

Abstract
In patients with chronic fibrosing interstitial lung disease (ILD), a progressive fibrosing phenotype (PF-ILD) may develop, but information on the frequency and characteristics of this population outside clinical trials is lacking.
We assessed the characteristics and outcomes of patients with PF-ILD other than idiopathic pulmonary fibrosis (IPF) in a real-world, single-centre clinical cohort. The files of all consecutive adult patients with fibrosing ILD (2010–2017) were examined retrospectively for pre-defined criteria of ≥10% fibrosis on high-resolution computed tomography and progressive disease during overlapping windows of 2 years. Baseline was defined as the date disease progression was identified. Patients receiving nintedanib or pirfenidone were censored from survival and progression analyses.
In total, 1395 patients were screened; 617 had ILD other than IPF or combined pulmonary fibrosis and emphysema, and 168 had progressive fibrosing phenotypes. In 165 evaluable patients, median age was 61 years; 57% were female. Baseline mean forced vital capacity (FVC) was 74±22% predicted. Median duration of follow-up was 46.2 months. Annualised FVC decline during the first year was estimated at 136±328 mL using a linear mixed model. Overall survival was 83% at 3 years and 72% at 5 years. Using multivariate Cox regression analysis, mortality was significantly associated with relative FVC decline ≥10% in the previous 24 months (p<0.05), age ≥50 years (p<0.01) and diagnosis subgroup (p<0.01).
In this cohort of patients with PF-ILD not receiving antifibrotic therapy, the disease followed a course characterised by continued decline in lung function, which predicted mortality.
Short abstract
In a real-world clinical cohort (PROGRESS), progressive fibrosing interstitial lung disease was characterised by continued lung function decline. Lung function decline, age and underlying diagnosis subgroup predicted mortality. https://bit.ly/2EB3OpF
Introduction
Interstitial lung diseases (ILDs) are a heterogeneous group of disorders [1] that encompass a wide range of conditions [1, 2]. In some patients with chronic fibrosing ILDs, a progressive phenotype develops comparable to that observed in idiopathic pulmonary fibrosis (IPF), including worsening respiratory symptoms, decline in lung function and early mortality despite conventional treatment [3–5]. IPF is the most common idiopathic fibrosing ILD with inexorable progression [2], with established patient registries and licensed treatments available [6–8]. Based on survey data from multiple countries, progressive fibrosis may develop in ∼18–32% of patients with ILDs other than IPF, with the time from symptom onset to death estimated at 61–80 months [9].
In clinical practice, monitoring disease progression includes various components, covering symptoms, patient-reported outcomes, exercise capacity, serial lung function testing, fibrosis measured by high-resolution computed tomography (HRCT) of the chest, serum biomarkers and need for supportive care [10]. The INBUILD trial demonstrated the efficacy of the antifibrotic drug nintedanib to slow disease progression, as assessed by lung function decline, in patients with non-IPF progressive fibrosing (PF)-ILD (also described as chronic fibrosing ILD with a progressive phenotype) [11], and has since been approved by the United States Food and Drugs Administration in this indication [12, 13]. Although data are available for individual types of PF-ILD (connective tissue disease ILD including systemic sclerosis (SSc)-ILD, hypersensitivity pneumonitis, unclassifiable ILD, etc.), data on PF-ILDs as a group and beyond clinical trials are scarce [11].
Here, we studied retrospective “real-life” data of patients with fibrosing ILDs other than IPF. We assessed the proportion of patients who developed a progressive phenotype and their characteristics, including the underlying disease aetiology. We also evaluated their outcomes and main determinants of mortality.
Materials and methods
Study population
A retrospective observational cohort study was conducted. All consecutive patients with ILD diagnosed by HRCT and hospitalised at a tertiary reference centre between January 1, 2010 and December 31, 2017 were identified using electronic hospital records and assessed for eligibility. Patients were aged ≥18 years and included elective short-term or day-care hospitalisations for diagnostic evaluation; this is standard procedure for the centre and allows for exhaustive identification of ILD cases through electronic hospital records.
Disease progression was considered at every hospital visit, using overlapping windows of 24 months prior to each hospital visit, until the first event meeting the definition criteria for progression was confirmed (after exclusion of all other possible causes; for example, pulmonary embolism, infection, pneumothorax or decompensated heart failure). Therefore, the date of “diagnosis” corresponded to the date of first meeting criteria for PF-ILD and the index date represents the date of first consultation, or January 1, 2010 if this occurred before 2010. The end of follow-up was defined as the date of last follow-up visit, death, lung transplantation or end of study period. Patients who received antifibrotic treatment for >3 months or had a lung transplant during the follow-up period were censored for the lung function analysis and for the survival analysis at the date of treatment initiation or transplant.
Patients were eligible if they had ≥10% fibrosis at baseline HRCT, and pre-defined criteria of disease progression for 2 years prior to the inclusion period (as defined in the INBUILD trial) [14], as follows: relative decline in forced vital capacity (FVC) ≥10% with or without clinical deterioration; relative decline in FVC between 5% and 10% associated with worsening respiratory symptoms; relative decline in FVC between 5% and 10% associated with an increased extent of fibrosis on chest HRCT; or increased extent of fibrosis on chest HRCT with worsening respiratory symptoms. Cases were excluded if they had pulmonary embolism, decompensated heart failure or lower respiratory tract infections linked to progression, IPF or combined pulmonary fibrosis and emphysema. In addition, patients who received antifibrotic treatment (nintedanib or pirfenidone) at baseline were excluded.
This study was conducted under Reference Methodology 004 [15] following the approval on November 9, 2018 (918 305) of the Comité National d'Informatique et Liberté, the committee dedicated to privacy, information technology and civil rights in France. This study was approved by the ethics committee of Hospices Civils de Lyon (Lyon, France).
Collected data
Information was collected on patients' characteristics (age, sex, smoking status) and clinical characteristics (diagnosis, medical history, radiological patterns, lung function, biomarkers, treatment). Gender, Age and Physiology (GAP) index values were calculated [16]. Data on medications were collected throughout the study, including immunosuppressive agents. HRCT scans were reviewed independently for percentage of fibrosis and usual interstitial pneumonia (UIP) pattern [14] by consensus of two experienced thoracic radiologists (SSM, DR), who were blinded to the patient clinical data. Discrepancies were adjudicated by consensus of one radiologist and two experienced pulmonologists (SSM, MN and VC).
Statistical analyses
Descriptive analyses of patients' characteristics were completed using standard summary statistics for distributions.
Change in FVC was analysed using a linear mixed model on raw measures, with a simple random-effect intercept and slope model, a fixed-effect intercept and a fixed-effect slope associated with an unstructured G-matrix covariance. Then, as the time intervals between measurements varied between subjects, we performed a sensitivity analysis using calculated time points for FVC and a similar linear mixed model as for the main analysis. For this analysis, time points were set at 12, 24, 36, 48, 60, 72 and 84 months, and FVC measurements were attributed to those time points using a ±6-month window, using the nearest measurement to the specific time point for the analysis.
Overall survival was defined as the time from the date of inclusion to the date of death due to any cause or the end of follow-up. Survival estimates were performed using the Kaplan–Meier method in the overall population and patient subgroups (by aetiology and FVC decline), and compared by log-rank test. Patients were censored at the time of last clinic visit, lung transplantation or initiation of antifibrotic treatment. As a sensitivity analysis, patients were all censored at the time of last clinic visit or death date, irrespective of initiation of antifibrotic treatment. Exploratory multivariate analyses were performed using a Cox proportional hazard model to determine factors associated with mortality among the following: FVC % predicted at inclusion, decline in FVC ≥10% pred, decline in FVC (5–10% pred) combined with symptoms and extent of fibrosis, diagnosis group, categorised age, sex, diffusing capacity of the lung for carbon monoxide (DLCO) at inclusion and UIP pattern. Proportional hazard assumption was checked for each covariate using Schoenfeld residuals [17], and all prognostic factors that demonstrated associations with overall survival in univariate analyses (p<0.25) were included in the multivariate model. The level of significance was set at p<0.05. Data were analysed using SAS (version 9.4; SAS Institute, Cary, NC, USA).
Results
Selected patient population
Out of 1395 patients identified (figure 1), 617 had fibrosing ILD other than IPF. Among them, 168 (27.2%) had a progressive fibrosing phenotype, and 165 were included in the final dataset after exclusion of three patients receiving antifibrotic treatment at baseline.
FIGURE 1.

Patient flowchart. IPF: idiopathic pulmonary fibrosis; CPFE: combined pulmonary fibrosis and emphysema; ILD: interstitial lung disease.
Two-thirds (66.1%) of patients had progressive disease based on a relative FVC decline ≥10% pred, 24.8% of patients based on a relative FVC decline of 5–10% pred plus worsening of respiratory symptoms or increased extent of fibrosis on HRCT and 9.1% of patients due to worsening of respiratory symptoms and increased extent of fibrotic changes on HRCT, all within 24 months. The mean time interval between ILD diagnosis and meeting the criteria for disease progression was 2.0 (interquartile range (IQR) 0–3.3) years (data were missing for 27 patients), with the longest time being 20.8 years (table 1).
TABLE 1.
Patient cohort characteristics at baseline
| Patients | 165 |
| Female | 94 (57.0) |
| Age years median (range) | 61 (18–83) |
| Former or current smoker | 69 (41.8) |
| GAP index+ | |
| Stage I | 81 (64.3) |
| Stage II | 39 (31.0) |
| Stage III | 6 (4.8) |
| UIP-like fibrotic pattern on HRCT | 46 (27.9) |
| Criteria for disease progression in previous 24 months | |
| Relative FVC decline of ≥10% pred | 109 (66.1) |
| Relative FVC decline of 5–<10% pred plus worsening of respiratory symptoms or increased extent of fibrosis on HRCT | 41 (24.8) |
| Worsening of respiratory symptoms and increased extent of fibrosis on HRCT | 15 (9.1) |
| FVC | |
| Volume mL# | 2333±935 |
| % predicted¶ | 74±22 |
| DLCO# % predicted | 44±18 |
| Time from diagnosis to first meeting criteria for progression# years mean (IQR) | 2.0 (0–3.3) |
Data are presented as n, n (%) or mean±sd, unless otherwise stated. GAP: Gender, Age and Physiology; UIP: usual interstitial pneumonia; HRCT: high-resolution computed tomography; FVC: forced vital capacity; DLCO: diffusing capacity of the lung for carbon monoxide; IQR: interquartile range. +: n=126; #: n=135; ¶: n=138.
Baseline characteristics
Patient characteristics are described in table 1. The median (range) age at inclusion was 61 (18–83) years and 57% of patients were female. Mean±sd FVC at baseline was 74±22% pred (n=138); 125 (90.6%) had FVC ≥45% pred, 82 (59.4%) had FVC ≥70% pred and 58 (42.0%) had FVC ≥80% pred. The mean±sd DLCO was 44±18% pred (n=135); 66.7% (84 out of 126) of patients had both an FVC ≥45% and DLCO 30–80%. The median (IQR) GAP index was 3.0 (2.0–4.0). 27.9% (46 out of 165) of patients had a UIP-like fibrotic pattern on HRCT at baseline (table 1).
Underlying lung disease characteristics
The largest categories by aetiology were autoimmune ILDs (46.7%), unclassifiable ILD (31.5%), chronic fibrosing hypersensitivity pneumonitis (8.5%) and idiopathic interstitial pneumonia (IIP) (7.3%) (table 2).
TABLE 2.
Underlying clinical diagnoses
| Patients | 165 |
| Chronic fibrosing hypersensitivity pneumonitis | 14 (8.5) |
| Idiopathic interstitial pneumonia | 12 (7.3) |
| Unclassifiable ILD | 52 (31.5) |
| Interstitial pneumonitis with autoimmune features | 2 (1.2) |
| Autoimmune ILD | 77 (46.7) |
| Rheumatoid arthritis-ILD | 7 (4.2) |
| Systemic sclerosis-ILD | 43 (26.1) |
| Dermatomyositis-ILD | 12 (7.3) |
| Mixed connective tissue disease-ILD | 10 (6.1) |
| Other autoimmune ILD# | 5 (3.0) |
| Other ILDs¶ | 10 (6.1) |
Data are presented as n or n (%). ILD: interstitial lung disease. #: Sjögren syndrome ILD (n=1), systemic lupus erythematosus (n=1), others (n=3); ¶: exposure-related ILD (n=2), other fibrosing ILD (n=5), sarcoidosis (n=3).
Several patients had concurrent cardiorespiratory conditions (supplementary table S1), including ischaemic heart disease (8.5%) and pulmonary hypertension (7.3%). Other medical conditions occurring in >5% of patients included hypertension (29.7%), gastro-oesophageal reflux disease (15.8%) and diabetes mellitus (20.6%).
Consistent with the underlying disease, a range of autoimmune autoantibodies were detected in the patient cohort (supplementary table S2), with the most frequent being rheumatoid factor (17.6%), anti-Scl-70 (14.5%) and anti-neutrophil cytoplasmic antibodies (10.9%).
Treatment
Almost all patients (98.8%) received at least one treatment (table 3). The most frequent treatment received overall was glucocorticoids (89.0%), and the most frequently used immunosuppressive agent was mycophenolate mofetil (38.7%).
TABLE 3.
Treatments received by patients in the cohort at any time during the course of the study period
| Patients with ≥1 treatment | 163 |
| Treatment received | |
| Azithromycin | 6 (3.7) |
| N-Acetylcysteine | 12 (7.4) |
| Oxygen therapy | 58 (35.6) |
| Glucocorticoids | 145 (89.0) |
| Immunosuppressive agents | |
| Mycophenolate mofetil | 63 (38.7) |
| Cyclophosphamide | 39 (23.9) |
| Rituximab | 29 (17.8) |
| Azathioprine | 24 (14.7) |
| Methotrexate | 21 (12.9) |
Data are presented as n or n (%).
Lung function evolution
Results from the linear mixed model on raw measures showed that mean±sd FVC decline from baseline was –136±328 mL at 12 months (n=114), –183±449 mL at 24 months (n=92), –304±492 mL at 48 months (n=54) and –570±606 mL at 84 months of follow-up (n=16). As a sensitivity analysis, evolution of FVC was analysed with a linear mixed model estimation on raw FVC (mL) at calculated time points, as shown in figure 2. The model allowed estimates of FVC evolution, taking account of variability in both the number of measurements and time intervals between them. The results were consistent with the decline observed in the main analysis, with a mean±sd FVC decline from baseline of –105±13 mL at 12 months, –209±25 mL at 24 months, –419±51 mL at 48 months and –733±89 mL at 84 months of follow-up.
FIGURE 2.

Linear mixed model estimation and 95% confidence intervals on raw forced vital capacity (FVC) (mL) associated with means (95% CI) of FVC (%) at calculated time points. For FVC decline, as the time intervals between measurements varied between subjects, time points were set at 12, 24, 36, 48, 60, 72 and 84 months, and FVC measurements were attributed to those time points using a ±6-month window, using the nearest measurement to the specific time point for analysis. Baseline number of patients at risk (n=135) indicates number of patients with values for pulmonary function tests. Change in FVC was analysed using a linear mixed model on raw measures and calculated time points, defined with a simple random-effect intercepts and slopes model with a fixed-effect intercept and a fixed-effect slope (β1), as well as a random-effect intercept (bi0) and random-effect slope (bi1), associated with an unstructured G-matrix covariance. The mixed model allowed estimates of FVC evolution, taking account of variability in both the number of measurements and time intervals between them.
At 12 months’ follow-up, mean±sd FVC decline was –105±340 mL in patients with non-UIP-like fibrotic pattern at baseline and –220±282 mL in patients with a UIP-like pattern; however, there was no significant difference between the subgroups, since confidence intervals overlapped. Furthermore, decline was very close among the two groups and at subsequent time points (supplementary figure S1).
Acute exacerbations
23 (13.9%) patients had at least one acute exacerbation, with a total of 26 acute exacerbations; three patients each reported two events.
Survival
The median (IQR) duration of follow-up was 46.2 (25.3–73.3) months. At last follow-up, no patient had received a lung transplant and 39 (23.0%) out of 165 patients had died: nine deaths were from unknown causes, three from nonrespiratory and 27 from respiratory causes. Overall survival at 12, 48 and 84 months was 99.3% (95% CI 95.4–99.9%), 77.1% (68.6–83.6%) and 65.0% (54.7–73.5%), respectively (figure 3a). Because >50% of the population survived, the median overall survival was not reached.
FIGURE 3.
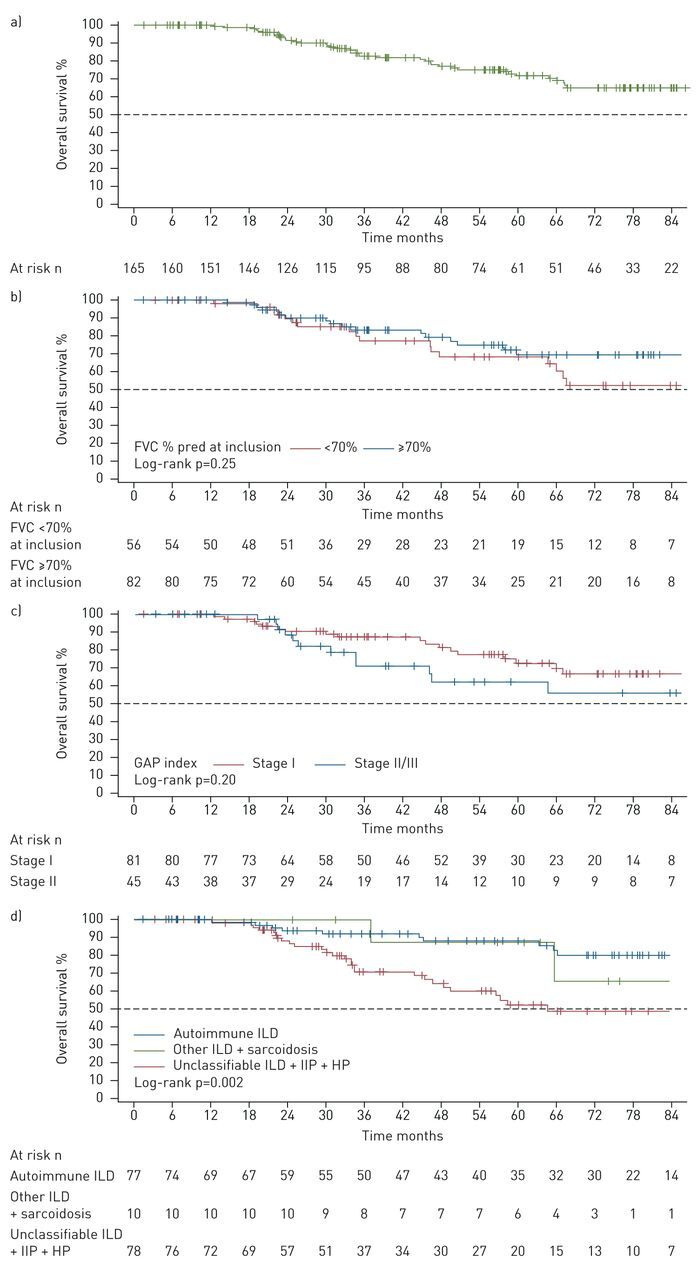
Overall survival. a) Overall patient cohort; b) overall survival according to forced vital capacity (FVC) percentage at inclusion with a 70% threshold; c) overall survival according to Gender, Age and Physiology (GAP) index; d) overall survival according to disease subgroups. Survival estimates were performed using the Kaplan–Meier method in a) the overall population and b) patient subgroups by FVC decline, c) GAP index stage and d) disease subgroups compared by log-rank test. Baseline was defined as the date of disease progression. Patients were censored at the time of last clinic visit, lung transplantation for interstitial lung disease (ILD) or initiation of antifibrotic treatment. IIP: idiopathic interstitial pneumonia; HP: hypersensitivity pneumonitis.
For patients with available FVC % pred at inclusion, 17 (20.7%) out of 82 patients with FVC ≥70% pred had died; and 17 (30.4%) out of 56 patients with FVC <70% had died. A log-rank test found no significant difference in overall survival according to FVC at inclusion with a ≥70% threshold (p=0.25), although overall survival was numerically higher in the group with ≥70% at all time points from 12 to 84 months (figure 3b and supplementary table S3).
At the end of follow-up, 18 (22.2%) out of 81 patients with GAP index stage I and 12 (26.7%) out of 45 patients with stage II/III had died. A log-rank test found no significant difference in overall survival according to GAP stage (p=0.20) (figure 3c and supplementary table S4).
When analysed by disease subgroup, the overall survival at 84 months was higher for patients with autoimmune ILD and patients with sarcoidosis or other ILDs compared with patients with unclassifiable ILD, IIP or hypersensitivity pneumonitis (figure 3d and supplementary table S5); there was a significant difference between groups (log-rank p=0.002).
When analysed by criteria for progression, 29 (26.6%) out of 109 patients with relative FVC decline ≥10% at baseline had died; nine (22.2%) out of 41 patients with relative decline of 5–<10% pred plus worsening of respiratory symptoms or increased extent of fibrosis on HRCT had died; and one (6.7%) out of 15 patients with worsening of respiratory symptoms and increased extent of fibrosis with a relative FVC decline of <5% had died. There was no significant difference in overall survival between the three groups (log-rank p=0.32) (supplementary figure S2a and supplementary table S6).
In total, 10 (21.7%) out of 46 patients with UIP-like fibrotic pattern on HRCT at baseline and 29 (24.4%) out of 119 patients with non-UIP-like pattern died. A log-rank test found no significant difference in overall survival according to UIP-like fibrotic pattern (p=0.95) (supplementary figure S2b and supplementary table S7).
Using factors pre-selected by a univariate analysis (supplementary table S8), the final multivariate Cox model (table 4) showed that mortality was significantly associated with FVC <70% at inclusion (hazard ratio (HR) 1.90, 95% CI 0.96–3.77; p=0.065), FVC decline ≥10% at baseline as an inclusion criterion (HR 2.43, 95% CI 1.06–5.61; p=0.04), diagnosis of unclassifiable ILD, IIP or hypersensitivity pneumonitis versus autoimmune ILD (HR 3.45, 95% CI 1.59–7.50; p=0.002) and age ≥50 years (HR 5.03, 95% CI 1.53–16.54; p=0.008).
TABLE 4.
Factors associated with mortality (multivariate Cox model)
| Parameter | Reference | Observations n | HR (95% CI) | p-value |
| FVC % at inclusion | <70% | 131 | 1.90 (0.96–3.77) | 0.065 |
| Decline in FVC of ≥10% at baseline | Yes | 131 | 2.43 (1.06–5.61) | 0.04 |
| Diagnosis | Unclassifiable ILD or IIP or HP versus autoimmune ILD | 131 | 3.45 (1.59–7.50) | 0.002 |
| Categorised age | ≥50 years | 131 | 5.03 (1.53–16.54) | 0.008 |
HR: hazard ratio; FVC: forced vital capacity; ILD: interstitial lung disease; IIP: idiopathic interstitial pneumonia; HP: hypersensitivity pneumonitis.
A sensitivity analysis was performed without censoring the 23 (14%) patients who initiated an antifibrotic therapy during the study period at the initiation of the therapy. All 23 patients met the criteria for disease progression before initiating therapy. None of these patients died, and therefore no event was added in the analysis. The impact on the factor estimates was very limited, leading to an overall robustness of results in terms of p-values and point estimates (supplementary table S9). Only the point estimate and p-value for FVC % was modified, with a weaker association but still a trend that remained close to significance. The assumption of proportionality of hazard was respected for all the variables included in the model.
Discussion
These results provide essential information on patients with PF-ILDs other than IPF. A progressive phenotype was observed in approximately one-quarter of patients with fibrosing ILDs other than IPF, and was associated with continued decline in lung function.
The baseline demographic characteristics of patients with PF-ILDs in the present PROGRESS clinical cohort were generally similar to those enrolled in the placebo arm of the INBUILD study [11]. However, a larger proportion of the patients in our cohort had SSc-ILD (26.1%) than in INBUILD (<7%) [11]. The relative lack of SSc-ILD in the INBUILD cohort may be due to other trials competing for enrolment of patients with SSc-ILD, including the SENSCIS [18], faSScinate [19] and focuSSced [20] trials. The proportion of patients with unclassifiable ILD was similar in PROGRESS and INBUILD, but proportions differed for all other underlying diseases.
The substantial decline in FVC in the PROGRESS clinical cohort after 52 weeks (–136 mL) was lower than the decline in FVC observed in the placebo groups in the INBUILD [11] and INPULSIS [21] trials (–187.8 mL and –207.3–239.9 mL, respectively), although there was no fixed time point for follow-up on pulmonary function tests in our study, given its retrospective nature. This difference may be because the majority of patients in this study received immunosuppressant treatment during the follow-up period and only a small proportion of patients had a UIP pattern of disease (although no difference in FVC decline was found according to the computed tomography pattern in PROGRESS). In addition, FVC % pred at baseline was higher in our cohort compared with INBUILD (74.0% versus 69.3%). Estimation of the FVC decline was limited by the relatively small sample size, and variability in both the number of measurements and time intervals between them. However, a sensitivity analysis was performed using recalculated time points. The results were consistent with those observed with the raw data, confirming the substantial FVC decline among the cohort. Furthermore, multivariate analyses found that FVC decline ≥10% was significantly associated with mortality.
Nearly 30% of patients had a UIP-like fibrotic pattern on HRCT imaging at baseline. In comparison, ∼62% of patients in the INBUILD trial had a UIP-like pattern at baseline. It is conceivable that patients with a UIP-like fibrotic pattern were more likely to be recommended for the INBUILD trial than their counterparts with non-UIP patterns. The extent of FVC decline over 12 months from baseline in patients with a UIP-like fibrotic pattern in the current study was similar to placebo-treated participants in the INBUILD trial [11], and to pooled data from the INPULSIS trials for patients that met a case definition for IPF (−220 mL, −211.1 mL and −223.5 mL per year, respectively). The rate of FVC decline in patients with non-UIP-like fibrotic patterns was lower than in those with UIP-like patterns (−105 mL), again similar to the findings of the INBUILD trial.
Patients in our study had less advanced disease and therefore a lower mortality rate compared with INBUILD. In PROGRESS, progression was assessed in all patients at the first visit where they fit the criteria for progression within the 8-year study period. In contrast, patients were selected for INBUILD based on eligibility criteria for progression when the trial was open, suggesting that patients with more progression were more likely to be referred, and irrespective of when the progression occurred.
Almost all patients in this study (98.8%) had received previous treatments (treatment type not assessed), and many received some medications throughout the study (table 3). In contrast, for the INBUILD trial no information was available regarding previous immunosuppressive treatments received, and concomitant medications were limited to oral glucocorticoids (prednisone ≤20 mg·day−1) or disease-modifying antirheumatic drugs for rheumatoid arthritis or connective tissue disease [11]. Patients in our cohort continued to progress, despite 91.4% receiving immunosuppressive treatment during follow-up, which underlines the need for alternative therapy. In INBUILD, nintedanib reduced the annual rate of FVC decline by 107.0 mL per year versus placebo [11]. In PROGRESS, patients were not receiving off-label antifibrotic therapy, and were censored for the survival and FVC analysis if they did. It remains to be explored how the previous treatments or immunosuppressive medications taken during the study period may have affected disease progression in the PROGRESS study cohort.
Median overall survival was not reached in this cohort, despite the long follow-up. 5-year overall survival was ∼73% in our cohort, compared with 70% in unclassifiable ILD and 25–30% in fibrotic hypersensitivity pneumonitis in other patient cohorts [22, 23]. There was some heterogeneity in the progression rate across subgroups according to underlying lung disease. Patients in the autoimmune and sarcoidosis or other ILD subgroups had 7-year overall survival of 80% and 86%, respectively, compared with only 50% of patients with unclassifiable ILD, IIP or hypersensitivity pneumonitis.
Numerically, there was reduced survival in patients with unclassifiable ILD and hypersensitivity pneumonitis, in those with relative FVC decline ≥10%, and in patients with baseline FVC <70% and GAP index stage II; presence or absence of UIP-like fibrotic pattern at baseline did not affect overall survival. In our exploratory analysis, multivariate modelling confirmed that FVC decline was predictive of mortality, similar to IPF [24]; therefore, changes in FVC are potentially clinically meaningful, providing a surrogate measure for disease progression and mortality. Sensitivity analyses performed without censoring patients at initiation of antifibrotic treatment were consistent with the results of the main analysis, producing estimates that were similar to the main analysis and highlighting the robustness of the results. In unclassifiable ILD, DLCO has already been shown as an independent predictor of disease progression and mortality [22]. The survival rate of unclassifiable ILD, with worse overall survival compared with autoimmune ILD, is in line with existing literature [25, 26]. However, a validation cohort would be required to confirm the above predictors of mortality.
Patients matching the third criterion for progression at baseline (worsening of respiratory symptoms and increased extent of fibrosis with a relative FVC decline of <5%) had numerically higher overall survival than patients who matched the first two criteria (91.7% at 8 years versus 62.3% and 69.0%, respectively). It cannot be excluded that the higher survival outcomes in this subgroup were related to difficulties in assessing symptom progression. Therefore, decline of FVC >5% appears to be important.
This study has several limitations related to its retrospective and monocentric design. However, all consecutive patients were identified using hospital electronic records, limiting selection bias, and there were no missing survival data. While data were routinely collected, information on patient end-points was not collected at fixed intervals. Our pre-defined criteria for disease progression and estimate of FVC decline could only be assessed against available patient data; however, there was a long follow-up with few missing data for FVC, still allowing robust statistical analysis. Some patients had disease progression and were excluded from the analysis due to off-label antifibrotic therapy, and in others progression did not meet the inclusion criteria, which may have led to underestimating the prevalence of PF-ILDs. Acute exacerbations are an important event associated with increased mortality in patients with fibrotic ILDs. However, the amount of missing data might be important due to the retrospective design, especially for patients who may have been hospitalised outside of our institution. Therefore, we focused on overall mortality, for which no patient was lost to follow-up.
In conclusion, the current study provides valuable information on a clinical cohort of patients with PF-ILDs in a real-world setting. These results show that, in this single-centre academic cohort, the course of disease in patients with PF-ILDs is characterised by continued progressive lung function decline, which along with age and underlying diagnosis subgroup was predictive of mortality. There remains an unmet need to slow disease progression in patients with PF-ILDs.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-02718-2020.SUPPLEMENT (451.3KB, pdf)
Shareable PDF
Acknowledgements
The authors wish to thank RaDiCo-PID and ERN-LUNG.
Footnotes
This article has supplementary material available from erj.ersjournals.com
This study is registered with ClinicalTrials.gov number NCT03858842.
Conflict of interest: M. Nasser received sponsorship for conference attendance from Boehringer Ingelheim and Hoffmann-La Roche, and received consultation fees from Boehringer Ingelheim.
Conflict of interest: S. Larrieu has nothing to disclose.
Conflict of interest: S. Si-Mohamed has nothing to disclose.
Conflict of interest: K. Ahmad reports relationships and activities from Roche and Boehringer Ingelheim, outside the submitted work.
Conflict of interest: L. Boussel has nothing to disclose.
Conflict of interest: M. Brevet has nothing to disclose.
Conflict of interest: L. Chalabreysse has nothing to disclose.
Conflict of interest: C. Fabre has nothing to disclose.
Conflict of interest: S. Marque has nothing to disclose.
Conflict of interest: D. Revel declares provision of scientific expertise under contract with IQVIA.
Conflict of interest: F. Thivolet-Bejui has nothing to disclose.
Conflict of interest: J. Traclet reports sponsorship for meeting attendance from Roche and Boehringer Ingelheim, outside the submitted work.
Conflict of interest: S. Zeghmar has nothing to disclose.
Conflict of interest: D. Maucort-Boulch has nothing to disclose.
Conflict of interest: V. Cottin reports personal fees for advisory board work and lectures, and non-financial support for meeting attendance from Actelion, grants, personal fees for consultancy and lectures, and non-financial support for meeting attendance from Boehringer Ingelheim and Roche, personal fees for advisory board and data monitoring committee work from Bayer/MSD, personal fees for advisory board work and lectures from Novartis, personal fees for lectures from Sanofi, personal fees for data monitoring and steering committee work from Promedior, personal fees for data monitoring committee work from Celgene and Galecto, and personal fees for advisory board and data monitoring committee work from Galapagos, outside the submitted work.
Support statement: The PROGRESS trial was funded by an unrestricted grant from Boehringer Ingelheim International GmbH (BI) to Hospices Civils de Lyon. IQVIA received financial support from Boehringer Ingelheim Santé Humaine France for the design, monitoring, data-management and statistical analysis of the study. Medical writing, editorial support and formatting assistance were provided by Helen Keyworth of Nucleus Global, which was contracted and funded by BI. The authors did not receive payment for the development of the manuscript. Funding information for this article has been deposited with the Crossref Funder Registry.
References
- 1.Travis WD, Costabel U, Hansell DM, et al. . An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013; 188: 733–748. doi: 10.1164/rccm.201308-1483ST [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valeyre D, Duchemann B, Nunes H, et al. . Interstitial lung diseases. In:Annesi-Maesano I, Lundbäck B, Viegi G, eds. Respiratory Epidemiology (ERS Monograph). Sheffield, European Respiratory Society, 2014; p. 79–87. [Google Scholar]
- 3.Wells AU, Brown KK, Flaherty KR, et al. . What's in a name? That which we call IPF, by any other name would act the same. Eur Respir J 2018; 51: 1800692. doi: 10.1183/13993003.00692-2018 [DOI] [PubMed] [Google Scholar]
- 4.Akira M, Inoue Y, Arai T, et al. . Long-term follow-up high-resolution CT findings in non-specific interstitial pneumonia. Thorax 2011; 66: 61–65. doi: 10.1136/thx.2010.140574 [DOI] [PubMed] [Google Scholar]
- 5.Kim EJ, Elicker BM, Maldonado F, et al. . Usual interstitial pneumonia in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J 2010; 35: 1322–1328. doi: 10.1183/09031936.00092309 [DOI] [PubMed] [Google Scholar]
- 6.Guenther A, Krauss E, Tello S, et al. . The European IPF registry (eurIPFreg): baseline characteristics and survival of patients with idiopathic pulmonary fibrosis. Respir Res 2018; 19: 141. doi: 10.1186/s12931-018-0845-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Behr J, Kreuter M, Hoeper MM, et al. . Management of patients with idiopathic pulmonary fibrosis in clinical practice: the INSIGHTS-IPF registry. Eur Respir J 2015; 46: 186–196. doi: 10.1183/09031936.00217614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wuyts WA, Dahlqvist C, Slabbynck H, et al. . Baseline clinical characteristics, comorbidities and prescribed medication in a real-world population of patients with idiopathic pulmonary fibrosis: the PROOF registry. BMJ Open Respir Res 2018; 5: e000331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wijsenbeek M, Kreuter M, Olson A, et al. . Progressive fibrosing interstitial lung diseases: current practice in diagnosis and management. Curr Med Res Opin 2019; 35: 2015–2024. doi: 10.1080/03007995.2019.1647040 [DOI] [PubMed] [Google Scholar]
- 10.Cottin V. Treatment of progressive fibrosing interstitial lung diseases: a milestone in the management of interstitial lung diseases. Eur Respir Rev 2019; 28: 190109. doi: 10.1183/16000617.0109-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flaherty KR, Wells AU, Cottin V, et al. . Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med 2019; 381: 1718–1727. doi: 10.1056/NEJMoa1908681 [DOI] [PubMed] [Google Scholar]
- 12.U.S. Food & Drug Administration. FDA Approves First Treatment for Group of Progressive Interstitial Lung Diseases. www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-group-progressive-interstitial-lung-diseasesDate last accessed: March 10, 2020.
- 13.European Medicines Agency. OFEV® (nintedanib): Summary of Product Characteristics. Date last accessed: September 24, 2020. www.ema.europa.eu/en/documents/product-information/ofev-epar-product-information_en.pdf
- 14.Flaherty KR, Brown KK, Wells AU, et al. . Design of the PF-ILD trial: a double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Respir Res 2017; 4: e000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Commission Nationale de l'Informatique et des Libertés. Délibération no 2018-155 du 3 Mai 2018 Portant Homologation de la Méthodologie de Référence Relative aux Traitements de Données à Caractère Personnel Mis en Oeuvre dans le Cadre des Recherches n'Impliquant pas la Personne Humaine, des Études et Évaluations dans le Domaine de la Santé (MR-004). [Deliberation No. 2018-155 of May 3, 2018 Approving the Reference Methodology Relating to the Processing of Personal Data Implemented in the Framework of Research not Involving Human Persons, Studies and Evaluations in the Health Field (MR-004)]. Journal Officiel de République Française, 2018. CNIL1818709X.
- 16.Ley B, Ryerson CJ, Vittinghoff E, et al. . A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med 2012; 156: 684–691. doi: 10.7326/0003-4819-156-10-201205150-00004 [DOI] [PubMed] [Google Scholar]
- 17.Schoenfeld D. Partial residuals for the proportional hazards regression model. Biometrika 1982; 69: 239–241. doi: 10.1093/biomet/69.1.239 [DOI] [Google Scholar]
- 18.Distler O, Highland KB, Gahlemann M, et al. . Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med 2019; 380: 2518–2528. doi: 10.1056/NEJMoa1903076 [DOI] [PubMed] [Google Scholar]
- 19.Khanna D, Denton CP, Jahreis A, et al. . Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet 2016; 387: 2630–2640. doi: 10.1016/S0140-6736(16)00232-4 [DOI] [PubMed] [Google Scholar]
- 20.Khanna D, Lin CJF, Goldin J, et al. . OP0245 Preservation of lung function observed in a phase 3 randomized controlled trial of tocilizumab for the treatment of early SSc. Ann Rheum Dis 2019; 78: Suppl. 2, 202–203. [Google Scholar]
- 21.Richeldi L, du Bois RM, Raghu G, et al. . Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2071–2082. doi: 10.1056/NEJMoa1402584 [DOI] [PubMed] [Google Scholar]
- 22.Ryerson CJ, Urbania TH, Richeldi L, et al. . Prevalence and prognosis of unclassifiable interstitial lung disease. Eur Respir J 2013; 42: 750–757. doi: 10.1183/09031936.00131912 [DOI] [PubMed] [Google Scholar]
- 23.Adegunsoye A, Oldham JM, Fernández Pérez ER, et al. . Outcomes of immunosuppressive therapy in chronic hypersensitivity pneumonitis. ERJ Open Res 2017; 3: 00016-2017. doi: 10.1183/23120541.00016-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collins BF, Raghu G. Antifibrotic therapy for fibrotic lung disease beyond idiopathic pulmonary fibrosis. Eur Respir Rev 2019; 28: 190022. doi: 10.1183/16000617.0022-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Traila D, Oancea C, Tudorache E, et al. . Clinical profile of unclassifiable interstitial lung disease: comparison with chronic fibrosing idiopathic interstitial pneumonias. J Int Med Res 2018; 46: 448–456. doi: 10.1177/0300060517719767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hyldgaard C, Bendstrup E, Wells AU, et al. . Unclassifiable interstitial lung diseases: clinical characteristics and survival. Respirology 2017; 22: 494–500. doi: 10.1111/resp.12931 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material ERJ-02718-2020.SUPPLEMENT (451.3KB, pdf)
This one-page PDF can be shared freely online.
Shareable PDF ERJ-02718-2020.Shareable (478.5KB, pdf)