

Abstract
The classification of idiopathic inflammatory myopathies (IIM) is based on clinical, serological and histological criteria. The identification of myositis‐specific antibodies has helped to define more homogeneous groups of myositis into four dominant subsets: dermatomyositis (DM), antisynthetase syndrome (ASyS), sporadic inclusion body myositis (sIBM) and immune‐mediated necrotising myopathy (IMNM). sIBM and IMNM patients present predominantly with muscle involvement, whereas DM and ASyS patients present additionally with other extramuscular features, such as skin, lung and joints manifestations. Moreover, the pathophysiological mechanisms are distinct between each myositis subsets. Recently, interferon (IFN) pathways have been identified as key players implicated in the pathophysiology of myositis. In DM, the key role of IFN, especially type I IFN, has been supported by the identification of an IFN signature in muscle, blood and skin of DM patients. In addition, DM‐specific antibodies are targeting antigens involved in the IFN signalling pathways. The pathogenicity of type I IFN has been demonstrated by the identification of mutations in the IFN pathways leading to genetic diseases, the monogenic interferonopathies. This constitutive activation of IFN signalling pathways induces systemic manifestations such as interstitial lung disease, myositis and skin rashes. Since DM patients share similar features in the context of an acquired activation of the IFN signalling pathways, we may extend underlying concepts of monogenic diseases to acquired interferonopathy such as DM. Conversely, in ASyS, available data suggest a role of type II IFN in blood, muscle and lung. Indeed, transcriptomic analyses highlighted a type II IFN gene expression in ASyS muscle tissue. In sIBM, type II IFN appears to be an important cytokine involved in muscle inflammation mechanisms and potentially linked to myodegenerative features. For IMNM, currently published data are scarce, suggesting a minor implication of type II IFN. This review highlights the involvement of different IFN subtypes and their specific molecular mechanisms in each myositis subset.
Keywords: anti‐synthetase syndrome, dermatomyositis, interferon, myositis, sporadic inclusion body myositis
Pathological role of interferon in dermatomyositis.
1. INTRODUCTION
Inflammatory idiopathic myopathies (IIM) are a group of rare autoimmune diseases involving muscle tissue but also other organs. They are categorised into four dominant subsets: dermatomyositis (DM), antisynthetase syndrome (ASyS), inclusion body myositis (IBM) and immune‐mediated necrotising myopathy (IMNM) based on clinical manifestations, the presence of myositis‐specific autoantibodies (MSA) and myopathological findings.
DM and ASyS are systemic diseases associated with skin, joints and/or lung involvement, in addition to myositis (1). DM is associated with a characteristic skin rash (Gottron’s papules/signs with heliotrope rash) as well as myopathological findings such as perifascicular atrophy and the presence of perivascular inflammatory infiltrates (2). Both muscular and cutaneous biopsies display vasculopathic features (loss of capillaries, C5b–9 immunoreactivity on capillaries) (3, 4).
ASyS also harbours a perifascicular pathology characterised by perifascicular necrosis, and the majority of patients have interstitial lung disease while this latter feature rarely occurs in DM patients. ASyS is defined by the presence of one of the anti‐histidyl RNA‐t‐synthetase antibodies (5, 6), and DM is associated with a different set of MSAs: anti‐Mi2 (7), anti‐SAE1 (8), anti‐TIF1γ (9), anti‐NXP2 (10) and anti‐MDA5 (11, 12, 13).
IBM and IMNM are muscle‐specific diseases as they are not substantially associated with extramuscular features. Myopathological features of IBM are characterised by the combination of muscle inflammation with endomysial CD8+ T cell infiltrates and distinctive degenerative muscle features (14, 15, 16, 17). In contrast, inflammatory infiltrates are usually mild in IMNM (18).
Transcriptomic and myopathological studies have demonstrated that interferons (IFNs) play important roles in the pathophysiology of IIM (19), but the IFN subtypes involved and their molecular mechanisms differ in each myositis subset.
Therefore, our objective was to review, clearly layout and summarise the role of IFNs in myositis. First, we describe the IFN family, including induction and signalling pathways, and then we discuss the concept of genetic interferonopathies in order to highlight the pathogenic role of IFNs in myositis. Finally, we focus on the different IFN pathways involved in each myositis subset: DM, ASyS, sIBM and IMNM.
2. INTERFERONS
2.1. IFN subtypes
IFNs have been discovered in 1957 by Isaacs and Lindenmann in a study entitled ‘virus interference’, where they identified a new factor able to prevent the virus from entering the cells and called it ‘interferon’ (20). To date, three types of IFNs have been identified, based on their specific receptors. In humans, the type I IFN (IFN‐I) family consists of 13 subtypes of IFNα and one subtype of IFNβ, ε, κ, ω each (21); type II IFN (IFN‐II) includes IFNγ only (22, 23), and type III IFN (IFN‐III) includes IFN‐λ1 (IL‐29), IFN‐λ2 (IL28A), IFN‐λ3 (IL‐28B) and IFN‐λ4 (24).
The IFN‐I receptor, the IFNα receptor (IFNAR), is a heterodimeric transmembrane receptor composed of IFNAR1 and IFNAR2 subunits. IFNAR1 is constitutively associated with the tyrosine kinase 2 (TYK2) and IFNAR2 with the Janus kinase 1 (JAK1). IFNγ binds to a distinct cell surface receptor composed of the IFNGR1 subunit, associated with JAK1 and the IFNGR2, constitutively associated with JAK2. Type III IFNs‐ also signal through a heterodimeric receptor composed of the IFNλR1 (high‐affinity chain) and IL‐10R2, a subunit shared with other cytokine receptors (24).
2.2. IFN‐secretion
2.2.1. IFN‐I
In humans, there is no, or only minimal, constitutive secretion of IFN‐I. Production of IFN‐I is induced by danger signals (pathogen‐associated molecular patterns; PAMP) released by viruses or microbial products. These PAMPs can either stimulate membrane receptors localised at the cellular or endosomal membranes (Toll‐Like Receptors, TLRs) (25), or cytosolic pattern recognition receptors including nucleotide sensors such as retinoic acid‐inducible gene I (RIG‐I)‐like receptors (RLRs) or DNA sensors to induce IFN production (26).
Endosomal TLRs recognise nucleic acids (double‐strand RNA and single‐strand RNA or double‐strand DNA by TLR3, 7/8, 9, respectively) (27). TLR activation leads to an activation of IFN response factor (IRF) (25, 28) and IFN production (e.g. IRF5 and IRF7 in plasmacytoid dendritic cells (pDCs) or IRF3 in monocytes) (29).
TLR‐independent pathways involve RIG‐I, melanoma differentiation‐associated protein 5 (MDA5) and stimulator of interferon genes protein (STING). RIG‐I and MDA5 recognise endogenous RNA (respectively single and double‐strand) (30) and induce IFN by IRF3 and IRF7 activation by a mitochondrial antiviral‐signalling protein (MAVS)‐dependent mechanism. STING is activated by the synthesis of cyclic GMP‐AMP (cGAMP) triggered by endogenous cytosolic double‐strand DNA (dsDNA) leading to IFN production by IRF3 activation (31).
While TLR‐dependent and ‐independent pathways are different, both lead to upregulation of IFN‐stimulated genes (ISG) including the IFN gene itself.
All cells harbour IFNR and may produce IFNs, but some are specialised cells like pDCs (32) or monocytes, and others like fibroblasts, dendritic cells and epithelial cells (33).
2.2.2. IFN‐II
IFNγ is a cytokine that is primarily produced by cells of the innate immune system, such as monocytes, macrophages and natural killer (NK) cells (23). IFN‐II can be induced by cytokines or following activation of pattern recognition receptors. In addition, CD4+ Th1 T lymphocytes and CD8+ cytotoxic T cells are able to produce IFNγ (34). IFN‐II has a key role in macrophage polarisation but also has homeostatic and anti‐tumoural effects (23).
2.2.3. IFN‐III
As type IFN‐I, also IFN‐III can be expressed in hematopoietic cells. Among nonhematopoietic cells, epithelial cells may also produce IFN‐III. Both IFN‐I and IFN‐III induce the same ISG, but IFN‐III primarily targets mucosal epithelial cells and protect against viral attacks (24).
2.3. IFN signalling, IFN‐stimulated genes and IFN effects
IFN receptors transduce the signal mainly through JAK‐signal transducer and activator of transcription (JAK‐STAT) signalling pathways (31, 35). There are four JAK (JAK 1, JAK2, JAK3 and TYK2) and seven different STAT proteins (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6) in humans (36). JAK activation enhances phosphorylation, dimerisation and nuclear translocation of STAT resulting in the upregulation of hundreds of ISGs (35). Even though IFN‐I, ‐II and ‐III have different receptors, their activation leads to induction of mostly the same set of ISGs (22). Nevertheless, some ISG promoters include a specific sequence: interferon‐stimulated response element that is only recognised by the ISGF3 complex (STAT1/STAT2/IRF9), which is only secreted by IFN‐I but not by IFN‐II (37).
ISGs have three major effects. First, ISGs have indirect anti‐viral effects. This major function was the first one described (20). Second, IFNs promote immune responses with an indirect anti‐viral effect by activation of inflammatory mediators (e.g. MxA, OAS, RNASEL) (31). Third, ISGs are also involved in immune regulation by downregulating the inflammatory responses (e.g., by suppression of cytokine signalling [SOCS]) (31,38).
3. THE PATHOGENIC ROLE OF IFNS: GENETIC INTERFERONOPATHIES SHARE COMMON FEATURES WITH DM
3.1. In humans
The monogenic forms of type I interferonopathies represent a recent group of diseases combining the characteristics of Mendelian transmission with a constitutive activation of the IFN‐I signalling pathway (39). Aicardi‐Goutières syndromes (AGS), chilblain lupus in the juvenile form, STING‐associated vasculopathy with onset in infancy (SAVI), PSMB8‐related diseases (like Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature [CANDLE]) and Singleton‐Merten syndrome (SMS) (39, 40), all of which basically occurring in children, are the main syndromes related to constitutive activation of IFN‐I signalling.
Five different genetic mechanisms leading to an overproduction of IFNs have been identified, so far (41). The first one is an abnormal accumulation of nucleic acids linked to a nuclease defect. For example, loss of function mutations in the exonuclease TREX1 (which normally downregulates STING activation), in the RNase H (2A, 2B and 2C) or the RNase SAMHD1, have been linked with AGS and familial chilblain lupus (39, 41). The second mechanism is a constitutive activation or enhanced sensitivity of an innate immune sensor or adaptive molecule. Gain of function mutations in IFIH1 (coding for MDA5), in DDX58 (coding for RIG‐I) or TMEM173 (coding for STING) were reported in AGS, SMS and SAVI, respectively (40, 42). A defect of IFN‐I regulation can lead to an interferonopathy. For example, loss of function mutations in ISG15 that normally decreases ISG activity by glycation (43) lead to interferonopathy (ADAR1 mutation found in some AGS). Finally, loss of function mutations in immunoproteasome subunits (e.g. PSMB8 for β5i subunit) impair its multicatalytic function causing cellular stress leading to sustained IFN‐I production (41, 44).
Several clinical features observed in genetic interferonopathies are very similar to those observed in DM (Table 1).
TABLE 1.
Overlapping features between DM and genetic interferonopathies
| AGS | Familial lupus | SAVI | CANDLE | Singleton‐merten | |
|---|---|---|---|---|---|
| Gene/pathway | TREX1, RNASEH2, SAMHD1, ADAR, IFIH1 | TREX1, SAMHD1 | TMEM173 | PSMB8 | IFIH1 DDX58 |
| Cutaneous manifestations | |||||
| Chilblain‐like lesions | + | + | + | + | + |
| Digital necrosis | + | + | + | + | − |
| Panniculitis | Occasional | − | − | − | + |
| Heliotrope rash | − | − | − | + | − |
| Telangiectasia | + | ||||
| Interstitial lung disease | − | − | + | − | − |
| Myositis | − | − | + | + | − |
| Arthritis | − | − | + | + | − |
| Constitutional fever | + | − | + | − | + |
Abbreviations: AGS, Aicardi Goutières syndrome, SAVI, sting‐associated vasculopathy with onset in infancy, CANDLE, Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature.
3.1.1. Skin manifestations
Chilblain and ulceration are present in AGS, and distal erythema and ulceration are observed in one‐third of patients with CANDLE (44). Erythematous oedema of the eyelids or panniculitis may be observed in genetic interferonopathies, which are also features encountered in some forms of DM. The genetic interferonopathy associated with the cutaneous phenotype having the most similarities with anti‐MDA5+ DM is SAVI. Indeed, telangiectatic erythema of the light‐exposed areas and skin ulcerations are a constant finding. Patients may also have distal ulcerative lesions. These lesions reflect underlying cutaneous vasculopathy and share similarities with the type of skin vasculopathy encountered in many patients with anti‐MDA5+ DM (45).
3.1.2. Muscle manifestations
Muscle inflammation has also been reported in genetic interferonopathies. Myositis may be observed in 80% and 25% of patients with CANDLE and SAVI, respectively (44, 45) although the precise characteristics may differ gradually and qualitatively.
Lung and joint involvements may occur both in DM and genetic interferonopathies. SAVI patients frequently develop interstitial lung disease and non‐erosive arthritis that may be severe (45). CANDLE patients may also have non‐erosive polyarthritis (44). Among DM, interstitial lung disease and synovitis are features commonly observed in anti‐MDA5+ DM (46).
3.2. Animal models
In vivo models of genetic interferonopathies (loss or gain of function mutations) confirm the presence of overlapping features with DM.
3.2.1. Skin manifestations
Cutaneous manifestations have been demonstrated in Trex1‐deficient mice, which develop inflammation of the skin (47, 48, 49). In normal condition, Trex1 degrades and metabolises endogenous DNA and is a specific negative regulator of STING‐dependent signalling. Trex1 −/− mice develop specific multiorgan inflammation with T cell infiltrates in the skin. STING antagonism attenuates pathological features of the autoinflammatory disease in these mice (49). Skin changes with vasculopathy were also demonstrated in a mouse model with a TMEM173 gain of function mutation that leads to the activation of STING. Two animal models (knock‐in with a V154M substitution and N153S) have been developed to reproduce SAVI (50). In the N153S model, mice developed skin ulceration with vasculopathy and interstitial lung disease. In the V154M knock‐in model, mice also developed interstitial pulmonary disease reproducing the phenotype of SAVI, similar to that in anti‐MDA5+ DM patients.
3.2.2. Muscle manifestations
Myositis and myocarditis have been demonstrated in some animal models. In Trex1 −/− mice, histological analysis showed T and B cells in striated and cardiac muscles (48). In IFNGR+/− mice with knockout of the IFN pathways regulator SOCS, histological analysis showed massive infiltration by T cells, macrophages and oeosinophils in the skeletal muscle and in the heart (51).
4. INTERFERON PATHWAY ACTIVATION IN DM
4.1. IFN pathway and DM‐specific autoantibodies
Antigenic targets of DM‐specific autoantibodies are involved in the IFN pathways. The most striking example is the anti‐MDA5 antibody (occurring in 10% of Caucasian DM patients (1)) associated with a subset of amyopathic DM with potentially severe interstitial lung disease (46). As mentioned above, MDA5 is an RLR playing a key role in IFN secretion (30).
Anti‐TIF1γ antibodies (present in 40% of DM (52)) are associated with DM and a high risk of malignancy in adults (53). TIF1γ (TRIM33) plays a critical role in downregulating IFN‐Iβ production by macrophages (54). TRIM33 is involved in sumoylation (55), a process involved in post‐translational modification by activation of small‐ubiquitin‐like modifiers (SUMO) (8). Sumoylation has an important role in IFN regulation. In the animal model, sumoylation deficiency causes increased levels of IFN‐Iβ in skin and muscle tissues (55).
Small ubiquitin‐like modifier activating enzyme (SAE) activates small‐ubiquitin‐like modifier‐1 (8) in the process of sumoylation. SAE is targeted by DM‐specific autoantibodies in 10% of patients (1). Mi‐2/NuRD (56) complex and NXP‐2 (57) are both inhibitors of SUMO. Anti‐Mi‐2 and anti‐NXP‐2 autoantibodies are observed in 30‐35% and 20% of DM patients, respectively (1).
4.2. IFN pathway is activated in DM
In 1986, Isenberg et al. were the first to report the presence of IFN‐I and IFN‐II in muscle biopsies of patients with inflammatory myopathies (58).
4.2.1. Muscle tissue
First evidence of the involvement of an IFN pathway in DM was demonstrated by Emslie‐Smith et al. in juvenile DM (59). The authors showed an upregulation of ISG in juvenile DM muscle biopsies. Later Greenberg et al. reported on the activation of IFN pathways in muscle biopsies from adult DM patients (60). This IFN signature was not observed in other subsets of inflammatory myopathies such as polymyositis (60). To date, several studies confirm the presence of an IFN signature in the blood, muscle and skin of DM patients (43, 60, 61, 62, 63, 64, 65).
Muscle transcriptomic analyses showed a similarly upregulated IFN signature in all serological subsets of DM (19) including muscle biopsies from amyopathic anti‐MDA5 DM patients (66).
Detection of IFN‐stimulated proteins by immunostaining in DM muscle biopsies have permitted to demonstrate the translation of some ISG. Sarcoplasmic expression of ISG15, RIG‐I and Myxovirus resistance A (MxA) were observed in the perifascicular area of DM muscle biopsies (43, 67, 68, 69, 70). Of note, based on these findings, MxA immunostaining is now considered the most specific myopathological diagnostic marker of DM (68, 69).
4.2.2. Skin tissue
Of the 25 most highly overexpressed genes in the skin of DM patients, 21 were ISGs (3). Moreover, the intensity of the IFN signature is mainly correlated with serum levels of IFN‐I (IFN‐Iβ) and not with IFN‐II (IFNγ) (3). A similar topography of damage is seen in keratinocytes and myofibres in DM (71). Ono et al. showed a correlation between IFN signature in blood and skin and vasculopathy features. Furthermore, they confirmed IFN pathway involvement at a protein level with MxA staining in the blood vessels and interstitial fibroblasts of DM patients. Proteomic analysis of skin biopsies derived from DM patients showed an IFN signature leading to a reduction of collagen by dermal fibroblasts (72).
4.2.3. Blood
IFN pathway activation is not restricted to the muscular and skin compartments. IFN signature has been found in the peripheral mononuclear blood cells of DM patients (61, 64, 73).
Among the highly overexpressed ISGs, there are common genes overexpressed in DM and interferonopathies such as IFI27, IFI44L, IFIT1, ISG15, RSAD2 and SIGLEC1 (74). It should be noted that DM with anti‐MDA5 antibodies, which has a similar phenotype with SAVI, also has a very similar IFN signature. Nevertheless, the intensity of the IFN signature is less than in genetic interferonopathies (75).
Moreover, the level of IFN pathway activation (i.e. IFN score) correlates with disease activity (61, 73, 76, 77). Accordingly, blood IFN‐Iβ levels correlate with skin disease activity (62, 63), and IFN‐Iα levels correlate with disease severity in anti‐MDA5+ DM (78, 79).
4.3. IFN production in DM
The triggers of IFN pathway induction as well as the main sources of IFNs in dermatomyositis remain unclear.
4.3.1. Immune cells and IFN production
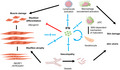
pDCs are known to produce large amounts of IFN‐I (32). IFN scores correlate with the pDCs density in the skeletal muscles and the skin of adult and juvenile DM (60, 80) (Figure 1). These cells can be found in the perifascicular and perivascular areas of muscle and skin biopsies (60, 81, 82, 83).
FIGURE 1.
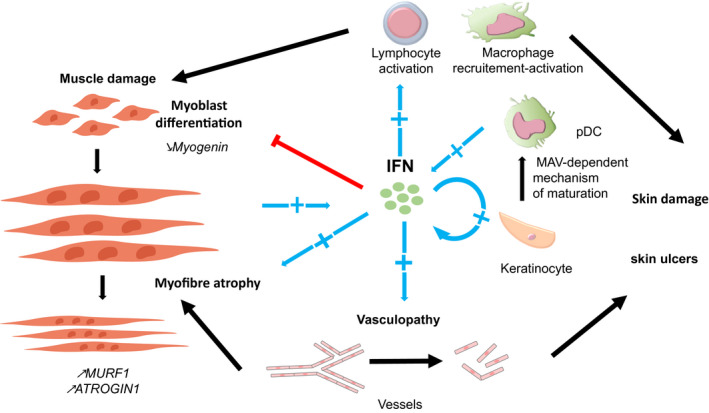
Pathologic effects of interferon in dermatomyositis. In vitro, type I interferon (IFN‐I) induces muscle damage. IFN‐I inhibits myoblast differentiation and decreases myogenin expression. IFN‐I induces myofibre atrophy and overexpression of muscle atrophy genes, ATROGIN1 (FBXO32) and TRIM63 (MURF1). In vitro, IFN‐I impairs angiogenesis of endothelial cells and disrupts the vascular network. Vasculopathy in dermatomyositis has been linked to indirect muscle pathology features and skin damage like ulcers. In vitro, IFN‐I‐stimulated keratinocytes produce IFN‐I and IFN‐III and activate plasmacytoid dendritic cells (pDC) as IFN‐I producers. pDCs are present in perifascicular and perivascular areas in muscle and skin tissues, but macrophages and muscle fibres may also participate in IFN‐I production. IFNs lead to the recruitment and activation of both innate and adaptive immune cells
4.3.2. Muscle cells
In muscle tissue, RIG‐I activation is responsible for an IFN‐Iβ secretion by human myotubes, resulting in an autocrine loop of activation (67). It has been shown that myogenic precursor cells from juvenile DM muscle produce higher levels of IFNs compared to those of healthy controls (84). Previous reports also showed that regenerating muscle fibres in addition to pDCs may produce IFNs in DM (85).
4.3.3. Keratinocytes
Keratinocytes can also produce IFN‐I and IFN‐III in DM in vitro (86). The IFN secretion in skin is associated with the recruitment of CXCR3+ lymphocytes and the expression of two ISGs, CXCL9 and CXCL10, correlating with capillary loss in juvenile DM (87). Another study showed a key role of immunoproteasome subunit PSMB9 that is up‐regulated in DM skin by proteomic analysis and could decrease collagen secretion when treated by IFN in vitro (72).
4.4. IFNs induce muscle damage
Ladislau et al. showed a pathogenic role of IFN‐I by impairing myoblast differentiation and by inducing atrophy of myotubes in vitro (88). Myotubes demonstrated an upregulation of two genes involved in the atrophy process, FBXO32 (ATROGIN1) and TRIM63 (MURF1).
IFN‐I pathway activation also disrupts the capillary network in vitro. Interestingly, the administration of JAK inhibitors improve IFN‐I‐induced damage. Additionally, Meyer et al. showed that IFN‐Iβ‐induced mitochondrial dysfunction contributes to the impairment of muscle function and persistent inflammation in DM (89).
Altogether, these data highlight the key role of the IFN pathways, especially IFN‐I, in the pathophysiology of DM.
5. INTERFERON PATHWAY ACTIVATION IN ASyS
DM and ASyS have previously been considered as part of the same disease spectrum as both conditions may present with similar skin features and a perifascicular pathology. Indeed, Mozaffar and Pestronk were the first to describe the myopathological features in anti‐Jo‐1+ ASyS patients demonstrating septal inflammation, and myofibre lesions restricted to the perifascicular area referred to as an “immune myopathy with perimysial pathology” (IMPP) (90). More recent studies, have demonstrated that ASyS muscle biopsies display perifascicular necrosis (91, 92, 93), while DM is associated with perifascicular atrophy (91), suggesting distinctly separate pathomechanisms.
5.1. IFN‐II pathway in muscle of ASyS
Overexpression of IFN‐I related proteins, such as MxA, ISG15 and RIG‐I, are absent in ASyS (69, 94), and perifascicular MHC class II (MHC‐II) expression is prominently observed in ASyS but not in DM (93, 95). Interestingly, in muscle biopsies from patients with ASyS, CD8+ T cells are located in close vicinity to MHC class‐II+ myofibres, suggesting the involvement of the IFN‐II pathway (96). Along that line, in vitro studies showed that IFN‐II can upregulate the MHC‐II expression on primary human myoblasts (97, 98).
Accordingly, RNA sequencing data from a large number of myositis muscle biopsies showed prominent IFN‐II activation in patients with ASyS (19), and conversely, IFN‐I‐induced genes were only mildly expressed in ASyS (19).
5.2. IFN‐II pathway in lungs of ASyS patients
The presence of histidine RNA‐Synthetase(HisRS)‐reactive CD4+ T cells in the bronchioalveolar lavage fluids as reported in ASyS patients (99). Along that line, shared oligoclonal CD4+ and CD8+ T cells were reported in the lungs and skeletal muscles associated with different myositis subsets (100).
In the bronchoalveolar lavage fluids, these cells display a Th1 phenotype and produce high levels of IFNγ (99). In addition, these Th1 cells are characterised by the expression of C‐C chemokine receptor type 5 (CCR5), involved in T cell homing. Interestingly, it was demonstrated that HisRS has chemotactic properties inducing tissue homing of CCR5‐positive cells (lymphocytes and antigen‐presenting cells) (101).
5.3. IFN pathways in blood of ASyS patients
In the peripheral blood of ASyS, the presence of HisRS‐reactive CD4+ T cells was also reported (99) even though the Th1 involvement was less significant in the lungs. Along that line, increased levels of IFNγ‐induced chemokines CXCL9 and CXCL10 are observed in the serum of patients with anti‐Jo‐1 antibodies (102).
Besides, the addition of ASyS patients’ serum induces the production of IFN in the blood from healthy donors (64). Indeed, it has been demonstrated that anti‐Jo‐1 antibody complexed with necrotic cells is able to stimulate peripheral blood mononuclear cells (PBMCs) from healthy donors to produce IFN‐Iα (103, 104).
5.4. Animal model of HisRS‐induced myositis‐associated IFN pathway
Mouse models have been developed and highlight the role of IFN pathway in ASyS (105).
Experimental autoimmune myositis induced by a single intramuscular immunisation with HisRS spontaneously resolved in few weeks. Persistent muscle inflammation and prolonged anti‐Jo‐1 production were observed only in mice stimulated by HisRS in the presence of a TLR7/8 agonist, but absent in IFNαβR‐null mice (105).
Altogether these data support the role of IFNs in ASyS. Myopathological findings and transcriptomic studies suggest the preferential involvement of IFN‐II, rather than of IFNI.
6. INTERFERON PATHWAY ACTIVATION IN sIBM
Sporadic inclusion body myositis (sIBM) is a slowly progressive skeletal muscle disease with degenerative features including rimmed vacuoles and related myonuclear degeneration, mitochondrial pathology and cytoplasmic protein aggregates within myofibres. Novel muscle histochemical degenerative biomarkers, such as p62, LC3 and TDP43, are involved in unfolded protein response, endoplasmic reticulum stress and altered autophagy. One of the pathological hallmarks of sIBM is the predominance of endomysial CD8+ T cells (and macrophages) surrounding and occasionally invading MHC‐I‐positive myofibres.
6.1. IFN‐II pathway in muscle tissue of sIBM
CD8+ T cells in sIBM muscles show a highly differentiated effector phenotype (106, 107, 108). Greenberg et al. found that muscle fibres are invaded by CD8+CD57+T cells (106). The proportion of invaded myofibres correlated positivity with the degree of muscle weakness (106). Microarray data from a large series of muscle biopsies identified killer cell lectin‐like receptor G1 (KLRG1) as a biomarker of these subpopulations (107). Accordingly, IFN‐II gene expression is higher in myofibres invaded by CD8+ T cells compared to expression in non‐invaded myofibres (109). These cytotoxic CD8+ T cells produce high levels of cytotoxic molecules such as perforin and granzymes (110, 111). Along that line, Th‐1 cytokines and chemokines such as IFN‐II, CXCL9, CCL5 and IL32 are increased in sIBM muscle tissue (107).
Analysis of proteomic profiling from muscle biopsies showed that 39% of upregulated proteins are involved in IFN‐I or mixed IFN‐I and IFN‐II pathways (112).
Accordingly, immunohistochemistry studies confirmed the presence of IFN‐related proteins such as ISG15 and IRF8 (112). Moreover, high expressions of CD74 and STAT1, as key molecules of IFN macrophage activation, were detected in sIBM (112). It was also demonstrated that IFN‐II can upregulate cell surface MHC‐I expression on myoblasts (113).
6.2. IFN‐II pathway in blood of sIBM
Expansion of highly differentiated T cells was also detected in blood of sIBM patients. sIBM patients have increased levels of differentiated populations of T cells such as CD8+CD28null (114), CD8+Tbet+ (115), CD8+CD57+ (106) and CD8+KLRG1+ T cells and high levels of Th1 chemokines and cytokines (CXCL9, CXCL10 and IL‐12) (114, 116). This specific T‐cell population is prone to produce higher levels of IFN‐II in sIBM compared to levels in healthy controls (114, 116). Moreover, this population is clonally expanded and shares the same oligoclonal repertoire that muscle‐infiltrating CD8+ T cells have (108). Greenberg et al. found that 58% of sIBM patients have abnormal populations of circulating large granular lymphocytes meeting diagnostic criteria of T cell large granular lymphocytic leukaemia (LGLL) (106). Highly differentiated clonal T cells in LGLL are resistant to apoptosis (117).
6.3. IFN‐II pathway and muscle degeneration
A growing body of evidence suggests that causality flows from autoimmunity to degeneration (15, 16). Abnormal sarcoplasmic accumulation of β‐amyloid may also be explained by the effect of inflammatory cytokines on muscle cells. Beta‐amyloid production may be induced in human primary myotubes by exposure to IL‐1β and IFN‐II (118). However, overexpression of β‐amyloid precursor protein in mouse muscle did not lead to any muscle inflammation (119).
Thus, analysis of the link between IFNs and protein aggregation in muscle cells is crucial to understand the pathogenesis of sIBM, and the immunoproteasome is a key element (Figure 2). Indeed, several studies showed that elements of the immunoproteasome are detected in muscle tissue of sIBM (107, 113). The immunoproteasome is an alternative isoform of the constitutive proteasome induced by inflammatory cytokines, in particular by IFNγ. The main role of the immunoproteasome is to process antigens for presentation on MHC‐I molecules to CD8+ T cells (120). Specific inhibitors of the immunoproteasome reduced surface expression of MHC‐I in myoblasts induced by IFNγ in vitro (113). Nakajo‐Nishimura syndrome (NNS) is a rare autosomal recessive autoinflammatory disorder caused by a homozygous mutation in PSMB8 that encodes the immunoproteasome subunit β5i. NNS is clinically characterised by a pernio‐like skin rash, periodic fever and myositis (121). Interestingly, myopathological analysis of small case series of NNS shows sarcolemmal MHC‐I expression, CD4+ and CD8+ T cell infiltrations, p62 and TDP43 aggregations and rimmed vacuoles in myofibres (122) similar to certain pathological features of sIBM muscle. Myeloid cell lines derived from pluripotent stem cell of NNS patients show dysfunction of proteasome activity and overproduction of inflammatory cytokines and chemokines such as IL‐6, CCL2 and CXCL10 with upregulation of reactive oxygen species under IFNγ and TNFα stimulation (123).
FIGURE 2.
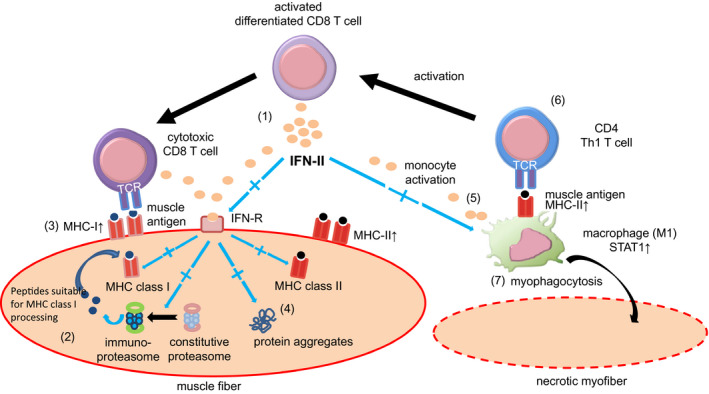
IFNs and IFN‐II in Inclusion body myositis. (1) Highly differentiated T cells secrete IFN‐II. (2) IFN‐II stimulates the expression of muscle antigens on MHC class I via the immunoproteasome in muscle fibres (3). Cytotoxic CD8+ T cells can be stimulated by the complex of muscle antigens and MHC class I and produce IFN‐II (4). IFN‐II may induce protein aggregates (5). IFN‐II also activates macrophages and MHC class II expression with muscle autoantigen (6). Activated macrophages stimulate CD4+ Th1 T cells and (7) are involved in myophagocytosis of necrotic muscle fibres
Altogether these data support the role of IFNs, in particular of IFN‐II, in sIBM. IFN‐II may be secreted by specific highly differentiated CD8+ T cells that are detectable in muscle tissue and blood of sIBM patients. Myodegenerative features in sIBM may be related to IFN‐II as well.
6.4. IFN and IMNM
IMNM represent another IIM subset associated with distinct clinical, serological and histological features. Different from sIBM, the presence of certain pathognomonic MSAs (anti‐SRP or anti‐HMGCR antibodies) are a major feature of the disease. Recent data point towards a direct pathogenic effect of these autoantibodies, potentially through the activation of the classical complement cascade (124).
Although the IFN signalling pathway does not seem to be the central mechanism of IMNM, transcriptomic data showed a modest overexpression of IFN‐inducible genes in muscle tissue compared to those in other IIM (19). The IFN signature appears more driven by IFN‐II rather than IFN‐I with IFI30 as the most upregulated IFN‐inducible gene (19). This predominant IFN‐II signature has been inconsistently identified with qRT‐PCR assays (96, 116, 125).
Indirect arguments are consistent with a potential role of IFN‐II. Although the inflammatory infiltrate is usually scarce in the muscle biopsies of IMNM, a decent amount of macrophages and T cells have been described in skeletal muscle tissues independent from myophagocytoses and necrosis. Macrophages harboured an M1 phenotype in the context of Th1 environment with upregulation of STAT1, IFNG, TNFA and IL12 (126). Double‐positive macrophages CD68+STAT1+ and CD68+NOS2+ were identified in muscle tissues by immunostainings confirming an M1‐polarised macrophage profile (125).
Moreover, mild to moderate sarcolemmal MHC‐I expression but the absence of sarcolemmal MHC class II is a characteristic myopathological feature observed in IMNM. As mentioned above, IFN‐II induces MHC‐I cell surface expression on myoblasts (109).
Only a few studies evaluated the IFN signalling pathway in IMNM suggesting a modest role of IFN, especially by IFN‐II.
6.5. Conclusion
Although IIMs are a heterogeneous group of autoimmune diseases, the IFN cytokine family plays an important role in its pathophysiology. DM is the subset where IFNs, and especially IFN‐I, have been highlighted as key pathogenic cytokines leading to consider DM as an acquired interferonopathy in analogy to the monogenic interferonopathies. In ASyS, both IFN‐I and II have been involved but transcriptomic analyses and pathological data showed a predominant role of IFN‐II. The key role of terminally differentiated CD8+ T cells in sIBM supports IFN‐II as an important cytokine, not only involved in muscle inflammation but also in muscle degenerative features, presumably mediated via the immunoproteasome. The role of IFN appears minor in IMNM compared to that in other myositis subsets, yet IFN‐II remains important for macrophages signalling.
CONFLICT OF INTEREST
The authors have declared no conflicts of interest for this article.
DATA AVAILABILITY STATEMENT
Not applicable.
This work is for a MINISYMPOSIUM on MUSCLE Diseases Edited by W. Stenzel & H. Goebel.
References
- 1.Wolstencroft PW, Fiorentino DF. Dermatomyositis clinical and pathological phenotypes associated with myositis‐specific autoantibodies. Curr Rheumatol Rep. 2018;20:28. [DOI] [PubMed] [Google Scholar]
- 2.Pinal‐Fernandez I, Casciola‐Rosen LA, Christopher‐Stine L, Corse AM, Mammen AL. The prevalence of individual histopathologic features varies according to autoantibody status in muscle biopsies from patients with dermatomyositis. J Rheumatol. 2015;42:1448–54. [PMC free article] [PubMed] [Google Scholar]
- 3.Wong D, Kea B, Pesich R, Higgs BW, Zhu W, Brown P, et al. Interferon and biologic signatures in dermatomyositis skin: specificity and heterogeneity across diseases. PloS One. 2012;7:e29161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gallay L, Gayed C, Hervier B. Antisynthetase syndrome pathogenesis: knowledge and uncertainties. Curr Opin Rheumatol. 2018;30:664–73. [DOI] [PubMed] [Google Scholar]
- 5.Hervier B, Devilliers H, Stanciu R, Meyer A, Uzunhan Y, Masseau A, et al. Hierarchical cluster and survival analyses of antisynthetase syndrome: phenotype and outcome are correlated with anti‐tRNA synthetase antibody specificity. Autoimmun Rev. 2012;12:210–7. [DOI] [PubMed] [Google Scholar]
- 6.Aggarwal R, Cassidy E, Fertig N, Koontz DC, Lucas M, Ascherman DP, et al. Patients with non‐Jo‐1 anti‐tRNA‐synthetase autoantibodies have worse survival than Jo‐1 positive patients. Ann Rheum Dis. 2014;73:227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Targoff IN, Reichlin M. The association between Mi‐2 antibodies and dermatomyositis. Arthritis Rheum. 1985;28:796–803. [DOI] [PubMed] [Google Scholar]
- 8.Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N. Identification of a novel autoantibody directed against small ubiquitin‐like modifier activating enzyme in dermatomyositis. Arthritis Rheum. 2007;56:3132–7. [DOI] [PubMed] [Google Scholar]
- 9.Trallero‐Araguás E, Rodrigo‐Pendás JÁ, Selva‐O’Callaghan A, Martínez‐Gómez X, Bosch X, Labrador‐Horrillo M, et al. Usefulness of anti‐p155 autoantibody for diagnosing cancer‐associated dermatomyositis: a systematic review and meta‐analysis. Arthritis Rheum. 2012;64:523–32. [DOI] [PubMed] [Google Scholar]
- 10.Albayda J, Pinal‐Fernandez I, Huang W, Parks C, Paik J, Casciola‐Rosen L, et al. Antinuclear matrix protein 2 autoantibodies and edema, muscle disease, and malignancy risk in dermatomyositis patients. Arthritis Care Res. 2017;69:1771–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sato S, Hirakata M, Kuwana M, Suwa A, Inada S, Mimori T, et al. Autoantibodies to a 140‐kd polypeptide, CADM‐140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum. 2005;52:1571–6. [DOI] [PubMed] [Google Scholar]
- 12.Li L, Wang Q, Yang F, Wu C, Chen S, Wen X, et al. Anti‐MDA5 antibody as a potential diagnostic and prognostic biomarker in patients with dermatomyositis. Oncotarget. 2017;8:26552–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Narang NS, Casciola‐Rosen L, Li S, Chung L, Fiorentino DF. Cutaneous ulceration in dermatomyositis: association with anti‐melanoma differentiation‐associated gene 5 antibodies and interstitial lung disease. Arthritis Care Res. 2015;67:667–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lloyd TE, Mammen AL, Amato AA, Weiss MD, Needham M, Greenberg SA. Evaluation and construction of diagnostic criteria for inclusion body myositis. Neurology. 2014;83:426–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benveniste O, Allenbach Y. Inclusion body myositis: accumulation of evidence for its autoimmune origin. Brain J Neurol. 2019;142:2549–51. [DOI] [PubMed] [Google Scholar]
- 16.Greenberg SA. Inclusion body myositis: clinical features and pathogenesis. Nat Rev Rheumatol. 2019;15:257–72. [DOI] [PubMed] [Google Scholar]
- 17.Benveniste O, Stenzel W, Hilton‐Jones D, Sandri M, Boyer O, van Engelen BGM. Amyloid deposits and inflammatory infiltrates in sporadic inclusion body myositis: the inflammatory egg comes before the degenerative chicken. Acta Neuropathol. 2015;129:611–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allenbach Y, Mammen AL, Benveniste O, Stenzel W, Immune‐Mediated Necrotizing Myopathies Working Group . 224th ENMC International Workshop: clinico‐sero‐pathological classification of immune‐mediated necrotizing myopathies Zandvoort, The Netherlands, 14‐16 October 2016. Neuromuscul Disord. 2018;28(1):87–99. [DOI] [PubMed] [Google Scholar]
- 19.Pinal‐Fernandez I, Casal‐Dominguez M, Derfoul A, Pak K, Plotz P, Miller FW, et al. Identification of distinctive interferon gene signatures in different types of myositis. Neurology. 2019;93:e1193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. 1957;147:258–67. [PubMed] [Google Scholar]
- 21.Pestka S, Krause CD, Walter MR. Interferons, interferon‐like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. [DOI] [PubMed] [Google Scholar]
- 22.Platanias LC. Mechanisms of type‐I‐ and type‐II‐interferon‐mediated signalling. Nat Rev Immunol. 2005;5:375–86. [DOI] [PubMed] [Google Scholar]
- 23.Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. 2018;18(9):545–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wack A, Terczyńska‐Dyla E, Hartmann R. Guarding the frontiers: the biology of type III interferons. Nat Immunol. 2015;16:802–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll‐like receptors and cytosolic pattern‐recognition receptors. Nat Rev Immunol. 2006;6:644–58. [DOI] [PubMed] [Google Scholar]
- 26.Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22(2):240–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawasaki T, Kawai T. Toll‐like receptor signaling pathways. Front Immunol. 2014;5:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Honda K, Taniguchi T. Toll‐like receptor signaling and IRF transcription factors. IUBMB Life. 2006;58(5–6):290–5. [DOI] [PubMed] [Google Scholar]
- 29.Muskardin TLW, Niewold TB. Type I interferon in rheumatic diseases. Nat Rev Rheumatol. 2018;14:214–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reikine S, Nguyen JB, Modis Y. Pattern recognition and signaling mechanisms of RIG‐I and MDA5. Front Immunol. 2014;5:342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider WM, Chevillotte MD, Rice CM. Interferon‐stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rönnblom L, Eloranta M‐L, Alm GV. Role of natural interferon‐alpha producing cells (plasmacytoid dendritic cells) in autoimmunity. Autoimmunity. 2003;36:463–72. [DOI] [PubMed] [Google Scholar]
- 33.Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6:975–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon‐gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–89. [DOI] [PubMed] [Google Scholar]
- 35.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levy DE, Darnell JE. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. [DOI] [PubMed] [Google Scholar]
- 37.Mowen K, David M. Role of the STAT1‐SH2 domain and STAT2 in the activation and nuclear translocation of STAT1. J Biol Chem. 1998;273:30073–6. [DOI] [PubMed] [Google Scholar]
- 38.Cheon H, Borden EC, Stark GR. Interferons and their stimulated genes in the tumor microenvironment. Semin Oncol. 2014;41:156–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci. 2011;1238:91–8. [DOI] [PubMed] [Google Scholar]
- 40.Munoz J, Rodière M, Jeremiah N, Rieux‐Laucat F, Oojageer A, Rice GI, et al. Stimulator of interferon genes‐associated vasculopathy with onset in infancy: a mimic of childhood granulomatosis with polyangiitis. JAMA Dermatol. 2015;151:872. [DOI] [PubMed] [Google Scholar]
- 41.Kretschmer S, Lee‐Kirsch MA. Type I interferon‐mediated autoinflammation and autoimmunity. Curr Opin Immunol. 2017;49:96–102. [DOI] [PubMed] [Google Scholar]
- 42.Crow YJ, Casanova J‐L. STING‐associated vasculopathy with onset in infancy–a new interferonopathy. N Engl J Med. 2014;371:568–71. [DOI] [PubMed] [Google Scholar]
- 43.Salajegheh M, Kong SW, Pinkus JL, Walsh RJ, Liao A, Nazareno R, et al. Interferon‐stimulated gene 15 (ISG15) conjugates proteins in dermatomyositis muscle with perifascicular atrophy. Ann Neurol. 2010;67:53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Torrelo A. CANDLE syndrome as a paradigm of proteasome‐related autoinflammation. Front Immunol. 2017;8:927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014;371:507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allenbach Y, Uzunhan Y, Toquet S, Leroux G, Gallay L, Marquet A, et al. Different phenotypes in dermatomyositis associated with anti‐MDA5 antibody: Study of 121 cases. Neurology. 2020;95:e70–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gall A, Treuting P, Elkon KB, Loo Y‐M, Gale M, Barber GN, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon‐dependent autoimmune disease. Immunity. 2012;36:120–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morita M, Stamp G, Robins P, Dulic A, Rosewell I, Hrivnak G, et al. Gene‐targeted mice lacking the Trex1 (DNase III) 3’–>5’ DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol. 2004;24:6719–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, et al. Targeting STING with covalent small‐molecule inhibitors. Nature. 2018;559:269–73. [DOI] [PubMed] [Google Scholar]
- 50.Bouis D, Kirstetter P, Arbogast F, Lamon D, Delgado V, Jung S, et al. Severe combined immunodeficiency in stimulator of interferon genes (STING) V154M/wild‐type mice. J Allergy Clin Immunol. 2019;143(2):712–25.e5. [DOI] [PubMed] [Google Scholar]
- 51.Metcalf D, Di Rago L, Mifsud S, Hartley L, Alexander WS. The development of fatal myocarditis and polymyositis in mice heterozygous for IFN‐gamma and lacking the SOCS‐1 gene. Proc Natl Acad Sci U S A. 2000;97:9174–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bolko L, Gitiaux C, Allenbach Y. [Dermatomyositis: new antibody, new classification]. Med Sci. 2019;35(2):18–23. [DOI] [PubMed] [Google Scholar]
- 53.Fiorentino DF, Chung LS, Christopher‐Stine L, Zaba L, Li S, Mammen AL, et al. Most patients with cancer‐associated dermatomyositis have antibodies to nuclear matrix protein NXP‐2 or transcription intermediary factor 1γ. Arthritis Rheum. 2013;65:2954–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferri F, Parcelier A, Petit V, Gallouet A‐S, Lewandowski D, Dalloz M, et al. TRIM33 switches off Ifnb1 gene transcription during the late phase of macrophage activation. Nat Commun. 2015;6:8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Decque A, Joffre O, Magalhaes JG, Cossec J‐C, Blecher‐Gonen R, Lapaquette P, et al. Sumoylation coordinates the repression of inflammatory and anti‐viral gene‐expression programs during innate sensing. Nat Immunol. 2016;17:140–9. [DOI] [PubMed] [Google Scholar]
- 56.Gong Z, Brackertz M, Renkawitz R. SUMO modification enhances p66‐mediated transcriptional repression of the Mi‐2/NuRD complex. Mol Cell Biol. 2006;26:4519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rosendorff A, Sakakibara S, Lu S, Kieff E, Xuan Y, DiBacco A, et al. NXP‐2 association with SUMO‐2 depends on lysines required for transcriptional repression. Proc Natl Acad Sci U S A. 2006;103:5308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Isenberg DA, Rowe D, Shearer M, Novick D, Beverley PC. Localization of interferons and interleukin 2 in polymyositis and muscular dystrophy. Clin Exp Immunol. 1986;63:450–8. [PMC free article] [PubMed] [Google Scholar]
- 59.Emslie‐Smith AM, Arahata K, Engel AG. Major histocompatibility complex class I antigen expression, immunolocalization of interferon subtypes, and T cell‐mediated cytotoxicity in myopathies. Hum Pathol. 1989;20:224–31. [DOI] [PubMed] [Google Scholar]
- 60.Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, et al. Interferon‐alpha/beta‐mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005;57:664–78. [DOI] [PubMed] [Google Scholar]
- 61.Greenberg SA, Higgs BW, Morehouse C, Walsh RJ, Kong SW, Brohawn P, et al. Relationship between disease activity and type 1 interferon‐ and other cytokine‐inducible gene expression in blood in dermatomyositis and polymyositis. Genes Immun. 2012;13:207–13. [DOI] [PubMed] [Google Scholar]
- 62.Huard C, Gullà SV, Bennett DV, Coyle AJ, Vleugels RA, Greenberg SA. Correlation of cutaneous disease activity with type 1 interferon gene signature and interferon β in dermatomyositis. Br J Dermatol. 2017;176:1224–30. [DOI] [PubMed] [Google Scholar]
- 63.Liao AP, Salajegheh M, Nazareno R, Kagan JC, Jubin RG, Greenberg SA. Interferon β is associated with type 1 interferon‐inducible gene expression in dermatomyositis. Ann Rheum Dis. 2011;70:831–6. [DOI] [PubMed] [Google Scholar]
- 64.Ekholm L, Vosslamber S, Tjärnlund A, de Jong TD, Betteridge Z, McHugh N, et al. Autoantibody specificities and type i interferon pathway activation in idiopathic inflammatory myopathies. Scand J Immunol. 2016;84:100–9. [DOI] [PubMed] [Google Scholar]
- 65.Bilgic H, Ytterberg SR, Amin S, McNallan KT, Wilson JC, Koeuth T, et al. Interleukin‐6 and type I interferon‐regulated genes and chemokines mark disease activity in dermatomyositis. Arthritis Rheum. 2009;60:3436–46. [DOI] [PubMed] [Google Scholar]
- 66.Allenbach Y, Leroux G, Suárez‐Calvet X, Preusse C, Gallardo E, Hervier B, et al. Dermatomyositis with or without anti‐melanoma differentiation‐associated gene 5 antibodies: common interferon signature but distinct NOS2 expression. Am J Pathol. 2016;186:691–700. [DOI] [PubMed] [Google Scholar]
- 67.Suárez‐Calvet X, Gallardo E, Pinal‐Fernandez I, De Luna N, Lleixà C, Díaz‐Manera J, et al. RIG‐I expression in perifascicular myofibers is a reliable biomarker of dermatomyositis. Arthritis Res Ther. 2017;19:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Uruha A, Nishikawa A, Tsuburaya RS, Hamanaka K, Kuwana M, Watanabe Y, et al. Sarcoplasmic MxA expression: a valuable marker of dermatomyositis. Neurology. 2017;88:493–500. [DOI] [PubMed] [Google Scholar]
- 69.Uruha A, Allenbach Y, Charuel J‐L, Musset L, Aussy A, Boyer O, et al. Diagnostic potential of sarcoplasmic MxA expression in subsets of dermatomyositis. Neuropathol Appl Neurobiol. 2019;45(5):513–522. [DOI] [PubMed] [Google Scholar]
- 70.Preuße C, Allenbach Y, Hoffmann O, Goebel H‐H, Pehl D, Radke J, et al. Differential roles of hypoxia and innate immunity in juvenile and adult dermatomyositis. Acta Neuropathol Commun. 2016;4:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Greenberg SA, Fiorentino D. Similar topology of injury to keratinocytes and myofibres in dermatomyositis skin and muscle. Br J Dermatol. 2009;160:464–5. [DOI] [PubMed] [Google Scholar]
- 72.Nakamura K, Jinnin M, Kudo H, Inoue K, Nakayama W, Honda N, et al. The role of PSMB9 upregulated by interferon signature in the pathophysiology of cutaneous lesions of dermatomyositis and systemic lupus erythematosus. Br J Dermatol. 2016;174:1030–41. [DOI] [PubMed] [Google Scholar]
- 73.Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis. 2011;70:2029–36. [DOI] [PubMed] [Google Scholar]
- 74.Rice GI, Melki I, Frémond M‐L, Briggs TA, Rodero MP, Kitabayashi N, et al. Assessment of type I interferon signaling in pediatric inflammatory disease. J Clin Immunol. 2017;37:123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rodero MP, Decalf J, Bondet V, Hunt D, Rice GI, Werneke S, et al. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. J Exp Med. 2017;214(5):1547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baechler EC, Bauer JW, Slattery CA, Ortmann WA, Espe KJ, Novitzke J, et al. An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol Med. 2007;13(1–2):59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Walsh RJ, Kong SW, Yao Y, Jallal B, Kiener PA, Pinkus JL, et al. Type I interferon‐inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum. 2007;56:3784–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Horai Y, Koga T, Fujikawa K, Takatani A, Nishino A, Nakashima Y, et al. Serum interferon‐α is a useful biomarker in patients with anti‐melanoma differentiation‐associated gene 5 (MDA5) antibody‐positive dermatomyositis. Mod Rheumatol. 2015;25:85–9. [DOI] [PubMed] [Google Scholar]
- 79.Zhang L, Wu G, Gao D, Liu G, Pan L, Ni L, et al. Factors associated with interstitial lung disease in patients with polymyositis and dermatomyositis. A systematic review and meta‐analysis. PLoS ONE. 2016; 11(5). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4865124/. [cité 2 juin 2019]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.López de Padilla CM, Vallejo AN, McNallan KT, Vehe R, Smith SA, Dietz AB, et al. Plasmacytoid dendritic cells in inflamed muscle of patients with juvenile dermatomyositis. Arthritis Rheum. 2007;56:1658–68. [DOI] [PubMed] [Google Scholar]
- 81.Nagaraju K, Rider LG, Fan C, Chen Y‐W, Mitsak M, Rawat R, et al. Endothelial cell activation and neovascularization are prominent in dermatomyositis. J Autoimmune Dis. 2006;3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shrestha S, Wershil B, Sarwark JF, Niewold TB, Philipp T, Pachman LM. Lesional and nonlesional skin from patients with untreated juvenile dermatomyositis displays increased numbers of mast cells and mature plasmacytoid dendritic cells. Arthritis Rheum. 2010;62:2813–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liao AP, Salajegheh M, Morehouse C, Nazareno R, Jubin RG, Jallal B, et al. Human plasmacytoid dendritic cell accumulation amplifies their type 1 interferon production. Clin Immunol Orlando Fla. 2010;136:130–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gitiaux C, Latroche C, Weiss‐Gayet M, Rodero MP, Duffy D, Bader‐Meunier B, et al. Myogenic progenitor cells exhibit type i interferon‐driven proangiogenic properties and molecular signature during juvenile dermatomyositis. Arthritis Rheumatol. 2018;70:134–45. [DOI] [PubMed] [Google Scholar]
- 85.Tournadre A, Lenief V, Miossec P. Expression of Toll‐like receptor 3 and Toll‐like receptor 7 in muscle is characteristic of inflammatory myopathy and is differentially regulated by Th1 and Th17 cytokines. Arthritis Rheum. 2010;62:2144–51. [DOI] [PubMed] [Google Scholar]
- 86.Zahn S, Rehkämper C, Kümmerer BM, Ferring‐Schmidt S, Bieber T, Tüting T, et al. Evidence for a pathophysiological role of keratinocyte‐derived type III interferon (IFNλ) in cutaneous lupus erythematosus. J Invest Dermatol. 2011;131:133–40. [DOI] [PubMed] [Google Scholar]
- 87.Fall N, Bove KE, Stringer K, Lovell DJ, Brunner HI, Weiss J, et al. Association between lack of angiogenic response in muscle tissue and high expression of angiostatic ELR‐negative CXC chemokines in patients with juvenile dermatomyositis: possible link to vasculopathy. Arthritis Rheum. 2005;52:3175–80. [DOI] [PubMed] [Google Scholar]
- 88.Ladislau L, Suárez‐Calvet X, Toquet S, Landon‐Cardinal O, Amelin D, Depp M, et al. JAK inhibitor improves type I interferon induced damage: proof of concept in dermatomyositis. Brain J Neurol. 2018;141:1609–21. [DOI] [PubMed] [Google Scholar]
- 89.Meyer A, Laverny G, Allenbach Y, Grelet E, Ueberschlag V, Echaniz‐Laguna A, et al. IFN‐β‐induced reactive oxygen species and mitochondrial damage contribute to muscle impairment and inflammation maintenance in dermatomyositis. Acta Neuropathol. 2017;134:655–66. [DOI] [PubMed] [Google Scholar]
- 90.Mozaffar T, Pestronk A. Myopathy with anti‐Jo‐1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry. 2000;68:472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mescam‐Mancini L, Allenbach Y, Hervier B, Devilliers H, Mariampillay K, Dubourg O, et al. Anti‐Jo‐1 antibody‐positive patients show a characteristic necrotizing perifascicular myositis. Brain J Neurol. 2015;138(Pt 9):2485–92. [DOI] [PubMed] [Google Scholar]
- 92.Uruha A, Suzuki S, Suzuki N, Nishino I. Perifascicular necrosis in anti‐synthetase syndrome beyond anti‐Jo‐1. Brain J Neurol. 2016;139(Pt 9):e50. [DOI] [PubMed] [Google Scholar]
- 93.Stenzel W, Preuße C, Allenbach Y, Pehl D, Junckerstorff R, Heppner FL, et al. Nuclear actin aggregation is a hallmark of anti‐synthetase syndrome‐induced dysimmune myopathy. Neurology. 2015;84:1346–54. [DOI] [PubMed] [Google Scholar]
- 94.Inoue M, Tanboon J, Okubo M, Theerawat K, Saito Y, Ogasawara M, et al. Absence of sarcoplasmic myxovirus resistance protein A (MxA) expression in antisynthetase syndrome in a cohort of 194 cases. Neuropathol Appl Neurobiol. 2019;45:523–4. [DOI] [PubMed] [Google Scholar]
- 95.Aouizerate J, De Antonio M, Bassez G, Gherardi RK, Berenbaum F, Guillevin L, et al. Myofiber HLA‐DR expression is a distinctive biomarker for antisynthetase‐associated myopathy. Acta Neuropathol Commun. 2014;2:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rigolet M, Hou C, Baba Amer Y, Aouizerate J, Periou B, Gherardi RK, et al. Distinct interferon signatures stratify inflammatory and dysimmune myopathies. RMD Open. 2019;5:e000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hohlfeld R, Engel AG. Induction of HLA‐DR expression on human myoblasts with interferon‐gamma. Am J Pathol. 1990;136:503–8. [PMC free article] [PubMed] [Google Scholar]
- 98.Mantegazza R, Gebbia M, Mora M, Barresi R, Bernasconi P, Baggi F, et al. Major histocompatibility complex class II molecule expression on muscle cells is regulated by differentiation: implications for the immunopathogenesis of muscle autoimmune diseases. J Neuroimmunol. 1996;68(1–2):53–60. [DOI] [PubMed] [Google Scholar]
- 99.Galindo‐Feria AS, Albrecht I, Fernandes‐Cerqueira C, Notarnicola A, James EA, Herrath J, et al. Proinflammatory histidyl‐transfer RNA synthetase‐specific CD4+ T cells in the blood and lungs of patients with idiopathic inflammatory myopathies. Arthritis Rheumatol. 2020;72:179–91. [DOI] [PubMed] [Google Scholar]
- 100.Englund P, Wahlström J, Fathi M, Rasmussen E, Grunewald J, Tornling G, et al. Restricted T cell receptor BV gene usage in the lungs and muscles of patients with idiopathic inflammatory myopathies. Arthritis Rheum. 2007;56:372–83. [DOI] [PubMed] [Google Scholar]
- 101.Howard OMZ, Dong HF, Yang D, Raben N, Nagaraju K, Rosen A, et al. Histidyl‐tRNA synthetase and asparaginyl‐tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J Exp Med. 2002;196:781–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Richards TJ, Eggebeen A, Gibson K, Yousem S, Fuhrman C, Gochuico BR, et al. Characterization and peripheral blood biomarker assessment of anti‐Jo‐1 antibody‐positive interstitial lung disease. Arthritis Rheum. 2009;60:2183–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ascherman DP. Role of Jo‐1 in the Immunopathogenesis of the Anti‐synthetase Syndrome. Curr Rheumatol Rep. 2015;17:56. [DOI] [PubMed] [Google Scholar]
- 104.Eloranta M‐L, Barbasso Helmers S, Ulfgren A‐K, Rönnblom L, Alm GV, Lundberg IE. A possible mechanism for endogenous activation of the type I interferon system in myositis patients with anti‐Jo‐1 or anti‐Ro 52/anti‐Ro 60 autoantibodies. Arthritis Rheum. 2007;56:3112–24. [DOI] [PubMed] [Google Scholar]
- 105.Sciorati C, Monno A, Doglio MG, Rigamonti E, Ascherman DP, Manfredi AA, et al. Exacerbation of murine experimental autoimmune myositis by toll‐like receptor 7/8. Arthritis Rheumatol. 2018;70:1276–87. [DOI] [PubMed] [Google Scholar]
- 106.Greenberg SA, Pinkus JL, Amato AA, Kristensen T, Dorfman DM. Association of inclusion body myositis with T cell large granular lymphocytic leukaemia. Brain J Neurol. 2016;139(Pt 5):1348–60. [DOI] [PubMed] [Google Scholar]
- 107.Greenberg SA, Pinkus JL, Kong SW, Baecher‐Allan C, Amato AA, Dorfman DM. Highly differentiated cytotoxic T cells in inclusion body myositis. Brain J Neurol. 2019;142:2590–604. [DOI] [PubMed] [Google Scholar]
- 108.Dimitri D, Benveniste O, Dubourg O, Maisonobe T, Eymard B, Amoura Z, et al. Shared blood and muscle CD8+ T‐cell expansions in inclusion body myositis. Brain J Neurol. 2006;129(Pt 4):986–95. [DOI] [PubMed] [Google Scholar]
- 109.Ivanidze J, Hoffmann R, Lochmüller H, Engel AG, Hohlfeld R, Dornmair K. Inclusion body myositis: laser microdissection reveals differential up‐regulation of IFN‐γ signaling cascade in attacked versus nonattacked myofibers. Am J Pathol. 2011;179:1347–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Orimo S, Koga R, Goto K, Nakamura K, Arai M, Tamaki M, et al. Immunohistochemical analysis of perforin and granzyme A in inflammatory myopathies. Neuromuscul Disord NMD. 1994;4:219–26. [DOI] [PubMed] [Google Scholar]
- 111.Schmidt J, Rakocevic G, Raju R, Dalakas MC. Upregulated inducible co‐stimulator (ICOS) and ICOS‐ligand in inclusion body myositis muscle: significance for CD8+ T cell cytotoxicity. Brain J Neurol. 2004;127(Pt 5):1182–90. [DOI] [PubMed] [Google Scholar]
- 112.Roos A, Preusse C, Hathazi D, Goebel H‐H, Stenzel W. Proteomic profiling unravels a key role of specific macrophage subtypes in sporadic inclusion body myositis. Front Immunol. 2019;10:1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bhattarai S, Ghannam K, Krause S, Benveniste O, Marg A, de Bruin G, et al. The immunoproteasomes are key to regulate myokines and MHC class I expression in idiopathic inflammatory myopathies. J Autoimmun. 2016;75:118–29. [DOI] [PubMed] [Google Scholar]
- 114.Allenbach Y, Chaara W, Rosenzwajg M, Six A, Prevel N, Mingozzi F, et al. Th1 response and systemic treg deficiency in inclusion body myositis. PloS One. 2014;9:e88788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Dzangué‐Tchoupou G, Mariampillai K, Bolko L, Amelin D, Mauhin W, Corneau A, et al. CD8+ T‐bet+ cells as a predominant biomarker for inclusion body myositis. Autoimmun Rev. 2019;18(4):325–333. [DOI] [PubMed] [Google Scholar]
- 116.Knauss S, Preusse C, Allenbach Y, Leonard‐Louis S, Touat M, Fischer N, et al. PD1 pathway in immune‐mediated myopathies. Neurol Neuroimmunol Neuroinflammation. 2019;6(3). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6467687/. [cité 13 févr 2021]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leblanc F, Zhang D, Liu X, Loughran TP. Large granular lymphocyte leukemia: from dysregulated pathways to therapeutic targets. Future Oncol Lond Engl. 2012;8:787–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schmidt J, Barthel K, Wrede A, Salajegheh M, Bähr M, Dalakas MC. Interrelation of inflammation and APP in sIBM: IL‐1 beta induces accumulation of beta‐amyloid in skeletal muscle. Brain J Neurol. 2008;131(Pt 5):1228–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Luo Y‐B, Johnsen RD, Griffiths L, Needham M, Fabian VA, Fletcher S, et al. Primary over‐expression of AβPP in muscle does not lead to the development of inclusion body myositis in a new lineage of the MCK‐AβPP transgenic mouse. Int J Exp Pathol. 2013;94:418–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Basler M, Kirk CJ, Groettrup M. The immunoproteasome in antigen processing and other immunological functions. Curr Opin Immunol. 2013;25:74–80. [DOI] [PubMed] [Google Scholar]
- 121.Kanazawa N. Nakajo‐Nishimura syndrome: an autoinflammatory disorder showing pernio‐like rashes and progressive partial lipodystrophy. Allergol Int. 2012;61:197–206. [DOI] [PubMed] [Google Scholar]
- 122.Ayaki T, Murata K, Kanazawa N, Uruha A, Ohmura K, Sugie K, et al. Myositis with sarcoplasmic inclusions in Nakajo‐Nishimura syndrome: a genetic inflammatory myopathy. Neuropathol Appl Neurobiol. 2020;46:579–87. [DOI] [PubMed] [Google Scholar]
- 123.Honda‐Ozaki F, Terashima M, Niwa A, Saiki N, Kawasaki Y, Ito H, et al. Pluripotent stem cell model of nakajo‐nishimura syndrome untangles proinflammatory pathways mediated by oxidative stress. Stem Cell Rep. 2018;10(6):1835–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Allenbach Y, Benveniste O, Stenzel W, Boyer O. Immune‐mediated necrotizing myopathy: clinical features and pathogenesis. Nat Rev Rheumatol. 2020;16(12):689–701. [DOI] [PubMed] [Google Scholar]
- 125.Allenbach Y, Arouche‐Delaperche L, Preusse C, Radbruch H, Butler‐Browne G, Champtiaux N, et al. Necrosis in anti‐SRP+ and anti‐HMGCR+myopathies: role of autoantibodies and complement. Neurology. 2018;90:e507–17. [DOI] [PubMed] [Google Scholar]
- 126.Preuße C, Goebel HH, Held J, Wengert O, Scheibe F, Irlbacher K, et al. Immune‐mediated necrotizing myopathy is characterized by a specific Th1‐M1 polarized immune profile. Am J Pathol. 2012;181:2161–71. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.