

Abstract
Since the description of the synthetic chemical clofibrate in 1962, various derivatives of fibrates with a diversity of chemical structures have been developed. Several of these are used clinically to treat dyslipidemia because they are generally effective in lowering elevated plasma triglycerides and cholesterol. Studies suggest that several biochemical mechanisms underlie fibrate-mediated modulation of lipoprotein and related metabolites. These mechanisms are: 1) induced lipoprotein lipolysis; 2) induced hepatic fatty acid uptake and reduced hepatic triglyceride formation; 3) amplified removal of low density lipoprotein (LDL) particles; 4) reduced neutral lipid (cholesteryl ester and triglyceride) exchange between very low density lipoprotein (VLDL) and high density lipoprotein (HDL) resulting from decreased plasma levels of triglyceride-rich lipoprotein (TRL); and 5) increased HDL production and stimulation of reverse cholesterol transport. Recent studies of structure-based inhibitor design strategy revealed that an independent enzyme, aldose reductase (AR), is a target of fibrate activity, an additional biochemical mechanism. AR has been implicated as a major player in the development of diabetes and diabetic complications because of its ability to catalyze the conversion of glucose to sorbitol. This article discusses various targets of fibrate action, biochemical pathways and commonalities in potential molecular interactions.
Keywords: Aldose reductase, fibrates, glucose, mechanism, polyol pathway, peroxisome proliferation, sorbitol
Peroxisome proliferation is a pleiotropic cellular response to a range of compounds that are chemically diverged derivatives of clofibrate [1]. This group of compounds, known as peroxisome proliferators and which includes fibrates, induces numerous alterations in hepatic lipid metabolism. There are several comprehensive review articles on various aspects of peroxisome proliferation [2]. This article focuses on five mechanisms that the fibrates are known to undergo in the light of the aldose reductase (AR) mediated pathway, which is recently identified as an independent target of action [3]. Furthermore the binding signatures of fibrates that may be a common determinant among targets are highlighted.
CLINICAL BENEFITS OF FIBRATES
Fibrates are synthetic molecules [4] and, as a group, have diverse chemical structures (Fig. 1). In general, fibrates have a phenoxyisobutyrate moiety that results in a hydrophilic and a hydrophobic group in close proximity. The parachloro moiety is present in many fibrates but not in gemfibrozil and ciprofibrate. Gemfibrozil also differs from other fibrates because of the methylene groups between the phenoxy oxygen atom and the phenyl ring. Addition of a second phenyl ring, in the case of fenofibrate and bezafibrate, seems to provide a higher specificity for the molecule as revealed by a 5–10-fold gain in relative lipid-lowering potency compared to that of the prototype of this class of compounds, clofibrate [5]. Although fibrates are generally compounds with an ester moiety, the term fibrate is used more broadly to refer to the class of fibric acids, their esters and their derivatives with divergent chemical structures.
Fig. (1). Chemical structures of fibrates.
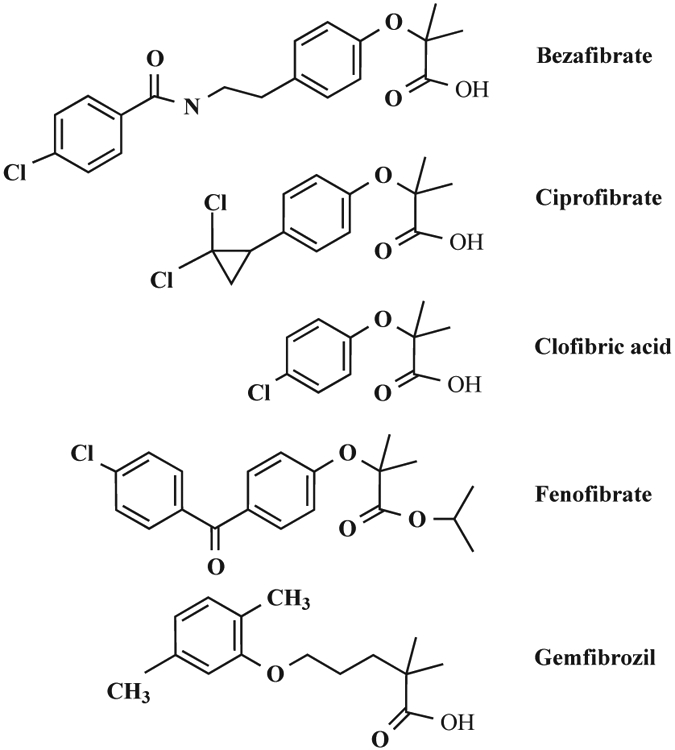
bezafibrate (2-[4-[2-(4-chloro-benzamiso)ethyl] phenoxy]-2-methyl-propionic acid), ciprofibrate (2-[p-(2,2-dichlorocyclopropyl) phenoxy]-2-methylpropanoic acid), clofibric acid (2-(4-chlorophenoxy)-2-methylpropionic acid), fenofibrate (Isopropyl 2-[4-(4-chlorobenzoyl) phenoxy]-2–methylpropionate) and gemfibrozil (2,2-dimethyl-5-[2,5-dimethylphenoxy]-pentanoic acid).
Fibrates are a part of a widely used class of lipid-lowering drugs that have been in utilization for more than thirty years for the treatment of hypertriglyceridemia or mixed hyperlipidemia [6]. Known pharmacological properties of fibrates are: reduced incidence of cardiovascular disease in patients with Type 2 diabetes, decreased serum triglycerides (via reduced hepatic production as well as increased triglyceride clearance by the peripheral tissues), lowered insulin resistance and fasting blood glucose levels in non-obese Japanese Type 2 diabetic patients, decreased concentrations of circulating, small, low density lipoproteins (LDL), increased high density lipoproteins (HDL) and improved glucose tolerance [7], resulting in favorable effects on blood coagulation and global fibrinolytic function. Fibrates may also be able to reduce the lipid content of intermediate density lipoprotein in Type 1 diabetes [8].
Fibrates can be used alone or in combination as therapies for various medical conditions. Accelerated coronary heart disease (CHD) is a major problem in the management of patients with renal disease [9]. The basis of lipid-lowering therapy in renal disease is the treatment with statins (3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors). Statins are a well-established therapy in coronary artery disease. For example, atorvastatin reduces LDL and triglycerides by 26% in patients with renal transplants [10]. However, statins have only a limited potential for reducing triglycerides in type V hyperlipidemia, although some evidence exists for the benefit in hypertriglyceridemia through an action dependent on the LDL-lowering potential and baseline triglycerides [11]. Also statins prevent coronary events in patients who have had myocardial infarction, unstable angina and hypercholesterolemia without clinical coronary disease. The benefits of statins are derived primarily by reducing elevated low-density lipoprotein cholesterol (LDL-C) [12]. However, the benefits of monotherapy are diminished when the baseline level of LDL-C is low [12]. Statin-fibrate combination therapy—a low frequency of fibrate administration coupled with daily treatment of statin—results in a 49% reduction in total cholesterol and 82% reduction in triglycerides along with a 52% increase in HDL [13].
INTERACTIONS WITH FIBRATES
Peroxisome proliferator-activated receptors (PPARs) are a subgroup of nuclear receptors that are thought to play important roles in both lipid and glucose homeostasis [14]. Members of this family of transcription factors show structural and functional conservation [15], with the most conserved domains being the DNA binding domain and the C-terminus, which encompass the ligand binding domain [16]. The C-terminus is also required for homo- and heterodimerization, as well as interaction with transcriptional cofactors and components of the general transcriptional assembly. Although the conserved amino acid sequence among ligand binding domains is less than 25%, the overall three-dimensional structure of different ligand binding domains is highly conserved [17].
In all of the PPAR-dependent mechanisms, fibrates bind with PPAR and activate them to form a complex with another transcription factor, retinoid X receptor (RXR) and cofactor (Fig. 2). Transcriptional regulation by PPAR is achieved through a PPAR-RXR heterodimer in complex with a cofactor, interacting with DNA motifs termed peroxisome proliferative response elements (PPREs), in the promoters of target genes [18].
Fig. (2). Mechanisms of fibrate action.
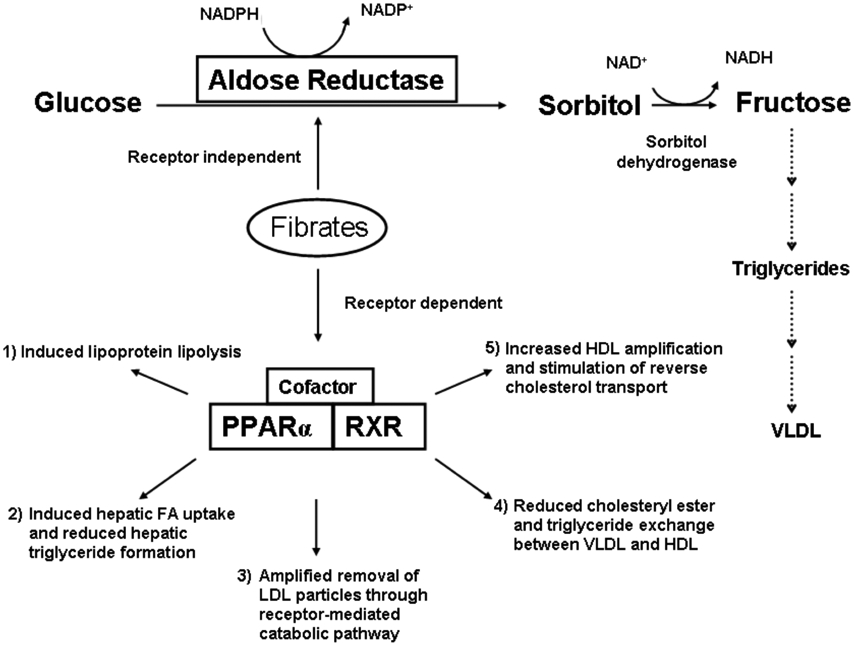
Fibrates are purported to act through five major mechanisms that are dependent on the PPAR receptor. The polyol pathway that goes through AR in the catalysis of sorbitol formation in the presence of NADPH is an additional pathway in the modulation of triglyceride and lipoproteins levels.
The ligand binding domain of PPARα is made up of a three-layer arrangement of 13 α-helixes and a small four-stranded β sheet [19, 20]. With the exception of an extra helix, this architecture is very similar to that of other nuclear receptors. Three long helices (H3, H7 and H10/H11) form the two outer layers (Fig. 3). The middle layer of helices (H4, H5, H8, H9) occupies the bottom half of the domain and is absent from the top half, thereby creating a very large cavity for ligand binding (Fig. 3). This cavity has a distinct three-arm Y-shape architecture allowing PPARs to bind ligands with multiple branches or multiple conformations of ligands with single branches. On its upper half, one side of the ligand binding domain is sealed by a two-stranded β sheet and the other side by the short C-terminal α-helix (H12) that constitutes the ligand-dependent transcriptional activation region of the receptor. GW409544, a human PPARα-specific agonist binds in the ligand-binding pocket of the receptor through a collection of amino acid residues [20]. Hydrogen bonds are formed between the carboxyl moiety and the residues His323, Tyr473 and His449 (Fig. 4). The stacking interaction between residue His323 and Tyr473 in this complex may play a role in stabilizing the H12. This interaction may help stabilize H12 and, thus, may affect the basal activity of PPARα.
Fig. (3). Three-dimensional structure of PPAR.

Ribbon drawing of PPARα ligand binding domain. Ligand is shown with surface representation in the binding cavity. H1 – H12 refer to the helices of the ligand binding domain of PPARα.
Fig. (4). Ligand interactions.

Ligand binding in PPARs involves conserved Tyr and His residues. Two out of four His and Try residues form hydrogen bonding interactions with the ligand. For clarity side chain for only residues His323, Tyr327, Tyr473 and His449 are shown. Hydrogen bonding interactions are marked with yellow dash lines with their bonding distance in Å.
MECHANISMS OF FIBRATE ACTION
Based on studies in rodents and in humans, five major mechanisms have been proposed [21, 22] in which lipoprotein phenotypes are modulated by fibrates through the activation of the PPAR-dependent pathway (Fig. 2): induced lipoprotein lipolysis [22]; induced hepatic fatty acid (FA) uptake and reduced hepatic triglyceride formation [23]; amplified removal of LDL particles through receptor-mediated catabolic pathway [24]; reduced neutral lipid (cholesteryl ester and triglyceride) exchange between very low density lipoprotein (VLDL) and HDL [25]; and increased HDL production and amplification of the reverse cholesterol transport through the formation of apolipoprotein (apo) A-I and apo A-II in liver [7].
INDUCED LIPOPROTEIN LIPOLYSIS
Fibrates increase the expression of the enzyme lipoprotein lipase (LPL) [26], which enhances the accessibility of triglyceride-rich lipoproteins (TRLs) for lipolysis and lowers TRL apo C-III content [27]. LPL plays a central role in lipid metabolism by hydrolyzing TRLs and releasing FAs in adipocytes and non-adipocytes. The initial step in the catabolism of plasma TRLs occurs through the hydrolysis of triglycerides by LPL. However, proper hydrolysis of these triglyceride-rich particles (chylomicrons and VLDL) depends not only on LPL but also on their relative contents of various C apolipoproteins. The ratio of apo C-III to apo C-II has been implicated as a determinant for efficient lipid metabolism, with low ratios favoring lipolysis [28]. Fibrate therapy results in the induction of liver LPL expression and a down-regulation of apo C-III expression [29]. These changes contribute to the increase in both lipolysis and receptor-mediated removal of remnant lipoproteins and results in the clinical effects observed after fibrate treatment. Fibrates also decrease gene expression of apo C-II and apo C-III, but not apo C-I, in rat and human hepatocytes. This decreased gene expression, together with a lowered apo C-III to apo C-II ratio, should result in an improved clearance of triglyceride-rich remnant lipoproteins from plasma without hampering triglyceride lipolysis by LPL [30]. The role of apo C-III is due to the transcriptional suppression of hepatic nuclear factor (HNF)-4 as well as the displacement of HNF-4 from the apo C-III promoter. The HNF-4 displacement exerted by peroxisome proliferators and hypolipidemic amphipathic carboxylates is mediated by PPAR [29].
INDUCED HEPATIC FA UPTAKE AND REDUCED HEPATIC TRIGLYCERIDE FORMATION
Uncharged molecules and weak acids such as FA can cross membranes rapidly due to their lipid solubility. Intracellular FA concentrations are, in part, determined by the regulation of an import/export system that is controlled by two key proteins, namely FA transport protein (FATP) [23] and acyl-CoA synthetase (ACS) [31]. FATP acts in the transport of FA, whereas ACS prevents efflux of the newly imported FA by their esterification with coenzyme A. These proteins enhance FA flux towards the β-oxidation pathway, which results in a lower availability of FA for triglycerides synthesis.
FAs are important cellular components that can function both as metabolic substrates or as signaling molecules, by operating as second messengers and triggering signal transduction pathways. Alternatively, FAs may directly activate transcription factors such as PPAR. The liver is one of the major organs susceptible to peroxisomal proliferation, and treatment with fibrates strongly induces FATP gene transcription. Peroxisome proliferation caused by fibrates is, at least in part, the result of a transcriptional induction of the β-oxidation pathway enzymes [32]. The peroxisomal β-oxidation system is composed of three proteins: fatty acyl-CoA oxidase, which catalyzes the dehydrogenation of fatty acyl-CoA leading to the production of H2O2; peroxisomal enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase bifunctional enzyme, which catalyzes the second and third reactions of the β-oxidation cycle, yielding 3-ketoacyl-CoA from enoyl-CoA: and 3-ketoacyl-CoA thiolase, which catalyzes the last reaction of the β-oxidation cycle, forming acetyl-CoA. Reduced FA synthesis by fibrates leads to lower triglyceride synthesis. This phenomenon is enhanced by fibrate-induced inhibition of hormone-sensitive lipase in adipose tissue. Administration of fibrates to rats induced liver and intestinal expression of the acyl CoA oxidase gene, the rate-limiting enzyme for peroxisomal β-oxidation of FAs [29].
Amplified removal of LDL particles through receptor-mediated catabolic pathway
Fibrates lower plasma triglycerides by inhibiting VLDL synthesis and secretion as a result of decreased free FA flux to the liver [33]. Furthermore, they are able to suppress hepatic cholesterol synthesis [34] and stimulate LDL receptor activity [35]. The effect that fibrates have on LDL varies and depends on the initial plasma triglyceride level. LDL is the major transport vehicle for cholesterol in human plasma. Several human cell types, including fibroblasts [36], arterial smooth muscle cells [37], lymphoid cells [38], and endothelial cells [39] share a specific LDL degradation pathway. This consists of an autoregulated, ordered sequence of events in which LDL binds first to a high affinity receptor on the cell surface and is then endocytosed and delivered to lysosomes. In lysosomes, the lipoprotein-associated cholesteryl esters are hydrolyzed to release free cholesterol for cellular use [40]. Fenofibrate not only promotes LDL catabolism through a receptor-mediated pathway, but also lowers plasma triglyceride levels to inhibit the formation of slowly-metabolized LDL particles [24]. Fibrate therapy results in both qualitative and quantitative normalization of the atherogenic-dense LDL particle profile characteristic of type IIB hyperlipoproteinemia. Fibrate therapy also induces a shift in the peak of the density profile from small, dense LDL toward larger, buoyant LDL receptor-active LDL particles, typical of normo-lipidemic subjects [41].
REDUCED NEUTRAL LIPID (CHOLESTERYL ESTER AND TRIGLYCERIDE) EXCHANGE BETWEEN VLDL AND HDL
Nascent HDL picks up cholesterol from cells in the periphery and converts it to cholesteryl ester-rich HDL through the action of lecithin-cholesterol acyltransferase (LCAT). In normal lipoprotein metabolism, HDL-cholesteryl ester can be transferred to triglyceride-rich lipoproteins through the plasma cholesteryl ester transfer protein (CETP) [42]. CETP promotes the net transfer of lipids among lipoproteins [43]. Because of its unique capacity to mediate net exchange of triglyceride and cholesteryl ester between triglyceride-rich VLDL and cholesteryl ester-rich lipoproteins such as LDL and HDL, CETP plays an essential role in regulating plasma cholesterol levels. By altering lipoprotein core lipid composition, CETP participates in the catabolism of VLDL to LDL [44] and affects the level of LDL subfractions [45].
Fibrates increase excretion of hepatic cholesterol in bile and endogenous hepatic cholesterol synthesis may be decreased [46]. Also, fibrates reduce the activity of CETP either by inhibiting it or reducing its expression and decreasing the conversion of HDL to VLDL, intermediate-density lipoprotein (IDL) leading to delayed catabolism of HDL-cholesteryl ester with consequent increase in the concentration of HDL cholesterol. This reduced conversion causes rapid reduction in the concentration of cholesteryl ester and triglycerides. Fenofibrate not only reduces absolute concentrations of circulating VLDL but also induces a reduction in the size of VLDL particles of hepatic origin. This phenomenon is a consequence of the intravascular remodeling of such VLDLs due to receptor-active LDLs that are formed, thereby resulting in an increase in the proportion of plasma LDLs that are catabolized by the nonatherogenic LDL receptor pathway [47]. CETP-facilitated transfer and exchange of neutral lipids between HDL and apo B-containing lipoproteins (VLDL, IDL, and LDL) play a major role in the determination of the apo A-I and apo B-containing lipoprotein subspecies profile [48]. Fibrates can induce significant reductions in both the elevated total plasma CETP-dependent cholesteryl ester transfer (CET) activity and in CET from HDL to VLDL in combined hyperlipidemia, where such total transfer activity is strongly correlated to plasma CETP mass [42].
INCREASED HDL PRODUCTION AND AMPLIFICATION OF THE REVERSE CHOLESTEROL TRANSPORT THROUGH THE FORMATION OF APO A-I AND APO A-II IN LIVER
The hypotriglyceridemic action of fibrates is attributable to both an enhanced catabolism of plasma triglyceride-rich particles and an inhibition of secretion of triglyceride-rich particles from the liver. Reverse cholesterol transport may contribute to the increase in plasma HDL concentrations and an increase in glucose tolerance [7]. Reverse cholesterol transport also describes the pathway and an important anti-atherogenic function of HDL, namely, the HDL-mediated efflux of cholesterol from non-hepatic cells and its subsequent delivery to the liver and steroidogenic organs in which it is used for the synthesis of lipoproteins, bile acids, vitamin D, and steroidal hormones [49]. Furthermore, reverse cholesterol transport involves the trafficking of excess cholesterol from peripheral cells to the liver by nascent HDL. HDL-bound cholesterol is taken up by the liver for biliary secretion and fecal excretion. The ATP-binding cassette transporter A1 (ABCA1) is critical for the efflux of excess cellular cholesterol to apo acceptors such as apo A-I in reverse cholesterol transport.
Cholesterol efflux, the first step in reverse cholesterol transport, occurs either via passive diffusion or via transmembrane transporters such as the scavenger receptor (SR) class B type 1 and the ABCA1 proteins. In human macrophages, PPARα- and PPARγ-activators induce protein levels of SR class B type 1 [50], whereas ligands of all the three PPAR isotypes induce the expression of ABCA1 [51]. PPARα and PPARγ induce ABCA1 by an indirect mechanism via induction of the nuclear liver X receptor (LXR)α [52]. However, the molecular mechanism of ABCA1 induction by PPARβ/γ activators appears to be independent of LXRα [51]. The increased ABCA1 expression results in a higher cholesterol efflux from macrophages. In addition, the HDL-associated free cholesterol would then be esterified in the extracellular compartment by LCAT, thereby increasing the cholesteryl ester content of the HDL. This increased amount of ester could then be delivered selectively to the liver by the mechanism described above without concomitant uptake of apo A-I. Thus, an HDL molecule or some parts of the HDL could function through several cycles for the transfer of cholesterol from the periphery to the liver [53].
AR MEDIATED FIBRATE MECHANISM IN THE REGULATION OF TRIGLYCERIDES SYNTHESIS
Fibrates are thought to reduce plasma triglycerides through the activation of PPAR by five major mechanisms [22]. In addition to the above five PPAR dependent mechanisms, some fibrates (bezafibrate, ciprofibrate, clofibrate, fenofibrate and gemfibrozil) (Fig. 1) have been found to inhibit AR activity independent of the PPAR pathway [3]. The catalysis by AR mediates the formation of sorbitol, which eventually gets converted to triglycerides under hyperglycemic conditions via fructose [54]. This biochemical process is the first step of the polyol pathway, which could possibly be the sixth mechanism by which fibrates regulate plasma triglycerides in the hepatic cells.
Sorbitol, formed from glucose by the catalysis of AR, is converted to fructose by sorbitol dehydrogenase (Fig. 2). This leads to an overflow of products of the polyol pathway, causing depletion in NADPH and NAD+ and a concomitant increase in the ratio of NADH:NAD+, modifying the redox state of the cells and leading to the production of superoxide anions [55]. Under hyperglycemic conditions, non-enzymatic glycation of structural proteins is enhanced and advanced glycation end-products accumulate in diabetic tissues. As a glycation agent, fructose is more potent than glucose [56], and the formation of fructose is augmented due to the accelerated flux of glucose through the polyol pathway. Fructose is more lipogenic than glucose because it stimulates a greater exchange of acetate to FAs [57] and increases rates of de novo lipogenesis [58]. Furthermore, fructose provides carbon atoms for both the glycerol and the acyl motifs of acylglycerol molecules [59] and is implicated as a major contributor of hyperlipidemia [60].
The hepatic metabolism of fructose has important effects on both glucose and lipid metabolism. Therefore, fructose is more lipogenic than glucose, an effect that might be exacerbated in subjects with existing hyperlipidemia [61] or Type 2 diabetes [62]. The inhibition of the polyol pathway enzyme, AR, under ischemic conditions in the hearts of diabetic and non-diabetic rats resulted in (i) ATP preservation, (ii) reduction in ischemic injury as well as the lowering of the abnormally elevated cytosolic redox state (NADH/NAD+), (iii) normalization of glycolysis, and (iv) increased glycolytic substrate availability for incorporation into the tricarboxylic acid (TCA) cycle [63]. Hence, it is conceivable that the inhibition of AR by fibrates may result in the regulation of plasma triglycerides in hepatic cells.
The structural architecture and the interactions, involving Tyr and His residues, that networks with the carboxyl moiety of ligands in PPAR are very similar to those found in AR inhibitor complexes. The active site of AR is defined by residues Trp20, Lys21, Val47, Glu49, Tyr48, His110, Trp111, Phe121, Phe122, Pro218, Trp219, Cys298, Leu300, Leu301 and Ser302. Among these active site residues His110 and Tyr48 are capable of forming hydrogen binding interactions with the carboxyl moiety of the ligands. In the crystal structure of the holoenzyme complexed with the inhibitor, sorbinil [64] binding involves the formation of hydrogen bonds with the catalytic residues and hydrophobic contacts within the residues of the active site pocket. Specifically, one of the carbonyl oxygen atoms of sorbinil forms a hydrogen bond with the OH group of the Tyr48 [64] in AR. Furthermore in the AR inhibitor IDD594 complex, the carboxyl group forms hydrogen bonding interactions with Tyr48 and His110 [65]. This implies that the electronic environment and structural arrangements found in AR are conducive to the unique features that are required for fibrate binding to PPARs.
CONCLUSIONS AND OUTLOOK
Physiological role of fibrates is dictated by their chemical interactions with the biological targets. Binding of fibrates to AR may be similar to that found to be formed by ionic and hydrophobic interactions with PPARs. However, the most intriguing features of AR and PPAR are the arrangement and the availability of Tyr and His residues in the active site and ligand binding cavity, respectively. In order to modulate the AR-related pathway in a manner different from the PPAR-mediated pathway, more specific derivatives of fibrates are required. Alternatively, when fibrates are administered based on requirements of the PPAR-mediated pathway, AR will be a target of fibrates action as well. This is mainly due to the flexibility of AR and PPAR, their broader substrate/ligand specificities and their ability to undergo induced fit among other factors. Therefore, to avoid cross-recognition, fibrate derivatives are required that regulate PPAR uniquely and are unable to bind to AR. Future studies such as knock-out at the cellular, tissue and animal level are required to demonstrate that AR is an independent target for fibrates action. Also, despite numerous attempts over many years to develop effective AR inhibitors to delay or prevent diabetic complications have not proven their efficacy and bioavailability [66]. Since fibrates have preferred drug-like absorption profiles as demonstrated by their clinical application, fibrate-like molecules may have advantages in drug discovery efforts to inhibit AR activity.
ACKNOWLEDGEMENTS
This work was supported by funding from the American Diabetes Association. We thank Dr. Kristine Justus for editorial assistance.
REFERENCES
- [1].Reddy JK, Krishnakantha TP. Hepatic peroxisome proliferation: induction by two novel compounds structurally unrelated to clofibrate. Science 1975; 190: 787–9. [DOI] [PubMed] [Google Scholar]
- [2].Peters JM, Cheung C, Gonzalez FJ. Peroxisome proliferator-activated receptor-alpha and liver cancer: where do we stand? J Mol Med 2005; 83: 774–85. [DOI] [PubMed] [Google Scholar]
- [3].Balendiran GK, Rajkumar B. Fibrate inhibit aldose reductase in the forward and reverse reactions. Biochem Pharmacol 2005; 70: 1653–63. [DOI] [PubMed] [Google Scholar]
- [4].Throp JM. Experimental evaluation of an orally active combination of androsterone with ethyl chlorophenoxy-isobutyrate. Lancet 1962; 1: 1323–6. [DOI] [PubMed] [Google Scholar]
- [5].Hunninghake DB, Peters JR. Effect of fibric acid derivatives on blood lipid and lipoprotein levels. Am J Med 1987; 83: 44–9. [DOI] [PubMed] [Google Scholar]
- [6].Forcheron F, Cachefo A, Thevenon S, Pinteur C, Beylot M. Mechanisms of the triglyceride- and cholesterol-lowering effect of fenofibrate in hyperlipidemic type 2 diabetic patients. Diabetes 2002; 51: 3486–91. [DOI] [PubMed] [Google Scholar]
- [7].Meade T, Zuhrie R, Cook C, Cooper J. Bezafibrate in men with lower extremity arterial disease: randomised controlled trial. BMJ 2002; 325: 1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Winocour PH, Durrington PN, Bhatagnar D, et al. The effect of bezafibrate on very low density lipoprotein (VLDL), intermediate density lipoprotein (IDL), and low density lipoprotein (LDL) composition in type 1 diabetes associated with hypercholesterolaemia or combined hyperlipidaemia. Atherosclerosis 1992; 93: 83–94. [DOI] [PubMed] [Google Scholar]
- [9].Levin A, Djurdjev O, Barrett B, et al. Cardiovascular disease in patients with chronic kidney disease: getting to the heart of the matter. Am J Kidney Dis 2001; 38: 1398–407. [DOI] [PubMed] [Google Scholar]
- [10].Romero R, Calvino J, Rodriguez J, Sanchez-Guisande D. Short-term effect of atorvastatin in hypercholesterolaemic renal-transplant patients unresponsive to other statins. Nephrol Dial Transplant 2000; 15: 1446–9. [DOI] [PubMed] [Google Scholar]
- [11].Wierzbicki AS, Lumb PJ, Chik G, Crook MA. High-density lipoprotein cholesterol and triglyceride response with simvastatin versus atorvastatin in familial hypercholesterolemia. Am J Cardiol 2000; 86: 547–9, A9. [DOI] [PubMed] [Google Scholar]
- [12].Badawy O, Wierzbicki AS, Hilton R. Combination fibrate-statin therapy for the treatment of severe hypertriglyceridaemia in renal disease. Int J Clin Pract 2003; 57: 249–51. [PubMed] [Google Scholar]
- [13].Sacks FM. The relative role of low-density lipoprotein cholesterol and high-density lipoprotein cholesterol in coronary artery disease: evidence from large-scale statin and fibrate trials. Am J Cardiol 2001; 88: 14N–18N. [DOI] [PubMed] [Google Scholar]
- [14].Spiegelman BM. PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes 1998; 47: 507–14. [DOI] [PubMed] [Google Scholar]
- [15].Escriva H, Safi R, Hanni C, et al. Ligand binding was acquired during evolution of nuclear receptors. Proc Natl Acad Sci USA 1997; 94: 6803–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Glass CK, Rose DW, Rosenfeld MG. Nuclear receptor coactivators. Curr Opin Cell Biol 1997; 9: 222–32. [DOI] [PubMed] [Google Scholar]
- [17].Tanenbaum DM, Wang Y, Williams SP, Sigler PB. Crystallographic comparison of the estrogen and progesterone receptor's ligand binding domains. Proc Natl Acad Sci USA 1998; 95: 5998–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schoonjans K, Staels B, Auwerx J. Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J Lipid Res 1996; 37: 907–25. [PubMed] [Google Scholar]
- [19].Nolte RT, Wisely GB, Westin S, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998; 395: 137–43. [DOI] [PubMed] [Google Scholar]
- [20].Xu HE, Lambert MH, Montana VG, et al. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA 2001; 98: 13919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gonzalez FJ. The peroxisome proliferator-activated receptor alpha (PPARa1-pha): role in hepatocarcinogenesis. Mol Cell Endocrinol 2002; 193: 71–9. [DOI] [PubMed] [Google Scholar]
- [22].Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998; 98: 2088–93. [DOI] [PubMed] [Google Scholar]
- [23].Martin G, Schoonjans K, Lefebvre AM, Staels B, Auwerx J. Coordinate regulation of the expression of the fatty acid transport protein and acyl-CoA synthetase genes by PPARalpha and PPARgamma activators. J Biol Chem 1997; 272: 28210–7. [DOI] [PubMed] [Google Scholar]
- [24].Chapman MJ, Guerin M, Bruckert E. Atherogenic, dense low-density lipoproteins. Pathophysiology and new therapeutic approaches. Eur Heart J 1998; 19(Suppl A): A24–30. [PubMed] [Google Scholar]
- [25].Mann CJ, Yen FT, Grant AM, Bihain BE. Mechanism of plasma cholesteryl ester transfer in hypertriglyceridemia. J Clin Invest 1991; 88: 2059–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Heller F, Harvengt C. Effects of clofibrate, bezafibrate, fenofibrate and probucol on plasma lipolytic enzymes in normolipaemic subjects. Eur J Clin Pharmacol 1983; 25: 57–63. [DOI] [PubMed] [Google Scholar]
- [27].Malmendier CL, Lontie JF, Delcroix C, Dubois DY, Magot T, De Roy L. Apolipoproteins C-II and C-III metabolism in hypertriglyceridemic patients. Effect of a drastic triglyceride reduction by combined diet restriction and fenofibrate administration. Atherosclerosis 1989; 77: 139–49. [DOI] [PubMed] [Google Scholar]
- [28].Carlson LA, Ballantyne D. Changing relative proportions of apolipoproteins CII and CIII of very low density lipoproteins in hypertriglyceridaemia. Atherosclerosis 1976; 23: 563–8. [DOI] [PubMed] [Google Scholar]
- [29].Staels B, Vu-Dac N, Kosykh VA, et al. Fibrates downregulate apolipoprotein C-III expression independent of induction of peroxisomal acyl coenzyme A oxidase. A potential mechanism for the hypolipidemic action of fibrates. J Clin Invest 1995; 95: 705–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Andersson Y, Majd Z, Lefebvre AM, et al. Developmental and pharmacological regulation of apolipoprotein C-II gene expression. Comparison with apo C-I and apo C-III gene regulation. Arterioscler Thromb Vasc Biol 1999; 19: 115–21. [DOI] [PubMed] [Google Scholar]
- [31].Schoonjans K, Watanabe M, Suzuki H, et al. Induction of the acyl-coenzyme A synthetase gene by fibrates and fatty acids is mediated by a peroxisome proliferator response element in the C promoter. J Biol Chem 1995; 270: 19269–76. [DOI] [PubMed] [Google Scholar]
- [32].Reddy JK, Goel SK, Nemali MR, et al. Transcription regulation of peroxisomal fatty acyl-CoA oxidase and enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase in rat liver by peroxisome proliferators. Proc Natl Acad Sci USA 1986; 83: 1747–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kissebah AH, Adams PW, Harrigan P, Wynn V. The mechanism of action of clofibrate and tetranicotinoylfructose (Bradilan) on the kinetics of plasma free fatty acid and triglyceride transport in type IV and type V hypertriglyceridaemia. Eur J Clin Invest 1974; 4: 163–74. [DOI] [PubMed] [Google Scholar]
- [34].Berndt J, Gaumert R, Still J. Mode of action of the lipid-lowering agents, clofibrate and BM 15075, on cholesterol biosynthesis in rat liver. Atherosclerosis 1978; 30: 147–52. [DOI] [PubMed] [Google Scholar]
- [35].Stewart JM, Packard CJ, Lorimer AR, Boag DE, Shepherd J. Effects of bezafibrate on receptor-mediated and receptor-independent low density lipoprotein catabolism in type II hyperlipoproteinaemic subjects. Atherosclerosis 1982; 44: 355–65. [DOI] [PubMed] [Google Scholar]
- [36].Goldstein JL, Brown MS. Binding and degradation of low density lipoproteins by cultured human fibroblasts. Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. J Biol Chem 1974; 249: 5153–62. [PubMed] [Google Scholar]
- [37].Goldstein JL, Brown MS. Lipoprotein receptors, cholesterol metabolism, and atherosclerosis. Arch Pathol 1975; 99: 181–4. [PubMed] [Google Scholar]
- [38].Ruiu G, Pinach S, Gambino R, et al. Influence of cyclosporine on low-density lipoprotein uptake in human lymphocytes. Metabolism 2005; 54: 1620–5. [DOI] [PubMed] [Google Scholar]
- [39].Rinninger F, Kaiser T, Mann WA, Meyer N, Greten H, Beisiegel U. Lipoprotein lipase mediates an increase in the selective uptake of high density lipoprotein-associated cholesteryl esters by hepatic cells in culture. J Lipid Res 1998; 39: 1335–48. [PubMed] [Google Scholar]
- [40].Kruth HS. Lipoprotein cholesterol and atherosclerosis. Curr Mol Med 2001; 1: 633–53. [DOI] [PubMed] [Google Scholar]
- [41].Guerin M, Le Goff W, Frisdal E, et al. Action of ciprofibrate in type IIb hyperlipoproteinemia: modulation of the atherogenic lipoprotein phenotype and stimulation of high-density lipoprotein-mediated cellular cholesterol efflux. J Clin Endocrinol Metab 2003; 88: 3738–46. [DOI] [PubMed] [Google Scholar]
- [42].Paromov VM, Morton RE. Lipid transfer inhibitor protein defines the participation of high density lipoprotein subfractions in lipid transfer reactions mediated by cholesterol ester transfer protein (CETP). J Biol Chem 2003; 278: 40859–66. [DOI] [PubMed] [Google Scholar]
- [43].Tall A Plasma lipid transfer proteins. Annu Rev Biochem 1995; 64: 235–57. [DOI] [PubMed] [Google Scholar]
- [44].Deckelbaum RJ, Eisenberg S, Oschry Y, Butbul E, Sharon I, Olivecrona T. Reversible modification of human plasma low density lipoproteins toward triglyceride-rich precursors. A mechanism for losing excess cholesterol esters. J Biol Chem 1982; 257: 6509–17. [PubMed] [Google Scholar]
- [45].Lagrost L, Gambert P, Lallemant C. Combined effects of lipid transfers and lipolysis on gradient gel patterns of human plasma LDL. Arterioscler Thromb 1994; 14: 1327–36. [DOI] [PubMed] [Google Scholar]
- [46].Guerin M, Bruckert E, Dolphin PJ, Turpin G, Chapman MJ. Fenofibrate reduces plasma cholesteryl ester transfer from HDL to VLDL and normalizes the atherogenic, dense LDL profile in combined hyperlipidemia. Arterioscler Thromb Vasc Biol 1996; 16: 763–72. [DOI] [PubMed] [Google Scholar]
- [47].Caslake MJ, Packard CJ, Gaw A, et al. Fenofibrate and LDL metabolic heterogeneity in hypercholesterolemia. Arterioscler Thromb 1993; 13: 702–11. [DOI] [PubMed] [Google Scholar]
- [48].Guerin M, Dolphin PJ, Chapman MJ. Preferential cholesteryl ester acceptors among the LDL subspecies of subjects with familial hypercholesterolemia. Arterioscler Thromb 1994; 14: 679–85. [DOI] [PubMed] [Google Scholar]
- [49].Curtiss LK, Valenta DT, Hime NJ, Rye KA. What is so special about apolipoprotein AI in reverse cholesterol transport? Arterioscler Thromb Vasc Biol 2006; 26: 12–9. [DOI] [PubMed] [Google Scholar]
- [50].Chinetti G, Gbaguidi FG, Griglio S, et al. CLA-1/SR-BI is expressed in atherosclerotic lesion macrophages and regulated by activators of peroxisome proliferator-activated receptors. Circulation 2000; 101: 2411–7. [DOI] [PubMed] [Google Scholar]
- [51].Oliver WR Jr., Shenk JL, Snaith MR, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci USA 2001; 98: 5306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chawla A, Boisvert WA, Lee CH, et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell 2001; 7: 161–71. [DOI] [PubMed] [Google Scholar]
- [53].Pittman RC, Steinberg D. Sites and mechanisms of uptake and degradation of high density and low density lipoproteins. J Lipid Res 1984; 25: 1577–85. [PubMed] [Google Scholar]
- [54].Hellerstein MK. Carbohydrate-induced hypertriglyceridemia: modifying factors and implications for cardiovascular risk. Curr Opin Lipidol 2002; 13: 33–40. [DOI] [PubMed] [Google Scholar]
- [55].Yabe-Nishimura C Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacol Rev 1998; 50: 21–33. [PubMed] [Google Scholar]
- [56].Stevens VJ, Vlassara H, Abati A, Cerami A. Nonenzymatic glycosylation of hemoglobin. J Biol Chem 1977; 252: 2998–3002. [PubMed] [Google Scholar]
- [57].Baker N, Chaikoff IL, Schusdek A. Effect of fructose on lipogenesis from lactate and acetate in diabetic liver. J Biol Chem 1952; 194: 435–43. [PubMed] [Google Scholar]
- [58].Schwarz JM, Neese RA, Turner SM, Nguyen C, Hellerstein MK. Effect of fructose ingestion on glucose production (GP) and de novo lipogenesis (DNL) in normal and hyperinsulinemic obese humans. Diabetes 1994; 43: 52A. [Google Scholar]
- [59].Mayes PA. Intermediary metabolism of fructose. Am J Clin Nutr 1993; 58: 754S–765S. [DOI] [PubMed] [Google Scholar]
- [60].Kelley GL, Allan G, Azhar S. High dietary fructose induces a hepatic stress response resulting in cholesterol and lipid dysregulation. Endocrinology 2004; 145: 548–55. [DOI] [PubMed] [Google Scholar]
- [61].Jeppesen J, Chen YI, Zhou MY, Schaaf P, Coulston A, Reaven GM. Post-prandial triglyceride and retinyl ester responses to oral fat: effects of fructose. Am J Clin Nutr 1995; 61: 787–91. [DOI] [PubMed] [Google Scholar]
- [62].Kanarek RB, Orthen-Gambill N. Differential effects of sucrose, fructose and glucose on carbohydrate-induced obesity in rats. J Nutr 1982; 112: 1546–54. [DOI] [PubMed] [Google Scholar]
- [63].Trueblood N, Ramasamy R. Aldose reductase inhibition improves altered glucose metabolism of isolated diabetic rat hearts. Am J Physiol 1998; 275: H75–83. [DOI] [PubMed] [Google Scholar]
- [64].Urzhumtsev A, Tete-Favier F, Mitschler A, et al. A 'specificity' pocket inferred from the crystal structures of the complexes of aldose reductase with the pharmaceutically important inhibitors tolrestat and sorbinil. Structure 1997; 5: 601–12. [DOI] [PubMed] [Google Scholar]
- [65].Howard EI, Sanishvili R, Cachau RE, et al. Ultrahigh resolution drug design I: details of interactions in human aldose reductase-inhibitor complex at 0.66 Å. Proteins 2004; 55: 792–804. [DOI] [PubMed] [Google Scholar]
- [66].Obrosova IG, Van Huysen C, Fathallah L, Cao XC, Greene DA, Stevens MJ. An aldose reductase inhibitor reverses early diabetes-induced changes in peripheral nerve function, metabolism, and antioxidative defense. FASEB J 2002; 16: 123–5. [DOI] [PubMed] [Google Scholar]