expressed in the endothelial cells of the vasculature. Double transgenic embryos were imaged for 55 hours from approximately 30 somite stage to visualize vascular development in the head and body (Figure 3 and Supplementary Movie S1) using a 10x objective with 0.45 NA. Two embryos/session were imaged with z-stacks and time-lapse in a loop so that after imaging the first embryo at two different wavelengths, the second one was subsequently imaged and then the first one again. Figure 3 shows the intersegmental vessel (ISV) sprouting, development of subintestinal vessels and head vasculature, and caudal vein plexus condensation together with trunk extension. Imaging of the whole embryo with the vasculature shows that ISV sprouting starts in between somites and grows dorsally up to the point of the neural tube, where the ISVs takes a different path and sprouts in a direction over to the next anterior somite boundary.
Figure 3: Visualization of the development of vasculature.

Cross of Tg(kdr1: EGFP)mitfab692/b692 and Ubi-zebrabow imaged from about 30 somite stage for 55 h on a confocal microscope. Vasculature in green and all other cells in red. Scale bar 500 μm.
Next, embryos of a cross of Tg(mnx:GFP)mitfab692/b629 and Ubi-zebrabow were imaged for 55 hours approximately from the 30 somite stage to visualize motorneuron development (Figure 4 and Supplementary Movie S2). Tg(mnx:GFP) had first been crossed to mitfab692/b692 (both from Zebrafish International Resource Center at the University of Oregon, OR) to produce Tg(mnx:GFP)mitfab692/b629. The motorneuron axons sprout from the ventral neural tube over the somites towards the ventral side of the embryo. Unlike the intersegmental vessels that sprout in between the somites, the axons sprout over the middle over the chevron-formed somites. The sprouting starts towards the anterior end of the neural tube, in a straight angle from the neural tube, and spreads posterior. By the somite/yolk interface, sprouting changes direction to anterior and posterior sprouting. Note the innervation of the developing heart by the anterior neurites.
Figure 4: Visualization of the development of neurons and neurite sprouting.

Cross of Tg(mnx:GFP)mitfab692/b629 and Ubi-zebrabow imaged from about 30 somite stage for 55 h on a confocal microscope. Motorneurons in green, all other cells in red. Scale bar 500 μm.
Also, embryos of a cross of Tg(kdr:enl.memRFP)mitfab692/b692 (cross of Tg(kdr:enl.memRFP and mitfab692/b692) and Tg(mnx:GFP)mitfab692/b692 were imaged. In the former embryos, the red vascular fluorescence was not visible until 36 hours post fertilization (hpf), and therefore we started the imaging at a later time point at 2 dpf. We imaged a part of the trunk, dorsally to the yolk sac extension, using higher magnification (20x objective). This movie shows that the embryos lay still enough for good imaging quality at higher magnification. The co-development of the dorsal sprouting of motorneuron axons in relation to the position of intersegmental vessels was followed (Figure 5 and Supplementary Movie S3). In these movies, the finer details of ventral axon sprouting are visible, as well as dorsal axon sprouting from the neural tube to a point where neuronal axons and intersegmental vessels co-migrate. The caudal vein plexus condensation is also clearly shown. Note that as a neurite sprout is missing toward the posterior end of the tail (in the position of the 12th neurite from the right side), the closest posterior neuron extends toward the anterior part of the embryo to cover the area between neurites 11 and 13.
Figure 5: Visualization of the co-development of neurons and vasculature.

Cross of Tg(kdr:enl.memRFP)mitfab692/b692 and Tg(mnx:GFP)mitfab692/b692 imaged for 55 hours from about 2 dpf on a confocal microscope. Vasculature in red, motorneurons in green. Scale bar 500 μm.
Finally, a cross of HGn39b12,13 and Ubi-zebrabow was imaged. HGn39b is a zTRAP line with the GFP insert in the activator of heat shock protein ATPase homolog 1 (AHA1) gene (http://kawakami.lab.nig.ac.jp/) and it expresses GFP in the muscles of the somites (Figure 6 and Supplementary Movie S4). As somite numbers increase, the somites also extend in length and width. This movie also nicely shows heart development in red fluorescence from the Ubi-zebrabow fish line.
Figure 6: Visualization of muscle GFP expression in somites.
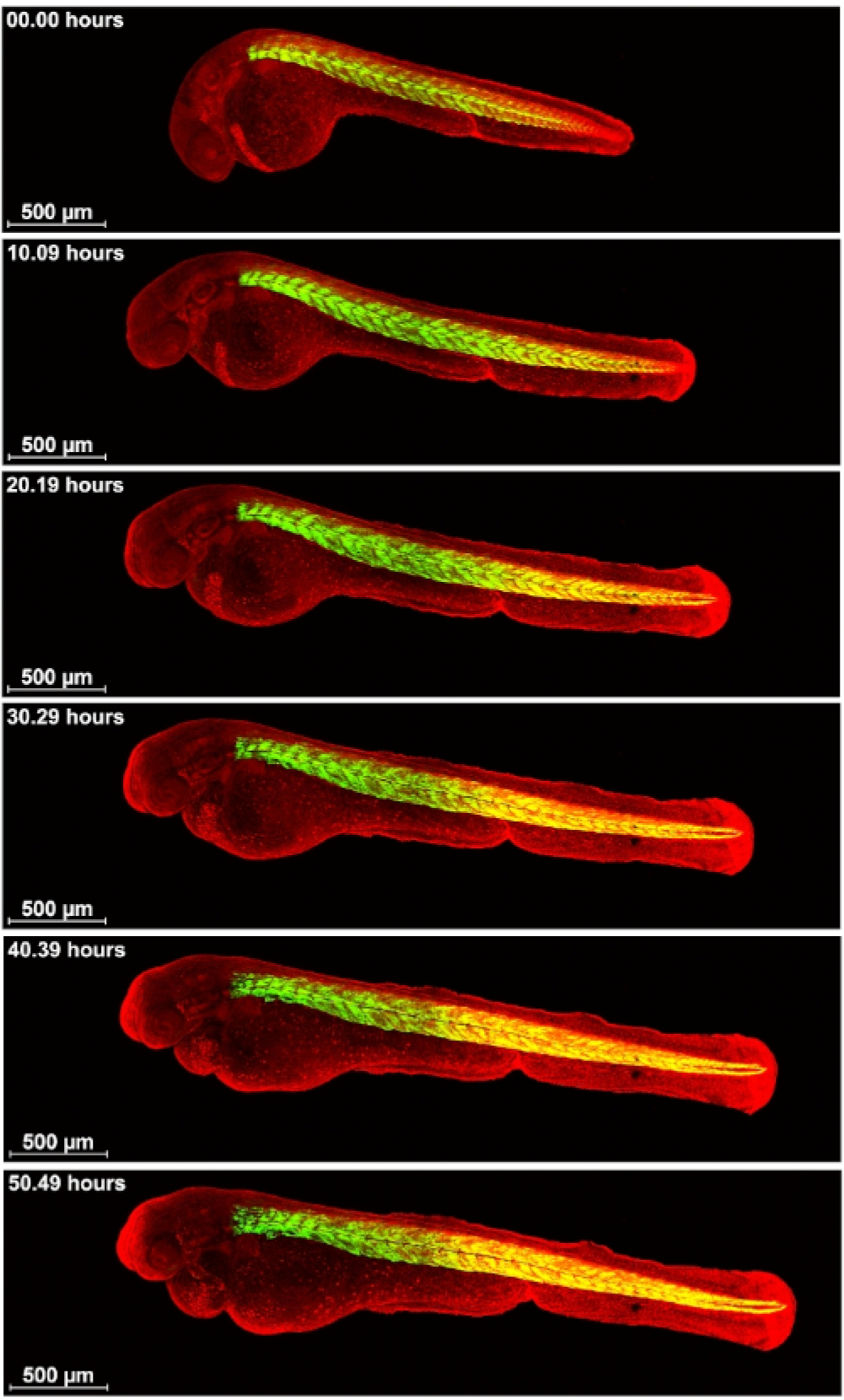
Cross of HGn39b and Ubi-zebrabow were imaged on a confocal microscope for 55 h from about 30 somite stage. Muscle in green, all other cells in red. Scale bar 500 μm
Discussion
A mounting method for extended time-lapse confocal microscopy of whole zebrafish embryos is described here. The most critical step for the mounting method is to identify the optimal concentration of agarose that will allow for unrestricted zebrafish embryo growth, and at the same time keep the embryos in a completely fixed position for confocal imaging. Because the optimal concentration of agarose is very narrow, this value is very sensitive to the errors in measurement of the weight of agarose and the volume of E3 during preparation of the solution. The optimal concentration may also depend on temperature and stage of the embryo. Thus, the optimal concentration will need to be re-defined for each new batch of agarose solution through repetition of tests of different concentrations.
Another critical step is in the mounting method is the addition of the second layer of agarose. The second layer holds the cover glass in place. It must be added carefully to the dish a little at a time so that it does not cause the cover glass to move. The second layer also serves as a permeable barrier for E3. Without E3, the embryos will dry out during the imaging. Without the second layer of agarose, the cover glass and the embryo will start floating.
A limitation of the proposed mounting method is that while it works well for inverted microscopes, it does not work for upright microscopes. Several attempts were made to perform time-lapse imaging using an upright microscope, by filling the glass bottom dish with E3, sealing it with parafilm, and turning it upside down. However, often this resulted in that the mounting collapsed halfway through the imaging. This might have been caused by increased heat in the sample after the long exposure to laser light, which causes the solidified agarose layer to melt.
By the end of the time-lapse microscopy the embryos started to show pericardial edema. Varying between different experiments, edema was observed between 35 and 50 hours of imaging. Whether this was caused by embryo immobilization, or an effect of anesthetics, is currently not known. Tricaine is known to suppress the contraction of skeletal and cardiac muscles. Consequently, Tricaine affects heart rate in adult fish14 and embryos15. Other studies have also reported that Tricaine treatment causes pericardial edema in zebrafish embryos2,16. In this article, a concentration of Tricaine (0.016–0.020%) was used that is commonly used in zebrafish research; still, the pericardial edema occurred by the end of our imaging period. The pericardial edema constituted a main restriction for enabling imaging of the embryos for even longer periods of time. Potential ways to decrease this cardiac toxicity are combining two different anesthetics, such as Tricaine with eugenol, or using α-bungarotoxin mRNA injection for anesthetics16; beneficial effects of combinatorial or alternative anesthetizing compounds need to be further investigated for extended time-lapse imaging of zebrafish development.
In conclusion, the described mounting method is fast, easy, cost-effective and works on any inverted microscope. Regular glass bottom dishes and low melting agarose can be used, and no special molds, equipment or instrumentation are required. The layered mounting method allows for embryo growth while at the same time keeping the embryos in a fixed position. By using extended time-lapse imaging of whole organism new knowledge of tissue development can be obtained.
Supplementary Material
Supplementary Movie S1: Movie of an embryo of a cross of Tg(kdr1:EGFP)mitfab692/b692 and Ubi-zebrabow imaged from about 30 somite stage for 55 h on a confocal microscope using a 10x objective.
Supplementary Movie S3: Movie of an embryo of a cross of Tg(kdr:enl.memRFP)mitfab692/b692 and Tg(mnx:GFP)mitfab692/b692 imaged from about 2 dpf for 55 h on a confocal microscope using a 20x objective.
Supplementary Movie S2: Movie of an embryo of a cross of Tg(mnx:GFP)mitfab692/b629 and Ubi-zebrabow imaged from about 30 somite stage for 55 h on a confocal microscope using a 10x objective.
Supplementary Movie S4: Movie of an embryo of a cross of HGn39b and Ubi-zebrabow imaged from about 30 somite stage for 55 h on a confocal microscope using a 10x objective.
Acknowledgments
We thank Albert Pan and Arndt Sieakmann for gifts of transgenic fish. We thank Koichi Kawakami at National Institute of Genetics, the National BioResource Project from the Ministry of Education, Culture, Sports, Science and Technology of Japan for the gift of the transgenic zebrafish HGn39b. We also thank Fatima Merchant and Kathleen Gajewski for assistance on confocal microscopy, and Tracey Theriault for photographs.
This work was supported by grants from the National Institute of Environmental Health Sciences of the National Institutes of Health (grant number P30ES023512 and contract number HHSN273201500010C). SU was supported by a fellowship from the Keck Computational Cancer Biology program (Gulf Coast Consortia CPRIT grant RP140113) and by Hugh Roy and Lillie Endowment Fund. J-Å G was supported by the Robert A. Welch Foundation (E-0004).
Footnotes
Disclosures
The authors have nothing to disclose.
Video Link
The video component of this article can be found at https://www.jove.com/video/60321/
References
- 1.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF Stages of embryonic development of the zebrafish. Developmental Dynamics. 203 (3), 253–310 (1995). [DOI] [PubMed] [Google Scholar]
- 2.Kaufmann A, Mickoleit M, Weber M, Huisken J Multilayer mounting enables long-term imaging of zebrafish development in a light sheet microscope. Development. 139 (17), 3242–3247 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Schmid B et al. High-speed panoramic light-sheet microscopy reveals global endodermal cell dynamics. Nature Communications. 4, 2207 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Megason SG In toto imaging of embryogenesis with confocal time-lapse microscopy. Methods in Molecular Biology. 546, 317–32 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masselink W, Wong JC, Liu B, Fu J Currie PD Low-cost silicone imaging casts for zebrafish embryos and larvae. Zebrafish. 11 (1), 26–31 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Wittbrodt JN, Liebel U Gehrig, J. Generation of orientation tools for automated zebrafish screening assays using desktop 3D printing. BMC Biotechnology. 14, 36 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weijts B, Tkachenko E, Traver D Groisman A A Four-Well Dish for High-Resolution Longitudinal Imaging of the Tail and Posterior Trunk of Larval Zebrafish. Zebrafish. 14 (5) 489–491 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirsinger E, Steventon B A Versatile Mounting Method for Long Term Imaging of Zebrafish Development. Journal of Visualized Experiment. 119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Avdesh A et al. , Regular care and maintenance of a zebrafish (Danio rerio) laboratory: an introduction. Journal of Visualized Experiment. (69), e4196 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCollum CW et al. Embryonic exposure to sodium arsenite perturbs vascular development in zebrafish. Aquatic Toxicology. 152, 152–63 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Pan YA et al. Zebrabow: multispectral cell labeling for cell tracing and lineage analysis in zebrafish. Development. 140 (13), 2835–46 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asakawa K et al. Genetic dissection of neural circuits by Tol2 transposon-mediated Gal4 gene and enhancer trapping in zebrafish. Proceedings of the National Academy of Science U. S. A, 105 (4), 1255–60 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagayoshi S et al. Insertional mutagenesis by the Tol2 transposon-mediated enhancer trap approach generated mutations in two developmental genes: tcf7 and synembryn-like. Development. 135 (1), 159–69 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Huang WC et al. Combined use of MS-222 (tricaine) and isoflurane extends anesthesia time and minimizes cardiac rhythm side effects in adult zebrafish. Zebrafish. 7 (3), 297–304 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Craig MP, Gilday SD, Hove JR, Dose-dependent effects of chemical immobilization on the heart rate of embryonic zebrafish. Laboratory Animal (NY). 35 (9), 41–7 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Swinburne IA, Mosaliganti KR, Green AA, Megason SG, Improved Long-Term Imaging of Embryos with Genetically Encoded alpha-Bungarotoxin. PLoS One. 10 (8) e0134005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Movie S1: Movie of an embryo of a cross of Tg(kdr1:EGFP)mitfab692/b692 and Ubi-zebrabow imaged from about 30 somite stage for 55 h on a confocal microscope using a 10x objective.
Supplementary Movie S3: Movie of an embryo of a cross of Tg(kdr:enl.memRFP)mitfab692/b692 and Tg(mnx:GFP)mitfab692/b692 imaged from about 2 dpf for 55 h on a confocal microscope using a 20x objective.
Supplementary Movie S2: Movie of an embryo of a cross of Tg(mnx:GFP)mitfab692/b629 and Ubi-zebrabow imaged from about 30 somite stage for 55 h on a confocal microscope using a 10x objective.
Supplementary Movie S4: Movie of an embryo of a cross of HGn39b and Ubi-zebrabow imaged from about 30 somite stage for 55 h on a confocal microscope using a 10x objective.