

Abstract
Background
Accumulating data indicate that higher rifampicin doses are more effective and shorten tuberculosis (TB) treatment duration. This study evaluated the safety, tolerability, pharmacokinetics, and 7- and 14-day early bactericidal activity (EBA) of increasing doses of rifampicin. Here we report the results of the final cohorts of PanACEA HIGHRIF1, a dose escalation study in treatment-naive adult smear-positive patients with TB.
Methods
Patients received, in consecutive cohorts, 40 or 50 mg·kg−1 rifampicin once daily in monotherapy (day 1–7), supplemented with standard dose isoniazid, pyrazinamide and ethambutol between days 8 and 14.
Results
In the 40 mg·kg−1 cohort (n=15), 13 patients experienced a total of 36 adverse events during monotherapy, resulting in one treatment discontinuation. In the 50 mg·kg−1 cohort (n=17), all patients experienced adverse events during monotherapy, 93 in total; 11 patients withdrew or stopped study medication. Adverse events were mostly mild/moderate and tolerability rather than safety related, i.e. gastrointestinal disorders, pruritis, hyperbilirubinaemia and jaundice. There was a more than proportional increase in the rifampicin geometric mean area under the plasma concentration–time curve from time 0 to 12 h (AUC0–24 h) for 50 mg·kg−1 compared with 40 mg·kg−1; 571 (range 320–995) versus 387 (range 201–847) mg·L−1·h, while peak exposures saw proportional increases. Protein-unbound exposure after 50 mg·kg−1 (11% (range 8–17%)) was comparable with lower rifampicin doses. Rifampicin exposures and bilirubin concentrations were correlated (Spearman's ρ=0.670 on day 3, p<0.001). EBA increased considerably with dose, with the highest seen after 50 mg·kg−1: 14-day EBA −0.427 (95% CI −0.500– −0.355) log10CFU·mL−1·day−1.
Conclusion
Although associated with an increased bactericidal effect, the 50 mg·kg−1 dose was not well tolerated. Rifampicin at 40 mg·kg−1 was well tolerated and therefore selected for evaluation in a phase IIc treatment-shortening trial.
Short abstract
While bactericidal activity continues to increase with dose, for the first time we identified dose-limiting intolerability for rifampicin dosed at 50 mg·kg−1; 40 mg·kg−1 seems the optimal tolerable dose for evaluation in TB treatment-shortening trials https://bit.ly/37dUIuB
Introduction
In 1971 the US Food and Drug Administration approved the pivotal tuberculosis (TB) drug rifampicin at a dose of 10 mg·kg−1. The recommended dose was chosen on the basis that it was effective at the lowest cost and limited by fear of adverse effects [1]. A dose-finding study with an assessment of a maximum tolerated dose (MTD) had not been performed.
In vitro and mouse models have since revealed that higher doses of rifampicin are associated with improved bactericidal and sterilising activity, indicating a possibility of shorter treatment for pulmonary TB [2–4]. The End TB Strategy has set targets for treatment coverage as high as ≥90% by 2025, which will increase the number of patients diagnosed with TB, and the number that will receive rifampicin as part of their TB regimen, underlining the urgency for dose optimisation of this pivotal drug [5]. Overall, rifampicin is expected to continue to play a fundamental role in TB treatment.
In the PanACEA HIGHRIF1 study in African patients with pulmonary TB, it was shown that doses up to 35 mg·kg−1 given for 2 weeks resulted in a nine-fold increase in average exposure compared with 10 mg·kg−1 [6], and were safe and well tolerated. Pharmacokinetic/pharmacodynamic modelling demonstrates that increased rifampicin exposure is likely to be associated with increased early bactericidal activity (EBA) [7, 8]. In a larger study of 365 patients, high-dose rifampicin (35 mg·kg−1) combined with isoniazid, pyrazinamide and ethambutol, when administered for a longer period of 3 months, was able to reduce time to sputum culture conversion in pulmonary TB [9].
Given these findings there is an urgent need to assess the safety, tolerability, pharmacokinetics and EBA of increasing doses of rifampicin to establish the optimum dose. To complete this task we extended the HIGHRIF1 study (ClinicalTrials.gov: NCT01392911) by including participants treated with 40 and 50 mg·kg−1 rifampicin.
Methods
Study design and participants
We performed an open-label, phase II, multiple-dose-ranging study to evaluate safety, tolerability, pharmacokinetics, and 7- and 14-day early EBA of 40 and 50 mg·kg−1 rifampicin. Adults (age 18–65 years) with newly diagnosed, previously untreated, drug-susceptible and sputum smear-positive pulmonary TB, and without medical contraindications, were included in the study. Patients were hospitalised in one of two study sites in Cape Town, South Africa. We recruited consecutive cohorts of 15 participants who received monotherapy of rifampicin for 7 days, supplemented with standard doses of isoniazid (5 mg·kg−1), pyrazinamide (25–30 mg·kg−1) and ethambutol (15–20 mg·kg−1) on days 8–14. Patients then continued TB treatment with standard doses of all drugs. Study medication was weight banded (supplementary figure E1), and was taken in the morning with a light breakfast and a glass of water. After completion of each of the cohorts, a safety review was performed by the Trial Steering Committee (TSC) to assess whether a dose increase was possible or whether the MTD was assessed. The MTD was predefined as the dose level below that producing unacceptable but reversible toxicity and is considered the upper limit of patient tolerance. The study protocol was approved by the applicable ethical review boards and by the South African Health Products Regulatory Authority, and was conducted according to international and South African Good Clinical Practice guidelines. Details on eligibility criteria and results of prior HIGHRIF1 cohorts have been published elsewhere [6].
Safety and tolerability
Symptom assessments and physical examinations, including vital signs, were performed daily. Haematological renal and liver function tests, glucose, uric acid and urinalysis, as well as an ECG were performed at baseline and on days 1, 3, 6, 10, 14 and 21. Adverse events were graded according to the US National Institute of Health Common Terminology Criteria for Adverse Events version 4.0 and were assessed as unrelated, possibly or definitely related to study therapy by site investigators [6]. A serious adverse event was defined as any untoward medical occurrence that in the opinion of the investigator results in death, is life threatening, requires (prolongation of) hospitalisation, results in persistent or significant disability/incapacity, or is a medically important event. A meeting of the TSC to discuss the continuation or termination of the study would take place if two subjects experiencing a grade 3 adverse event assessed as probably or definitely related to administration of high-dose rifampin, or one subject experiencing a grade 4 or 5 adverse event assessed as definitely related to rifampin, would occur in one dose group.
Pharmacokinetics
Blood samples were taken pre-dose and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12 and 24 h post-rifampicin intake with a standardised meal on days 7 and 14 to obtain full pharmacokinetic profiles. Rifampicin total (protein-bound plus protein-unbound) and protein-unbound plasma concentrations were measured after each cohort. Plasma samples of the 40 mg·kg−1 dose group were analysed using the same validated ultra-performance liquid chromatography method with ultraviolet detection as in the preceding cohorts [6]. For the 50 mg·kg−1 group, total concentrations of anti-TB drug were analysed using an extensively validated liquid chromatography-mass spectrometry (LC-MS/MS) multidrug assay. The assay accuracy for rifampicin quantification was 94.24–102.06% dependent on concentration level; the within-run imprecision ranged from 0.9% to 4.89%. Protein-unbound determination of rifampicin occurred via ultrafiltration as previously described [10]. Noncompartmental pharmacokinetic analysis was performed with Phoenix WinNonlin version 6.4 (Certara, Princeton, NJ, USA) as previously described [11]. The unbound fraction in the 50 mg·kg−1 cohort was calculated by dividing the unbound area under the plasma concentration–time curve from time 0 to 12 h (AUC0–24 h) by the total AUC0–24 h for all subjects in this group. A full description of the bioanalytical and pharmacokinetic analyses is provided in the supplementary material.
Antimycobacterial activity
Pooled overnight (16 h) sputum samples were collected at baseline, daily to day 7, and on days 9 and 14. Samples were processed for culture on selective Middlebrook 7H10S agar plates and in liquid broth using the BACTEC MGIT 960 (BD, Franklin Lakes, NJ, USA) mycobacterial growth indicator tube system. Spot sputum samples were collected before enrolment, at day 19 and 12 weeks after starting study therapy, and were prepared for auramine O-stained direct microscopy and rapid resistance testing via the Xpert MTB/RIF (Cepheid, Sunnyvale, CA, USA) assay. All microbiological testing was performed at the Dept of Medical Biochemistry, Faculty of Medicine and Health Sciences, Stellenbosch University (Cape Town, South Africa) as previously described [12].
Statistical analyses
This was a descriptive study with no inferential statistics or hypothesis testing [6].
The planned sample size of 15 patients in each group is in keeping with other trials of this type and accounts for the estimate of three dropouts per group. The distribution of time to positivity (TTP) on MGIT was positively skewed, with log-transformed TTP more closely following a symmetric normal distribution. Mixed effects models with visit day as a discrete random effect were used to estimate the mean log10CFU and log10TTP in each treatment arm at each visit to describe the data. As in our previous study [6], we found an unexpectedly high number of negative cultures for the short treatment duration. To include these censored observations, Tobit regression [13] was used to estimate the 14-day EBA, accounting for negatives cultures using a lower limit of detection censoring for log10CFU of 1 and an upper limit for TTP of 42 days. Separate models were fitted for each patient with parameter estimates summarised by treatment group using a random effects model accounting for within- and between-patient variability. We were concerned that data from patients without cultures after day 5 inflated the estimates of the fall in CFU or increase in TTP over time. Given these early withdrawals, data from these patients were excluded to account for the loss to follow-up in the 50 mg·kg−1 cohort in an additional analysis. Sensitivity analyses included analysis of the full data as planned and a one-stage mixed effects model, allowing for censoring of negative cultures, to assess robustness of the results.
The safety population consisted of all participants who took at least one dose of trial medication. Associations between exposure and liver laboratory assessments were made using Spearman rank correlation. In addition, dose–exposure–tolerability relationships during the monotherapy phase were evaluated post hoc with an ordered categorical model estimating the probability of having none, one, two, or three or more adverse effects given the rifampicin dose and exposure. In these analyses, total exposure in plasma (AUC0–24 h) at day 7 was used as a measure for rifampicin exposure in the study. All system organ classes with tolerability-related adverse events were included in the analysis (see supplementary table E2 for details).
Analyses were undertaken using Stata version 15.1 (StataCorp, College Station, TX, USA) and NONMEM version 7.4 (Icon Development Solutions, Ellicott City, MD, USA).
Results
Patients
15 culture-positive patients with pulmonary TB were enrolled in the 40 mg·kg−1 cohort, of whom 14 patients completed the study. One patient was withdrawn because of raised liver enzymes. 17 patients were enrolled in the 50 mg·kg−1 cohort. Recruitment was temporarily suspended in this cohort for TSC review of interim data. Nine patients (53%) withdrew early from the 50 mg·kg−1 group: seven during monotherapy and two during combination therapy. One additional patient stopped treatment from day 11 onwards but completed study visits (excluding pharmacokinetic assessment) awaiting outcome of the interim TSC review and one patient had a 3-day dose interruption from day 10 to 12. Patient characteristics are shown in table 1.
TABLE 1.
Demographic and baseline characteristics of study participants in HIGHRIF1 [6]
| Rifampicin dose group | All | |||||||
| 10 mg·kg−1 | 20 mg·kg−1 | 25 mg·kg−1 | 30 mg·kg−1 | 35 mg·kg−1 | 40 mg·kg−1 | 50 mg·kg−1 | ||
| Patients | 8 | 15 | 15 | 15 | 15 | 15 | 17 | 100 |
| Age years | 28 (20–49) | 28 (18–47) | 26 (20–47) | 40 (20–60) | 38 (21–60) | 35 (23–58) | 25 (20–55) | 31 (18–60) |
| Weight kg | 57 (47–65) | 52 (42–63) | 53 (40–68) | 54 (46–84) | 57 (41–74) | 59 (47–65) | 53 (43–64) | 53 (40–84) |
| BMI kg·m−2 | 21 (16–26) | 18 (17–26) | 19 (15–25) | 21 (16–31) | 19 (15–25) | 19 (17–25) | 18 (16–23) | 19 (15–31) |
| Male | 6 (75) | 11 (73) | 10 (67) | 11 (73) | 10 (67) | 11 (73) | 15 (88) | 74 (74) |
| Race | ||||||||
| Black | 3 (38) | 7 (47) | 4 (27) | 9 (60) | 5 (33) | 10 (67) | 8 (47) | 46 (46) |
| Coloured | 5 (63) | 8 (53) | 11 (73) | 6 (40) | 10 (67) | 5 (33) | 8 (47) | 53 (53) |
| Caucasian | 1 (6) | 1 (1) | ||||||
| HIV positive | 0 | 0 | 0 | 3 (20) | 1 (7) | 1 (7) | 0 | 5 (5) |
| Baseline log10CFU·mL−1 | 5.4 (4.0–6.4) | 5.0 (2.6–7.3) | 6.5 (5.4–7.6) | 6.4 (5.3–7.4) | 5.8 (1.0–7.2) | 6.5 (5.1–7.3) | 6.4 (4.8–8.5) | 6.1 (1.0–8.5) |
| Baseline TTP days | 4.0 (3.3–5.3) | 4.7 (3.0–9.1) | 4.0 (3.4–6.6) | 3.9 (2.9–6.0) | 3.9 (2.6–19.3) | 4.0 (2.2–7.6) | 4.4 (2.7–7.3) | 4.0 (2.2–19.3) |
Data are presented as n, median (range) or n (%). BMI: body mass index; TTP: time to positivity. Due to an analysis error, the baseline bacterial load was previously [6] erroneously reported ~1 log10CFU lower and ~2 days TTP higher in the 10–35 mg·kg−1 dose groups than we report now. The number of HIV-positive participants was also incorrect. Both of these errors have been corrected in the current article.
Safety and tolerability
Of the 15 patients starting on 40 mg·kg−1 rifampicin, 13 (87%) reported adverse events during monotherapy (36 events in total). All 17 patients in the 50 mg·kg−1 cohort reported adverse events during monotherapy (93 events in total). See table 2 for an overview of the adverse events and their severity during the monotherapy and combination therapy periods in the 40 and 50 mg·kg−1 groups, and previous dosing groups [6]. Adverse events in both the 40 and 50 mg·kg−1 cohorts were mostly mild/moderate, i.e. >97% of all adverse events during monotherapy were grade 1/2 in both cohorts. In addition, adverse events were tolerability rather than safety related, i.e. gastrointestinal disorders, pruritis, hyperbilirubinaemia and jaundice. No grade 4 or 5 adverse events occurred in either cohort.
TABLE 2.
Total number of adverse events per severity, dose group and treatment period
| Rifampicin dose group | ||||
| 10–30 mg·kg−1 | 35 mg·kg−1 | 40 mg·kg−1 | 50 mg·kg−1 | |
| Monotherapy | ||||
| Patients | 53 | 15 | 15 | 17 |
| Adverse events | ||||
| Total | 46 | 25 | 36 | 93# |
| Not specified | 0 | 0 | 0 | 10 |
| Unrelated | 13 | 4 | 9 | 10 |
| Possibly related | 28 | 19 | 19 | 24 |
| Related | 5 | 2 | 8 | 49 |
| Grade 1 | 38 | 18 | 26 | 60 |
| Grade 2 | 6 | 7 | 9 | 21 |
| Grade 3 | 2 | 0 | 1 | 1 |
| Serious adverse event | 0 | 0 | 0 | 0 |
| Combination therapy | ||||
| Patients | 53 | 15 | 15 | 10 |
| Adverse events | ||||
| Total | 62 | 32 | 24 | 34 |
| Not specified | 0 | 0 | 0 | 0 |
| Unrelated | 23 | 13 | 10 | 11 |
| Possibly related | 38 | 19 | 13 | 14 |
| Related | 1 | 0 | 1 | 9 |
| Grade 1 | 52 | 22 | 19 | 28 |
| Grade 2 | 8 | 9 | 1 | 6 |
| Grade 3 | 2 | 1 | 4¶ | 0 |
| Serious adverse event | 1 | 0 | 2 | 0 |
Data are presented as n. #: 11 adverse events were not graded and for one event severity was not indicated; ¶: in one of the four patients with a grade 3 adverse event developing in the combination phase this was defined as unrelated to high-dose rifampicin (hyperuricaemia) and in three of the four patients this was defined as possibly related (n=1 hyperuricaemia and n=2 hepatic enzyme increased).
The most common adverse events in the 40 and 50 mg·kg−1 cohorts were gastrointestinal disorders (grade 1–2), hyperbilirubinaemia (grade 1–3), pruritis (grade 1–2) and jaundice (50 mg·kg−1 rifampicin only, grade 1–2), all expected from rifampicin. Overall, the 50 mg·kg−1 cohort contained more cases with gastrointestinal disorders (12 (71%) versus seven (47%) patients during monotherapy), hyperbilirubinaemia (10 (59%) versus four (27%) patients) and jaundice (nine (53%) versus zero patients). Elevations in bilirubin (grade 1–3) peaked around day 3–4 after rifampicin start (supplementary figure E6). See supplementary tables E2 and E3 for an overview of the incidence of adverse events during monotherapy and combination therapy, respectively, per system organ class in both groups.
During 40 mg·kg−1 combination therapy, four patients developed a grade 3 adverse event. In one of the four patients this was defined as unrelated to high-dose rifampicin (hyperuricaemia) and in three of the four patients this was defined as possibly related (n=1 hyperuricaemia and n=2 hepatic enzyme increased). TSC evaluation of these adverse events considered that these grade 3 adverse events were either not typical for high-dose rifampicin (n=2 hyperuricaemia) and/or considered unrelated or only possibly related to high-dose rifampicin (increased transaminases only developed after introduction of combination therapy). Nonetheless, the increased transaminases had been classified as “serious adverse events” and one patient was withdrawn, even though there was no immediate life-threatening risk. All grade 3 adverse events resolved.
While no serious adverse events occurred in the 50 mg·kg−1 cohort, there were nine early withdrawals. Four were withdrawn from the study by the investigator because of adverse events (e.g. grade 2 elevated bilirubin) and five withdrew consent because of social/personal reasons, which were hypothesised by investigators and TSC to be related to experienced intolerability. In addition, one patient was withheld from study treatment between day 10 and 12 because of intolerability.
Based on the high incidence of adverse effects and the many withdrawals in the 50 mg·kg−1 cohort, the TSC assessed that the 50 mg·kg−1 dose was not tolerable and that the 40 mg·kg−1 dose was to be regarded as the MTD. The TSC considered that the safety profile of 40 mg·kg−1 rifampicin was acceptable, mostly mild/moderate and reversible, and therefore 40 mg·kg−1 was considered the MTD. In addition, the safety profile was considered to be comparable to that of 35 mg·kg−1, a dose that also has been found to be safe and effective when given for 12 weeks in a randomised controlled trial [9].
Pharmacokinetics
The geometric mean AUC0–24 h and peak plasma concentration (Cmax) values of rifampicin at day 7 and 14 are presented in table 3. On adding 25% of the dose of rifampicin from 40 to 50 mg·kg−1, the geometric mean AUC0–24 h increased ∼50%, which reflects a more than dose-proportional increase of exposure of a similar magnitude as previously observed (supplementary figure E2) [6]. In contrast, rifampicin peak exposure increased proportional with the dose. Of note, large interindividual variability in AUC0–24 h and Cmax was observed, with exposures between groups overlapping considerably (table 3 and supplementary figure E2). Protein-unbound rifampicin exposure, or free fraction, was comparable to other (lower) doses of rifampicin [14]. For rifampicin pharmacokinetic profiles and pharmacokinetic parameters of other study drugs, see supplementary figure E3 and supplementary table E1, respectively.
TABLE 3.
Rifampicin pharmacokinetics during monotherapy (day 7) and combination therapy (day 14)
| Rifampicin dose group | ||
| 40 mg·kg−1 | 50 mg·kg−1 | |
| Monotherapy | ||
| Patients | 15 | 10 |
| AUC0–24 h mg·L−1·h | 387 (201–847) | 571 (320–995) |
| Cmax mg·L−1 | 53.9 (40.0–80.8) | 63.4 (42.3–85.0) |
| CL/F L·h−1 | 5.9 (3.2–9.7) | 4.6 (2.6–6.6) |
| Vd/F L | 30.9 (18.5–50.7) | 32.4 (23.9–45.2) |
| T1/2 h | 3.7 (1.7–4.1) | 4.9 (2.5–8.5) |
| AUC free fraction % (average) | NA | 10.8 (8.4–16.9) |
| Combination therapy (steady-state)# | ||
| Patients | 14 | 7 |
| AUC0–24 h mg·L−1·h | 257 (173–349) | 370 (231–559) |
| Cmax mg·L−1 | 41.4 (26.4–56.6) | 53.2 (39.7–73.6) |
| CL/F L·h−1 | 8.7 (7.0–11.7) | 6.9 (5.4–9.1) |
| Vd/F L | 31.6 (18.5–45.3) | 26.3 (20.0–39.5) |
| T1/2 h | 5.6 (2.1–9.5) | 2.6 (2.0–5.1) |
| AUC free fraction % (average) | NA | 10.6 (8.8–13.2) |
Data are presented as n or geometric mean (range), unless otherwise stated. AUC0–24 h: area under the plasma concentration–time curve from time 0 to 12 h; Cmax: peak plasma concentration; CL: clearance; F: bioavailability fraction; Vd: volume of distribution; T1/2: half-life; NA: not available. #: rifampicin clearance increases during multiple-dose therapy due to its known induction of hepatic enzymes, which leads to autoinduction of its own metabolism.
Antimycobacterial activity
One patient from the 50 mg·kg−1 group had consistent negative cultures at baseline and throughout treatment, and was therefore removed from all analyses. Figure 1 summarises the change in viable bacterial load in sputum over 7 and 14 days expressed as fall of CFU and increase of TTP for patients from all cohorts. The 7-day EBA shows that the fall in bacterial load in the first week is due to rifampicin alone and is extended in the second week. In a post hoc analysis, six patients in the 50 mg·kg−1 group without cultures after day 5 were excluded because they only contributed data up to day 4, thereby making it challenging to estimate change over a 14-day period (supplementary figure E4). This was a post hoc analysis not anticipated in the statistical analysis plan, but was conservative, resulting in smaller estimates of slope than our planned primary analysis. Overall, bactericidal activity as measured on both solid and liquid media increased over the 10–50 mg·kg−1 cohorts, with the highest 14-day activity seen in the highest dose cohort in all analyses. Sensitivity analyses using a one-stage mixed effect model showed consistent results when all negative cultures were imputed with the lower limit of detection of the respective culture method, when the first negative culture was imputed with the lower limit of detection and when negative cultures were ignored, supporting the robustness of our findings. For the differences in bacterial load in CFU and TTP compared with baseline over time, see supplementary figure E5.
FIGURE 1.
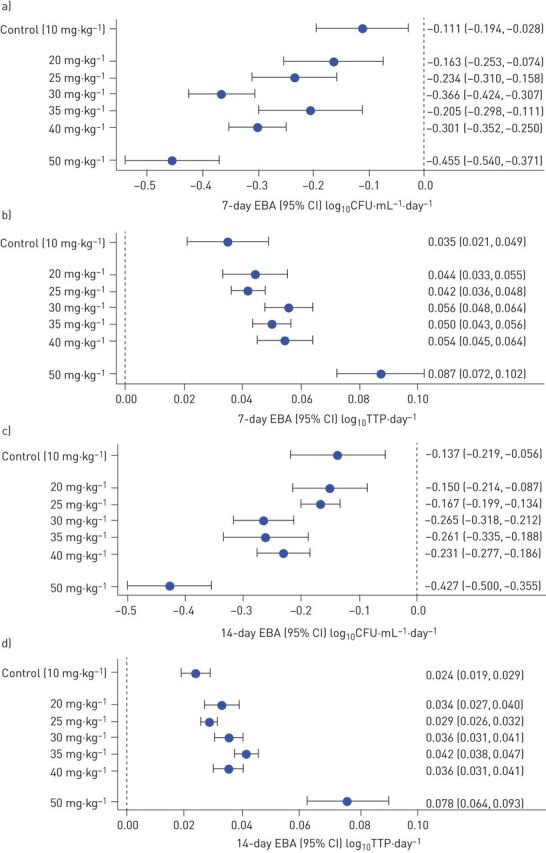
a, b) 7- and c, d) 14-day early bactericidal activity (EBA) (95% CI) of rifampicin based on a, c) CFU and b, d) time to positivity (TTP). Data from all patients were included with the exception of data from one patient from each of the 20 and 50 mg·kg−1 groups that had consistent negative cultures at baseline and throughout. The slight change in the estimates for 10 and 20 mg·kg−1 compared with previously [6] are because of data corrections (four CFU cultures were recorded as negative while they were actually missing).
Exposure–safety analyses
Figure 2 shows the relationship between rifampicin AUC0–24 h at day 7 and total serum bilirubin for all scheduled safety visits across HIGHRIF1 cohorts (n=93). Rifampicin exposures and bilirubin concentrations were correlated (Spearman's ρ=0.670 on day 3, p<0.001). Alanine transaminase and aspartate transaminase concentrations were not correlated with rifampicin exposure (supplementary figures E7 and E8).
FIGURE 2.

Total serum bilirubin per day on rifampicin plotted against rifampicin total exposures (area under the plasma concentration–time curve from time 0 to 12 h (AUC0–24 h) at day 7) in all patients with pharmacokinetic results (n=93) in the HIGHRIF1 study. The lines represent linear regressions (for illustration; statistical testing described in the text). The y-axis is capped at 70 µmol·L−1 for readability, excluding three outlying points (day 3 in one patient in the 40 mg·kg−1 group with AUC0–24 h 338 mg·L−1·h and bilirubin 111 µmol·L−1; day 7 and 10 in one patient in the 50 mg·kg−1 group with AUC0–24 h 980 mg·L−1·h and bilirubin 100 and 175 µmol·L−1, respectively).
With respect to the dose–exposure–tolerability evaluation, a linear relation on a logit scale described the data appropriately (goodness-of-fit pots are provided in supplementary figure E9). Dose and exposure (AUC0–24 h at day 7) were both separately strong predictors of the probability of developing tolerability adverse events (likelihood ratio test, p<0.0001 for both). A 50 mg·kg−1 dose was associated with 76% (90% CI 56–88%) risk of three or more tolerability-related (rather than safety-related) adverse events, for 40 and 10 mg·kg−1 the corresponding risks were 47% (90% CI 29–64%) and 2.0% (90% CI 0.1–5.4%), respectively. The relationships with associated uncertainty are illustrated in figure 3.
FIGURE 3.

Probability of tolerability-related adverse events during the first week of rifampicin monotherapy related to the rifampicin a) dose or b) exposure (area under the plasma concentration–time curve from time 0 to 12 h (AUC0–24 h) at day 7). The shaded areas represent 90% confidence intervals based on the estimated parameter uncertainty
Discussion
More than 40 years after the introduction of rifampicin, during which time it has become the most important drug for the treatment of TB, we have now identified a MTD. In our first reports of the HIGHRIF1 study, we showed that high-dose rifampicin up to 35 mg·kg−1 was safe and well tolerated, exposure increased more than proportional with dose, and there was greater EBA at higher exposures [6–8]. We have now shown continued increases in drug exposures and extended EBA in the 40 and 50 mg·kg−1 cohorts. The 40 mg·kg−1 cohort was in line with previous cohorts, with adverse events of only mild to moderate severity. Rifampicin dosed at 50 mg·kg−1 once daily, however, was poorly tolerated, with a sharp increase in frequency and severity of adverse events as well as subject withdrawals compared with 40 mg·kg−1. Thus, we consider that rifampicin dosed at 40 mg·kg−1 is the MTD.
Overall, experienced adverse events were mild or moderate and mostly tolerability-related, i.e. gastrointestinal disorders (grade 1–2), pruritis (grade 1–2) and jaundice (grade 1–2, in the 50 mg·kg−1 group only, related to hyperbilirubinaemia). It was the large number rather than the severity of individual adverse events that caused poor tolerability and withdrawals in the 50 mg·kg−1 group.
While minor bilirubin elevations were common in all groups, a remarkably high incidence of hyperbilirubinaemia (grade 1–3) was observed in the highest dose group. The elevations in bilirubin peaked around day 3–4 after start of rifampicin, were exposure dependent and were not associated with other liver enzyme elevations (figure 2 and supplementary figures E6–E8). Strikingly, normalisation of bilirubin levels in the 50 mg·kg−1 arm was slower compared with other arms (supplementary figure E6). McColl et al. [15] found that in healthy subjects unconjugated bilirubin increases after starting rifampicin, which we now believe is because of inhibition of bilirubin hepatocellular uptake via organic anion transport protein (OATP) and/or glucuronidation by UDP glucuronosyltransferase family 1 member A1 (UGTA1A1) [16]. Bilirubin levels then decline to less than pre-treatment values upon rifampicin continuation, suggestive of induction of net bilirubin clearance [15]. In contrast, increased bilirubin levels were all conjugated (direct) in the subset of patients tested in the 50 mg·kg−1 arm (supplementary figure E10), suggesting reduced biliary clearance by multidrug resistance protein 2 (MRP2) after intracellular conjugation in the liver [17]. In line with this, we may anticipate other liver transporters to also be inhibited by rifampicin at these high intracellular exposures [17].
Rifampicin exposures following 40 and 50 mg·kg−1 were high, but within the expected range based on previous results and modelling predictions [7], and again without a ceiling effect as reported in the pharmacokinetics of rifapentine [18]. More importantly, apart from average exposures, the lowest observed AUC0–24 h and Cmax values also increased with almost every dose step (supplementary figure E2). These low exposures may cause treatment failures and relapses, and create conditions for the emergence of resistance [19, 20]. Strikingly, no saturation of plasma proteins occurred at high exposure levels, as the unbound protein fraction (free, active drug) was comparable with other reports [14, 21].
Early-phase clinical TB studies usually only include small numbers of patients selected on the basis of very strict criteria who are treated for only a short period of time. Our EBA findings, therefore, need to be confirmed in phase II studies with less narrow inclusion criteria and adequate patient numbers. However, even though this study was not powered or designed to test statistical differences between groups, the presented EBA results are striking. There is a clear increase in EBA with dose and exposure, with the highest EBA so far seen in the 50 mg·kg−1 cohort. The broad trend of increasing EBA with dose was seen on both liquid and solid media; there was a suggestion that the increase up to 40 mg·kg−1 was less conspicuous on liquid than solid media, although our study was too small to draw definitive conclusions. Inclusion in the analysis of the six patients who withdrew early from the 50 mg·kg−1 arm may have artificially inflated the estimate of the 14-day bactericidal activity; our sensitivity analysis excluding these patients did show slightly lower 14-day activity, although it was still clearly higher than any other dose group. We chose to retain the analysis including all patients since this was an observational, hypothesis-generating study and we were keen to include all the data in the primary analysis. In addition, we cannot exclude that participants withdrawing early may also have had increased EBA because of elevated rifampicin exposures, possibly explaining tolerability-related early withdrawal from the study. In general, our EBA results are in line with findings from other high-dose rifampicin studies in pulmonary TB [8, 9, 22, 23] and in TB mouse models [4, 24]. In our previous study in patients with pulmonary TB, high-dose rifampicin at 35 mg·kg−1 for 12 weeks was found to be safe and reduce the time to culture conversion, an intermediate clinical end-point [9]. Our current work further supports that higher rifampicin doses perform better, and thus have the potential to improve clinical outcome, decrease relapse rates, reduce the emergence of rifampicin resistance and reduce treatment duration.
To further optimise the rifampicin dose and its dosing strategy, multiple approaches could be considered. The most promising strategy from a programmatic point of view would be to start therapy with lower rifampicin doses, allowing the body to get used to rifampicin in terms of gastrointestinal tolerance while also facilitating induction of rifampicin and bilirubin clearance. After this initial period, a higher dose of rifampicin could be introduced in all patients. Personalised medicine with titration for individual maximum exposures is promising because of the increase in variation of exposures found with higher doses, and higher rifampicin exposures, in turn, are associated with improved bactericidal activity and culture conversion [8, 22]. Unfortunately, a maximal effect has not yet been identified and as such there are no clear exposure targets. A low-cost point-of-care device to estimate rifampicin concentrations in real-time could support the implementation of high-dose rifampicin treatment in programmatic settings [25]. Finally, our group recently reported that weight-band dosing yields a small and nonclinically relevant decrease in variability of AUC0–24 h compared with flat dosing [26]. This supports the use of flat dosing and we are planning to implement this in our follow-up trial.
For now we have sufficient data to move high-dose rifampicin forward to a study with less narrow inclusion criteria, increased patient numbers, longer treatment duration and clinical end-points, i.e. a so-called phase IIc Selection Trial with Extended Post-treatment follow-up (STEP) design [27]. In this trial, the experimental regimen is given for the duration for which it will be studied in phase III (presently 3 or 4 months), and patients are followed for clinical outcomes of treatment failure and relapse for a total of 12 months from randomisation. Generated data will provide valuable information about the likelihood of success of high-dose rifampicin-containing regimens in a future phase III trial.
Currently, one phase III study with the objective to reduce treatment duration by increasing the dose of rifampicin is enrolling (RIFASHORT; ClinicalTrials.gov: NCT02581527). The primary end-points are treatment failure and relapse after 12 months. Based on the data presented here, the rifampicin dose increase evaluated in RIFASHORT, i.e. 1200 or 1800 mg of rifampicin corresponding to around 20–30 mg·kg−1, may seem modest [28, 29], but will still provide important input to support the ability of higher doses to prevent failure and relapse.
In conclusion, rifampicin dosed at 50 mg·kg−1 once daily, introduced at once at the start of TB therapy, was poorly tolerated. It was associated with a remarkably improved decrease in bacterial load compared with other dosages. The 40 mg·kg−1 dose was safe, tolerable and associated with improved bactericidal effect, and is therefore the appropriate dose to be evaluated in a follow-up phase IIc trial investigating the treatment-shortening potential of high-dose rifampicin. Such a study also provides the opportunity to study tolerability of 40 mg·kg−1 in a larger population when given for a longer duration.
Our research concludes a journey that started in the 1960s. We need to move forward with confirmative clinical trials to further inform implementation of high-dose rifampicin in programmes and guidelines.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00955-2020.SUPPLEMENT (2MB, pdf)
Shareable PDF
Acknowledgements
The authors thank Peter R. Donald (Dept of Paediatrics and Child Health, Stellenbosch University, Cape Town, South Africa) and Helmuth Reuter (Division of Clinical Pharmacology, Stellenbosch University, Cape Town, South Africa).
Footnotes
Author contributions: All authors substantially contributed to the conception/design of the work and/or participated in either the acquisition, analysis or interpretation of data for the work; helped in drafting the work or revising it critically; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
This article has supplementary material available from erj.ersjournals.com
This study is registered at ClinicalTrials.gov with identifier number NCT01392911.
Conflict of interest: L.H.M. te Brake has nothing to disclose.
Conflict of interest: V. de Jager has nothing to disclose.
Conflict of interest: K. Narunsky has nothing to disclose.
Conflict of interest: N. Vanker has nothing to disclose.
Conflict of interest: E.M. Svensson has nothing to disclose.
Conflict of interest: P.P.J. Phillips reports grants from Ludwig Maximilian University of Munich, during the conduct of the study.
Conflict of interest: S.H. Gillespie reports grants from the European and Developing Countries Clinical Trials Partnership and TB Alliance, outside the submitted work.
Conflict of interest: N. Heinrich reports grants from the European and Developing Countries Clinical Trials Partnership and German Ministry for Education and Research, during the conduct of the study; other (paid presentation) from AstraZeneca, outside the submitted work.
Conflict of interest: M. Hoelscher has nothing to disclose.
Conflict of interest: R. Dawson has nothing to disclose.
Conflict of interest: A.H. Diacon has nothing to disclose.
Conflict of interest: R.E. Aarnoutse has nothing to disclose.
Conflict of interest: M.J. Boeree has nothing to disclose.
Support statement: This publication was produced by PanACEA, which is part of the European and Developing Countries Clinical Trials Partnership (EDCTP) 1 programme (project code IP.2007.32011.012 (HIGHRIF)) and the EDCTP2 programme supported by the European Union (grant number TRIA2015-1102-PanACEA). The funding source had no influence on study design; in the collection, analysis and interpretation of data; or in the writing of the report. Publication of the article is defined as a deliverable under the grant agreement. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.van Ingen J, Aarnoutse RE, Donald PR, et al. Why do we use 600 mg of rifampicin in tuberculosis treatment? Clin Infect Dis 2011; 52: e194–e199. doi: 10.1093/cid/cir184 [DOI] [PubMed] [Google Scholar]
- 2.Jayaram R, Gaonkar S, Kaur P, et al. Pharmacokinetics–pharmacodynamics of rifampin in an aerosol infection model of tuberculosis. Antimicrob Agents Chemother 2003; 47: 2118–2124. doi: 10.1128/AAC.47.7.2118-2124.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenthal IM, Tasneen R, Peloquin CA, et al. Dose-ranging comparison of rifampin and rifapentine in two pathologically distinct murine models of tuberculosis. Antimicrob Agents Chemother 2012; 56: 4331–4340. doi: 10.1128/AAC.00912-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Steenwinkel JE, Aarnoutse RE, de Knegt GJ, et al. Optimization of the rifampin dosage to improve the therapeutic efficacy in tuberculosis treatment using a murine model. Am J Respir Crit Care Med 2013; 187: 1127–1134. doi: 10.1164/rccm.201207-1210OC [DOI] [PubMed] [Google Scholar]
- 5.World Health Organization . Global Tuberculosis Report 2018. Geneva, WHO, 2018. [Google Scholar]
- 6.Boeree MJ, Diacon AH, Dawson R, et al. A dose-ranging trial to optimize the dose of rifampin in the treatment of tuberculosis. Am J Respir Crit Care Med 2015; 191: 1058–1065. doi: 10.1164/rccm.201407-1264OC [DOI] [PubMed] [Google Scholar]
- 7.Svensson RJ, Aarnoutse RE, Diacon AH, et al. A population pharmacokinetic model incorporating saturable pharmacokinetics and autoinduction for high rifampicin doses. Clin Pharmacol Ther 2018; 103: 674–683. doi: 10.1002/cpt.778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Svensson RJ, Svensson EM, Aarnoutse RE, et al. Greater early bactericidal activity at higher rifampicin doses revealed by modeling and clinical trial simulations. J Infect Dis 2018; 218: 991–999. doi: 10.1093/infdis/jiy242 [DOI] [PubMed] [Google Scholar]
- 9.Boeree MJ, Heinrich N, Aarnoutse R, et al. High-dose rifampicin, moxifloxacin, and SQ109 for treating tuberculosis: a multi-arm, multi-stage randomised controlled trial. Lancet Infect Dis 2017; 17: 39–49. doi: 10.1016/S1473-3099(16)30274-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ewijk-Beneken Kolmer EWJ, Teulen MJA, van den Hombergh ECA, et al. Determination of protein-unbound, active rifampicin in serum by ultrafiltration and ultra performance liquid chromatography with UV detection. A method suitable for standard and high doses of rifampicin. J Chromatogr B 2017; 1063: 42–49. doi: 10.1016/j.jchromb.2017.08.004 [DOI] [PubMed] [Google Scholar]
- 11.Ruslami R, Nijland HM, Alisjahbana B, et al. Pharmacokinetics and tolerability of a higher rifampin dose versus the standard dose in pulmonary tuberculosis patients. Antimicrob Agents Chemother 2007; 51: 2546–2551. doi: 10.1128/AAC.01550-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donald PR, Sirgel FA, Venter A, et al. Early bactericidal activity of antituberculosis agents. Expert Rev Anti Infect Ther 2003; 1: 141–155. doi: 10.1586/14787210.1.1.141 [DOI] [PubMed] [Google Scholar]
- 13.Long JS. Regression Models for Categorical and Limited Dependent Variables. Thousand Oaks, Sage, 1997. [Google Scholar]
- 14.Litjens CHC, Aarnoutse RE, van Ewijk-Beneken Kolmer EWJ, et al. Protein binding of rifampicin is not saturated when using high-dose rifampicin. J Antimicrob Chemother 2019; 74: 986–990. doi: 10.1093/jac/dky527 [DOI] [PubMed] [Google Scholar]
- 15.McColl KE, Thompson GG, el Omar E, et al. Effect of rifampicin on haem and bilirubin metabolism in man. Br J Clin Pharmacol 1987; 23: 553–559. doi: 10.1111/j.1365-2125.1987.tb03091.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiou WJ, de Morais SM, Kikuchi R, et al. OATP1B1 and OATP1B3 inhibition is associated with observations of benign clinical unconjugated hyperbilirubinemia. Xenobiotica 2014; 44: 276–282. doi: 10.3109/00498254.2013.820006 [DOI] [PubMed] [Google Scholar]
- 17.te Brake LHM, Russel FG, van den Heuvel JJ, et al. Inhibitory potential of tuberculosis drugs on ATP-binding cassette drug transporters. Tuberculosis 2016; 96: 150–157. doi: 10.1016/j.tube.2015.08.004 [DOI] [PubMed] [Google Scholar]
- 18.Savic RM, Lu Y, Bliven-Sizemore E, et al. Population pharmacokinetics of rifapentine and desacetyl rifapentine in healthy volunteers: nonlinearities in clearance and bioavailability. Antimicrob Agents Chemother 2014; 58: 3035–3042. doi: 10.1128/AAC.01918-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srivastava S, Pasipanodya JG, Meek C, et al. Multidrug-resistant tuberculosis not due to noncompliance but to between-patient pharmacokinetic variability. J Infect Dis 2011; 204: 1951–1959. doi: 10.1093/infdis/jir658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pasipanodya JG, Srivastava S, Gumbo T. Meta-analysis of clinical studies supports the pharmacokinetic variability hypothesis for acquired drug resistance and failure of antituberculosis therapy. Clin Infect Dis 2012; 55: 169–177. doi: 10.1093/cid/cis353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.te Brake LHM, Ruslami R, Later-Nijland H, et al. Exposure to total and protein-unbound rifampin is not affected by malnutrition in Indonesian tuberculosis patients. Antimicrob Agents Chemother 2015; 59: 3233–3239. doi: 10.1128/AAC.03485-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Svensson EM, Svensson RJ, te Brake LHM, et al. The potential for treatment shortening with higher rifampicin doses: relating drug exposure to treatment response in patients with pulmonary tuberculosis. Clin Infect Dis 2018; 67: 34–41. doi: 10.1093/cid/ciy026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Velasquez GE, Brooks MB, Coit JM, et al. Efficacy and safety of high-dose rifampin in pulmonary tuberculosis. A randomized controlled trial. Am J Respir Crit Care Med 2018; 198: 657–666. doi: 10.1164/rccm.201712-2524OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Pertinez H, Ortega-Muro F, et al. Optimal doses of rifampicin in the standard drug regimen to shorten tuberculosis treatment duration and reduce relapse by eradicating persistent bacteria. J Antimicrob Chemother 2018; 73: 724–731. doi: 10.1093/jac/dkx467 [DOI] [PubMed] [Google Scholar]
- 25.De Jager V, Le Roux S, Mnunu M, et al. Transcutaneous rifampicin concentration monitoring. Abstract presented a the 10th International Workshop on Clinical Pharmacology of Tuberculosis Drugs, Atlanta, GA, 2017.
- 26.Susanto BO, Svensson RJ, Svensson EM, et al. Rifampicin can be given as flat-dosing instead of weight-band dosing. Clin Infect Dis 2020; 71: 3055–3060. doi: 10.1093/cid/ciz1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Phillips PP, Dooley KE, Gillespie SH, et al. A new trial design to accelerate tuberculosis drug development: the phase IIC Selection Trial with Extended Post-treatment follow-up (STEP). BMC Med 2016; 14: 51. doi: 10.1186/s12916-016-0597-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aarnoutse RE, Kibiki GS, Reither K, et al. Pharmacokinetics, tolerability and bacteriological response of 600, 900 and 1200 mg rifampicin daily in patients with pulmonary TB. Antimicrob Agents Chemother 2017; 61: e01054-17. doi: 10.1128/AAC.01054-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.te Brake LHM, Boeree MJ, Aarnoutse RE. Conflicting findings on an intermediate dose of rifampicin for pulmonary tuberculosis. Am J Respir Crit Care Med 2019; 199: 1166–1167. doi: 10.1164/rccm.201811-2101LE [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-00955-2020.SUPPLEMENT (2MB, pdf)
This one-page PDF can be shared freely online.
Shareable PDF ERJ-00955-2020.Shareable (391.1KB, pdf)