

Abstract
Upon exposure to blood, a corona of proteins adsorbs to nanocarrier surfaces to confer a biological identity that interfaces with the immune system. While the nanocarrier surface chemistry has long been the focus of protein corona formation, the influence of nanostructure has remained unclear despite established influences on biodistribution, clearance, and inflammation. Here, combinations of nanocarrier morphology and surface chemistry are engineered to i) achieve compositionally distinct protein coatings in human blood and ii) control protein-mediated interactions with the immune system. A library of nine PEGylated nanocarriers differing in their combination of morphology (spheres, vesicles, and cylinders) and surface chemistry (methoxy, hydroxyl, and phosphate) are synthesized to represent properties of therapeutic and biomimetic delivery vehicles. Analysis by quantitative label-free proteomic techniques reveal that specific surface chemistry and morphology combinations adsorb unique protein signatures from human blood, resulting in differential complement activation and elicitation of distinct proinflammatory cytokine responses. Furthermore, nanocarrier morphology is shown to primarily influence uptake and clearance by human monocytes, macrophages, and dendritic cells. This comprehensive analysis provides mechanistic insights into rational design choices that impact the immunological identity of nanocarriers in human blood, which can be leveraged to engineer drug delivery vehicles for precision medicine and immunotherapy.
Keywords: immunotherapy, morphology, nanocarrier, protein adsorption, rational design, surface chemistry
1. Introduction
Nanoscale carriers, i.e., nanocarriers, serve as delivery vehicles for therapeutic and diagnostic agents and are frequently customized to possess a wide range of application-specific physicochemical properties.[1,2] The fate of nanocarriers following in vivo administration is highly dependent on their interactions with cells of the immune system,[3] which is of significant interest for therapeutic strategies involving immunomodulation.[1,2,4] In these applications, nanocarriers are utilized with the intent of altering the host’s immune system by eliciting either a pro- or anti-inflammatory response through targeting various immune cell subsets. While the cellular target may vary based on the specific application or disease state, the cells of the mononuclear phagocyte system (MPS), namely dendritic cells, macrophages, and monocytes, have garnered significant attention due to their importance in maintaining homeostasis and contributions to inflammation and autoimmunity.[5] A paradoxical scenario is presented by the diverse cellular functions of the MPS, where these cells represent both desirable targets for the administered therapy, as well as sources of nonspecific uptake capable of quickly clearing the nanocarrier from circulation before it can complete its desired function.[2,6] As such, understanding how nanocarriers can be rationally designed to avoid or target specific components of the MPS has been an area of active investigation. These efforts have included both active[7] and passive[8] targeting strategies that typically involve the respective surface display of targeting ligands or the modulation of either surface chemistry or morphology of the nanocarrier.[9–12]
After introducing nanocarriers into the biological milieu, the once meticulously designed and characterized synthetic identity of the nanocarrier surface is immediately replaced by a new immunological identity defined by an adsorbed protein corona.[13] This dynamic shell of adsorbed biomolecules at the nanocarrier-fluid interface is the actual surface that cells interact with and plays a central role in defining nanocarrier interactions with immune cells and thus their inflammatory potential and circulation time.[14] A variety of physicochemical characteristics of the biomaterial employed, including morphology,[15] surface chemistry,[13,16–18] surface charge,[17,19] size,[13,15,19–21] radius of curvature,[22] and porosity,[23] influence both the amount and identity of the absorbed protein. These sometimes subtle compositional changes within the protein corona have been attributed to both the promotion[24] and suppression[24,25] of nanocarrier internalization by MPS cells.[26] Furthermore, these protein coatings eventually dictate how effective the engineered construct is at reaching and interacting with its cellular target.[26,27] Therefore, rationally designing the nanocarrier chassis (i.e., combined surface chemistry and morphology framework) to govern the composition of the protein corona, rather than to prevent its formation, is a potential strategy for tailoring cellular uptake.
Poly(ethylene glycol)-block-poly(propylene sulfide) (PEG-b-PPS) is an established block copolymer (BCP) that has been employed to produce a variety of nanocarrier chassis types designed for immunomodulatory applications.[17,28–39] The chassis morphology is dictated by the BCP composition and hydrophilic mass fraction (fPEG), allowing the formation of PEG-b-PPS spherical micelles (MCs),[40–43] vesicular polymersomes (PSs),[17,37,38,44,45] and cylindrical filomicelles (FMs).[33,35,36] For the PEG-b-PPS BCP system, morphological complexity is inversely related to fPEG, where fPEG < 0.12 results in bicontinuous nanospheres (BCNs), 0.22 < fPEG < 0.35 results in PSs, 0.37 < fPEG < 0.39 results in FMs, and fPEG > 0.45 forms MCs.[31,33,36,38,39] This characteristic of self-assembled systems allows diverse morphologies to be prepared with the same chemical identity, and thereby permits the evaluation of morphology-driven variation in nanocarrier chassis performance. Previous studies have highlighted that PEG-b-PPS nanocarrier morphology is instrumental in governing material-host interactions, as evidenced by the altered organ- and cellular-level biodistributions achieved by these nanocarriers.[33,37,46] However, attempts to further tailor cellular interactions of PEG-b-PPS nanocarriers by combining the influence of morphology with surface chemistry have been limited.[33]
Here, we systematically investigate the relationship between material rational design choices for the nanocarrier chassis and their consequences on the biological performance of delivery vehicles in human blood. We synthesize PEG-b-PPS BCPs with fPEG ratios that self-assemble into MC, FM, and PS morphologies, and terminate PEG with either methoxy (MeO), hydroxy (OH), or phosphate (Phos) chemical groups. This BCP library results in a total of nine nanocarrier formulations that vary in their combination of morphology (sphere, vesicle and cylinder) and surface chemistry (MeO, OH, and Phos). These morphology choices include clinically-relevant spherical delivery vehicles (micelles and vesicles),[47] as well as a cylindrical micelle chassis that has gained attention in preclinical studies for its longer circulation time and versatile release properties.[33,35,36,48,49] Regarding surface chemistry, our objective was to include functional groups with greatest relevance to systemic delivery applications. Previous studies involving PEGylated nanocarriers and drugs have largely utilized MeO- or OH-functionalization for neutral surfaces,[47,50,51] necessitating their inclusion in our investigation. PEG contains an OH group at its terminus and provides a model hydroxylated surface that is prone to complement activation due to its potential for covalent attachment to C3 protein.[52–55] The MeO-functionalization provides a relatively inert surface that is commonly employed in FDA-approved liposomal formulations to minimize protein adsorption and overcome complement activation issues.[56–59] Our library also included anionic surfaces as a charged alternative surface chemistry. The phosphate group is an anionic, biomimetic surface chemistry that we previously reported to modulate nanocarrier uptake by MPS cells.[17] A cationic surface was not pursued in our investigation, due to its greater propensity for nonspecific and often toxic interactions with negatively charged biological membranes[60] and lower bioavailability compared to neutral or anionic surfaces.[61] We employ label-free proteomics and immunological assays to assess protein adsorption, complement activation, immune cell interactions, and cytokine responses after introducing this nanocarrier library to human blood samples. By linking the combined morphological and chemical properties of nanocarriers with the immunological identity of the resulting protein corona, we provide design principles to guide the future design and development of soft nanocarriers for applications in drug delivery and immunotherapy.
2. Results
2.1. Synthesis and Physicochemical Characterization of the PEG-b-PPS Nanocarrier Library
Phos-, OH-, and MeO-functionalized BCPs were each synthesized with varying fPEG to prepare PS, FM, and MC exhibiting each of the three aforementioned surface chemistries (Figure 1A,B; Figures S1–S3, Supporting Information). CryoTEM micrographs (Figure 1C; Figure S4, Supporting Information) confirmed that each BCP, irrespective of the end terminus chemistry, self-assembled into its anticipated morphology as predicted by the fPEG. The size characteristics of each nanocarrier formulation in the library (Figure 1D) was assessed by DLS and SAXS (Figure 1E; Figure S5, Supporting Information). Zeta potential was assessed via ELS (Figure 1F).
Figure 1.
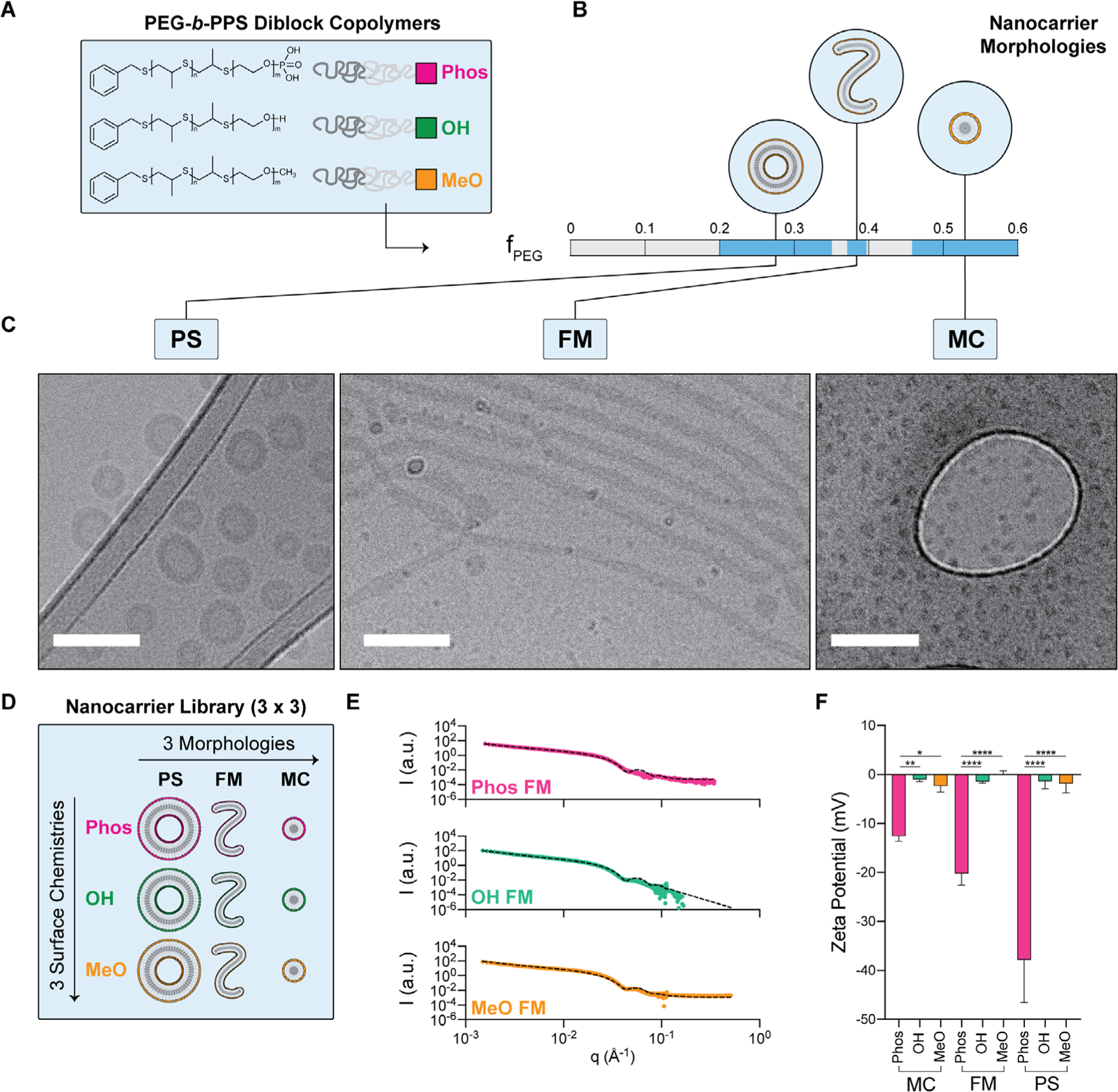
PEG-b-PPS nanocarrier chassis library development and physicochemical characterization A) Schematic of the different functionalized BCPs terminated with phosphate-, hydroxyl-, or methoxy- chemical groups. B) The BCP hydrophilic mass fraction (fPEG) was varied to promote the favorable self-assembly of three morphologies: polymersomes (PS), filomicelles (FM), and micelles (MC). C) Cryogenic TEM micrographs of PS, FM, and MC nanocarrier morphologies (Scale bar = 100 nm). The OH-functionalized examples are displayed as an example. D) Illustration of the complete library of nanocarriers chassis (i.e., distinct combinations of morphology and surface chemistry). E) SAXS profiles of PEG-b-PPS FMs overlaid with corresponding fit for a flexible cylinder model (χ2 = 0.004, 0.0439, 0.777 for MeO-, OH-, and Phos-FMs, respectively). F) Zeta potential of each nanocarrier formulation (n = 3). Significance was determined with Tukey’s multiple comparison test. *p < 0.05, **p < 0.01, ****p < 0.0001. Error bars represent s.d.
PS, FM, and MC nanostructures exhibiting either hydroxyl or methoxy surface functionalities had zeta potentials ranging from −2 to 0 mV, whereas the zeta potential of nanocarriers self-assembled from Phos-functionalized BCPs ranged from −38 to −13 mV. The zeta-potential of Phos-functionalized nanocarriers increased with the fPEG of the BCPs (i.e., the smaller the fPEG the more negative the zeta potential). This may arise from morphology-specific molecular packing differences that result in variations in the surface area per BCP,[62] thereby manifesting as zeta potential differences between the Phos nanocarrier types. Additionally, morphological differences, pertaining to both size[63] and shape,[64,65] have been implicated in influencing particle electrophoretic mobility in solution. This latter effect may contribute to the differences observed between Phos MC and Phos FM, since the orientation of high aspect ratio structures, like the FM morphology, in the direction of the electric field can increase electrophoretic mobility.[64,65] Table S1 in the Supporting Information displays physicochemical characteristics for both spherical and cylindrical nanocarriers. We note that while size influences the protein adsorption process,[13,15,18,19,21] our investigation examines the relationship between morphology and surface chemistry for self-assembling polymeric nanocarriers of defined size without further manipulation to their physical properties.
2.2. Overview of Human Plasma Protein Adsorption to Distinct Nanocarrier Chassis Types
2.2.1. The amount of Adsorbed Protein Decreases with Time for All Chassis Types Except for the Methoxy-Terminated PS
We first assessed protein adsorption to the nine nanocarrier types and investigated whether their physicochemical properties alter the composition of the protein coronas formed in human plasma (Figure 2A). Our past studies demonstrate that PEG-b-PPS nanocarriers exhibit good serum and in vivo stability,[17,28,33,34,66,67] and polymersomes of MeO, OH, and Phos surface chemistry are stable in complex with serum albumin, serum protein mixtures, and in blood protein exchange experiments.[17] Here, we first developed and validated an ultracentrifugation-based procedure for isolating all PEG-b-PPS nanocarrier chassis types following corona formation (Figure 2A). Ultracentrifugation is the most commonly used method for protein corona isolation, and is more commonly employed than size exclusion chromatography (SEC) and magnetic separation techniques.[68] The Pierce A660 assay was used to determine adsorbed protein concentration, since this assay exhibits the highest specificity for protein with lowest interference from PEG-b-PPS copolymer (Figure S6, Supporting Information). In the present study, SEC was ruled out as a viable separation technique since it is unable to resolve micelles and free protein (Figure S7, Supporting Information). Iterative rounds of ultracentrifugation (at 100000 × g for 45 min at 4 °C) and washing was capable of isolating micelles without substantial changes to diameter or PDI (Figure S8, Supporting Information). Furthermore, human plasma proteins did not sediment at detectable levels using our optimized conditions (Figure S9, Supporting Information), which allowed identification of adsorbed protein while minimizing false positives.
Figure 2.

A) Overview of proteomic analysis of human plasma proteins adsorbed to each chassis type after 2 and 24 h. Total adsorbed protein measured in nanocarrier-protein complexes isolated after incubation with human plasma for B) two hours and C) 24 h. D) Difference in total adsorbed protein at the 24 h timepoint relative to the 2 h timepoint. For all cases, error bars represent s.e.m., n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. E) Silver stained adsorbed proteins separated by SDS-PAGE grouped by micelle (left gel), filomicelle (middle gel), and polymersome (right gel) morphologies. 1 µg of total protein was run per lane for each sample. An orange arrow highlights the high molecular weight region where there are the greatest clear differences across nanocarrier types. F) The total number of each protein corona identified by label-free quantitation. G,H) Venn diagrams depicting common and shared constituents identified in each protein corona. Morphological comparisons within each surface chemistry are displayed in (G), whereas (H) displays surface chemistry comparisons within each morphology.
Nanocarriers (5 mg mL−1 polymer concentration) were incubated 1:1 with human plasma pooled from healthy donors to form nanocarrier-protein complexes (Figure S10, Supporting Information). Significant changes in total adsorbed protein were found between different surface chemistries within a specified morphology at 2 h (Figure 2B) and 24 h (Figure 2C) time points. For MC and FM morphologies, the lowest level of protein adsorption was consistently observed for MeO-functionalized nanocarriers (Figure 2B,C). This result suggested the methoxy group renders nanocarriers most resistant to protein adsorption. A departure from this trend was observed for the PS chassis at 24 h, where the minimum adsorption was found for OH PS, rather than MeO PS (Figure 2C). Interestingly, MeO-functionalized PS was the only chassis type that accumulated protein with time, whereas adsorption decreased over time for all other nanocarriers (Figure 2D). Time-dependent decreases in protein adsorption were observed in eight of the nine formulations (Figure 2D), as observed by the lower levels of adsorbed protein at the 24 h timepoint compared to 2 h (Figure 2B,C). This observation suggests the total amount of adsorbed protein decreases as the protein coatings equilibrate with time (Figure 2B–D). In summary, polar surface chemistries were generally more prone to protein adsorption, whereas the MeO surface chemistry minimized protein adsorption. Neutral surfaces (OH, MeO) had the most stable level of adsorbed protein between timepoints, whereas polar surfaces were more dynamic (Figure 2D).
2.2.2. The Adsorption of 43–154 Human Plasma Proteins Differs between Chassis Types
We next examined whether the bulk composition and relative abundance of adsorbed proteins differed depending on morphology, surface chemistry, and plasma incubation time. To this end, the adsorbed protein was separated by SDS-PAGE and protein species were detected by silver-staining (Figure 2E). The PEG-b-PPS BCPs are undetectable by silver staining and do not interfere with this analysis (Figure S11, Supporting Information). Differences in the number and relative intensity of protein bands were observed across each chassis type (Figure 2E). Interestingly, high molecular weight proteins (>180 kDa) adsorbed to all MC and FM formulations, yet were much more variable within the PS morphology (Figure 2E). In particular, proteins of >200 kDa were not detected in the PS corona by silver staining, regardless of PS surface chemistry (Figure 2E). We used label-free proteomics techniques to identify protein constituents of each corona formed in human plasma, and quantified their relative abundances.
Our analysis identified a total of 171 proteins using a 1% false discovery rate (Figure S12, Supporting Information). As expected, the majority of identified proteins had a molecular weight below 100 kDa (Figure S13, Supporting Information), which is consistent with the corresponding distributions observed by SDS-PAGE (Figure 2E). The number of identified proteins ranged between 43–154 protein species, depending on the nanocarrier morphology and surface chemistry, as well as the timepoint examined (Figure 2F). The MeO FM corona contained the fewest number of adsorbing proteins (57 proteins at 2 h and 43 proteins at 24 h), compared to greater than 100 proteins for the eight other chassis types (Figure 2F). For surface chemistry groupings (Phos, OH, MeO) and morphology groupings (MC, FM, PS), the number of adsorbing species was not significantly different when considered individually as bulk groups (Figure S14, Supporting Information).
Trends in the time-dependent protein corona composition differed between surface chemistry (Figure 2G) and morphology (Figure 2H). Neutral surfaces enriched a smaller subset of shared proteins, as observed by the decrease in shared constituents for OH and MeO nanocarriers, while the number of shared adsorbing species increased from 119 proteins (2 h) to 137 proteins (24 h) for anionic Phos surfaces (Figure 2G). With regard to morphology, the number of shared constituents increased for spherical nanostructures (MC, PS), whereas a loss in shared constituents was observed for the cylindrical FM morphology (Figure 2H). The overall shape of the relative abundance distributions further demonstrates bulk compositional differences between nanocarrier formulations (Figure S15, Supporting Information). These distributions differed from that of the pooled human plasma input sample (Figure S15, Supporting Information). Protein relative abundance generally did not correlate across formulations (Figures S16 and S17, Supporting Information). Two blood proteins were found as the most abundant protein corona constituent in all cases: serum albumin (ALB) and immunoglobulin G (IgG) (Figure S18, Supporting Information). IgG dominated at 2 h, whereas albumin displaced IgG as the most abundant constituent of the OH FM and MeO PS protein coronas at 24 h (Figure S18, Supporting Information). The serum amyloid P-component (APCS), apolipoprotein A-I (APOA1), complement C1q subcomponent subunit C (C1QC; P02747), and complement C3 (C3; P01024) were also found in high abundance.
To understand the inherent tendency of each protein corona to aggregate in blood, we examined the population-level aggregation propensity of each protein coating using the Tango[69–71] statistical mechanics algorithm. Significant differences in aggregation propensity were not found between the time-dependent protein coronas and plasma control (Figure S19A,B, Supporting Information). Furthermore, the aggregation propensity of the 2 h protein corona was not significantly different than the corona formed at 24 h (Figure S19C, Supporting Information). Similar results were obtained when analyzing the distribution of aggregation propensities weighted by the relative abundance determined by our proteomic studies (Figure S19D–F, Supporting Information). These analyses suggest the nanocarriers do not enrich aggregation-prone proteins, and the aggregation propensity of the protein corona does not change between the timepoints examined.
Collectively, these results suggest polar surface chemistries generally render soft nanocarrier surfaces more susceptible to protein adsorption than those terminated with methoxy groups. The number of adsorbing species, trends in shared versus unique protein corona constituents, and their relative abundances differed depending on the combination of nanocarrier morphology and surface chemistry.
2.3. The Adsorption of 121 Plasma Proteins Differs Significantly between Nanocarrier Chassis Types After 2 h and Suggest Differential Immune Responses
2.3.1. Complement and Inflammation-Related Proteins Differentially Adsorb to Distinct Chassis Types after 2 h in Human Blood
Since the majority of intravenously administered nanocarriers are cleared systemically within a few hours of injection,[17,33,61,72,73] the protein corona that forms during this early time period represents a critical biological identity that strongly influences nanocarrier fate and performance. We therefore examined high level gene ontology (GO) terms associated with the set of differentially enriched proteins across all nine nanocarriers at the 2 h absorption timepoint following incubation with human blood (Figure 3A,B). Significant differences in protein corona relative abundances were found for a set of 121 proteins. This set of proteins is significantly associated with stress responses, immune system processes, and transport (Figure 3B). A more detailed network analysis reveals significant involvement of these 121 differentially adsorbed proteins in blood coagulation and complement activation, as well as wounding and inflammatory responses (Figure 3C).
Figure 3.

The biological identity of soft polymeric nanocarriers in human plasma at the 2 h timepoint is dependent on the combination of morphology and surface chemistry. A) Illustrative overview. B,C) Biological process gene ontology (GO) analysis of the set of 121 differentially enriched proteins across all nine NCs (ANOVA, q < 0.01). B) The top ten overrepresented GO slim terms (high level nodes). C) Network analysis of overrepresented protein corona constituents. The network was constructed to only display enriched terms of greatest significance (i.e., by p-value rank). The ten most significant nodes are annotated (corrected p-value < 1e-20). D) Hierarchical clustering of the 56 shared constituents detected in all nine coronas. Hierarchical clustering (Euclidean distance metric) was performed using the average linkage method to represent cluster proximity. Protein-encoding gene names are displayed due to their ease of interpretation. Protein clusters are organized into ten groups, labeled i–x.
Compositional differences were found in many different protein groupings, including common adsorbing proteins (serum albumin, fibrinogen, fibronectin), as well as apolipoproteins, protein aggregation and complement-responsive chaperones, coagulation enzymes, and major complement components (Figure S20, Supporting Information). Fibrinogen had a higher relative abundance than albumin at 2 h, and fibronectin was only found in micellar (MC, FM) coronas with the exception of OH PS (Figure S20, Supporting Information). Interestingly, C1QC, APCS, and C3 were dominant constituents of the respective MeO MC, Phos PS, and OH PS coronas, suggesting strong interactions with these surfaces (Figure S20, Supporting Information). The biological chemistry that underlies the C3 observation is particularly interesting. The polar surfaces (Phos, OH) included in this investigation contain hydroxyl groups capable of nucleophilic attack on C3, whereas the nonpolar MeO surface lacks this capability. This observation may suggest the polar nanocarrier surfaces may arise from covalent attachment between C3 and the hydroxyl groups of the OH PS and Phos PS polymers.
2.3.2. The biological Identity of All Nanocarriers at 2 h in Human Blood is Defined by 56 Adsorbed Proteins
Biological identities are complex fingerprints defined by both the constituency and relative abundance of numerous adsorbing plasma proteins. To compare the similarity of protein coronas formed on PEG-b-PPS nanocarriers at 2 h, we performed hierarchical clustering on two different subsets of proteins: the set of all shared protein corona constituents (Figure S21A, Supporting Information) and the inclusive list of all 171 proteins identified by our proteomic investigation (Figure S21B, Supporting Information). In this analysis, nanocarrier protein coronas that cluster together are more similar than those that do not. The dendrogram branching patterns were identical for both cases (Figure S21A,B, Supporting Information). Thus, differences in the relative abundance of 56 proteins that were common to the corona of all nine chassis types is sufficient to capture the defining features of each biological identity at 2 h.
The major difference was that MeO FM did not cluster with the other chassis types (Figure S21A,B, Supporting Information). This suggests the biological identity of MeO FM is the most unique, which is supported by our initial, more crude analysis of protein corona constituency (Figure 2F). For the remaining eight nanocarrier types, MeO MC clustered with polar vesicular structures, whereas the polar spherical and filamentous micelle structures (Phos MC, OH MC, Phos FM, and OH FM) clustered with the nonpolar MeO PS vesicular structures (Figure S21A,B, Supporting Information). The former analysis therefore suggests nonpolar micelles have a similar biological identity to polar vesicles, whereas the latter analysis suggests the biological identity of polar micellar spheres and cylinders are most similar to that of nonpolar vesicles.
Clustering the 56 shared protein constituents revealed ten distinct protein groupings (Figure 3D, groups i–x). Inspecting the protein functions within these groupings may shed light on the nanocarrier fate in human blood. Among these proteins, were the abundant blood proteins fibrinogen (various chain types) and transferrin, as well as C1Q, C8 complement components and immunoglobulin chains (Figure 3D). Many of these proteins serve as molecular tags that flag circulating foreign objects for removal, including drug delivery vehicles. Filamentous micelles are known to exhibit long circulation times compared to their spherical micelle counterparts.[48] For MeO-terminated PEG-b-PPS nanocarriers, MeO FM are cleared at a slower rate than MeO MC in mice.[33] Our analysis thus suggests a role for the protein surface coatings on the clearance rates of nanocarrier chassis types. Compared to the MeO FM protein corona, the representation of clearance-mediating proteins of group vi-vii is much greater on MeO MC surfaces (Figure 3D), which likely would accelerate MC removal. Furthermore, a high relative abundance of these same group vi-vii proteins resulted in MeO MC clustering together with the polar PS (OH PS, Phos PS) (Figure 3D). But these PS types accumulated more C3 than MeO MC (Figure S20, Supporting Information), presumably through their chemical potential for covalent attachment to C3. These considerations suggest polar PS will be more immunogenic than MeO MC, despite the overall similarities in their protein coating compositions. Lastly, high levels of group i proteins largely accounted for compositional similarities between polar MC, polar FM, and MeO PS chassis types, which deviated from MeO MC, OH PS, and Phos PS (Figure 3D).
2.4. Assessment of the Inherent Inflammatory Activity of PEG-b-PPS Nanocarriers in Human Blood Samples
2.4.1. Only Polar Vesicle Surfaces Activate Complement, which is via C4a and/or C5a without Significant C3a Induction
Having defined the biological identity of each PEG-b-PPS nanocarrier chassis type formed shortly after introduction to blood, we next directly examined their potential for evoking complement activation and cytokine responses (Figure 4). We hypothesized PEG-b-PPS nanocarriers with polar surfaces (OH or Phos) would be more prone to activating complement and downstream proinflammatory events, since these surface chemistries accumulated greater levels of C3 protein (Figure S20, Supporting Information) and hydroxylated surfaces are known to react with C3b thioester linkage.[53–55] Per guidelines issued by the United States Food and Drug Administration (FDA) on the use of International Standard ISO 10993–1 for assessing blood-circulating device hemocompatibility, it is recommended that complement activation by a test article is compared against that induced by a negative control. In this assay, significant complement activation is defined as a statistically significant increase in anaphylatoxin concentration in the nanocarrier treatment group compared to the negative control group. We therefore assessed complement activation after incubating each of the nine nanocarriers, or PBS negative control, with pooled human sera. The concentration of C3a, C4a, and C5a peptide cleavage products (anaphylatoxins) was determined in collected serum.
Figure4.

PEG-b-PPS nanocarriers exhibit low inherent immunostimulation in human blood, despite differential complement activation by polymersomes with polar surfaces. A–C) The serum concentrations of A) C3a, B) C3a, and C) C3a anaphylatoxins were determined (n = 3). Error bars represent s.e.m. Significance was determined with Tukey’s multiple comparison test. **p < 0.01, ****p < 0.0001. Treatment with PBS is denoted by the “vehicle” condition. D) Illustration of cytokine profiling experiment. Cytokine levels were assessed after incubating nanocarriers in human whole blood samples obtained from individual donors (n = 3). E) Heatmap depicting changes in PBMC cytokine secretion levels induced by nanocarrier incubation with whole blood for 4 h. F) Hierarchical clustering of nanocarriers based on C3a, C4a, C5a, IL-8 responses. Each response was normalized to a 0.0–1.0 scale. Normalization procedures included PBS negative control. Hierarchical clustering was performed using the Euclidean distance metric and average linkage methods.
Interestingly, while no statistically significant differences were observed for the serum concentration of C3a (Figure 4A), significant increases in C4a (Figure 4B) and C5a (Figure 4C) were observed. Phos-functionalized surfaces consistently induced greater increases in C4a concentration than OH and MeO surfaces (Figure 4B). However, the C4a induction was only significant in the case of Phos PS (Figure 4B). Charge is a particularly strong contributing factor for classical pathway activation, but it does not completely explain the greater C4a generation. Phos PS, as Phos FM and Phos MC were also highly negatively charged but did not elicit significant responses (Figures 4A–C). C5a is the most potent of the three anaphylatoxins and is a downstream product of all three arms of the complement cascade. Only Phos- and OH-functionalized PSs induced statistically significant increases in C5a serum concentration (Figure 4C). The C5a concentrations that result from incubation with Phos PS and OH PS correspond to increases of approximately 150% and 270%, respectively (Figure 4C). Despite differential complement activation, we note that the C3a, C4a, and C5a activation induced by PEG-b-PPS was low (<10 ng mL−1; Figure 4A–C). The absolute concentrations reported here are orders of magnitude lower than those reported elsewhere in the literature for Zymosan, an established activator of the complement system, which elicited average C3a, C4a, and C5a concentrations in the range of 150–200, 150–200, and 60–80 ng mL−1, respectively.[74] Interpreted in the context of FDA guidelines, we conclude that only PEG-b-PPS PS with polar surfaces invoke significant complement activation in human blood. In contrast, all PEG-b-PPS MC and FM nanocarrier chassis types, and MeO-terminated PS, do not significantly activate complement.
2.4.2. PEG-b-PPS Nanocarriers do not Elicit Consistent Proinflammatory Cytokine Responses in Human Whole Blood Obtained from Single Patient Donors
To assess proinflammatory responses, nanocarriers were incubated with whole blood from three healthy donors (Table S2 and Figure S22, Supporting Information) and PBMC-secreted cytokine profiles were measured after 4 h (Figure 4D,E), or 20 h (Figures S22 and S23, Supporting Information). We hypothesized that anaphylatoxin-inducing nanocarriers would elicit significantly higher secretion of proinflammatory cytokines from PBMCs. Plasma was collected after incubation and was analyzed for proinflammatory cytokines, including interleukin (IL)-6, tumor necrosis factor (TNF)-α, interferon (IFN)-γ, IFN-α, IL-1β, IL-12p70, IL-17A, IL-18, and IL-33, proinflammatory chemokines monocyte chemoattractant protein (MCP)-1 and IL-8, and the anti-inflammatory cytokines IL-10 and IL-23. PEG-b-PPS nanocarriers failed to induce a statistically significant alteration in the levels of secreted cytokines when compared to PBS treatment or lipopolysaccharide (LPS) and R848 (Resiquimod) positive controls (Figure 4E).
Compared to PBS control, OH-functionalized PSs induced substantial increases in IL-8 concentration in Donor 1 following plasma incubation times of 4 and 20 h (Figure 4E; Figures S22 and S23, Supporting Information). Interestingly, IL-8 secretion in whole blood was greatest in response to Phos PS and OH PS at 4 h (Figure 4E). A hierarchical clustering analysis of the normalized, average anaphylatoxin and IL-8 responses suggests a link between the C5a and IL-8 responses and the greater immunostimulation of OH PS observed across multiple experiments (Figure 4F). We acknowledge that the IL-8 increase resulted from a particularly strong response from one individual donor, rather than a consistent uptick in chemokine secretion amongst all donors (Figure 4E). The general variability among the three donor’s responses, where in some instances converse responses for the same cytokine are observed, highlight the necessity of exploring a personalized approach for the development of patient-specific platforms. However, further development of such an approach extends outside the scope of this work. Hierarchical clustering further suggests the combination of cytokine responses evoked by OH PS is unique (Figure 4F). Aside from OH PS, clustering nanocarriers by complement and IL-8 responses revealed similar responses for all nanocarrier chassis possessing MeO surface chemistry despite their differences in morphology (Figure 4F). This clustering also revealed the similarity of large Phos-functionalized FM and PS that induced greater C4a responses (Figure 4B). The immunogenicity of polar MCs was most similar to that of OH FM (Figure 4F). These results indicate the surface chemistry of soft nanocarriers is the strongest physicochemical determinant of nanocarrier complement activation and potentially adverse cytokine responses in our studies.
2.5. The Composition of Protein Coronas Adsorbed to PEG-b-PPS Nanocarriers is Dynamic
2.5.1. The Nanocarrier Immunological Identity at 24 h is Distinct for Each Chassis Type with Greater Influence by Less Abundant Protein Subsets
Hierarchical clustering was performed on the shared (Figure 5A) and complete (Figure 5B) protein corona constituents to examine nanocarrier immunological identity at 24 h. The corona of MeO FM was the most distinct (Figure 5A,B), which is similar to our observations at 2 h (Figure S21A,B, Supporting Information). This uniqueness of MeO FM may be due to the substantially fewer constituents that comprise its protein corona (Figure S21B, Supporting Information). The MeO PS immunological identity at 24 h was also distinct from other chassis types (Figure 5A,B). The shared protein corona of polar (OH, Phos) MC clustered together at 24 h, as did FM and PS of identical surface chemistry (Figure 5A). In contrast to our analysis at 2 h (Figure S21A,B, Supporting Information), differences in dendrograms were observed for the shared and complete biological identities at 24 h (Figure 5A,B). When considering the presence of proteins that were unique to one or more nanocarriers (Figure 5B), the corona formed on the nonpolar MeO MC surface became more similar to that of Phos FM (Figure 5B). This suggests the unique signatures of the Phos FM corona contributes to a divergence in its biological identity from those of polar PS and the OH FM (Figure 5B). Furthermore, the complete polar PS biological identities were similar (Figure 5B), and the inclusion of less common constituents differentiated these nanostructures from FM counterparts of identical surface chemistry (Figure 5A).
Figure 5.

Vroman effect analysis demonstrates the unshared protein corona constitutes strongly define PEG-b-PPS nanocarrier biological identities at 24 h. A,B) Hierarchical clustering of nanocarrier biological identity defined by the A) shared 43 proteins adsorbed to all nine nanocarriers at 24 h, and B) the complete set of protein corona constituents quantified at 24 h. C) Vroman effect illustration. D) Percent change in albumin relative abundance at 24 h. E–G) Volcano plots displaying proteins with significant changes in relative abundance at 24 h. The −log10 of the p-value is plotted against the log2 fold change (FC). Proteins with a significant ≥ twofold change (FC) in relative abundance at 24 h (compared to 4 h) are displayed in blue (≥ twofold decrease in relative abundance) and red (≥ twofold increase in relative abundance). The abbreviations of the genes that encode the quantified proteins are displayed as space permits. Gene names are used in place of protein accession numbers for ease of interpretation. Significance was determined by t-test (*p < 0.05).
2.5.2. MC and PS Morphologies Accumulate Stealth and Clearance-Related Molecular Signatures, Respectively
The “Vroman effect” refers to the exchange of absorbed protein constituents that occurs with time, due to the replacement of early-adsorbing higher concentration proteins with proteins that have a greater affinity for the nanocarrier surface. We therefore sought to specifically investigate changes in protein corona constituents between 2 h versus 24 h that may impact nanocarrier clearance from the blood by circulating- and organ-resident innate immune cells. We first examined changes in the highest concentration protein in the blood—-serum albumin. When adsorbed to nanocarrier surfaces, albumin adsorption is considered to have a stealth effect, helping to prolong circulation time in the blood.[75] The relative abundance of albumin in the MC corona increased with time (Figure 5D). For FM and PS nanostructures, use of the MeO surface chemistry increased albumin concentration as well, whereas anionic surfaces enhanced albumin dissociation and hydroxylated surfaces typically maintained constant levels of albumin (Figure 5D). Statistically significant differences in relative abundance were determined for all proteins between the 2 and 24 h (Figures 5E–G; Figure S24, Supporting Information). Two patterns stood out that may influence PS circulation time. First, alpha-2-HS-glycoprotein (AHSG), an endocytosis-promoting protein with opsonic properties, increased in an abundance with time on all PS surfaces yet its levels did not change on micellar (MC, FM) nanostructures (Figure 5G; Figure S24, Supporting Information). Second, a variety of complement proteins accumulated on the surface of neutral PS surfaces (OH, MeO) with time as well, but not on MC structures (Figure 5G; Figure S24, Supporting Information). Collectively, these analyses further demonstrate that changes in immunological identity over time depend on the combination of nanocarrier morphology and surface chemistry. The accumulation of AHSG on all PS surfaces, and select complement proteins on neutral PS surfaces, demonstrate these vesicular drug delivery vehicles have clearance-accelerating molecular obstacles that do not burden micellar carriers. In contrast, the spherical MC consistently accumulated albumin, which can promote circulation time in some cases, whereas molecular-level trends in clearance-mediating molecules were not as clear for FMs.
2.6. Uptake of PEG-b-PPS Nanocarriers by Human MPS Cells
Modulating the structure of proteins adsorbed to soft nanocarriers is one mechanism for modifying soft nanocarrier biodistribution and clearance by the MPS,[17] yet leveraging steady-state compositional differences for this purpose has not been explored in detail. We sought to determine how immune cells of the human MPS interact with the bare synthetic surface of each nanocarrier chassis type. We further examined how these cellular interactions are modulated by the compositionally distinct, time-dependent blood protein coatings formed on each nanocarrier morphology and surface chemistry combination (Figures 2–3 and 5). We hypothesized that the rate of nanocarrier uptake by human MPS cells would be responsive to the immunological identity, since this protein coating is what directly interfaces with cell surfaces. To this end, we assessed how changes to the protein corona influence nanocarrier interactions with representative innate immune cell populations of the MPS: monocyte-like THP-1 cells, macrophage-like differentiated THP-1 cells (denoted MΦ), and monocyte derived immature dendritic cells (DC) (Figure 6A).
Figure 6.

Blood protein coatings formed at 2 and 24 h modulate PEG-b-PPS nanocarrier interactions with human MPS cells involved in clearance and/or antigen presentation. A) Schematic depicting process flow for nanocarrier uptake studies. Flow cytometric analysis of nanocarrier (DiI) uptake within B) THP-1 monocytes (Mono; n = 10; except OH-PSs with 24 h plasma incubation where n = 9), C) THP-1 differentiated macrophages (MΦ; n = 10), and D) immature dendritic cells (DC; n = 4). All radar plots depict the percentage of nanocarrier positive cells (%NC+). E) Fold change in %NC+ cells after human plasma protein adsorption. The fold change was calculated w.r.t. the uptake of pristine nanocarriers.
2.6.1. PEG-b-PPS Nanocarriers are Nontoxic to Human MPS Cells In Vitro
We first assessed the cytotoxicity of each nanocarrier formulation. Monocyte-like THP-1 cells were incubated with nanocarriers (0.1–1.0 mg mL−1) for 24 h. Irrespective of the chassis type, the viability of treated THP-1 cells exceeded 80% for all material concentrations tested (Figure S25, Supporting Information). The concentration-dependent differences in cell viability were not significant within the examined range (Figure S25, Supporting Information). The noncytotoxic nature of PEG-b-PPS nanocarriers observed here is consistent with previously published studies.[17,37,45] To ensure that cytotoxicity would not influence our ensuing cell uptake studies, we used a BCP concentration of 0.5 mg mL−1. Cell viability exceeded 85% for all formulations administered at this concentration (Figure S25, Supporting Information).
2.6.2. Adsorbed Human Blood Proteins Generally Enhance PS Clearance and Minimize FM Clearance by THP-1 Monocytes
Nanocarrier uptake studies were performed in serum-free media to focus only on uptake differences mediated by adsorbed human plasma proteins without confounding effects from other protein sources (e.g., fetal bovine serum). Formulations were administered under three separate conditions: i) no protein exposure (i.e., pristine nanocarriers) or after a ii) 2 h or iii) 24 h pre-incubation with human plasma. The 24 h timepoint was included since previous studies involving PEG-b-PPS nanocarriers demonstrate circulation times on the scale of days can be achieved following intravenous administration.[33] Flow cytometry was used to determine the percentage of cells that internalized and were positive for nanocarriers (%NC+ cells) after a 4 h incubation period (Figure 6B–D). Furthermore, we quantified the fold change in nanocarrier uptake that is mediated by the protein corona formed at 2 or 24 h (Figure 6E).
Nanocarrier uptake was first assessed in THP-1 monocyte cells.[76] Between the three morphologies, MC nanocarriers generally were taken up less by monocytes than either PS or FM under all conditions (Figure 6B; Figure S26A, Supporting Information). The protein corona formed at 2 and 24 h enhanced monocyte uptake of polar PS surface chemistries although changes were much more subtle for MeO PS (Figure 6B,E; Figure S26A, Supporting Information). For the FM chassis types, the 2 h protein corona altered monocyte uptake of cylindrical soft structures, although the magnitude of this effect varied (Figure 6B,E). In contrast, the 24 h protein corona of FM nanocarriers decreased uptake by monocytes with respect to the pristine condition (Figure 6B,E). Previous studies investigating the endocytosis of metallic and solid core nanoparticles in THP-1 cells similarly observed decreases in nanocarrier internalization when serum proteins are present.[77,78] These studies suggested that the reduced uptake resulted from decreases in nanocarrier zeta potential (more negative) upon protein adsorption. Here, significant correlations were not found between soft nanocarrier uptake and zeta potential for any of the condition groupings (Figure S27, Supporting Information). These uptake studies suggest the effects of blood protein coatings on monocyte-mediated clearance are mostly uniform within nanocarrier morphology groupings, and surface chemistry is less influential.
Lastly, the FM morphology develops a protein corona at 2 h that has little effect on their rate of clearance by monocytes, whereas the 24 h protein corona decreases monocyte uptake. The latter observation is particularly interesting, as it suggests that the adsorbed protein coating that forms on FMs at later timepoints contribute to their known longer circulation times.[48] Since this trend was observed regardless of surface chemistry, these results suggest that a subset of the 43 proteins that are common to all FM surfaces at 24 h (Figure 2F,H) increases the circulation time of these filamentous nanocarriers through an inhibitory effect on monocytes. This result further suggests that developing protein coatings that mimic the shared biological identity of FMs may prove useful for improving the circulation time of drug delivery vehicles.
2.6.3. MC Protein Coronas Enhance Clearance by Human Macrophages
Next, we investigated nanocarrier uptake by macrophages differentiated from THP-1 cells. These macrophages are preferable to peripheral blood mononuclear cell (PBMC)-derived macrophages on the basis of their higher phagocytic capacity.[79] As expected, dTHP-1 cells displayed an increased affinity for PEG-b-PPS nanocarriers (Figure 6C) compared to their monocyte-like counterparts (Figure 6B). General trends in nanocarrier association and uptake varied based on nanocarrier morphology. For PS and MC chassis types, the protein coatings increased uptake for all except for MeO-functionalized PS (Figure 6C,E). Protein adsorption resulted in subtle increases in macrophage uptake of PS with polar surfaces and decreased MeO PS uptake, but these differences were not statistically significant (Figure 6C; Figure S26B, Supporting Information). In contrast, significant protein-mediated increases were observed for MC internalization (Figure 6E; Figure S26B, Supporting Information), despite their accumulation of albumin that may promote evasion to some extent (Figure 5D). Cylindrical FM nanocarriers displayed no major trends in uptake following plasma exposure. On the basis of surface chemistry, plasma exposure increased macrophage uptake of all Phos- and OH-functionalized nanocarriers (except Phos-functionalized FM incubated with plasma for 2 h). MC aside, plasma exposure decreased macrophage uptake of MeO-functionalized nanocarriers (Figure 6C,E). We conclude that the immunological identities of MC, regardless of composition perturbations by surface chemistry, significantly enhance their clearance rate by human macrophages.
2.6.4. The Protein Corona of PS Enhances Uptake by Human Dendritic Cells
Their phagocytic capacity, efficient processing and presentation of antigens, and ability to initiate T cell immunity make dendritic cells (DCs) a desirable cellular target for biomaterial-based delivery systems designed for vaccination.[80] Our studies performed using immature DCs revealed several notable trends relevant to such applications (Figure 6D). Under the pristine condition, PEG-b-PPS nanocarriers were endocytosed less by DCs compared to monocyte-like or macrophage-like dTHP-1 cells (Figures 6B–D). Generally, nanocarrier internalization and association decreased with increasing BCP fPEG. For example, PS exhibited increased uptake compared to FM nanocarriers, which exhibited increased uptake compared to MC nanocarriers (Figure 6D). All chassis types were taken up at a greater rate by DCs when coated with human plasma proteins (Figure 6D,E), but these increases were generally only significant for the PS morphology (Figure S26C, Supporting Information). The PS result is highly significant in the context of our past work examining MPS uptake preferences of MeO MC, MeO PS, and MeO FM nanostructures.[33] Those studies demonstrated that the PS morphology has a greater specificity for targeting DCs in vivo compared to MC or FM,[33] but the mechanism for this effect was not clear. In the present work, our results suggest the protein corona formed on the surface of PS nanostructures to be the source of this targeting specificity.
3. Discussion
We conducted a systems level investigation of the relationship between protein adsorption and the biological performance of soft nanocarriers in human-derived samples. Using nine model soft nanocarriers and label-free proteomic techniques, we demonstrate that the combination of morphology and surface chemistry defines the immunological identity of a nanocarrier in human blood (selected features are summarized in Figure 7). We introduce the term “chassis” to describe the unique combination of these two physicochemical design parameters, which were found to influence the adsorption of protein subsets that influence interactions with the MPS and subsequent complement and inflammatory responses. The chassis morphology primarily modulated nanocarrier cellular uptake, which was shown to be dependent on the specific MPS cell type. In contrast, the surface chemistry-driven differences influenced complement C3 accumulation and the production of downstream anaphylatoxins.
Figure 7.

Summary of morphology and surface chemistry combinations on nanocarrier interactions with human blood and immune cells. The columns present the nine nanocarrier chassis types investigated. Each nanocarrier chassis type is defined by a unique combination of morphology and surface chemistry. Each row depicts the relative magnitude of the mean value for the specified effect across nanocarrier chassis types. The diameter of each dot corresponds to its relative magnitude compared to the other values in that row.
Our protein adsorption studies show polar surface chemistries accumulate larger amounts of adsorbed protein and generally enrich complement proteins to a greater extent than nanocarriers with nonpolar methoxy surfaces. The methoxy surface chemistry has been used extensively by FDA-approved nanocarriers since Doxil,[51] and our results indicate terminal methoxy groups resist blood protein interactions regardless of nanocarrier morphology. Examining time-dependent trends in composition revealed a group of 56 blood proteins were common to the protein corona of all chassis types at 2 h, whereas protein exchange on the nanocarrier surfaces developed more unique protein fingerprints by 24 h. Hierarchical clustering identified a variety of interesting trends, the most notable of which was the consistent uniqueness of the MeO FM protein coatings at 2 and 24 h. This chassis type interacted with the fewest protein species and formed the most distinct protein coatings.
Complement activation assays and cytokine profiling revealed a handful of trends that can be explained by biological chemistry. Phosphate- and hydroxyl-functionalized PS enriched complement C3, and induced serum anaphylatoxin concentrations that were significantly higher than the other nanocarrier types. Interestingly, only the hydroxyl-functionalized PS elicited a proinflammatory response by PBMCs in human blood. The IL-8 response to OH PS is likely a consequence of enhanced C5a production, since this anaphylatoxin has been shown to amplify IL-8 secretion.[81] The more distinct immunogenicity of OH PS and high albumin content of its protein corona suggest this chassis type will be most prone to rapid removal from the blood by the innate immune system. These results are consistent with our past demonstration of albumin-denaturing effects and class A1 scavenger receptor clearance of OH PS.[17]
We provide mechanistic insights into the role of protein coatings in nanocarrier-MPS interactions and in driving multiple morphology-specific phenomena in drug delivery. The FM immunological identities formed by 24 h significantly decreased their rate of clearance by monocytes, suggesting these protein coatings have an inhibitory effect that promotes longer circulation times in human blood. Furthermore, monocytes were generally unresponsive to the plasma protein coatings formed on spherical MC. In contrast, the MC immunological identities most significantly enhanced the rate of clearance by human macrophages in vitro, suggesting that their rapid removal from the blood is not only attributable to their small size, but also the composition of their adsorbed protein coatings. The PS protein coronas significantly enhanced their internalization by DCs, thereby providing a molecular mechanism to explain our prior finding that PS are most preferrable for targeting this therapeutically advantageous immune cell type.[33] The protein corona that evolves with time on polar vesicle surfaces renders these structures more prone to internalization by monocytes. In contrast, the MeO PS surface develops a protein corona that has small yet significant effects on monocyte uptake. Our results suggest that replicating influential blood protein corona compositions may provide a means to bias the cellular-level biodistribution of nanocarriers in meaningful ways to meet challenges in drug delivery and vaccine development.
In closing, the multiscale biological relationships established here can be leveraged to rationally design drug delivery vehicles for personalized nanomedicine and targeted immunomodulation. The immunological identity that is unique to the specific nanocarrier chassis type dictates its molecular and cellular interactions in blood. Therefore, engineering both the nanocarrier morphology and surface chemistry together may provide greater control over drug biodistribution, adverse effects, and clinical outcomes.
4. Experimental Section
Protein Identification and Label-Free Quantification:
Prior to protein identification, samples were briefly electrophoresed following established procedures. A total of 5 µg of adsorbed protein was prepared in Laemelli buffer and β-mercaptoethanol, loaded into a 4–20% Tris-glycine polyacrylamide gel, and were briefly electrophoresed at 120 V for roughly 5 min to achieve 3–5 mm separation from the well. A 1 cm x 1 cm band containing the mixed adsorbed protein population was excised and processed further for label free quantification (LFQ). After in-gel trypsin digestion, samples were analyzed using a Q Exactive HF Hybrid Quadrupole-Orbitrap mass spectrometer (ThermoFisher Scientific). Data was collected from two technical replicates. The data were searched against the UniProt Homo sapiens database using the MaxQuant search engine and LFQ was performed based on the MS1 peptide intensity. Proteins were identified using a threshold of >1 unique peptide and a 1% FDR. A total of 171 proteins were identified and quantified by these procedures.
Statistical Analysis of Proteomic Data:
The LFQ intensity was log2 transformed and normalized by median subtraction. Imputation was applied to fill in missing values from a normal distribution, which is a recommended procedure for LFQ analyses. Statistically significant differences in protein relative abundance across the nine nanocarrier formulations were determined by ANOVA. Within each formulation, significant differences in protein relative abundance at 2 h versus 24 h were determined by pairwise t-tests. The p-values were calculated using an FDR-based permutation procedure.
Assessment of Nanocarrier-Induced Cytokine Secretion:
De-identified human whole blood samples were purchased from Research Blood Components, LLC (Boston, MA). Whole blood samples were collected from healthy donors by trained phlebotomists under the direction of registered nurses and licensed physicians. An IRB approved consent form was obtained from each donor that gives Research Blood Components, LLC permission to collect their blood and use or sell it at their discretion, for research purposes. Whole blood samples containing ethylenediaminetetraacetic acid (EDTA) collected from three individual, healthy donors were acquired from Research Blood Components, LLC. Nanocarrier formulations (10 mg mL−1) were added 1:20 to human whole blood from each of the donors for 4 or 20 h at 37 °C with a 5% CO2 atmosphere. Lipopolysaccharide (LPS) (InvivoGen) and R848 (InvivoGen) were included as positive controls and were tested at final concentrations of 1.0 and 10 µg mL−1, respectively. After incubation, plasma was collected via centrifugation. Samples were subsequently prepared and analyzed following the manufacturer protocols. Data acquisition was completed using a BD LSRFortessa flow cytometer. Cytobank software was used to analyze the data.
Assessment of Nanocarrier-Induced Complement Activation:
Nanocarrier formulations (10 mg mL−1) were added to pooled complement human serum (Innovative Research) in a 1:20 ratio and incubated at 37 °C, 200 rpm for 1 h. Afterward, samples were treated with 5 × 10−3 m EDTA to halt complement activation and were placed on ice. The C3a, C4a, and C5a human anaphylatoxin concentrations were quantitatively assessed using a BD™ CBA Human Anaphylatoxin Kit. The C3a, C4a, and C5a standards and were prepared following the manufacturer’s protocol. Data acquisition was completed using a BD LSRFortessa. Analysis proceeded using Cytobank software.
Supplementary Material
Acknowledgements
M.P.V. gratefully acknowledges support from the Ryan Fellowship and the International Institute for Nanotechnology at Northwestern University. The authors acknowledge Jonathan Remis (Structural Biology Facility, NU) for assisting with cryoTEM and Dr. Matt Clutter (High Throughput Analysis Laboratory, NU) for assisting with the acquisition of data used in the nanocarrier immunogenicity and complement activation analyses. The authors acknowledge Dr. Young Ah Goo (NU Proteomic Facility) and Dr. Byoungkyu Cho (NU Proteomics Facility) for their support and assistance with the proteomic data acquisition. The authors acknowledge staff and instrumentation support from the Structural Biology Facility at Northwestern University, the Robert H Lurie Comprehensive Cancer Center of Northwestern University and NCI CCSG P30 CA060553. The Gatan K2 direct electron detector was purchased with funds provided by the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust. SAXS experiments were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of the Advanced Photon Source (APS). DND-CAT was supported by Northwestern University, E.I. DuPont de Nemours & Co., and The Dow Chemical Company. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. This work made use of the EPIC facility of Northwestern University’s NUANCE Center, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the MRSEC program (NSF DMR-1121262) at the Materials Research Center; the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois, through the IIN. This work made use of the IMSERC at Northwestern University, which has received support from the NSF (CHE-1048773); Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the State of Illinois and International Institute for Nanotechnology (IIN). This work was supported by the Northwestern University – Flow Cytometry Core Facility supported by Cancer Center Support Grant (NCI CA060553). The authors further acknowledge proteomics services provided by the Northwestern Proteomics Core Facility, generously supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center, instrumentation award (S10OD025194) from NIH Office of Director, and the National Resource for Translational and Developmental Proteomics supported by P41 GM108569. This research was supported by the National Science Foundation grant 1453576, the National Institutes of Health Director’s New Innovator Award no. 1DP2HL132390–01 and the 2014 McCormick Catalyst Award.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Michael P. Vincent, Department of Biomedical Engineering, Northwestern University, Evanston, IL 60208, USA
Nicholas B. Karabin, Department of Biomedical Engineering, Northwestern University, Evanston, IL 60208, USA
Sean D. Allen, Interdisciplinary Biological Sciences, Northwestern University, Evanston, IL 60208, USA
Sharan Bobbala, Department of Biomedical Engineering, Northwestern University, Evanston, IL 60208, USA.
Molly A. Frey, Interdisciplinary Biological Sciences, Northwestern University, Evanston, IL 60208, USA
Sijia Yi, Department of Biomedical Engineering, Northwestern University, Evanston, IL 60208, USA.
Yufan Yang, Department of Biomedical Engineering, Northwestern University, Evanston, IL 60208, USA.
Evan A. Scott, Department of Biomedical Engineering, Northwestern University, Evanston, IL 60208, USA; Interdisciplinary Biological Sciences, Northwestern University, Evanston, IL 60208, USA; Chemistry of Life Processes Institute, Northwestern University, Evanston, IL 60208, USA; Simpson Querrey Institute, Robert H. Lurie Medical Research Center, Northwestern University, Chicago, IL 60611, USA; Robert H. Lurie Comprehensive Cancer Center, Northwestern University, Chicago, IL 60611, USA
Data Availability Statement
Relevant materials and/or data will be made available by the authors upon request.
References
- [1].Allen S, Liu Y-G, Scott E, Regener. Eng. Transl. Med 2016, 2, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Scott EA, Karabin NB, Augsornworawat P, Annu. Rev. Biomed. Eng 2017, 19, 57. [DOI] [PubMed] [Google Scholar]
- [3].Dobrovolskaia MA, Aggarwal P, Hall JB, McNeil SE, Mol. Pharmaceutics 2008, 5, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Frey M, Bobbala S, Karabin N, Scott E, Nanomedicine 2018, 13, 1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chow A, Brown BD, Merad M, Nat. Rev. Immunol 2011, 11, 788. [DOI] [PubMed] [Google Scholar]
- [6].Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H, Nano. Today 2015, 10, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bazak R, Houri M, El Achy S, Kamel S, Refaat T, J. Cancer Res. Clin. Oncol 2015, 141, 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Perry JL, Reuter KG, Luft JC, Pecot CV, Zamboni W, DeSimone JM, Nano Lett 2017, 17, 2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Su G, Jiang H, Xu B, Yu Y, Chen X, Mol. Pharm 2018, 15, 5019. [DOI] [PubMed] [Google Scholar]
- [10].Salvati A, Pitek AS, Monopoli MP, Prapainop K, Bombelli FB, Hristov DR, Kelly PM, Åberg C, Mahon E, Dawson KA, Nat. Nanotechnol 2013, 8, 137. [DOI] [PubMed] [Google Scholar]
- [11].Mirshafiee V, Kim R, Park S, Mahmoudi M, Kraft ML, Biomaterials 2016, 75, 295. [DOI] [PubMed] [Google Scholar]
- [12].Modak M, Frey MA, Yi S, Liu Y, Scott EA, Curr. Opin. Biotechnol 2020, 66, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Walkey CD, Olsen JB, Guo H, Emili A, Chan WCW, J. Am. Chem. Soc 2012, 134, 2139. [DOI] [PubMed] [Google Scholar]
- [14].Walczyk D, Bombelli FB, Monopoli MP, Lynch I, Dawson KA, J. Am. Chem. Soc 2010, 132, 5761. [DOI] [PubMed] [Google Scholar]
- [15].García-Álvarez R, Hadjidemetriou M, Sánchez-Iglesias A, Liz-Marzán LM, Kostarelos K, Nanoscale 2018, 10, 1256. [DOI] [PubMed] [Google Scholar]
- [16].Chen D, Ganesh S, Wang W, Amiji M, Nanoscale 2019, 11, 8760. [DOI] [PubMed] [Google Scholar]
- [17].Vincent MP, Bobbala S, Karabin NB, Frey M, Liu Y, Navidzadeh JO, Stack T, Scott EA, Nat. Commun 2021, 12, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA, Proc. Natl. Acad. Sci. USA 2008, 105, 14265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tenzer S, Docter D, Rosfa S, Wlodarski A, Kuharev J, Rekik A, Knauer SK, Bantz C, Nawroth T, Bier C, Sirirattanapan J, Mann W, Treuel L, Zellner R, Maskos M, Schild H, Stauber RH, ACS Nano 2011, 5, 7155. [DOI] [PubMed] [Google Scholar]
- [20].Lundqvist M, Augustsson C, Lilja M, Lundkvist K, Dahlbäck B, Linse S, Cedervall T, PLoS One 2017, 12, 0175871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hu Z, Zhang H, Zhang Y, Wu R, Zou H, Colloids Surf., B 2014, 121, 354. [DOI] [PubMed] [Google Scholar]
- [22].Clemments AM, Botella P, Landry CC, ACS Appl. Mater. Interfaces 2015, 7, 21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vidaurre-Agut C, Rivero-Buceta E, Romaní-Cubells E, Clemments AM, Vera-Donoso CD, Landry CC, Botella P, ACS Omega 2019, 4, 8852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ritz S, Schöttler S, Kotman N, Baier G, Musyanovych A, Kuharev J, Landfester K, Schild H, Jahn O, Tenzer S, Mailänder V, Biomacromolecules 2015, 16, 1311. [DOI] [PubMed] [Google Scholar]
- [25].Schöttler S, Becker G, Winzen S, Steinbach T, Mohr K, Landfester K, Mailänder V, Wurm FR, Nat. Nanotechnol 2016, 11, 372. [DOI] [PubMed] [Google Scholar]
- [26].Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, McNeil SE, Adv. Drug Delivery Rev 2009, 61, 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yan Y, Gause KT, Kamphuis MMJ, Ang C-S, O’BrienSimpson NM, Lenzo JC, Reynolds EC, Nice EC, Caruso F, ACS Nano 2013, 7, 10960. [DOI] [PubMed] [Google Scholar]
- [28].Allen SD, Bobbala S, Karabin NB, Modak M, Scott EA, ACS Appl. Mater. Interfaces 2018, 10, 33857. [DOI] [PubMed] [Google Scholar]
- [29].Allen S, Osorio O, Liu Y-G, Scott E, J. Controlled Release 2017, 262, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Allen S, Vincent M, Scott E, J. Visualized Exp 2018, 57793. [DOI] [PMC free article] [PubMed]
- [31].Bobbala S, Allen SD, Scott EA, Nanoscale 2018, 10, 5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bobbala S, Allen SD, Yi S, Vincent M, Frey M, Karabin NB, Scott EA, Nanoscale 2020, 12, 5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yi S, Allen SD, Liu Y-G, Ouyang BZ, Li X, Augsornworawat P, Thorp EB, Scott EA, ACS Nano 2016, 10, 11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yi S, Zhang X, Sangji MH, Liu Y, Allen SD, Xiao B, Bobbala S, Braverman CL, Cai L, Hecker PI, DeBerge M, Thorp EB, Temel RE, Stupp SI, Scott EA, Adv. Funct. Mater 2019, 29, 1904399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yi S, Karabin NB, Zhu J, Bobbala S, Lyu H, Li S, Liu Y, Frey M, Vincent M, Scott EA, Front. Bioeng. Biotechnol 2020, 8, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Karabin NB, Allen S, Kwon H-K, Bobbala S, Firlar E, Shokuhfar T, Shull KR, Scott EA, Nat. Commun 2018, 9, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Scott EA, Stano A, Gillard M, Maio-Liu AC, Swartz MA, Hubbell JA, Biomaterials 2012, 33, 6211. [DOI] [PubMed] [Google Scholar]
- [38].Cerritelli S, Velluto D, Hubbell JA, Biomacromolecules 2007, 8, 1966. [DOI] [PubMed] [Google Scholar]
- [39].Cerritelli S, O’Neil CP, Velluto D, Fontana A, Adrian M, Dubochet J, Hubbell JA, Langmuir 2009, 25, 11328. [DOI] [PubMed] [Google Scholar]
- [40].Velluto D, Demurtas D, Hubbell JA, Mol. Pharmaceutics 2008, 5, 632. [DOI] [PubMed] [Google Scholar]
- [41].Shang S, Kats D, Cao L, Morgun E, Velluto D, He Y, Xu Q, Wang C-R, Scott EA, Front. Immunol 2018, 9, 2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Stack T, Vahabikashi A, Johnson M, Scott E, J. Biomed. Mater. Res. A 2018, 106, 1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Stack T, Vincent M, Vahabikashi A, Li G, Perkumas KM, Stamer WD, Johnson M, Scott E, Small 2020, 16, 2004205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Stano A, Scott EA, Dane KY, Swartz MA, Hubbell JA, Biomaterials 2013, 34, 4339. [DOI] [PubMed] [Google Scholar]
- [45].Dowling DJ, Scott EA, Scheid A, Bergelson I, Joshi S, Pietrasanta C, Brightman S, Sanchez-Schmitz G, Van Haren SD, Ninkovic J, Kats D, Guiducci C, de Titta A, Bonner DK, Hirosue S, Swartz MA, Hubbell JA, Levy O, J. Allergy Clin. Immunol 2017, 140, 1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Yu SS, Lau CM, Thomas SN, Jerome WG, Maron DJ, Dickerson JH, Hubbell JA, Giorgio TD, Int. J. Nanomed 2012, 7, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Choi YH, Han H-K, J. Pharm. Invest 2018, 48, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE, Nat. Nanotechnol 2007, 2, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Oltra NS, Swift J, Mahmud A, Rajagopal K, Loverde SM, Discher DE, J. Mater. Chem. B 2013, 1, 5177. [DOI] [PubMed] [Google Scholar]
- [50].Gabizon A, Catane R, Uziely B, Kaufman B, Safra T, Cohen R, Martin F, Huang A, Barenholz Y, Cancer Res 1994, 54, 987. [PubMed] [Google Scholar]
- [51].Barenholz YC, J. Controlled Release 2012, 160, 117. [DOI] [PubMed] [Google Scholar]
- [52].Gadjeva M, Dodds AW, Taniguchi-Sidle A, Willis AC, Isenman DE, Law SK, J. Immunol 1998, 161, 985. [PubMed] [Google Scholar]
- [53].Tang L, Liu L, Elwing HB, J. Biomed. Mater. Res 1998, 41, 333. [DOI] [PubMed] [Google Scholar]
- [54].Kidane A, Park K, J. Biomed. Mater. Res 1999, 48, 640. [DOI] [PubMed] [Google Scholar]
- [55].Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O’Neil CP, Lee LK, Swartz MA, Hubbell JA, Nat. Biotechnol 2007, 25, 1159. [DOI] [PubMed] [Google Scholar]
- [56].Malmsten M, J. Colloid Interface Sci 1995, 172, 106. [Google Scholar]
- [57].Bradley AJ, Devine DV, Ansell SM, Janzen J, Brooks DE, Arch. Biochem. Biophys 1998, 357, 185. [DOI] [PubMed] [Google Scholar]
- [58].Malmsten M, Colloids Surf., A 1999, 159, 77. [Google Scholar]
- [59].Moghimi SM, Szebeni J, Prog. Lipid Res 2003, 42, 463. [DOI] [PubMed] [Google Scholar]
- [60].Leroueil PR, Hong S, Mecke A, Baker JR, Orr BG, Banaszak Holl MM, Acc. Chem. Res 2007, 40, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Arvizo RR, Miranda OR, Moyano DF, Walden CA, Giri K, Bhattacharya R, Robertson JD, Rotello VM, Reid M, Mukherjee P, PLoS One 2011, 6, 24374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Nagarajan R, Langmuir 2002, 18, 31. [Google Scholar]
- [63].Roy MT, Gallardo M, Estelrich J, J. Colloid Interface Sci 1998, 206, 512. [DOI] [PubMed] [Google Scholar]
- [64].Tseng S, Yeh P-H, Hsu J-P, Langmuir 2014, 30, 8177. [DOI] [PubMed] [Google Scholar]
- [65].Grossman PD, Soane DS, Anal. Chem 1990, 62, 1592. [DOI] [PubMed] [Google Scholar]
- [66].Modak M, Bobbala S, Lescott C, Liu Y-G, Nandwana V, Dravid VP, Scott EA, ACS Appl. Mater. Interfaces 2020, 12, 55584. [DOI] [PubMed] [Google Scholar]
- [67].Allen SD, Liu Y-G, Bobbala S, Cai L, Hecker PI, Temel R, Scott EA, Nano Res 2018, 11, 5689. [Google Scholar]
- [68].Böhmert L, Voß L, Stock V, Braeuning A, Lampen A, Sieg H, Nanoscale Adv 2020, 2, 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Rousseau F, Schymkowitz J, Serrano L, Curr. Opin. Struct. Biol 2006, 16, 118. [DOI] [PubMed] [Google Scholar]
- [70].Fernandez-Escamilla A-M, Rousseau F, Schymkowitz J, Serrano L, Nat. Biotechnol 2004, 22, 1302. [DOI] [PubMed] [Google Scholar]
- [71].Linding R, Schymkowitz J, Rousseau F, Diella F, Serrano L, J. Mol. Biol 2004, 342, 345. [DOI] [PubMed] [Google Scholar]
- [72].Senior JH, Crit. Rev. Ther. Drug Carrier Syst 1987, 3, 123. [PubMed] [Google Scholar]
- [73].Cullis PR, Chonn A, Semple SC, Adv. Drug Delivery Rev 1998, 32, 3. [DOI] [PubMed] [Google Scholar]
- [74].Escamilla-Rivera V, Solorio-Rodríguez A, Uribe-Ramírez M, Lozano O, Lucas S, Chagolla-López A, Winkler R, De Vizcaya-Ruiz A, Int. J. Nanomed 2019, 14, 2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Sato H, Nakhaei E, Kawano T, Murata M, Kishimura A, Mori T, Katayama Y, Langmuir 2018, 34, 2324. [DOI] [PubMed] [Google Scholar]
- [76].Bosshart H, Heinzelmann M, Ann. Transl. Med 2016, 4, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Lunov O, Syrovets T, Loos C, Beil J, Delacher M, Tron K, Nienhaus GU, Musyanovych A, Mailänder V, Landfester K, Simmet T, ACS Nano 2011, 5, 1657. [DOI] [PubMed] [Google Scholar]
- [78].Kettler K, Giannakou C, de Jong WH, Hendriks AJ, Krystek P, J. Nanopart. Res 2016, 18, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Shiratori H, Feinweber C, Luckhardt S, Linke B, Resch E, Geisslinger G, Weigert A, Parnham MJ, Mol. Immunol 2017, 88, 58. [DOI] [PubMed] [Google Scholar]
- [80].Reddy ST, Swartz MA, Hubbell JA, Trends Immunol 2006, 27, 573. [DOI] [PubMed] [Google Scholar]
- [81].Turner MD, Nedjai B, Hurst T, Pennington DJ, Biochim. Biophys. Acta 2014, 1843, 2563. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Relevant materials and/or data will be made available by the authors upon request.