

Abstract
Trichothiodystrophy (TTD) is a rare hereditary neurodevelopmental disorder defined by sulfur-deficient brittle hair and nails and scaly skin, but with otherwise remarkably variable clinical features. The photosensitive TTD (PS-TTD) forms exhibits in addition to progressive neuropathy and other features of segmental accelerated aging and is associated with impaired genome maintenance and transcription. New factors involved in various steps of gene expression have been identified for the different non-photosensitive forms of TTD (NPS-TTD), which do not appear to show features of premature aging. Here, we identify alanyl-tRNA synthetase 1 and methionyl-tRNA synthetase 1 variants as new gene defects that cause NPS-TTD. These variants result in the instability of the respective gene products alanyl- and methionyl-tRNA synthetase. These findings extend our previous observations that TTD mutations affect the stability of the corresponding proteins and emphasize this phenomenon as a common feature of TTD. Functional studies in skin fibroblasts from affected individuals demonstrate that these new variants also impact on the rate of tRNA charging, which is the first step in protein translation. The extension of reduced abundance of TTD factors to translation as well as transcription redefines TTD as a syndrome in which proteins involved in gene expression are unstable.
Introduction
Trichothiodystrophy (TTD) is a multi-system recessive disorder whose most characteristic features are brittle hair and nails and ichthyosis. Brittle hair in TTD is associated with low cysteine content and shows characteristic alternating light and dark bands (‘tiger tail’ banding) by polarized light microscopy. The spectrum and severity of additional clinical symptoms is extremely broad. It includes mental and growth retardation, microcephaly, ocular and skeletal abnormalities, recurrent infections, impaired sexual development and a range of other features. The clinical heterogeneity of TTD is paralleled by an increasing number of functionally diverse causative genes, which may provide hints for disentangling the intricate genotype–phenotype relationships.
About half of TTD patients show hypersensitivity to sunlight combined with progressive neurological decline and premature aging features (1,2). All photosensitive TTD cases [photosensitive TTD (PS-TTD), OMIM 601675, 606390 and 616395] carry bi-allelic mutations within three genes encoding distinct subunits of the multi-protein transcription initiation factor IIH (TFIIH) complex (3,4). TFIIH is essential for both transcription initiation and nucleotide excision repair (NER) (5). NER impaired by these TTD mutations causes the photosensitive phenotype since this repair process is the only pathway in human cells which is able to remove lesions induced by solar ultraviolet (UV) radiation. Tissue-specific transcription that is hampered by the affected TFIIH is likely to be the cause of the additional complicated pathophysiology associated with PS-TTD (6,7). The associated premature aging features are most likely caused by the accelerated accumulation of DNA lesions, which interfere with transcription and replication, resulting in functional decline and increased cell death. This occurs particularly in post-mitotic differentiated tissues (e.g. neurons and hair follicles), which cannot dilute DNA damage by proliferation and accrue transcription stress. This explains why post-mitotic tissues are progressively affected in PS-TTD (8).
The remaining half of TTD patients, who are NER-proficient and categorized as non-photosensitive TTD (NPS-TTD, OMIM 234050, 616943, 300953, 618546), display extensive genetic and clinical heterogeneity. Thus far, five affected genes have been identified for NPS-TTD, which encode M-phase-specific PLK1 interacting protein (MPLKIP/TTDN1) (9,10), general transcription factor IIE subunit 2 (GTF2E2/TFIIEβ) (11,12), the X-linked ring finger protein 113A (RNF113A) (13,14) and the recently identified cysteinyl-tRNA synthetase 1 (CARS1) and threonyl-tRNA synthetase 1 (TARS1) (15,16).
Although no clear function for the protein encoded by the NPS-TTD-causative gene MPLKIP/TTDN1 has been documented so far, the identification of NPS-TTD-related mutations in GTF2E2 initially supported the idea that part of the TTD-specific features derives from transcriptional impairment. GTF2E2 encodes the beta subunit of the basal transcription factor TFIIE that interacts with and supports TFIIH function in transcription initiation. Strikingly, the identified pathological alterations within TFIIEβ result in decreased stability and consequently reduced cellular abundance of the entire tetrameric TFIIE (α2β2) complex and reduced TFIIH-mediated TFIIEα phosphorylation (11). Furthermore, reprogramming of induced pluripotent stem cells from GTF2E2-mutated fibroblasts highlighted the transcription defects in late-stage differentiated erythroid cells (12). Recently, GTF2E2 mutations have been reported to disrupt in vitro recruitment of the TFIIH kinase module at gene promoters, potentially affecting early transcriptional events (17). RNF113A was also shown to function in the transcriptional process, albeit in the subsequent step of transcript maturation and splicing (18,19). However, proteins encoded by the other recently identified genes causing NPS-TTD are not involved in the transcriptional process and thus seem to challenge the concept of transcription-related problems as the underlying cause of TTD-specific features. CARS1 and TARS1 encode the cysteinyl-tRNA synthetase (CysRS) and threonyl-tRNA synthetase (ThrRS), respectively (15,16). CysRS and ThrRS belong to the family of aminoacyl-tRNA synthetases (aaRS) whose function is to load each amino acid onto its specific cognate tRNA through aminoacylation reactions.
The functional heterogeneity of the genes causing NPS-TTD complicates genotype–phenotype relationships. Moreover, each of these five genes accounts for a limited number of cases that overall explain less than 30% of the NPS-TTD individuals. Here, we report the identification of new, rare missense variants in two additional aaRS coding genes, alanyl-tRNA synthetase 1 (AARS1) and methionyl-tRNA synthetase 1 (MARS1), that lead to the NPS-TTD phenotype. AARS1 and MARS1 encode alanyl-tRNA synthetase (AlaRS) and methionyl-tRNA synthetase (MetRS), respectively.
Results
We performed whole-exome sequencing (WES) or whole-genome sequencing (WGS) in a group of 34 unsolved cases with multi-system phenotypes, each characterized by the presence of the hair alterations that are typical of TTD and by the absence of reported photosensitivity in the clinical presentations, combined with normal NER activity in cultured cells. Sequence data were aligned with the human reference genome GRCh37-hg19, and lists of sequence variants were obtained. We identified two cases (TTD236AM and TTD1GL) carrying potentially pathogenic variants in the AARS1 gene and one case (TTD17PV) with variants in the MARS1 gene.
Identification of AARS1 mutations in two NPS-TTD cases
TTD236AM was the third daughter of Indian non-consanguineous parents. The couple had one miscarriage and she had an older sibling who was similarly affected and a normal 7-year-old sibling. The mother was treated throughout the pregnancy with carbamazepine (400 mg per day) as she was an epileptic. The infant was born by normal delivery and her birth weight was 3 kg. Hypothyroidism was detected on day 22 of life and she was treated with 25 μg of eltroxin. The child could smile at 3 months of age and recognized her mother at 5 months. On the first examination at 8 months, she only showed partial head control, her weight was 5.8 kg (third percentile), height was 66 cm (third percentile) and head circumference was 37.5 cm (<third percentile). Neurological investigations revealed normal reflexes and mild spasticity. MRI showed delayed myelination (Fig. 1A) and periventricular diffusion restriction at the region of optic radiation. She had a fair complexion and was dysmorphic with hypotelorism, bilateral infraorbital creases, left convergent squint, long philtrum, thin upper lip and prominent nose (Fig. 1B). In addition, the infant presented mild skin laxity, sparse wooly hair (Fig. 1C) and sparse eyebrows. Hair analysis revealed the low sulfur content typical of TTD (Supplementary Material, Fig. S1A), and electron microscopic examination showed trichorrhexis nodosa and fractures (Supplementary Material, Fig. S1B). On follow-up visit at 18 months, all her growth parameters were still below the third percentile (weight: 7 kg, height: 71 cm and head circumference: 39 cm) and she could only sit by herself and crawl. Photosensitivity was not reported, which was in agreement with the normal response to UV irradiation of TTD236AM primary skin fibroblasts. Following UV irradiation, levels of both unscheduled DNA synthesis (UDS) and cell survival were similar to those in normal cells (Fig. 1D and E). Unfortunately, we lost contact with the family for follow-up. WGS identified two heterozygous missense variants in AARS1, c.2096 T > C resulting in p.Ile699Thr and c.2702G > A resulting in p.Cys901Tyr (GenBank: NM_001605.3 and NP_001596.2), which were both confirmed by Sanger sequencing (Fig. 1F).
Figure 1 .
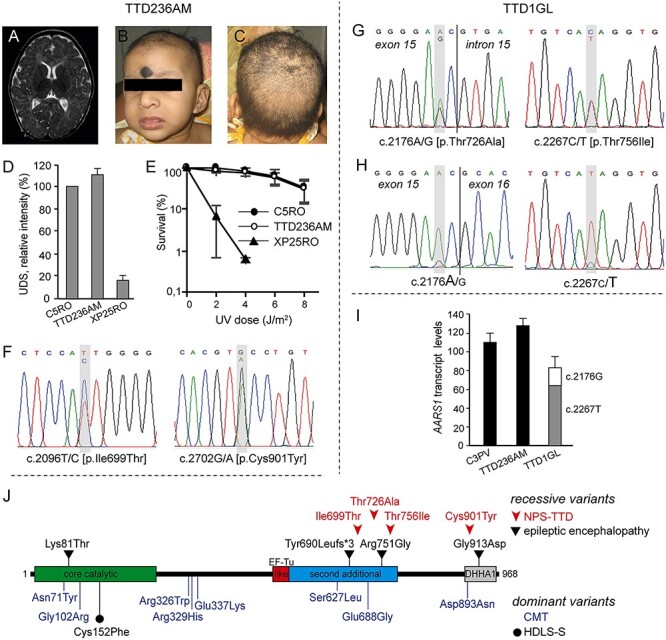
AARS1-defective NPS-TTD cases. (A–C) Clinical features of TTD236AM. MRI of the brain showing delayed myelination (A); frontal face appearance (B); view of the scalp with sparse hair (C) and (D) DNA repair capacity in control (C5RO), TTD236AM and NER-deficient xeroderma pigmentosum group A (XP-A; XP25RO) primary fibroblasts. DNA repair capacity was measured as UV-induced DNA repair synthesis (UDS), using EdU incorporation after UV irradiation and visualization by fluorescence-conjugated azide (Click-iT assay). Mean fluorescence intensities of at least 50 nuclei were expressed as percentage of those of control cells analyzed in parallel. Bars indicate standard deviation (SD) of three independent experiments. (E) Clonogenic UV-survival assay to measure UV sensitivity in C5RO (closed circles), TTD236AM (open circles) and XP25RO (closed triangles) primary fibroblasts. One day after seeding, fibroblasts were irradiated with different doses of UV and cultured for 2 weeks. Survival was plotted as the percentage of colonies obtained after treatment compared with the mock-treated fibroblasts (set at 100%). Bars indicate SD of three independent experiments. (F) Sanger sequencing traces of TTD236AM AARS1 genomic DNA. TTD236AM shows compound heterozygosity for c.2096C > T and c.2702G > A. (G, H) Sanger sequencing traces of TTD1GL AARS1 genomic DNA (G) and cDNA (H). TTD1GL shows compound heterozygosity for c.2176A > G and c.2267C > T (G) and reduced expression of the c.2176A > G carrying AARS1 allele (H). (I) Cellular levels of AARS1 transcripts assessed by RT-qPCR. Total AARS1 transcript levels were first normalized to the levels of GAPDH mRNA and then expressed as percentages of the corresponding value in the normal C3PV cells. The relative percentage of the mutant AARS1 transcripts in TTD1GL cells (c.2176A > G and c.2267C > T) was assessed by allele-specific RT-qPCR. The reported values are the means of two independent experiments, each done in triplicate. Bars indicate SD. (J) Schematic representation of the domain structure of the AlaRS protein and location of the identified variants. Recessive and dominant variants are shown over and under the protein scheme, respectively. EF-Tu-like, elongation factor Tu-like; DHHA1, desert hedgehog homolog-associated domain.
The older sister from whom samples could not be obtained was affected similarly. This child was born by normal delivery and her birth weight was 3 kg. Jitteriness was observed from the day after birth. She could smile at 1 year of age and showed head steadiness at 2 years of age. On examination at 8 years, her weight was 9.1 kg (<third percentile), height was 91.5 cm (<third percentile) and head circumference was 40.5 cm (<third percentile). She presented with dystonia of the face and self-mutilation behavior. She had no meaningful language, but she could understand commands to some extent. She could sit up from a lying position, but she showed dystonic posturing with severe stiffness of limbs and ankle clonus. In addition, she had soft wooly hair. Hearing and vision were normal and she was not photosensitive. She died aged 9 years.
TTD1GL was a Scottish male first reported in 1984 at the age of 4 years with a severe clinical phenotype characterized by trichocutaneous alterations and neurodevelopmental delay (case 1 in (20)). Follow-up at age 13 years recorded profound degeneration of his physical and neurological states and the absence of clinical photosensitivity, which is consistent with the normal DNA repair efficiency in TTD1GL skin fibroblasts (21,22). No later records of this case are available. The c.2176A > G (p.Thr726Ala) and c.2267C > T (p.Thr756Ile) variants in AARS1 were identified by WGS and were then confirmed by Sanger sequencing (Fig. 1G). The c.2176A > G change is located in the second base from the 3′ end of exon 15, and it is computationally predicted to slightly decrease the strength of the donor splice-site for intron 15 (score from 9.5 to 8.2, Human Splicing Finder). However, no aberrantly spliced AARS1 transcripts were detected by PCR amplification of AARS1 cDNA, but poor expression of the c.2176A > G containing AARS1 allele was revealed by the cDNA sequencing traces (Fig. 1H). Real-time quantitative polymerase chain reaction (RT-qPCR) analysis demonstrated a slight reduction in the total amount of AARS1 transcripts in TTD1GL compared with the control and TTD236AM cells. Further investigations by allele-specific assays showed that only 20% of AARS1 transcripts are produced by the c.2176A > G allele in TTD1GL fibroblasts, whereas the remaining mRNAs contain the c.2267C > T change (Fig. 1I).
The four AARS1 variants identified in the two affected individuals are all ultra-rare in the human population, in particular the c.2176A > G (p.Thr726Ala) and c.2702G > A (p.Cys901Tyr) changes are not found in the aggregate 150 000 human genomes available from the Genome Aggregation Database (gnomAD). Variants c.2096 T > C (p.Ile699Thr) and c.2267C > T (p.Thr756Ile) are present with low frequency (0.000003991 and 0.00001194, respectively), and no homozygotes have been detected. They all affect conserved amino acid residues located in functional domains of the AlaRS protein (Supplementary Material, Fig S2A and Fig. 1L) and are predicted to be damaging because of their high CADD_phred v 1.5 scores (26.2 for Ile699Thr, 32 for Thr726Ala, 29.3 for Thr756Ile and 22.7 for Cys901Tyr). Variants in AARS1 have been previously implicated in the dominant peripheral neuropathy Charcot Marie Tooth (CMT) disease (23,24) and in the dominant Swedish type hereditary diffuse leukoencephalopathy with spheroids (HDLS-S, (25) as well as in the recessive epileptic encephalopathy (26,27). Thus far, none of the variants displayed by the cases of NPS-TTD investigated here have been found to be associated with other AARS1-related disorders (Fig. 1L).
Identification of a homozygous MARS1 mutation in one NPS-TTD case
TTD17PV is a female born in 1987 to Italian parents with no known consanguinity. She has a healthy older brother. Fetal growth arrest was reported after the sixth gestational month, and she was delivered by cesarean section performed at 36 weeks. At birth her weight was 1200 g (<third percentile). During infancy, she showed slow weight gain and delayed neuromotor development. On the basis of hair analysis, she was clinically diagnosed with TTD at the age of 3 years. Hair amino acid content analysis showed a low amount of cysteine (9.9% compared with 20.8% in healthy donors), as reported in all PS- and NPS-TTD cases. In addition, methionine was undetectable in TTD17PV hair (Supplementary Material, Fig. S1C). Polarized light microscopy examination revealed a moderate tiger-tail pattern and showed hair fractures (Supplementary Material, Fig. S1D). At a recent evaluation (age: 32 years), her height was 142 cm (<third percentile), her weight was 48.3 kg and her head circumference was 49.5 cm (<third percentile). She showed facial asymmetry (Fig. 2A) owing to congenital paresis of her left facial nerve. In addition, she had bilateral epicanthus and high and narrow palate leading to dental malocclusion. She presented with intellectual disability, reduced tendon reflexes and ataxia. Further clinical signs were brachydactyly of the hands and feet (Fig. 2B and C), joint laxity, brittle nails and ungueal dystrophy. Her eyebrows and hair were sparse and she had a white hair tuft on her forehead. Her parents revealed that she had her menarche at the age of 16 years, but after three periods, she stopped menstruating. At the age of 20 years, she was diagnosed with type 2 diabetes (treated with metformin). Eye examination did not reveal any alteration, and echocardiography was in the normal range. She presented with follicular keratosis and ichthyosis that was especially marked on her underarms (Fig. 2D). She was not sun-sensitive, and investigations on her primary skin fibroblasts confirmed normal repair of UV-induced DNA damage (Fig. 2E and F). WES analysis detected the homozygous variant c.1201G > A in MARS1 that results in the substitution p.Val401Met (GenBank: NM_004990.4 and NP_004981.2). The presence and inheritance of the variant were confirmed by Sanger sequencing of the relevant genomic DNA region amplified from the subject and both parents’ blood samples, identifying them as carriers of the same sequence variant (Fig. 2G). The steady-state level of the c.1201G > A variant transcript in patient cells is similar to that of the wild-type transcript in control cells (Fig. 2H). This allele is very rare (gnomAD frequency 0.00001414) and, consistent with a pathogenic effect, it has a high CADD_phred v 1.5 score (28.5) and affects a highly conserved residue located in the core catalytic domain of the MetRS protein (Fig. S2B and Fig. 2I). Thus far, no homozygosity has been reported for this c.1201G > A allele, and this variant has never been associated with any disease (Fig. 2I), even though several other MARS1 variants have been related to dominant CMT (28,29) or recessive presentations, including interstitial lung and liver disease/pulmonary alveolar proteinosis (ILLD/PAP, (30,31)) and spastic paraplegia 70 (SPG70, (32)). Of note, the typical TTD hair alterations could not be observed on polarized light microscopy examination in the hair from a PAP case carrying bi-allelic variants in the MARS1 gene (Supplementary Material, Fig. S1E).
Figure 2 .
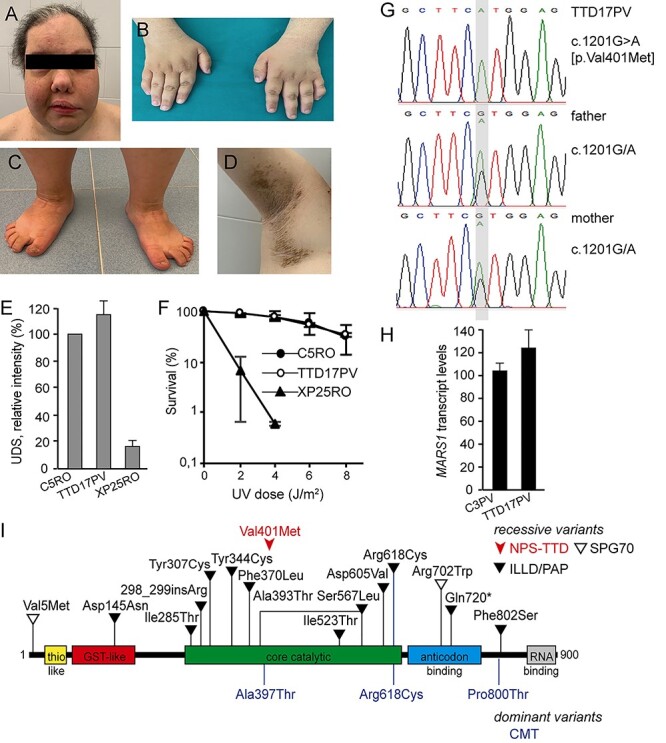
MARS1-defective NPS-TTD case. (A–D) Clinical features of TTD17PV. Facial asymmetry (A); brachydactyly of the hands and feet (B, C); follicular keratosis and ichthyosis on patient underarm (D). (E) DNA repair capacity in control (C5RO), TTD17PV and NER-deficient xeroderma pigmentosum group A (XP-A; XP25RO) primary fibroblasts. DNA repair capacity was measured as UV-induced DNA repair synthesis (UDS) using EdU incorporation after UV irradiation and visualization by fluorescence-conjugated azide (Click-iT assay). Mean fluorescence intensities of at least 50 nuclei were expressed as percentage of those of control cells analyzed in parallel. Bars indicate the SEM of three independent experiments. (F) Clonogenic UV-survival assay to measure UV sensitivity in C5RO (closed circles), TTD17PV (open circles) and XP25RO (closed triangles) primary fibroblasts. One day after seeding, fibroblasts were irradiated with different doses of UV and were cultured for 2 weeks. Survival was plotted as the percentage of colonies obtained after treatment compared with the mock-treated fibroblasts (set at 100%). Bars indicate SD of three independent experiments. (G) Sanger sequencing traces of MARS1 genomic DNA in TTD17PV and her parents. TTD17PV is homozygous for c.1201G > A. (H) Cellular levels of MARS1 transcripts assessed by RT-qPCR. Total MARS1 transcript levels were first normalized to the levels of GAPDH mRNA and then expressed as percentages of the corresponding value in the normal C3PV cells. The reported values are the means of two independent experiments, each done in triplicate. Bars indicate SD. (I) Schematic representation of the domain structure of the MetRS protein and location of the identified variants. Recessive and dominant variants are shown over and under the protein scheme, respectively.
Following identification of the AARS1 and MARS1 variants, using Sanger sequencing, we screened the coding regions of AARS1 and MARS1 cDNAs in 21 additional still unsolved individuals who had a clinical diagnosis of TTD and were NER-proficient. No further potentially pathogenic variant was identified, indicating that, in our combined cohort of 55 unsolved NPS-TTD affected individuals (from Dutch, French, Italian and UK laboratories), alterations in AARS1 and MARS1 accounted for about 2–4% of the cases.
Cellular amount and aminoacylation activity of mutant AlaRS and MetRS proteins
To investigate the consequences of the identified variants in AARS1 and MARS1 genes, we first evaluated the cellular amounts of the AlaRS and MetRS proteins in primary fibroblasts from the affected individuals. Immunoblot analysis of whole-cell lysates revealed a notable reduction of the MetRS content in TTD17PV cells carrying the MARS1 variant (to 30% of the control amount) (Fig. 3A and B) as well as of the AlaRS content in the cells from the two cases carrying AARS1 variants (to about 30 and 15% of the control amount in TTD236AM and TTD1GL, respectively) (Fig. 3C and D). These observations indicate that NPS-TTD-associated variants in MARS1 and AARS1 drastically reduce the MetRS and AlaRS cellular concentrations. Since the reduced MetRS and AlaRS amounts are not caused by reduced transcript levels (Figs 1I and 2H), it is likely that these missense mutations affect protein stability, as commonly observed among variant polypeptides encoded by TTD-causative mutations in TFIIH-related genes (ERCC3/XPB, ERCC2/XPD and GTF2H5/TTDA) or in the other NPS-TTD causative genes (GTF2E2, RNF113A, CARS1 and TARS1) (11–13,15,16,33–35).
Figure 3 .
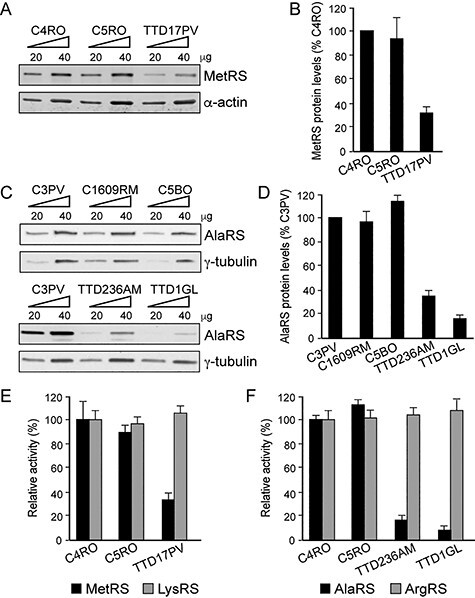
MetRS or AlaRS protein levels and aminoacylation activity in primary fibroblasts from TTD17PV, TTD236AM and TTD1GL. (A) Immunoblot of whole-cell lysates from TTD17PV and two healthy donors (C4RO and C5RO) probed with anti-MetRS antibody. α-actin was used as loading control. (B) Quantification of MetRS protein levels. The amount of MetRS was first expressed as the mean value of the levels observed in the two increasing concentrations of the cell lysate and was normalized to the α-actin content. For each cell strain, the MetRS level was then expressed as percentage of the corresponding value in C4RO cells. The reported values are the means of two independent experiments. Bars indicate SD. (C) Immunoblot of whole-cell lysates from TTD236AM, TTD1GL and three healthy donors (C3PV, C1609RM and C5BO) probed with anti-AlaRS antibody. γ-tubulin was used as loading control. (D) Quantification of AlaRS protein levels carried out as in (B). The AlaRS amount was normalized to γ-tubulin. For each cell strain, the AlaRS level was expressed as percentage of the corresponding value in C3PV cells. The reported values are the means of two independent experiments. Bars indicate SD. (E, F) Cytosolic fractions from TTD17PV, TTD236AM and TTD1GL and two healthy donor fibroblasts (C4RO and C5RO) were used to determine MetRS (E) or AlaRS (F) amino acid charging on tRNA. LysRS (E) or ArgRS (F) charging in control fibroblast C4RO was set at 100%, and error bars indicate SD of three independent experiments.
The function of each aaRS is to charge a specific tRNA molecule with its cognate amino acid (aminoacylation of tRNA) via a two-step enzymatic reaction that includes activation (adenylation) of the amino acid and transfer of the aminoacyl group to the tRNA. To verify whether the identified variants in MARS1 and AARS1 impair tRNA charging, we performed steady-state aminoacylation reactions on protein lysates from TTD17PV (MetRS-defective), TTD236AM and TTD1GL (AlaRS-defective) fibroblasts and from control fibroblasts (C4RO and C5RO) that were analyzed in parallel. In all analyzed cell strains, similar aminoacylation activities were observed for arginyl-tRNA synthetase (ArgRS) and lysyl-tRNA synthetase (LysRS) used as internal controls. In contrast, a 65% decrease in the MetRS activity was found in lysates from TTD17PV fibroblasts compared with the activities measured in control lysates. AlaRS activities in lysates from TTD236AM and TTD1GL fibroblasts were even more severely decreased (Fig. 3E and F), corresponding to only 10–20% activity measured in control lysates. These data are consistent with a loss-of-function effect in tRNA aminoacylation activity, which may be partly derived from the observed reduced levels of AlaRS and MetRS in these NPS-TTD cells.
Discussion
Impact of AARS1 and MARS1 TTD mutations
A total of 37 aaRS coding genes exist in the human genome, and variants in nearly all of them lead to a variety of inherited pathological conditions (36). Here, we identify novel bi-allelic variants in the AARS1 and MARS1 genes associated with the rare NPS-TTD disorder, making tRNA synthetases, together with the previously identified CARS1 and TARS1 genes, the largest gene family within TTD. In addition, we provide evidence that these mutations cause a decrease in the cellular amount and may alter the enzymatic activities of AlaRS and MetRS, which may eventually impair the translational output in different ways. First, quantitatively reduced cellular concentrations of the mutated tRNA synthetases may cause translational insufficiency in specific stages of development, organs, tissues or circumstances with a high translational demand and/or shortage of the specific enzyme. Second, the altered enzymatic properties could qualitatively affect translation in all organs. Obviously, these two effects might synergize in certain conditions.
Quantitative aspects of tRNA synthetase mutations and the hallmark features of TTD
Our findings of reduced levels of both mutated tRNA synthetases extend previous observations of TTD mutations affecting protein amounts. Lower quantities of the TFIIH complex were observed in PS-TTD cells (33–35) and reduced amounts of TFIIE subunits, RNF113A protein, CysRS or ThrRS enzymes were found in NPS-TTD (11–13,15,16). The lower amounts appear owing to protein (and consequent complex) instability, most clearly revealed in the case of TFIIH and TFIIE mutations, which are associated with temperature sensitivity in cells. This is clinically apparent as fever-dependent aggravation of symptoms in several patients, such as sudden loss of all hair during episodes of pneumonia (12,37).
The extension of reduced abundance to TTD proteins not involved in transcription, but in other stages of gene expression (i.e. translation and splicing), firmly establishes protein instability in gene expression as a causal link with the TTD features. As originally proposed for transcription (6), the instability leads to premature depletion of the mutated factor interfering with the completion of terminal differentiation. For example, in the case of skin or hair tissues, premature exhaustion of the mutated gene expression factor reduces the expression of the very cysteine-rich proteins, which cross-link the keratin filaments at the end of terminal differentiation to strengthen skin or hair. Consequently, TTD hair is brittle and the skin is scaly. These findings may redefine TTD as a syndrome of instability in gene expression.
Qualitative aspects of tRNA synthetase mutations and the variable TTD symptoms
Apart from protein instability, culprit mutations may also exert qualitative effects on the function of the proteins implicated in different TTD variants. It is thus likely that the affected enzymatic activities are involved in the other heterogeneous clinical manifestations that are exhibited in different forms of TTD. This holds, for example, for the distinction between PS- and NPS-TTD, with photosensitivity caused by the DNA repair impairment. What are the specific features and enzymatic defects linked with the variants in AARS1 and MARS1? Other mutations in these genes have previously been associated with both dominant and recessive disorders (Figs 1L and 2I and Supplementary Material, Tables S1 and S2). Dominant variants are usually found in patients with peripheral neuropathies referred to as the CMT disease (36). In addition, a mono-allelic AARS1 variant has been recently described in a family presenting with the severe adult-onset HDLS-S (25). However, there is currently no evidence of symptoms suggestive of peripheral neuropathies or leukoencephalopathy in NPS-TTD parents, obligate heterozygous carriers of the AARS1 and MARS1 variants described here. The recessively inherited clinical presentations already linked to AARS1 and MARS1 pathogenic alleles are extremely rare and include a wide spectrum of symptoms. In the case of AARS1 mutations, there have been reports of three families with a total of five cases showing early-onset severe epileptic encephalopathy, progressive microcephaly and hypomyelination (26,27), which only partially overlap with the symptoms reported in NPS-TTD patients. Furthermore, there are no similarities between the previously reported MARS1-mutated phenotypes (ILLD/PAP and SPG70) and the NPS-TTD phenotype observed in the MARS1-mutated patient reported here.
Owing to the limited availability of AARS1- and MARS1-defective patients’ cells, loss-of-function effects for only some AARS1 and MARS1 variants have been shown either by the absence of complementation using heterologous complementation assays in yeast and/or by defective aminoacylation as determined by in vitro acetylation assays. Reduced cellular amounts of the AlaRS enzyme has been reported only in one epileptic encephalopathy case, whereas no alteration of MetRS stability has been found for the two MARS1 mutations analyzed so far. A summary of all reported cases with mutations in AARS1 or MARS1 gene is reported in Supplementary Material, Tables S1 and S2, respectively.
The ability of cells to maintain proteostasis requires, in addition to optimal gene expression by transcription and RNA maturation, strict control over accurate and efficient protein production, folding, conformational maintenance and degradation. It is highly likely that the reduced availability and enzymatic activity of aaRS may affect efficient and or proper tRNA charging, which eventually may cause reduced translation efficiency or misincorporation of amino acids during protein synthesis. Consequently, errors will be introduced in protein sequences which will likely cause impaired protein folding and, in turn, facilitate protein aggregation and/or degradation. Proteostasis dysfunction is at the root of many neurological and developmental diseases and is a hallmark of many age-related disorders (38). Of note, loss of proteostasis has been proposed as a possible pathogenic mechanism that contributes to the severe neurodevelopmental and progeroid Cockayne syndrome (CS), a TCR-defective disorder related to PS-TTD (39). It has been shown that CS cells contain high levels of misfolded proteins likely owing to imbalanced protein synthesis (40) as a consequence of transcription stress, which is caused by the inability to remove DNA damage in active genes (8). Mutant aaRS may similarly affect the translation efficiency and/or accuracy in aaRS-defective NPS-TTD cells. Terminally differentiated neuronal cells, but also terminally differentiated keratinocytes involved in cornified envelope or hair production, are likely to be more sensitive to subtle changes in transcription and translation efficiency and fidelity since the resulting misfolded proteins cannot be diluted by cell division and thus become potentially cytotoxic.
Concluding remarks
Our findings expand the genetic and phenotypic spectrum resulting from AARS1 and MARS1 mutations to include NPS-TTD and suggest that in addition to protein instability, defective enzyme activity may also contribute to the TTD-specific features. These observations further corroborate our previous hypothesis that part of the salient TTD features (e.g. brittle hair and nails and scaly skin) are a consequence of disturbed gene expression which is caused by mutations in factors involved either in transcription initiation (11,12,35), RNA splicing (13) or protein translation (15,16). The variable additional features associated to TTD, including neurodevelopmental abnormalities, are more likely related to the function of the causative gene.
However, the genes currently identified involved in NPS-TTD still account for only a proportion of the patients. Based on the concepts outlined here, we predict that the TTD cases that are still unsolved may carry mutations in other factors required for proper gene expression. Apart from other aaRS, these may involve transcription, RNP processing and transport, mRNA stability and various steps in translation and protein folding.
Materials and Methods
Study subjects
This research project was reviewed and approved by the Erasmus MC, Institutional Review Board/Medical Ethical Committee (MEC 2005-273). The WGS experiments were performed under the general Medical Ethical Committee approval (MEC 2011-253). This research is in line with the World Medical Association Declaration of Helsinki.
Patients and cell culture
Individuals investigated in this study followed the protocols approved by institute-specific ethical review boards, and informed consents were obtained for biological samples according to the Helsinki guidelines.
Primary skin fibroblasts from the NPS-TTD cases TTD236AM, TTD1GL, TTD17PV, TTD18PV, from XP25RO (XP-A) and from the genetically unrelated healthy donors C3PV, C1609RM, C5BO, C4RO and C5RO were used. Fibroblasts were cultured in DMEM medium (DMEM High Glucose with L-glutamine, EuroClone) or Ham’s F10 medium (Lonza) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin–streptomycin (EuroClone) at 37°C and 5% CO2.
Colony-forming ability/survival following UV irradiation
Cells were plated in 10 cm dishes (1500 cells/dish), in triplicate. After 24 h, cells were irradiated with different doses of UV-C irradiation (0–8 J/m2) and were incubated for approximately 2 weeks. Colonies were fixed and stained with 0.1% Brilliant Blue R (Sigma) and were counted (Gelcount, Oxford Optronix Ltd). Survival was plotted as the percentage of colonies obtained after treatment compared with the mean number of colonies from the mock-treated cells (set at 100%).
Whole-genome sequencing
Nanoball-based massive parallel sequencing (software version 2.5.0.44) was done as described previously (41). Image data analysis INCLUDED base calling, DNB mapping and sequence assembly. Reads were mapped to the National Center for Biotechnology Information (NCBI) reference genome, build 37. Variants were annotated using NCBI build 37 and dbSNP build 137. Data were provided as lists of sequence variants (SNPs and short indels) relative to the reference genome. Analysis and filtering of the massive parallel sequencing data was performed using Complete Genomics analysis tools (cga tools version 1.8.0 build 1; http://www.completegenomics.com/sequence-data/cgatools/) and TIBCO/Spotfire version 7.0.1 (http://spotfire.tibco.com/).
Whole-exome sequencing
Exonic DNA was captured using the Agilent SureSelect Human All Exon target enrichment kit (Agilent Technologies, Santa Clara, CA) and sequencing was performed with paired-end 100 bp reads on one-quarter of an Illumina Analyzer IIx (Illumina, San Diego, CA). About 50 M reads/individual were generated resulting in approximately 30–40× coverage of the targeted exome. The Genome Analysis Toolkit (GATK v2.5) was used to perform variant discovery and genotyping. SNPs and indels were called according to the GATK’s Best Practices. Variants were filtered assuming a recessive inheritance model. Given the rare incidence of TTD (estimated to be 1.2 per million live births in Western Europe) (42), only variants with a frequency ≤0.001 and CADD_phred scores >15 were considered.
Sanger sequencing
AARS1 and MARS1 variants identified in the WES/WGS analyses were validated by Sanger sequencing of either the cDNA (TTD1GL) or genomic DNA (TTD1GL, TTD236AM and TTD17PV), as previously described (43). Additional NPS-TTD cases were screened for candidate mutations in AARS1 and MARS1 by directed cDNA sequencing. Primer sequences are given in Supplementary Material, Table S3.
Real-time quantitative polymerase chain reaction
RT-qPCR was performed as previously described (44). Briefly, total RNA was extracted using RNeasy Mini Kit (Qiagen). Transcript levels of both AARS1 and the housekeeping GAPDH gene (for normalization) were measured on a LightCycler 480 (Roche). Primer sequences are given in Supplementary Material, Table S3.
Immunoblot analysis
Whole-cell extracts were prepared as previously described (34) by lysing fibroblasts in Laemmli buffer, and proteins were separated on Mini-Protean TGX gels (BioRad). Blots were incubated with antibodies to AlaRS (A303-473A Bethyl, 1:1000), MetRS (HPA004125, Atlas antibodies, 1:1000), γ-tubulin (T6557), Sigma Aldrich (1:10000) and α-actin (MAB1501R, Merck Millipore, 1:5000) and were analyzed using a ChemiDoc XRS imager (BioRad). The densities of the bands were quantified using the ImageJ 1.50i software (NIH).
Aminoacylation reactions
Aminoacylation was assessed by measuring the AlaRS or MetRS activity in cultured fibroblasts. Fibroblast lysates (cytosolic fraction) were incubated at 37°C for 10 min in a reaction buffer containing 50 mM Tris buffer pH 7.5, 12 mM MgCl2, 25 mM KCl, 1 mg/ml bovine serum albumin, 0.5 mM spermine, 1 mM ATP, 0.2 mM yeast total tRNA, 1 mM dithiothreitol, 0.3 mM of either [D3]-alanine and [15N2]-arginine for TTD236AM and TTD1GL or [13C,D3]-methionine and [D4]-lysine for TTD17PV. The reaction was terminated using trichloroacetic acid. After sample washing with trichloroacetic acid, ammonia was added to release the labeled amino acids from the tRNAs. [D4]-alanine and [13C6]-arginine were added as the internal standards for TTD236AM and TTD1GL. For TTD17PV, the internal standards used were [13C]-valine and [13C6]-arginine. The labeled amino acids were quantified by LC–MS/MS. Intra-assay and inter-assay variation were <15%. ArgRS (TTD236AM and TTD1GL) and LysRS activity (TTD17PV) were simultaneously detected as the control enzymes.
Supplementary Material
Acknowledgements
We are grateful to the families of patients for participation in this study. We also thank Dr Alice Hadchouel (Hôpital Universitaire Necker-Enfants Malades, Paris) for providing the hair sample from one PAP patient.
Funding
The work was supported by Associazione Italiana Ricerca sul Cancro (Id. 21737) to D.O.; Telethon Foundation (GEP13022) to E.B.; European Research Council Advanced grant (233424), Oncode Institute (partly financed by the Dutch Cancer Society), NIH grant (PO1 AG017242), Memorabel and Chembridge (ZonMW) and BBoL (NWO-ENW), EJP-RD project TC-NER RD20-113 and the Deutsche Forschungsgemeinschaft (SFB 829) to J.H.J.H.; European Research Council Advanced grant (340988), Dutch Science Organization (NWO) ZonMW division (912.12.132) and Oncode Institute (partly financed by the Dutch Cancer Society) to W.V.; Bioinformatrics support was partially supported by various H2020 Bigmedilytics, Moodstratification, ImmuneAID and TranSYS grants to P.J.v.d.S.
Contributor Information
Elena Botta, Istituto di Genetica Molecolare ‘Luigi Luca Cavalli-Sforza’ (IGM) CNR, Via Abbiategrasso 207, Pavia 27100, Italy.
Arjan F Theil, Department of Molecular Genetics, Oncode Institute, Erasmus MC, University Medical Center Rotterdam, Dr. Molewaterplein 40, Rotterdam 3015 GD, The Netherlands.
Anja Raams, Department of Molecular Genetics, Oncode Institute, Erasmus MC, University Medical Center Rotterdam, Dr. Molewaterplein 40, Rotterdam 3015 GD, The Netherlands.
Giuseppina Caligiuri, Istituto di Genetica Molecolare ‘Luigi Luca Cavalli-Sforza’ (IGM) CNR, Via Abbiategrasso 207, Pavia 27100, Italy.
Sarah Giachetti, Istituto di Genetica Molecolare ‘Luigi Luca Cavalli-Sforza’ (IGM) CNR, Via Abbiategrasso 207, Pavia 27100, Italy.
Silvia Bione, Istituto di Genetica Molecolare ‘Luigi Luca Cavalli-Sforza’ (IGM) CNR, Via Abbiategrasso 207, Pavia 27100, Italy.
Maria Accadia, Medical Genetics Service, Hospital ‘Cardinale G. Panico’, Via San Pio X Tricase 73039, Italy.
Anita Lombardi, Istituto di Genetica Molecolare ‘Luigi Luca Cavalli-Sforza’ (IGM) CNR, Via Abbiategrasso 207, Pavia 27100, Italy.
Desiree E C Smith, Metabolic Unit, Department of Clinical Chemistry, Amsterdam UMC, Vrije Universiteit Amsterdam, Amsterdam Neuroscience, Amsterdam Gastroenterology & Metabolism, Amsterdam 1081 HZ, The Netherlands.
Marisa I Mendes, Metabolic Unit, Department of Clinical Chemistry, Amsterdam UMC, Vrije Universiteit Amsterdam, Amsterdam Neuroscience, Amsterdam Gastroenterology & Metabolism, Amsterdam 1081 HZ, The Netherlands.
Sigrid M A Swagemakers, Department of Pathology, Clinical Bioinformatics Unit, Erasmus University Medical Center Rotterdam, Dr. Molewaterplein 40, Rotterdam 3015 GD, The Netherlands.
Peter J van der Spek, Department of Pathology, Clinical Bioinformatics Unit, Erasmus University Medical Center Rotterdam, Dr. Molewaterplein 40, Rotterdam 3015 GD, The Netherlands.
Gajja S Salomons, Metabolic Unit, Department of Clinical Chemistry, Amsterdam UMC, Vrije Universiteit Amsterdam, Amsterdam Neuroscience, Amsterdam Gastroenterology & Metabolism, Amsterdam 1081 HZ, The Netherlands; Laboratory of Genetic Metabolic Diseases, Amsterdam UMC, University of Amsterdam, Amsterdam 1105 AZ, The Netherlands.
Jan H J Hoeijmakers, Department of Molecular Genetics, Oncode Institute, Erasmus MC, University Medical Center Rotterdam, Dr. Molewaterplein 40, Rotterdam 3015 GD, The Netherlands; Oncode Institute, Princess Maxima Center for Pediatric Oncology, Utrecht 3584 CS, The Netherlands; Institute for Genome Stability in Ageing and Disease, CECAD Forschungszentrum, University of Cologne, Cologne 50931, Germany.
Dhanya Yesodharan, Department of Pediatric Genetics, Amrita Institute of Medical Sciences & Research Centre, AIMS Ponekkara PO, Cochin 682041, Kerala, India.
Sheela Nampoothiri, Department of Pediatric Genetics, Amrita Institute of Medical Sciences & Research Centre, AIMS Ponekkara PO, Cochin 682041, Kerala, India.
Tomoo Ogi, Department of Genetics, Research Institute of Environmental Medicine (RIEM), Nagoya University, Nagoya 464-8601, Japan; Department of Human Genetics and Molecular Biology, Graduate School of Medicine, Nagoya University, Nagoya 466-8550, Japan.
Alan R Lehmann, Genome Damage and Stability Centre, School of Life Sciences, University of Sussex, Famer, Brighton BN1 9RQ, UK.
Donata Orioli, Istituto di Genetica Molecolare ‘Luigi Luca Cavalli-Sforza’ (IGM) CNR, Via Abbiategrasso 207, Pavia 27100, Italy.
Wim Vermeulen, Department of Molecular Genetics, Oncode Institute, Erasmus MC, University Medical Center Rotterdam, Dr. Molewaterplein 40, Rotterdam 3015 GD, The Netherlands.
References
- 1.de Boer, J., Andressoo, J.-O., de Wit, J., Huijmans, J., Beems, R.B., van Steeg, H., Weeda, G., van der Horst, G.T.J., van Leeuwen, W., Themmen, A.P.N., Meradji, M. and Hoeijmakers, J.H. (2002) Premature aging in mice deficient in DNA repair and transcription. Science, 296, 1276–1279. [DOI] [PubMed] [Google Scholar]
- 2.Orioli, D. and Stefanini, M. Trichothiodystrophy. In Nishigori, C. and Sugasawa, K. (eds) (2019) DNA Repair Disorders—Clinical and Molecular Aspects. Springer, Japan. [Google Scholar]
- 3.Giglia-Mari, G., Coin, F., Ranish, J.A., Hoogstraten, D., Theil, A., Wijgers, N., Jaspers, N.G.J., Raams, A., Argentini, M., van der Spek, P.J. et al. (2004) A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A. Nat. Genet., 36, 714–719. [DOI] [PubMed] [Google Scholar]
- 4.Theil, A.F., Hoeijmakers, J.H.J. and Vermeulen, W. (2014) TTDA: big impact of a small protein. Exp. Cell Res., 329, 61–68. [DOI] [PubMed] [Google Scholar]
- 5.Compe, E. and Egly, J.M. (2016) Nucleotide excision repair and transcriptional regulation: TFIIH and beyond. Annu. Rev. Biochem., 85, 265–290. [DOI] [PubMed] [Google Scholar]
- 6.de Boer, J., de Wit, J., van Steeg, H., Berg, R.J., Morreau, H., Visser, P., Lehmann, A.R., Duran, M., Hoeijmakers, J.H. and Weeda, G. (1998) A mouse model for the basal transcription/DNA repair syndrome trichothiodystrophy. Mol. Cell, 1, 981–990. [DOI] [PubMed] [Google Scholar]
- 7.Marteijn, J.A., Lans, H., Vermeulen, W. and Hoeijmakers, J.H.J. (2014) Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol., 15, 465–481. [DOI] [PubMed] [Google Scholar]
- 8.Lans, H., Hoeijmakers, J.H.J., Vermeulen, W. and Marteijn, J.A. (2019) The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol., 20, 766–784. [DOI] [PubMed] [Google Scholar]
- 9.Nakabayashi, K., Amann, D., Ren, Y., Saarialho-Kere, U., Avidan, N., Gentles, S., MacDonald, J.R., Puffenberger, E.G., Christiano, A.M., Martinez-Mir, A. et al. (2005) Identification of C7orf11 (TTDN1) gene mutations and genetic heterogeneity in nonphotosensitive trichothiodystrophy. Am. J. Hum. Genet., 76, 510–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou, Y.-K., Yang, X.-C., Cao, Y., Su, H., Liu, L., Liang, Z. and Zheng, Y. (2018) A homozygous G insertion in MPLKIP leads to TTDN1 with the hypergonadotropic hypogonadism symptom. BMC Med. Genet., 19, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuschal, C., Botta, E., Orioli, D., Digiovanna, J.J., Seneca, S., Keymolen, K., Tamura, D., Heller, E., Khan, S.G., Caligiuri, G. et al. (2016) GTF2E2 mutations destabilize the general transcription factor complex TFIIE in individuals with DNA repair-proficient trichothiodystrophy. Am. J. Hum. Genet., 98, 627–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Theil, A.F., Mandemaker, I.K., van den Akker, E., Swagemakers, S.M.A., Raams, A., Wüst, T., Marteijn, J.A., Giltay, J.C., Colombijn, R.M., Moog, U. et al. (2017) Trichothiodystrophy causative TFIIEβ mutation affects transcription in highly differentiated tissue. Hum. Mol. Genet., 26, 4689–4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corbett, M.A., Dudding-Byth, T., Crock, P.A., Botta, E., Christie, L.M., Nardo, T., Caligiuri, G., Hobson, L., Boyle, J., Mansour, A. et al. (2015) A novel X-linked trichothiodystrophy associated with a nonsense mutation in RNF113A. J. Med. Genet., 52, 269–274. [DOI] [PubMed] [Google Scholar]
- 14.Tessarech, M., Gorce, M., Boussion, F., Bault, J.-P., Triau, S., Charif, M., Khiaty, S., Delorme, B., Guichet, A., Ziegler, A. et al. (2020) Second report of RING finger protein 113A (RNF113A) involvement in a Mendelian disorder. Am. J. Med. Genet. A, 182, 565–569. [DOI] [PubMed] [Google Scholar]
- 15.Kuo, M.E., Theil, A.F., Kievit, A., Malicdan, M.C., Introne, W.J., Christian, T., Verheijen, F.W., Smith, D.E.C., Mendes, M.I., Hussaarts-Odijk, L. et al. (2019) Cysteinyl-tRNA synthetase mutations cause a multi-system, recessive disease that includes microcephaly, developmental delay, and brittle hair and nails. Am. J. Hum. Genet., 104, 520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Theil, A.F., Botta, E., Raams, A., Smith, D.E.C., Mendes, M.I., Caligiuri, G., Giachetti, S., Bione, S., Carriero, R., Liberi, G. et al. (2019) Bi-allelic TARS mutations are associated with brittle hair phenotype. Am. J. Hum. Genet., 105, 434–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Compe, E., Genes, C.M., Braun, C., Coin, F. and Egly, J.M. (2019) TFIIE orchestrates the recruitment of the TFIIH kinase module at promoter before release during transcription. Nat. Commun., 10, 2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu, N.-Y., Chung, C.-S. and Cheng, S.-C. (2017) Role of Cwc24 in the first catalytic step of splicing and fidelity of 5′ splice site selection. Mol. Cell. Biol., 37:e00580–16. doi: 10.1128/MCB.00580-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shostak, K., Jiang, Z., Charloteaux, B., Mayer, A., Habraken, Y., Tharun, L., Klein, S., Xu, X., Duong, H.Q., Vislovukh, A. et al. (2020) The X-linked trichothiodystrophy-causing gene RNF113A links the spliceosome to cell survival upon DNA damage. Nat. Commun., 11, 1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.King, M.D., Gummer, C.L. and Stephenson, J.B. (1984) Trichothiodystrophy-neurotrichocutaneous syndrome of Pollitt: a report of two unrelated cases. J. Med. Genet., 21, 286–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehmann, A.R., Arlett, C.F., Broughton, B.C., Harcourt, S.A., Steingrimsdottir, H., Stefanini, M., Malcolm, A., Taylor, R., Natarajan, A.T. and Green, S. (1988) Trichothiodystrophy, a human DNA repair disorder with heterogeneity in the cellular response to ultraviolet light. Cancer Res., 48, 6090–6096. [PubMed] [Google Scholar]
- 22.Tolmie, J.L., de Berker, D., Dawber, R., Galloway, C., Gregory, D.W., Lehmann, A.R., McClure, J., Pollitt, R.J. and Stephenson, J.B. (1994) Syndromes associated with trichothiodystrophy. Clin. Dysmorphol., 3, 1–14. [PubMed] [Google Scholar]
- 23.Latour, P., Thauvin-Robinet, C., Baudelet-Méry, C., Soichot, P., Cusin, V., Faivre, L., Locatelli, M.-C., Mayençon, M., Sarcey, A., Broussolle, E. et al. (2010) A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-tooth disease. Am. J. Hum. Genet., 86, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weterman, M.A.J., Kuo, M., Kenter, S.B., Gordillo, S., Karjosukarso, D.W., Takase, R., Bronk, M., Oprescu, S., van Ruissen, F., Witteveen, R.J.W. et al. (2018) Hypermorphic and hypomorphic AARS alleles in patients with CMT2N expand clinical and molecular heterogeneities. Hum. Mol. Genet., 27, 4036–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sundal, C., Carmona, S., Yhr, M., Almström, O., Ljungberg, M., Hardy, J., Hedberg-Oldfors, C., Fred, Å., Brás, J., Oldfors, A., Andersen, O. and Guerreiro, R. (2019) An AARS variant as the likely cause of Swedish type hereditary diffuse leukoencephalopathy with spheroids. Acta Neuropathol. Commun., 7, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simons, C., Griffin, L.B., Helman, G., Golas, G., Pizzino, A., Bloom, M., Murphy, J.L.P., Crawford, J., Evans, S.H., Topper, S. et al. (2015) Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am. J. Hum. Genet., 96, 675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakayama, T., Wu, J., Galvin-Parton, P., Weiss, J., Andriola, M.R., Hill, R.S., Vaughan, D.J., el-Quessny, M., Barry, B.J., Partlow, J.N. et al. (2017) Deficient activity of alanyl-tRNA synthetase underlies an autosomal recessive syndrome of progressive microcephaly, hypomyelination, and epileptic encephalopathy. Hum. Mutat., 38, 1348–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gonzalez, M., McLaughlin, H., Houlden, H., Guo, M., Yo-Tsen, L., Hadjivassilious, M., Speziani, F., Yang, X.-L., Antonellis, A., Reilly, M.M., Züchner, S. and Inherited Neuropathy Consortium (INC) (2013) Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J. Neurol. Neurosurg. Psychiatry, 84, 1247–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gillespie, M.K., McMillan, H.J., Kernohan, K.D., Pena, I.A., Meyer-Schuman, R., Care4Rare Canada Consortium, Antonellis, A. and Boycott, K.M. (2019) A novel mutation in MARS in a patient with Charcot-Marie-Tooth disease, axonal, type 2U with congenital onset. J Neuromuscul. Dis., 6, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Meel, E., Wegner, D.J., Cliften, P., Willing, M.C., White, F.V., Kornfeld, S. and Cole, F.S. (2013) Rare recessive loss-of-function methionyl-tRNA synthetase mutations presenting as a multi-organ phenotype. BMC Med. Genet., 14, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alzaid, M., Alshamrani, A., Harbi, Al, A.S., Alenzi, A. and Mohamed, S. (2019) Methionyl-tRNA synthetase novel mutation causes pulmonary alveolar proteinosis. Saudi Med. J., 40, 195–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Novarino, G., Fenstermaker, A.G., Zaki, M.S., Hofree, M., Silhavy, J.L., Heiberg, A.D., Abdellateef, M., Rosti, B., Scott, E., Mansour, L. et al. (2014) Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science, 343, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vermeulen, W., Bergmann, E., Auriol, J., Rademakers, S., Frit, P., Appeldoorn, E., Hoeijmakers, J.H. and Egly, J.M. (2000) Sublimiting concentration of TFIIH transcription/DNA repair factor causes TTD-A trichothiodystrophy disorder. Nat. Genet., 26, 307–313. [DOI] [PubMed] [Google Scholar]
- 34.Botta, E., Nardo, T., Lehmann, A.R., Egly, J.M., Pedrini, A.M. and Stefanini, M. (2002) Reduced level of the repair/transcription factor TFIIH in trichothiodystrophy. Hum. Mol. Genet., 11, 2919–2928. [DOI] [PubMed] [Google Scholar]
- 35.Stefanini, M., Botta, E., Lanzafame, M. and Orioli, D. (2010) Trichothiodystrophy: from basic mechanisms to clinical implications. DNA Repair (Amst.), 9, 2–10. [DOI] [PubMed] [Google Scholar]
- 36.Kuo, M.E. and Antonellis, A. (2020) Ubiquitously expressed proteins and restricted phenotypes: exploring cell-specific sensitivities to impaired tRNA charging. Trends Genet., 36, 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vermeulen, W., Rademakers, S., Jaspers, N.G., Appeldoorn, E., Raams, A., Klein, B., Kleijer, W.J., Hansen, L.K. and Hoeijmakers, J.H. (2001) A temperature-sensitive disorder in basal transcription and DNA repair in humans. Nat. Genet., 27, 299–303. [DOI] [PubMed] [Google Scholar]
- 38.Klaips, C.L., Jayaraj, G.G. and Hartl, F.U. (2018) Pathways of cellular proteostasis in aging and disease. J. Cell Biol., 217, 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferri, D., Orioli, D. and Botta, E. (2019) Heterogeneity and overlaps in nucleotide excision repair disorders. Clin. Genet. 97, 12–24. [DOI] [PubMed] [Google Scholar]
- 40.Alupei, M.C., Maity, P., Esser, P.R., Krikki, I., Tuorto, F., Parlato, R., Penzo, M., Schelling, A., Laugel, V., Montanaro, L., Scharffetter-Kochanek, K. and Iben, S. (2018) Loss of proteostasis is a pathomechanism in Cockayne syndrome. Cell Rep., 23, 1612–1619. [DOI] [PubMed] [Google Scholar]
- 41.Drmanac, R., Sparks, A.B., Callow, M.J., Halpern, A.L., Burns, N.L., Kermani, B.G., Carnevali, P., Nazarenko, I., Nilsen, G.B., Yeung, G. et al. (2010) Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science, 327, 78–81. [DOI] [PubMed] [Google Scholar]
- 42.Kleijer, W.J., Laugel, V., Berneburg, M., Nardo, T., Fawcett, H., Gratchev, A., Jaspers, N.G.J., Sarasin, A., Stefanini, M. and Lehmann, A.R. (2008) Incidence of DNA repair deficiency disorders in western Europe: xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst.), 7, 744–750. [DOI] [PubMed] [Google Scholar]
- 43.Botta, E., Nardo, T., Orioli, D., Guglielmino, R., Ricotti, R., Bondanza, S., Benedicenti, F., Zambruno, G. and Stefanini, M. (2009) Genotype-phenotype relationships in trichothiodystrophy patients with novel splicing mutations in the XPD gene. Hum. Mutat., 30, 438–445. [DOI] [PubMed] [Google Scholar]
- 44.Arseni, L., Lanzafame, M., Compe, E., Fortugno, P., Afonso-Barroso, A., Peverali, F.A., Lehmann, A.R., Zambruno, G., Egly, J.M., Stefanini, M. and Orioli, D. (2015) TFIIH-dependent MMP-1 overexpression in trichothiodystrophy leads to extracellular matrix alterations in patient skin. Proc. Natl. Acad. Sci. U. S. A., 112, 1499–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.