

Abstract
Focal epilepsies are the largest epilepsy subtype and associated with significant morbidity. Somatic variation is a newly recognized genetic mechanism underlying a subset of focal epilepsies, but little is known about the processes through which somatic mosaicism causes seizures, the cell types carrying the pathogenic variants, or their developmental origin. Meanwhile, the inception of single cell biology has completely revolutionized the study of neurological diseases and has the potential to answer some of these key questions. Focusing on single cell genomics, transcriptomics, and epigenomics in focal epilepsy research, circumvents the averaging artifact associated with studying bulk brain tissue and offers the kind of granularity that is needed for investigating the consequences of somatic mosaicism. Here we have provided a brief overview of some of the most developed single cell techniques and the major considerations around applying them to focal epilepsy research.
Keywords: focal epilepsy, single cell genomics, somatic variant
Focal epilepsies are caused by somatic variation in a significant subset of the cases. Single cell genomics offers the kind of granularity that is needed for investigating the consequences of somatic mosaicism. This article provides a brief overview of some of the most developed single cell techniques and the major considerations around applying them to focal epilepsy research.
1. INTRODUCTION
Focal epilepsies are a heterogeneous group of disorders that are associated with significant morbidity and approximately one‐third of focal epilepsy patients do not respond to available anti‐seizure medications (1, 2, 3). The most common type of focal epilepsy, temporal lobe epilepsy (TLE), is notoriously pharmacoresistant and in roughly two‐thirds of the medically refractory cases requires surgical intervention, which is not always effective (4) and can have negative effects on cognition (5). In the pediatric population, malformations of cortical development (MCD) account for the majority of focal epilepsies and may need surgical resection if deemed medically refractory, although outcomes vary greatly based on pathology (6). Notably, the success rate is lower among patients who do not have a lesion visible on MRI (7, 8, 9, 10). One of the most important advances in the field over past two decades, is establishing a clear link between somatic variants and MCD. Somatic variants arise when spontaneous DNA damage escapes DNA repair machinery and most commonly gives rise to single nucleotide variants (SNVs) and indels (11, 12, 13), or to larger structural abnormalities such as copy number variants (CNVs) (14, 15). Historically, twin studies (16) and de novo variant discovery in genetic generalized epilepsies (17, 18) represented the first wave of investigation in epilepsy genetics, but naturally, the main focus was on identifying damaging germline variants that essentially excluded most focal epilepsies. Despite epilepsy surgery providing a unique opportunity to investigate the affected brain tissue directly (19), the recognition of somatic variation as a major contributor to focal epilepsies was delayed partly due to technical factors such as sequencing technology and partly as the result of the barriers to routinely testing surgical resections. Naturally, identifying somatic mosaicism in subtypes of focal epilepsies created a new wave of excitement as it explained the focality of lesions and/or neuronal circuit disruptions that are typically observed. This created a paradigm shift, moving us away from descriptive pathology to molecular genetics as the standard diagnostic approach to focal epilepsy.
While sequencing genomic DNA in blood or saliva is sufficient to diagnose germline variants and a small fraction of somatic variants (20), the recognition of somatic mosaicism in focal epilepsies brought to light the importance of studying the affected tissue directly. This is not only key for the detection of somatic variants with low variant allele fraction (VAF), but also to discern the impact of these variants in situ. In order to understand whether a somatic variant contributes to disease in a cell‐autonomous manner or if it acts through disruption of complex cellular networks, it is important to know which cells carry the variant and how they differ from their genotypically normal counterparts. In the next phase of discovery that ensued, targeted sequencing of the affected brain tissue helped identify a large set of somatic variants, the majority of which were associated with MCD (21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33). This experimental approach has not been as effective for non‐lesional cases, particularly most adult‐onset focal epilepsies, although these cases are not as well studied. Furthermore, even in “solved” cases, bulk sequencing is inherently blind to the specific cell types or clones carrying the pathogenic variants and rarely provides a mechanistic explanation for epileptogenesis. The absence of a clear correlation between genotype and clinical phenotype hinders the efforts to devise new targeted treatments. Unlike some germline genetic epilepsies where the genetic diagnosis is now informing treatment, the limited understanding of molecular mechanisms in focal epilepsies has hindered the use of genetic diagnosis in clinical decision making. While we grapple with these fundamental challenges in studying and treating focal epilepsies, the inception of single cell biology has completely revolutionized cancer research and treatment and is beginning to permeate the study of all other human diseases. Focusing on single cell genomics, transcriptomics, and epigenomics in focal epilepsy research, circumvents the averaging artifact associated with studying bulk brain tissue and offers the kind of granularity that is needed for investigating the consequences of somatic variants. Moreover, since most focal epilepsies are not yet associated with a causal gene variant, identifying defective transcriptional or epigenetic changes at the single cell level may shed light on the affected cell types/pathways and provide an opportunity for intervention. Here we will review some of the key single cell genomic techniques with their unique advantages and limitations, and highlight a few areas in focal epilepsy where they could be immediately applied.
2. SOMATIC MOSAICISM IN FOCAL EPILEPSIES AND THE NEED FOR SINGLE CELL GENOMIC APPROACHES
It is estimated that somatic variants develop at a rate of ~1–3 variants per division per cell during early embryonic development (11, 13, 34, 35). This suggests that a typical individual carries ~80 somatic single nucleotide variants (sSNVs) in ≥2% of cells with ~2% of these being exonic variants (34). Some striking estimates predict ~37%–45% of new exonic variants that have not yet undergone evolutionary selection to be damaging (34, 36, 37), which means about half the population carries ≥1 damaging exonic sSNV at ≥2% of cells (34). This number does not include the potentially toxic sSNVs in non‐coding parts of the genome that have also been linked to neurodevelopmental disorders such as autism (34, 38, 39, 40, 41, 42, 43). Nor does it take into account the effect of somatic structural variants such CNVs that are known causes of focal epilepsies (44, 45). In other words, the contribution of somatic variants to focal epilepsies is likely more far‐reaching than what is known to date. For example, only a handful of genes have been implicated in focal epilepsies so far, while larger scale investigation is underway that will certainly expand that list. Animal studies have demonstrated that cell type specific knock‐ins or deletions of ion channel or small molecule transporter genes such as SCN1A (46, 47, 48), SCN2A (49), SCN8A (50), KCNQ2 (51), and GLT1 (52) can generate a seizure phenotype, which supports the notion that a subset of focal epilepsies may be caused by somatic variation in these classic germline epilepsy gene families. It is important to note though, even in MCD where causal somatic variants have been identified in a significant number of cases, the downstream effects of these variants on cells and on neuronal circuits are largely unknown. This is partly due to the fact that epigenetic factors such as chromatin accessibility and DNA methylation heavily and dynamically regulate gene expression in each cell, making it very difficult to study the effects of somatic variation in bulk tissue. These interactions may be even more intricate in focal epilepsies that are associated with germline variants in genes such as TSC1, TSC2, DEPDC5, NPRL2, which require a somatic second‐hit (21, 32).
Focal cortical dysplasia type 2 (FCD II) is an example that highlights some of these challenges in assessing somatic variants. The majority of known somatic variants that cause FCD II, either directly or indirectly activate the PI3K/Akt/mTOR signaling cascade (21, 53). This is a crucial intracellular signaling pathway that is involved in a range of intracellular processes such as protein synthesis, gene transcription, autophagy, nutrient sensing, growth, etc (54). However, depending on the timing of mTOR overactivation, the specific part of the pathway affected, and the particular clones involved, cellular phenotypes could range from FCD, to neurodegeneration, to death (55). In FCD II, a simplistic explanation may be that mTOR overactivation during development disrupts normal neuro‐glial differentiation, migration, and integration into the neuronal circuit, which gives rise to epilepsy. While this hypothesis may be partially valid, it is unlikely to capture the full spectrum of mTOR functions, in particular the ongoing effects of mTOR overactivation on cellular excitability and altered inhibition in mature neuronal circuits (53). The fact that mTOR inhibitors improve seizure control in animal models of FCD (23), as well as patients with FCD and tuberous sclerosis complex (TSC) (56) is a clear demonstration of that point. Several RNA‐sequencing and whole‐genome methylation profiling studies have had modest success in characterizing some of the differentially expressed genes and unique methylation signatures in FCD (57, 58). For example, Kobow et al. identified unique methylation signatures that distinguished subtypes of FCD from TLE and non‐epileptic controls (57). However, these studies are inherently limited in scope and more granular investigation of single cell transcriptomes and epigenomes is necessary to push the study of FCD and other focal epilepsies to the next level.
3. TRANSCRIPTIONAL AND EPIGENETIC PROFILING OF SINGLE CELLS
3.1. Single cell (nucleus) RNA sequencing
mRNA is the molecule through which the identity of a cell, its activities, and function are determined—it is the direct link between genotype and phenotype in a single cell. Naturally, studying mRNA expression in the affected tissue can provide a great deal of information about the pathogenesis and downstream consequences of a diseased state. To study gene expression, we previously relied on reverse transcription (RT)‐PCR of specific genes or microarray analysis of the transcriptome, which are low throughput and riddled with technical limitations (59). But with the advent of RNA sequencing (60, 61, 62), high throughput and accurate identification, quantification, and discovery of new genes and their splicing isoforms became possible (59, 60, 61, 62). Development of reliable single cell transcriptome amplification (63, 64, 65) paved the way for single cell RNA sequencing (scRNA‐seq) (64, 65). In just over a decade since the first proof of concept scRNA‐seq experiment (65), there has been an explosion in the number of scRNA‐seq tools with significant improvements in the technology (66, 67, 68). Adaptation of single cell cDNA library preparation to single nuclei (69) facilitated the application of this important technology to human tissue where isolating whole cells is frequently not possible (70, 71). Additionally, the adoption of unique molecular identifiers (UMIs) allowed for absolute quantification of molecular counts, increasing accuracy, reducing cost, and improving throughput (72, 73).
scRNA‐seq methods can be divided based on two key features: single cell isolation and cDNA library preparation strategies. The initial step in all single cell molecular genetics techniques is assigning a unique identifier to each cell, either through physical separation of individual cells/nuclei or through single cell combinatorial indexing (SCI). Physical isolation of cells in individual wells of microtiter plates (74, 75, 76, 77) can be achieved through limiting dilution (71, 78), micromanipulation with a capillary pipette (79), fluorescent activated cell sorting (FACS) (74, 80), or laser capture microdissection (81). But most recent techniques use either microfluidic devices to isolate cells in nanoliter droplets (82, 83) or apply split‐pool barcoding to index cDNA molecules from each cell with a unique molecular tag without attempting physical separation (84) (Figure 1). The key distinction between cDNA library preparation techniques is whether full‐length transcripts are sequenced (66, 75) or if only the 3’‐ (82) or 5’‐ends (85) of the transcripts are captured.
FIGURE 1.
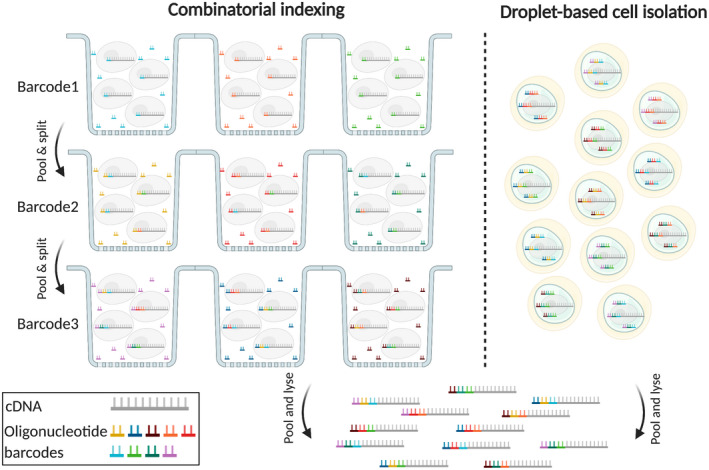
SCI‐ and droplet‐based cell isolation are the most popular barcoding strategies used by most high‐throughput single cell genomic applications. SCI uniquely tags the nucleic acid molecules in each cell through serial mixing, splitting, and barcoding. The higher the number of barcoding steps, the higher the number of cells that can be uniquely tagged in each experiment. Droplet‐based techniques rely on physical isolation of individual cells and engineered barcoded beads in nanoliter droplets, which limits their scalability but they produce less noisy results
Each scRNA‐seq strategy has its own advantages and drawbacks, which should be carefully considered for the specific application in mind (68). Full‐transcript scRNA‐seq technologies have lower transcript dropout rates, allow for isoform detection and RNA editing analysis, and are in general superior in capturing rare and lowly expressed transcripts (86). However, they require physical isolation of single cells in microtiter plates and rely on significant amounts of sequencing per cell to cover the relatively large cDNA library, which renders them labor‐intensive, inefficient, and expensive for most high‐throughput applications. The most popular single cell full‐transcript sequencing technique is Smart‐seq2 (75) that is commercially available and was recently updated to include UMIs for improved isoform quantification (Smart‐seq3 (73)). Microfluidic droplet‐based approaches are highly scalable, optimized for transcript quantification, and capture only one end of the transcript to reduce sequencing cost through smaller cDNA libraries (87). The main drawbacks are that only a fraction of the transcripts in each cell is captured, diminishing their efficiency in detecting low‐abundance transcripts, the 3’‐ or 5’‐bias significantly reduces their utility for isoform quantification or allele expression detection, and although cheaper than full‐transcript techniques in cost of sequencing per cell, they are still expensive. Recently, commercial droplet‐based scRNA‐seq technologies, such as the product by 10X Genomics, have become the standard approach for most scRNA‐seq applications. It is noteworthy that SCI‐based scRNA‐seq is gaining some traction due to theoretically unlimited scalability and lower cost. These techniques do not require physical separation of cells and are entirely performed at the benchtop (84), but until recently had lower transcript‐capture efficiency compared to droplet‐based approaches (84), which limited their utility (88). While not a standard application of the technique, it is noteworthy that RNA‐seq (89, 90) and scRNA‐seq (86, 91) data could also be used for germline and somatic variant discovery, although with a lot of limitations.
3.2. Single cell characterization of chromatin accessibility
Nucleosome, which consists of an octamer of histone proteins encircled by DNA, is the basic structural element of chromatin (92)—the complex responsible for packaging the long DNA molecule in eukaryotic cells. Nucleosomes, along with other chromatin binding structures, indirectly affect cellular function by restricting access to parts of DNA. For example, internucleosomal DNA is rich in gene regulatory elements (GREs) such as enhancers, promoters, insulators, as well as transcription factor binding sites (93, 94). Nucleosome occupancy is not a binary variable and it can dynamically change from closed and inaccessible chromatin to open and fully accessible chromatin (95). These features create an intricate and dynamic process through which gene expression is regulated in each cell. In other words, chromatin accessibility is a surrogate marker for transcription factor binding and regulatory potential of a given locus (92), and not only provides information about the current state of a cell, but it can also predict its future function (88, 96).
The most commonly used techniques for measuring chromatin accessibility across the genome are DNAse I hypersensitive site sequencing (DNAse‐seq) (97), assay for transposase‐accessible chromatin (ATAC‐seq) (98), and micrococcal nuclease sequencing (MNase‐seq) (99), which all rely on enzymatic cleavage of the DNA molecule to mark open regions of chromatin. Both DNAase‐seq and ATAC‐seq have be adapted for single cell applications (100, 101), but due to the ability of Tn5 transposase to easily tag cleaved oligonucleotide fragments in each nucleus, ATAC‐seq has been the most easily scalable and widely used. An array of commercial and non‐commercial technologies have been developed for scATAC‐seq that uses microfluidic capture (102), individually indexable wells of a nano‐well array (103), SCI (104), and droplet‐based microfluidic isolation (105). Similar to scRNA‐seq, combinatorial strategies offer excellent scalability, but the library complexity is lower than the microfluidic‐based approaches (92). Given that only ~10% of the DNAse I hypersensitive sites are detected via scATAC‐seq (101), this could appear as a major drawback of the technique. However, similar to scRNA‐seq, SCI‐based protocols are rapidly improving (88) and will likely be the standard in the future.
3.3. Single cell methylation profiling
DNA methylation is an important epigenetic modification that plays a key role in the regulation of transcription, X chromosome inactivation, genomic imprinting, and chromosomal stability through silencing of transposable elements (106, 107, 108, 109). 5‐methylcytosine is the most common methylated DNA base in vertebrates (110). Cytosine is generally methylated in the context of a CpG dinucleotide—cytosine followed by guanine on the same DNA strand—which clusters together in distinct genomic regions called CpG islands (111). CpG islands are frequently associated with promoters and enhancers of gene expression, so hypermethylation indicates repression of these GREs whereas DNA hypomethylation is a surrogate for active regulation of gene transcription (111, 112).
Even though DNA methylation has been an active area of scientific exploration for years (113), techniques for measurement of single cell genome‐wide methylation still face some technical hurdles. Recently whole‐genome bisulfide sequencing (WGBS) has established itself as the gold standard for bulk DNA methylation sequencing, covering as much as ~95% of the CpG sites (114). In WGBS, unmethylated cytosines are deaminated into uracil, while methylated cytosines remain unaltered (115). When combined with next‐generation sequencing, methylated cytosines can be detected as the single base resolution (116). To overcome the costs associated with deep whole‐genome sequencing required for WGBS, reduced representation bisulfite sequencing (RRBS) was developed that uses methylation‐insensitive restriction enzymes to generate smaller sequencing libraries (117, 118). The main drawback for RRBS is that it only covers ~10% of the total CpG sites, which means regions of low CpG density such as enhancers are not well‐represented (119). Several different single cell adaptations of both bisulfide‐based and restriction enzyme‐based methylation sequencing have been developed, but they have several key differences and should be chosen based on the biologic question in mind (115). The first iteration of single cell methylation profiling used a bisulfide‐based approach, but suffered from poor and inconsistent coverage across different cells (120). Some of these limitations are inherent to bisulfite conversation, as it causes DNA degradation (115), but using UMIs (121), bisulfite conversion prior to adapter ligation (122), and PCR amplification of the tagged fragments, have extended the coverage rate to ~18% of all CpG sites (scBS‐seq) (123). Further improvements in library preparation and read mapping have increased uniformity and reduced artifactual reads (snmC‐seq2) (124, 125). However, they are lower throughput compared to SCI‐based strategies (sci‐MET) (126) that are highly scalable at the cost of lower data quality. To circumvent the problems associated with bisulfite conversion, a small number of single cell methylation techniques utilize methylation‐sensitive restriction enzymes (127, 128), but their lower resolution and non‐quantitative design, limits their application. Overall, single cell methylation profiling is more challenging and less developed compared to other single cell molecular genetics tools, but if applied to the right biological question, it could be quite powerful—particularly when combined with scRNA‐seq or scATAC‐seq.
3.4. Combined transcriptional and epigenetic analysis of single cells
It has been demonstrated that DNA hypomethylated and DNAse I hypersensitive sites overlap at a high rate, suggesting that a combination of these signatures may reflect stages of enhancer activation (119, 129), Importantly though, GRE accessibility and DNA methylation state change independently during cell fate transitions with delayed loss of methylation in regions of open chromatin (129). This creates a complex dynamic between these important epigenetic regulatory mechanisms through which transcription is regulated. In other words, independent measurements of gene transcription, chromatin accessibility, and DNA methylation will not tell the full story on epigenetic regulation of gene expression at the single cell level. A combined assay could be quite impactful in unraveling how different cell types are affected by focal epilepsies and even predict their ongoing adaptative response to seizures.
Since most combined single cell approaches are derived from techniques that have already been described, we will not elaborate on each method, but rather list some of the more popular options available. The most robust co‐assays developed to date, profile RNA and chromatin accessibility in single cells (scRNA+ATAC‐seq). The first generation of these techniques were sci‐CAR (130), Paired‐seq (131), and SNARE‐seq (132), which used similar protocols, but relied on SCI vs droplet‐based barcoding of single cells. While an important achievement, these initial methods produced lower quality data compared to what could be generated by individual scRNA‐seq or scATAC‐seq assays (88). The second‐generation scRNA+ATAC‐seq techniques, which include SHARE‐seq (88) and a commercial product by 10X genomics, have closed that gap considerably with remarkable improvement in data quality. Other available co‐assays simultaneously profile single cell transcriptome and methylome (scM&T‐seq (133)), single cell nucleosome occupancy and DNA methylation (scNMOe‐seq (134)), and single cell transcriptome, chromatin accessibility, and DNA methylation (scNMT‐seq (135)), but they have lower throughput and are less developed compared to scRNA+ATAC‐seq. It is important to note that combined single cell techniques are in their nascency and have not been extensively applied to the study of human brain. In other words, they may have important limitations that are not yet known, however, a handful of examples offer a glimpse of their future potential in studying the human brain and focal epilepsies (88, 131, 136).
4. APPLICATION OF SOMATIC VARIANTS TO LINEAGE TRANCING IN NORMAL AND DISEASED BRAINS
During neurogenesis approximately 105 neurons are generated per minute (137, 138, 139), making the brain particularly susceptible to somatic variants that have been estimated to accumulate at rates as high rate as ~5.1 sSNVs per day per cerebral cortical progenitor (11). Different studies have estimated as many as ~300–900 somatic SNVs for a post‐zygotic neuron soon after birth (11, 140). Somatic CNVs are not thought to be as common—though more difficult to detect—but they reportedly happen in up to 41% of human neurons (141). Somatic transposon insertion events are an important and well‐studied cause of somatic subchromosomal CNVs that happen at rates significantly lower than sSNVs (15, 142). When discussing somatic mutagenesis, typically the primary focus is on its role in disease causation. Importantly though, somatic variants that are present in all the clones of a progenitor, can also serve as a lineage map to determine both their origin and timing of development (11). For example, when a neuroglial progenitor has spontaneous DNA damage that escapes the DNA repair machinery, it accumulates variants that are unique to that cell and are passed down to its progeny as a lineage barcode (Figure 2). If the lineage‐defining clonal variants in a given tissue are known—typically through bulk or synthetic bulk WGS (143)—it is possible to trace back the developmental origin of cortical neurons or glia by identifying the variants that they share. The cell types and the fraction of cells carrying a specific somatic variant could serve as surrogate markers to infer the developmental timing of when a mutation occurred (11, 144). This technique is particularly powerful when combined with RNA sequencing, as it can draw a connection between specific cell types, their developmental origins, and their timeline of differentiation (143). Lineage tracing in the human brain has so far been mostly limited to the study of normal tissue (11, 143, 144), but it is easy to imagine how this transformative technology could elucidate the mechanisms of genetic focal epilepsies. Lineage tracing could shed light on the timing of when pathogenic somatic variants arise and the conditions under which they cause disease. Beyond the obvious diagnostic and treatment implications, knowing when and how pathogenic somatic variants occur could provide clues about potential modifiable factors and may eventually lead to preventive measures.
FIGURE 2.
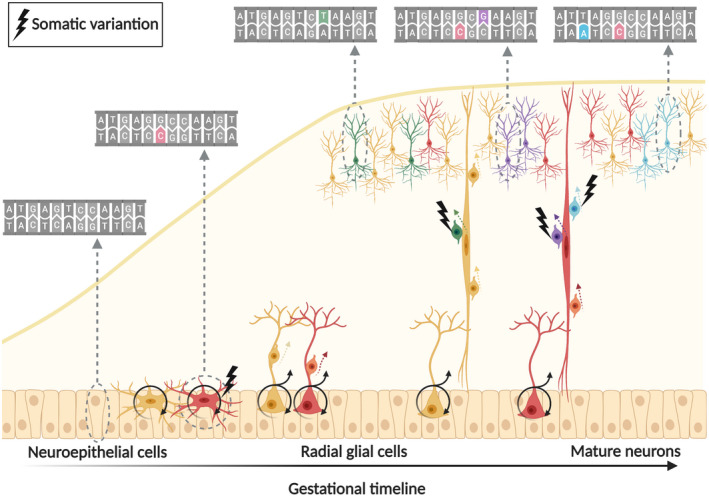
Somatic variants are spontaneously acquired during development. All the somatic variants in a progenitor cell are passed down to its daughter cells. The number of cells carrying a specific variant is an indirect marker for the developmental timepoint at which it was generated
Let's continue using FCD II as an example. One of the hallmarks of FCD II is the presence of dysmorphic neurons (DN) and balloon cells (BC, FCD IIb) (145). Naturally, understanding the developmental lineage of these aberrant cell types is an important step in deciphering how FCD lesions develop and how they give rise to epilepsy. One of the first attempts at lineage tracing in FCD, used single cell microdissection and an X‐androgen receptor (XAR) inactivation (146) to show disparate XAR CAG repeat lengths in single DNs and BCs, and proposed a possible role for random X‐inactivation in FCD (146). Several follow‐up studies have used more advanced techniques such as laser capture microdissection and SNP genotyping to detect the presence of somatic SNVs in individual DNs and BCs. One such study looking at a DEPDC5‐related FCD IIa, demonstrated that the second‐hit somatic DEPDC5 pathogenic variant was enriched in DNs compared to their normal‐appearing counterparts (32, 147). Another study that used a similar experimental approach in FCD IIb, showed enrichment of pathogenic MTOR and PIK3CA variants in DNs and BCs compared to morphologically normal‐appearing neurons and glial cells (32). While DNs typically have neuronal properties and BCs express some glial markers, there is a range of intermediate cellular phenotypes that share properties of both as well markers of immaturity (53, 145). Older studies relying on immunohistochemistry and cell type‐specific antibodies, have suggested that DNs and BCs arise from the telencephalic ventricular zone and neuroglial progenitors (148). Although, cytomegalic interneurons have also been reported (149), calling the developmental origin of these cells into question. In a more recent effort to characterize the developmental lineage of FCD II, D’Gama et al. used FACS to sort neurons vs non‐neuronal cells followed by scWGA and SNP genotyping to demonstrate an apparent enrichment of pathogenic variants in the neuronal lineage (21). However, due to technical limitations, they stopped short of characterizing the specific neuronal cell types. Animal studies have had modest success, honing in on the developmental timing of somatic mutagenesis in FCD (23, 27), but much is left to be desired. In the meanwhile, with the advent of single cell DNA sequencing (scDNA‐seq), single cell lineage tracing in normal human brain has been advancing rapidly. Application of this technology to FCD and other focal epilepsies could help answer many of these important questions.
4.1. Single cell whole‐genome amplification and sequencing
To better understand the advantages and drawbacks of scDNA‐seq technologies it is important to review some concepts in bulk DNA sequencing first. The gold standard for unbiased discovery of clonal somatic variants is unamplified bulk whole‐genome sequencing (bWGS) (15). However, the human genome is approximately 3 billion base pairs in size, which means bWGS can be prohibitively expensive at the high sequencing depth that is required for the detection of rare somatic variants (21, 139). To circumvent the enormous financial burden of bWGS, target capture/amplification techniques such whole‐exome sequencing (WES) and gene panel sequencing, have gained popularity in disease‐associated somatic variant discovery. These techniques though very efficient and high yield, suffer from major artifacts associated with PCR duplication errors (150) that reduce mosaic variant validation rate (151), and by definition miss any pathogenic variants outside the selected genomic regions. In broad terms, accuracy, resolution, and efficiency are competing interests in bulk DNA sequencing technologies. The same challenges persist in scDNA‐seq, but are even more magnified since the starting material is just one DNA molecule.
A normal diploid human genome contains about 6‐7 picograms of DNA (152), but several nanograms of input DNA are required for NGS. Logically, a DNA amplification step is necessary. All single cell whole‐genome amplification (scWGA) strategies are imperfect, and to that end, the utility of a specific scDNA‐seq approach is determined by the scWGA strategy applied (Table 1). Some common considerations include amplification bias with preferential amplification of specific genomic regions (uniformity), allelic dropouts (coverage), and nucleotide copy errors (false positive mutations) (153). The Holy Grail of single cell genomics is a scWGA technique that covers the entire genome, is uniform, has low copy errors, and is scalable. Since the first attempt at scWGA (154), there has been a lot of progress although two scWGA techniques have dominated the field so far. These methods that rely on non‐linear exponential amplification are degenerate oligonucleotide‐primed PCR (DOP‐PCR) (155) and multiple displacement amplification (MDA) (156). DOP‐PCR is fast, uniform, and readily accessible through popular commercial products such as GenomePlex (Sigma‐Aldrich) (157), but it has low coverage of the genome and is error prone (158). MDA is another popular and commercially available strategy for scWGA (159) that offers high coverage of the genome and low error rate, but it lacks uniformity (15). Given their highly uniform product, PCR‐based approaches are more suitable for CNV analysis, while high coverage and low error rate make MDA ideal for SNV detection (14, 144, 160). To minimize the random non‐uniform amplification associated with MDA, a semi‐linear scWGA technique, multiple annealing and looping‐based amplification cycles (MALBAC) (161) was created. MALBAC generates self‐annealing amplicons to facilitate several cycles of linear amplification. However, it requires exponential amplification during the final steps and has a higher false‐positive SNV rate compared to MDA (162). Linear amplification via transposon insertion (LIANTI) (163) took a major leap by solely utilizing linear amplification, offering more uniformity and improved coverage. LIANTI is not yet commercially available and it relies on a more complex protocol and Tn5 transposases so its adoption has been slow. A very recent development in scWGA is primary template‐directed amplification (PTA) (164) that similar to MDA uses the high‐fidelity Phi29 DNA polymerase, but unlike MDA it generates the majority of the copies from the primary DNA strand to achieve linear amplification. PTA is a commercial product and relatively expensive, but it achieves uniform, high coverage, low error rate scWGA through a simple protocol.
TABLE 1.
Comparison between common scWGA techniques
| Amplification strategy | Uniformity | Coverage | Error rate | |
|---|---|---|---|---|
| DOP‐PCR | Exponential |
|
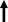
|

|
| MDA | Exponential |
|
|
|
| MALBAC | Semi‐linear |
|
|
|
| LIANTI | Linear |
|
|
|
| PTA | Linear |
|
|
|
Number of upward arrows reflects assay reliability in each represented category; one arrow depicts least and three arrows represents most reliable.
High coverage scWGS is not easily scalable due to prohibitive sequencing costs, limiting the study of SNVs to a small subset of cells (140, 144). On the other hand, low coverage sequencing is sufficient for large CNV detection, making it the primary high throughput application of scWGS (14). Many microfluidic‐based and droplet‐based adoptions of PCR‐ (165), MDA‐ (166, 167), MALBAC‐based (168) scWGA techniques exist. SCI has also been applied to scWGS (169) including a new technique that combinates SCI with linear amplification (170). But due to the inherent limitations outlined above, all of these techniques are only optimized for studying CNVs at a large scale, which is helpful if CNVs play a major role in the disease under investigation such as hemimegalencephaly (HME) (44). Another barrier to using scWGS for large‐scale lineage tracing experiments is limited access to transcriptional information to perform detailed cell type analysis, forcing us to resort to nuclear sorting for the determination of cellular identity. A handful of techniques have had modest success in combining scRNA‐seq and scDNA‐seq (G&T‐seq (171) and DR‐seq (172)), but the protocols are laborious and transcript dropout limits their utility. A recent method, parallel RNA and DNA analysis after sequencing (PRDD‐seq), used a microfluidic approach to simultaneously interrogate cell‐type‐specific cDNA and lineage‐informative sSNVs using single cell qPCR (143). This approach facilitated large‐scale lineage tracing with the incorporation of more granular cellular identities. Another promising new tool that has not been yet applied to lineage tracing, sci‐L3, utilizes SCI to perform single cell RNA and SNP‐genotyping in thousands of cells (170). Sci‐L3 does not require a priori knowledge about patterns of gene expression, which allows for the identification of new and rare cell types. While new and better techniques will be emerging in the near future, many of the available tools can be immediately used to study cellular lineage in focal epilepsies.
5. PRIVATE VARIANTS AND THE POTENTIAL FOR GENOTOXIC DAMAGE IN EPILEPSY
Surprisingly, even terminally differentiated neurons continue to accumulate private variants (somatic variants unique to each cell) at a rate of ~23 SNVs per year per neuron in the prefrontal cortex and at ~40 SNVs per year per neuron in the dentate gyrus (140). It is difficult to conceive, and it is unlikely that focal epilepsy is caused by these truly private variants. It is, however, plausible that cells that are exposed to chronic seizures acquire private somatic variants at an accelerated rate, due to increased oxidative stress and disruption of normal cellular homeostasis. If this were to be the case, a subset of these private somatic variants in exonic or regulatory regions may have direct function‐altering or toxic effects with deleterious consequences at the single cell level. It has been previously demonstrated that neurons in FCD and HME express abnormally high levels of phosphorylated tau (173, 174), which is typically a molecular feature of neurodegenerative diseases (175). Interestingly, patients with AD are at increased risk of having seizures (176), which has also been corroborated in animal models of AD (177). In other words, it is possible that genotoxic damage from chronic seizures plays a role in treatment‐refractory epilepsy and could be a plausible explanation for accelerated neurodegeneration seen in patients with epilepsy (178, 179).
6. THE CHALLENGE OF NON‐LESIONAL FOCAL EPILEPSIES AND OPPORTUNITY FOR SINGLE CELL INVESTIGATION
In order for somatic variants to be detected by the current diagnostic approaches, the variant allele fraction (VAF) should typically exceed 1% in the tested tissue (21). This may partly account for the fact that most somatic variants have been detected in MCD, where the affected tissue is easy to identify and the pathogenic variants arise mid‐gestation so they are expressed in a higher percentage of cells (53). Since some neurogenesis continues after birth (180, 181), clonal somatic variants may continue to be passed down to a small subset of daughter cells postnatally. While the percentage of cells harboring such variants is likely extremely small, it is nevertheless theoretically possible that these variants contribute to the pathogenesis of a subset of adolescent‐ and adult‐onset focal epilepsies such as TLE. At this juncture, this claim is purely theoretical with no experimental evidence to support it. Nevertheless, irrespective of whether somatic mosaicism contributes to the development of non‐lesional focal epilepsies, single cell DNA, RNA, and epigenomic sequencing will give us the opportunity to shed light on the specific cell types affected, the burden of clonal and private somatic variants, and the GREs involved in the disease process.
7. CONCLUDING REMARKS AND OUTLOOK
Epilepsy is one of the oldest diseases described in human literature and one of the most studied neurologic diseases, yet our approach to treating it has remained unchanged for centuries. Part of the challenge is that the limited clinical classification of seizures is not reflective of the great molecular heterogeneity underlying different seizure types. The growing influence of somatic mosaicism in the scientific discourse surrounding focal epilepsies has generated a novel, mechanistic framework that takes into account genetic diversity at the single cell level. In light of this, to adequately investigate the molecular mechanisms underlying focal epilepsies, it is obligatory that we take advantage of single cell genomic approaches. Here we have provided a brief overview of a few available single cell techniques and some major considerations around using them. It is important to note that despite their differences, many of these techniques have reached their maturity and can be immediately utilized to study focal epilepsies.
ACKNOWLEDGMENTS
S.K. is supported by NINDS (R25NS065743). C.A.W. is supported by NINDS (R01NS35129 and R01NS032457) and the NIMH (U01MH106883), and the Allen Discovery Center program, a Paul G. Allen Frontiers Group advised program of the Paul G. Allen Family Foundation. C.A.W. is an Investigator of the Howard Hughes Medical Institute. Figures were created with BioRender.com. Figure 2 is adapted from “Embryonic Migration of Neuronal Progenitors Relies on Astrocytic Scaffold”, by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender‐templates.
Dennis Lal and Christopher A. Walsh are contributed equally to this work.
Contributor Information
Dennis Lal, Email: lald@ccf.org, Email: dlal@broadinstitute.org.
Christopher A. Walsh, Email: christopher.walsh@childrens.harvard.edu.
REFERENCES
- 1.Berg AT, Vickrey BG, Testa FM, Levy SR, Shinnar S, DiMario F, et al. How long does it take for epilepsy to become intractable? A prospective investigation. Ann Neurol. 2006;60:73–9. [DOI] [PubMed] [Google Scholar]
- 2.Aaberg KM, Bakken IJ, Lossius MI, Lund Søraas C, Tallur KK, Stoltenberg C, et al. Short‐term seizure outcomes in childhood epilepsy. Pediatrics. 2018;141(6):e20174016. [DOI] [PubMed] [Google Scholar]
- 3.Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–9. [DOI] [PubMed] [Google Scholar]
- 4.Lamberink HJ, Otte WM, Blümcke I, Braun KP, Aichholzer M, Amorim I, et al. Seizure outcome and use of antiepileptic drugs after epilepsy surgery according to histopathological diagnosis: a retrospective multicentre cohort study. Lancet Neurol. 2020;19:748–57. [DOI] [PubMed] [Google Scholar]
- 5.Sherman EMS, Wiebe S, Fay‐McClymont TB, Tellez‐Zenteno J, Metcalfe A, Hernandez‐Ronquillo L, et al. Neuropsychological outcomes after epilepsy surgery: Systematic review and pooled estimates. Epilepsia. 2011;52:857–69. [DOI] [PubMed] [Google Scholar]
- 6.Barkovich AJ, Dobyns WB, Guerrini R. Malformations of cortical development and epilepsy. Cold Spring Harb Perspect Med. 2015;5(5):a022392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bien CG, Szinay M, Wagner J, Clusmann H, Becker AJ, Urbach H. Characteristics and surgical outcomes of patients with refractory magnetic resonance imaging‐negative epilepsies. Arch Neurol. 2009;66:1491–9. [DOI] [PubMed] [Google Scholar]
- 8.Sylaja PN, Radhakrishnan K, Kesavadas C, Sarma PS. Seizure outcome after anterior temporal lobectomy and its predictors in patients with apparent temporal lobe epilepsy and normal MRI. Epilepsia. 2004;45:803–8. [DOI] [PubMed] [Google Scholar]
- 9.Immonen A, Jutila L, Muraja‐Murro A, Mervaala E, Äikiä M, Lamusuo S, et al. Long‐term epilepsy surgery outcomes in patients with MRI‐negative temporal lobe epilepsy. Epilepsia. 2010;51:2260–9. [DOI] [PubMed] [Google Scholar]
- 10.Bulacio JC, Jehi L, Wong C, Gonzalez‐Martinez J, Kotagal P, Nair D, et al. Long‐term seizure outcome after resective surgery in patients evaluated with intracranial electrodes. Epilepsia. 2012;53:1722–30. [DOI] [PubMed] [Google Scholar]
- 11.Bae T, Tomasini L, Mariani J, Zhou BO, Roychowdhury T, Franjic D, et al. Different mutational rates and mechanisms in human cells at pregastrulation and neurogenesis. Science. 2018;359:550–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De S. Somatic mosaicism in healthy human tissues. Trends Genet. 2011;27:217–23. [DOI] [PubMed] [Google Scholar]
- 13.Ju YS, Martincorena I, Gerstung M, Petljak M, Alexandrov LB, Rahbari R, et al. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature. 2017;543:714–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cai X, Evrony GD, Lehmann HS, Elhosary PC, Mehta BK, Poduri A, et al. Single‐cell, genome‐wide sequencing identifies clonal somatic copy‐number variation in the human brain. Cell Rep. 2015;10:645. [DOI] [PubMed] [Google Scholar]
- 15.Evrony G, Cai X, Lee E, Hills L, Elhosary PC, Lehmann H, et al. Single‐neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell. 2012;151:483–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins: genetics of the major epilepsy syndromes. Ann Neurol. 1998;43:435–45. [DOI] [PubMed] [Google Scholar]
- 17.Heyne HO, Singh T, Stamberger H, Abou Jamra R, Caglayan H, Craiu D, et al. De novo variants in neurodevelopmental disorders with epilepsy. Nat Genet. 2018;50:1048–53. [DOI] [PubMed] [Google Scholar]
- 18.ElliCA PS, Berkovic SF. Epilepsy genetics: clinical impacts and biological insights. Lancet Neurol. 2020;19:93–100. [DOI] [PubMed] [Google Scholar]
- 19.Blumcke I, Spreafico R, Haaker G, Coras R, Kobow K, Bien CG, et al. Histopathological findings in brain tissue obtained during epilepsy surgery. N Engl J Med. 2017;377:1648–56. [DOI] [PubMed] [Google Scholar]
- 20.de Lange IM, Koudijs MJ, van't Slot R, Gunning B, Sonsma AC, van Gemert LJ, et al. Mosaicism of de novo pathogenic SCN1A variants in epilepsy is a frequent phenomenon that correlates with variable phenotypes. Epilepsia. 2018;59:690–703. [DOI] [PubMed] [Google Scholar]
- 21.D’Gama AM, Woodworth MB, Hossain AA, Bizzotto S, Hatem NE, LaCoursiere CM, et al. Somatic mutations activating the mTOR pathway in dorsal telencephalic progenitors cause a continuum of cortical dysplasias. Cell Rep. 2017;21:3754–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jansen LA, Mirzaa GM, Ishak GE, O'Roak BJ, Hiatt JB, Roden WH, et al. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain J Neurol. 2015;138:1613–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim JS, Kim WI, Kang HC, Kim SH, Park AH, Park EK, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. 2015;21:395–400. [DOI] [PubMed] [Google Scholar]
- 24.Nakashima M, Saitsu H, Takei N, Tohyama J, Kato M, Kitaura H, et al. Somatic mutations in the MTOR gene cause focal cortical dysplasia type IIb. Ann Neurol. 2015;78:375–86. [DOI] [PubMed] [Google Scholar]
- 25.Mirzaa GM, Campbell CD, Solovieff N, Goold CP, Jansen LA, Menon S, et al. Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol. 2016;73:836–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Møller RS, Weckhuysen S, Chipaux M, Marsan E, Taly V, Bebin EM, et al. Germline and somatic mutations in the MTOR gene in focal cortical dysplasia and epilepsy. Neurol Genet. 2016;2:e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim JS, Gopalappa R, Kim SH, Ramakrishna S, Lee M, Kim WI, et al. Somatic mutations in TSC1 and TSC2 cause focal cortical dysplasia. Am J Hum Genet. 2017;100:454–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.D'Gama AM, Geng Y, Couto JA, Martin B, Boyle EA, LaCoursiere CM, et al. Mammalian target of rapamycin pathway mutations cause hemimegalencephaly and focal cortical dysplasia. Ann Neurol. 2015;77:720–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao S, Li Z, Zhang M, Zhang L, Zheng H, Ning J, et al. A brain somatic RHEB doublet mutation causes focal cortical dysplasia type II. Exp Mol Med. 2019;51:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niestroj L‐M, May P, Artomov M, Kobow K, Coras R, Pérez‐Palma E, et al. Assessment of genetic variant burden in epilepsy‐associated brain lesions. Eur J Hum Genet. 2019;27:1738–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sim NS, Ko A, Kim WK, Kim SH, Kim JS, Shim KW, et al. Precise detection of low‐level somatic mutation in resected epilepsy brain tissue. Acta Neuropathol. 2019;138:901–12. [DOI] [PubMed] [Google Scholar]
- 32.Baldassari S, Ribierre T, Marsan E, Adle‐Biassette H, Ferrand‐Sorbets S, Bulteau C, et al. Dissecting the genetic basis of focal cortical dysplasia: a large cohort study. Acta Neuropathol. 2019;138:885–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Z, Gao K, Liu Q, Zhou J, Li X, Lang NA, et al. Somatic variants in new candidate genes identified in focal cortical dysplasia type II. Epilepsia. 2020;61:667–78. [DOI] [PubMed] [Google Scholar]
- 34.Rodin RE, Dou Y, Kwon M, Sherman MA, D'Gama AM, Doan RN, et al. The Landscape of mutational mosaicism in autistic and normal human cerebral cortex. bioRxiv. 2020. 10.1101/2020.02.11.944413. [DOI] [Google Scholar]
- 35.Ye AY, Dou Y, Yang X, Wang S, Huang AY, Wei L. A model for postzygotic mosaicisms quantifies the allele fraction drift, mutation rate, and contribution to de novo mutations. Genome Res. 2018;28:943–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neale BM, Kou Y, Liu LI, Ma’ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. 2007;80:727–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Short PJ, McRae JF, Gallone G, Sifrim A, Won H, Geschwind DH, et al. De novo mutations in regulatory elements in neurodevelopmental disorders. Nature. 2018;555:611–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams SM, An JY, Edson J, Watts M, Murigneux V, Whitehouse AJ, et al. An integrative analysis of non‐coding regulatory DNA variations associated with autism spectrum disorder. Mol Psychiatry. 2019;24:1707–19. [DOI] [PubMed] [Google Scholar]
- 40.Zhou J, Park CY, Theesfeld CL, Wong AK, Yuan Y, Scheckel C, et al. Whole‐genome deep‐learning analysis identifies contribution of noncoding mutations to autism risk. Nat Genet. 2019;51:973–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.An J‐Y, Lin K, Zhu L, Werling DM, Dong S, Brand H, et al. Genome‐wide de novo risk score implicates promoter variation in autism spectrum disorder. Science. 2018;362:eaat6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turner TN, Coe BP, Dickel DE, Hoekzema K, Nelson BJ, Zody MC, et al. Genomic patterns of de novo mutation in simplex autism. Cell. 2017;171:710–722.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turner TN, Hormozdiari F, Duyzend MH, McClymont SA, Hook PW, Iossifov I, et al. Genome sequencing of autism‐affected families reveals disruption of putative noncoding regulatory DNA. Am J Hum Genet. 2016;98:58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poduri A, Evrony G, Cai X, Elhosary P, Beroukhim R, Lehtinen M, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74:41–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niestroj LM, Perez‐Palma E, Howrigan DP, Zhou Y, Cheng F, Saarentaus E, et al. Epilepsy subtype‐specific copy number burden observed in a genome‐wide study of 17 458 subjects. Brain. 2020;143:2106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheah CS, Frank HY, Westenbroek RE, Kalume FK, Oakley JC, Potter GB, et al. Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A. 2012;109:14646–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dutton SB, Makinson CD, Papale LA, Shankar A, Balakrishnan B, Nakazawa K, et al. Preferential inactivation of Scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol Dis. 2013;49:211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tatsukawa T, Ogiwara I, Mazaki E, Shimohata A, Yamakawa K. Impairments in social novelty recognition and spatial memory in mice with conditional deletion of Scn1a in parvalbumin‐expressing cells. Neurobiol Dis. 2018;112:24–34. [DOI] [PubMed] [Google Scholar]
- 49.Ogiwara I, Ogiwara I, Miyamoto H, Tatsukawa T, Yamagata T, Nakayama T, et al. Nav1.2 haplodeficiency in excitatory neurons causes absence‐like seizures in mice. Commun Biol. 2018;1:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bunton‐Stasyshyn RK, Wagnon JL, Wengert ER, Barker BS, Faulkner A, Wagley PK, et al. Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain J Neurol. 2019;142:362–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soh H, Pant R, LoTurco JJ, Tzingounis AV. Conditional deletions of epilepsy‐associated KCNQ2 and KCNQ3 channels from cerebral cortex cause differential effects on neuronal excitability. J Neurosci. 2014;34:5311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, et al. Conditional deletion of the glutamate transporter GLT‐1 reveals that astrocytic GLT‐1 protects against fatal epilepsy while neuronal GLT‐1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci. 2015;35:5187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iffland PH, Crino PB. Focal cortical dysplasia: gene mutations, cell signaling, and therapeutic implications. Annu Rev Pathol. 2017;24:547–71. [DOI] [PubMed] [Google Scholar]
- 54.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lipton JO, Sahin M. The Neurology of mTOR. Neuron. 2014;84:275–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim JK, Lee JH. Mechanistic target of rapamycin pathway in epileptic disorders. J. Korean Neurosurg Soc. 2019;62:272–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobow K, Ziemann M, Kaipananickal H, Khurana I, Mühlebner A, Feucht M, et al. Genomic DNA methylation distinguishes subtypes of human focal cortical dysplasia. Epilepsia. 2019;60:1091–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dixit AB, Sharma D, Tripathi M, Srivastava A, Paul D, Prakash D, et al. Genome‐wide DNA methylation and RNAseq analyses identify aberrant signalling pathways in focal cortical dysplasia (FCD) type II. Sci Rep. 2018;8:17976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Z, Gerstein M, Snyder M. RNA‐Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA‐Seq. Nat Methods. 2008;5:621–8. [DOI] [PubMed] [Google Scholar]
- 61.Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single‐nucleotide resolution. Nature. 2008;453:1239–43. [DOI] [PubMed] [Google Scholar]
- 63.Kurimoto K, Yabuta Y, Ohinata Y, Saitou M. Global single‐cell cDNA amplification to provide a template for representative high‐density oligonucleotide microarray analysis. Nat Protoc. 2007;2:739–52. [DOI] [PubMed] [Google Scholar]
- 64.Lao KQ, Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, et al. mRNA‐sequencing whole transcriptome analysis of a single cell on the SOLiD system. J Biomol Tech. 2009;20:266–71. [PMC free article] [PubMed] [Google Scholar]
- 65.Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, et al. mRNA‐Seq whole‐transcriptome analysis of a single cell. Nat Methods. 2009;6:377–82. [DOI] [PubMed] [Google Scholar]
- 66.Chen G, Ning B, Shi T. Single‐cell RNA‐Seq technologies and related computational data analysis. Front Genet. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mereu E, Lafzi A, Moutinho C, Ziegenhain C, McCarthy DJ, Álvarez‐Varela A, et al. Benchmarking single‐cell RNA‐sequencing protocols for cell atlas projects. Nat Biotechnol. 2020;38:747–55. [DOI] [PubMed] [Google Scholar]
- 68.Ding J, Adiconis X, Simmons SK, Kowalczyk MS, Hession CC, Marjanovic ND, et al. Systematic comparison of single‐cell and single‐nucleus RNA‐sequencing methods. Nat Biotechnol. 2020;38:737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grindberg RV, Yee‐Greenbaum JL, McConnell MJ, Novotny M, O’Shaughnessy AL, Lambert GM, et al. RNA‐sequencing from single nuclei. Proc Natl Acad Sci U S A. 2013;110:19802–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Habib N, Avraham‐Davidi I, Basu A, Burks T, Shekhar K, Hofree M, et al. Massively parallel single‐nucleus RNA‐seq with DroNc‐seq. Nat Methods. 2017;14:955–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hochgerner H, Lönnerberg P, Hodge R, Mikes J, Heskol A, Hubschle H, et al. STRT‐seq‐2i: dual‐index 5ʹ single cell and nucleus RNA‐seq on an addressable microwell array. Sci Rep. 2017;7:16327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, et al. Quantitative single‐cell RNA‐seq with unique molecular identifiers. Nat Methods. 2014;11:163–6. [DOI] [PubMed] [Google Scholar]
- 73.Hagemann‐Jensen M, Ziegenhain C, Chen P, Ramsköld D, Hendriks GJ, Larsson AJ, et al. Single‐cell RNA counting at allele and isoform resolution using Smart‐seq3. Nat Biotechnol. 2020;38:708–14. [DOI] [PubMed] [Google Scholar]
- 74.Jaitin DA, Kenigsberg E, Keren‐Shaul H, Elefant N, Paul F, Zaretsky I, et al. Massively parallel single cell RNA‐Seq for marker‐free decomposition of tissues into cell types. Science. 2014;343:776–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Picelli S, Faridani OR, Björklund ÅK, Winberg G, Sagasser S, Sandberg R. Full‐length RNA‐seq from single cells using Smart‐seq2. Nat Protoc. 2014;9:171–81. [DOI] [PubMed] [Google Scholar]
- 76.Hashimshony T, Wagner F, Sher N, Yanai I. CEL‐Seq: single‐cell RNA‐Seq by multiplexed linear amplification. Cell Rep. 2012;2:666–73. [DOI] [PubMed] [Google Scholar]
- 77.Islam S, Kjällquist U, Moliner A, Zajac P, Fan JB, Lönnerberg P, et al. Characterization of the single‐cell transcriptional landscape by highly multiplex RNA‐seq. Genome Res. 2011;21:1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lefkovits I, Pernis B. Immunological Methods, III. Elsevier; 2014. [Google Scholar]
- 79.Brehm‐Stecher BF, Johnson EA. Single‐cell microbiology: tools, technologies, and applications. Microbiol Mol Biol Rev. 2004;68:538–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Julius MH, Masuda T, Herzenberg LA. Demonstration that antigen‐binding cells are precursors of antibody‐producing cells after purification with a fluorescence‐activated cell sorter. Proc Natl Acad Sci U S A. 1972;69:1934–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nichterwitz S, Chen G, Benitez JA, Yilmaz M, Storvall H, Cao M, et al. Laser capture microscopy coupled with Smart‐seq2 for precise spatial transcriptomic profiling. Nat Commun. 2016;7:12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Macosko E, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome‐wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Klein A, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, et al. Droplet barcoding for single‐cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, et al. Comprehensive single‐cell transcriptional profiling of a multicellular organism. Science. 2017;357:661–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Islam S, Kjällquist U, Moliner A, Zajac P, Fan JB, Lönnerberg P, et al. Highly multiplexed and strand‐specific single‐cell RNA 5’ end sequencing. Nat Protoc. 2012;7:813–28. [DOI] [PubMed] [Google Scholar]
- 86.Liu F, Zhang Y, Zhang L, Li Z, Fang Q, Gao R, et al. Systematic comparative analysis of single‐nucleotide variant detection methods from single‐cell RNA sequencing data. Genome Biol. 2019;20:242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ziegenhain C, Vieth B, Parekh S, Reinius B, Guillaumet‐Adkins A, Smets M, et al. comparative analysis of single‐cell RNA sequencing methods. Mol Cell. 2017;65:631–643.e4. [DOI] [PubMed] [Google Scholar]
- 88.Ma S, Zhang B, LaFave LM, Earl AS, Chiang Z, Hu Y, et al. Chromatin potential identified by shared single‐cell profiling of RNA and chromatin. Cell. 2020;183:1103–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sahraeian SM, Mohiyuddin M, Sebra R, Tilgner H, Afshar PT, Au KF, et al. Gaining comprehensive biological insight into the transcriptome by performing a broad‐spectrum RNA‐seq analysis. Nat Commun. 2017;8:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Piskol R, Ramaswami G, Li JB. Reliable identification of genomic variants from RNA‐Seq data. Am J Hum Genet. 2013;93:641–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fasterius E, Uhlén M, Al‐Khalili Szigyarto C. Single‐cell RNA‐seq variant analysis for exploration of genetic heterogeneity in cancer. Sci Rep. 2019;9:9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Klemm SL, Shipony Z, Greenleaf WJ. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet. 2019;20:207–20. [DOI] [PubMed] [Google Scholar]
- 93.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee C‐K, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome‐wide. Nat Genet. 2004;36:900–5. [DOI] [PubMed] [Google Scholar]
- 95.Poirier MG, Bussiek M, Langowski J, Widom J. Spontaneous access to DNA target sites in folded chromatin fibers. J Mol Biol. 2008;379:772–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ziffra RS, Kim CN, Wilfert A, Turner TN, Haeussler M, Casella AM, et al. Single cell epigenomic atlas of the developing human brain and organoids. bioRxiv. 2020;2019–12. 10.1101/2019.12.30.891549. [DOI] [Google Scholar]
- 97.Boyle AP, Davis S, Shulha HP, Meltzer P, Margulies EH, Weng Z, et al. High‐resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA‐binding proteins and nucleosome position. Nat Methods. 2013;10:1213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mieczkowski J, Cook A, Bowman SK, Mueller B, Alver BH, Kundu S, et al. MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat Commun. 2016;7:11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jin W, Tang Q, Wan M, Cui K, Zhang YI, Ren G, et al. Genome‐wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature. 2015;528:142–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, et al. Single‐cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Woodcock CLF, Safer JP, Stanchfield JE. Structural repeating units in chromatin: I. Evidence for their general occurrence. Exp Cell Res. 1976;97:101–10. [DOI] [PubMed] [Google Scholar]
- 103.Mezger A, Klemm S, Mann I, Brower K, Mir A, Bostick M, et al. High‐throughput chromatin accessibility profiling at single‐cell resolution. Nat Commun. 2018;9:3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, et al. Multiplex single‐cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015;348:910–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Satpathy AT, Granja JM, Yost KE, Qi Y, Meschi F, McDermott GP, et al. Massively parallel single‐cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol. 2019;37:925–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet. 1998;20:116–7. [DOI] [PubMed] [Google Scholar]
- 107.Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–5. [DOI] [PubMed] [Google Scholar]
- 108.Mohandas T, Sparkes RS, Shapiro LJ. Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation. Science. 1981;211:393–6. [DOI] [PubMed] [Google Scholar]
- 109.Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Culp LA, Dore E, Brown GM. Methylated bases in DNA of animal origin. Arch Biochem Biophys. 1970;136:73–9. [DOI] [PubMed] [Google Scholar]
- 111.Bird AP. CpG‐rich islands and the function of DNA methylation. Nature. 1986;321:209–13. [DOI] [PubMed] [Google Scholar]
- 112.Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, et al. DNA‐binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–5. [DOI] [PubMed] [Google Scholar]
- 113.Laird PW. Principles and challenges of genome‐wide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. [DOI] [PubMed] [Google Scholar]
- 114.Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, et al. Comparison of sequencing‐based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 2010;28:1097–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Karemaker ID, Vermeulen M. Single‐cell DNA methylation profiling: technologies and biological applications. Trends Biotechnol. 2018;36:952–65. [DOI] [PubMed] [Google Scholar]
- 116.Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high‐resolution DNA methylation analysis. Nucleic Acids Res. 2005;33:5868–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome‐scale DNA methylation profiling. Nat. Protoc. 2011;6:468–81. [DOI] [PubMed] [Google Scholar]
- 119.Schlesinger F, Smith AD, Gingeras TR, Hannon GJ, Hodges ED. novo DNA demethylation and noncoding transcription define active intergenic regulatory elements. Genome Res. 2013;23:1601–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Guo H, Zhu P, Wu X, Li X, Wen L, Tang F. Single‐cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res. 2013;23:2126–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang K, Li X, Dong S, Liang J, Mao F, Zeng C, et al. Q‐RRBS: a quantitative reduced representation bisulfite sequencing method for single‐cell methylome analyses. Epigenetics. 2015;10:775–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Miura F, Enomoto Y, Dairiki R, Ito T. Amplification‐free whole‐genome bisulfite sequencing by post‐bisulfite adaptor tagging. Nucleic Acids Res. 2012;40:e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, et al. Single‐cell genome‐wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods. 2014;11:817–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Luo C, Keown CL, Kurihara L, Zhou J, He Y, Li J, et al. Single‐cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science. 2017;357:600–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Luo C, Rivkin A, Zhou J, Sandoval JP, Kurihara L, Lucero J, et al. Robust single‐cell DNA methylome profiling with snmC‐seq2. Nat Commun. 2018;9:3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mulqueen RM, Pokholok D, Norberg SJ, Torkenczy KA, Fields AJ, Sun D, et al. Highly scalable generation of DNA methylation profiles in single cells. Nat Biotechnol. 2018;36:428–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kantlehner M, Kirchner R, Hartmann P, Ellwart JW, Alunni‐Fabbroni M, Schumacher A, et al. A high‐throughput DNA methylation analysis of a single cell. Nucleic Acids Res. 2011;39:e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Han L, Wu HJ, Zhu H, Kim KY, Marjani SL, Riester M, et al. Bisulfite‐independent analysis of CpG island methylation enables genome‐scale stratification of single cells. Nucleic Acids Res. 2017;45:e77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Barnett KR, Decato BE, Scott TJ, Hansen TJ, Chen B, Attalla J, et al. ATAC‐Me captures prolonged DNA methylation of dynamic chromatin accessibility loci during cell fate transitions. Mol Cell. 2020;77:1350–1364.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Cao J, Cusanovich DA, Ramani V, Aghamirzaie D, Pliner HA, Hill AJ, et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 2018;361:1380–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhu C, Yu M, Huang H, Juric I, Abnousi A, Hu R, et al. An ultra high‐throughput method for single‐cell joint analysis of open chromatin and transcriptome. Nat Struct Mol Biol. 2019;26:1063–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chen S, Lake BB, Zhang K. High‐throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat Biotechnol. 2019;37:1452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Angermueller C, Clark SJ, Lee HJ, Macaulay IC, Teng MJ, Hu TX, et al. Parallel single‐cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods. 2016;13:229–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pott S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. eLife. 2017;6:e23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Clark SJ, Argelaguet R, Kapourani CA, Stubbs TM, Lee HJ, Alda‐Catalinas C, et al. scNMT‐seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun. 2018;9:781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bakken TE, Jorstad NL, Hu Q, Lake BB, Tian W, Kalmbach BE, et al. Evolution of cellular diversity in primary motor cortex of human, marmoset monkey, and mouse. bioRxiv. 10.1101/2020.03.31.016972. [DOI] [Google Scholar]
- 137.Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, Leite RE, et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled‐up primate brain. J Comp Neurol. 2009;513:532–41. [DOI] [PubMed] [Google Scholar]
- 138.Workman AD, Charvet CJ, Clancy B, Darlington RB, Finlay BL. Modeling transformations of neurodevelopmental sequences across mammalian species. J Neurosci. 2013;33:7368–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.D’Gama AM, Walsh CA. Somatic mosaicism and neurodevelopmental disease. Nat Neurosci. 2018;21:1504–14. [DOI] [PubMed] [Google Scholar]
- 140.Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science. 2018;359:555–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing‐Zitron C, et al. Mosaic copy number variation in human neurons. Science. 2013;342:632–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Evrony GD, Lee E, Park PJ, Walsh CA. Resolving rates of mutation in the brain using single‐neuron genomics. eLife. 2016;5:e12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Huang AY, Li P, Rodin RE, Kim SN, Dou Y, Kenny CJ, et al. Parallel RNA and DNA analysis after deep sequencing (PRDD‐seq) reveals cell type‐specific lineage patterns in human brain. Proc Natl Acad Sci U S A. 2020;117:13886–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, Karger A, et al. Somatic mutation in single human neurons tracks developmental and transcriptional history. Science. 2015;350:94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Blu I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A, et al. The clinico‐pathological spectrum of Focal Cortical Dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia. 2011;52:158–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hua Y, Crino PB. Single cell lineage analysis in human focal cortical dysplasia. Cereb Cortex. 2003;13:693–9. [DOI] [PubMed] [Google Scholar]
- 147.Lee WS, Stephenson SE, Howell KB, Pope K, Gillies G, Wray A, et al. Second‐hit DEPDC5 mutation is limited to dysmorphic neurons in cortical dysplasia type IIA. Ann Clin Transl Neurol. 2019;6:1338–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lamparello P, Baybis M, Pollard J, Hol EM, Eisenstat DD, Aronica E, et al. Developmental lineage of cell types in cortical dysplasia with balloon cells. Brain. 2007;130:2267–76. [DOI] [PubMed] [Google Scholar]
- 149.André VM, Wu N, Yamazaki I, Nguyen ST, Fisher RS, Vinters HV, et al. Cytomegalic interneurons: a new abnormal cell type in severe pediatric cortical dysplasia. J Neuropathol Exp Neurol. 2007;66:491–504. [DOI] [PubMed] [Google Scholar]
- 150.Kebschull JM, Zador AM. Sources of PCR‐induced distortions in high‐throughput sequencing data sets. Nucleic Acids Res. 2015;43:e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dou Y, Kwon M, Rodin RE, Cortés‐Ciriano I, Doan R, Luquette LJ, et al. Accurate detection of mosaic variants in sequencing data without matched controls. Nat Biotechnol. 2020;38:314–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dolezel J, Bartos J, Voglmayr H, Greilhuber J. Nuclear DNA content and genome size of trout and human. Cytom Part J Int Soc Anal Cytol. 2003;51:127–8.Author reply 129. [DOI] [PubMed] [Google Scholar]
- 153.Van Loo P, Voet T. Single cell analysis of cancer genomes. Curr Opin Genet Dev. 2014;24:82–91. [DOI] [PubMed] [Google Scholar]
- 154.Zhang L, Cui X, Schmitt K, Hubert R, Navidi W, Arnheim N. Whole genome amplification from a single cell: implications for genetic analysis. Proc Natl Acad Sci U S A. 1992;89:5847–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Telenius H, Carter NP, Bebb CE, Nordenskjo¨ld M, Ponder BAJ, Tunnacliffe A, et al. Degenerate oligonucleotide‐primed PCR: general amplification of target DNA by a single degenerate primer. Genomics. 1992;13:718–25. [DOI] [PubMed] [Google Scholar]
- 156.Dean FB, Hosono S, Fang L, Wu X, Faruqi AF, Bray‐Ward P, et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U S A. 2002;99:5261–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Rusakova V, Nosek L. Amplification of genome‐representative DNA from limited sources with GenomePlex® WGA technology for use in genetic alterations studies. Nat Methods. 2006;3:i–ii. [Google Scholar]
- 158.Blagodatskikh KA, Kramarov VM, Barsova EV, Garkovenko AV, Shcherbo DS, Shelenkov AA, et al. Improved DOP‐PCR (iDOP‐PCR): A robust and simple WGA method for efficient amplification of low copy number genomic DNA. PLoS One. 2017;12:e0184507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Spits C, Le Caignec C, De Rycke M, Van Haute L, Van Steirteghem A, Liebaers I, et al. Whole‐genome multiple displacement amplification from single cells. Nat Protoc. 2006;1:1965–70. [DOI] [PubMed] [Google Scholar]
- 160.Treff NR, Su J, Tao X, Northrop LE, Scott RT. Single‐cell whole‐genome amplification technique impacts the accuracy of SNP microarray‐based genotyping and copy number analyses. Mol Hum Reprod. 2011;17:335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zong C, Lu S, Chapman AR, Xie XS. Genome‐wide detection of single‐nucleotide and copy‐number variations of a single human cell. Science. 2012;338:1622–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lasken RS. Single‐cell sequencing in its prime. Nat Biotechnol. 2013;31:211–2. [DOI] [PubMed] [Google Scholar]
- 163.Chen C, Xing D, Tan L, Li H, Zhou G, Huang L, et al. Single‐cell whole‐genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science. 2017;356:189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gawad C, Easton J, Gonzalez‐Pena V. Method for nucleic acid amplification. U.S. Patent Application 16/965,796; 2019. [Google Scholar]
- 165.Velazquez‐Villarreal EI, Maheshwari S, Sorenson J, Fiddes IT, Kumar V, Yin Y, et al. Single‐cell sequencing of genomic DNA resolves sub‐clonal heterogeneity in a melanoma cell line. Commun Biol. 2020;3:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Marcy Y, Ishoey T, Lasken RS, Stockwell TB, Walenz BP, Halpern AL, et al. Nanoliter reactors improve multiple displacement amplification of genomes from single cells. PLOS Genet. 2007;3:e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gole J, Gore A, Richards A, Chiu YJ, Fung HL, Bushman D, et al. Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells. Nat Biotechnol. 2013;31:1126–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yu Z, Lu S, Huang Y. Microfluidic whole genome amplification device for single cell sequencing. Anal Chem. 2014;86:9386–90. [DOI] [PubMed] [Google Scholar]
- 169.Vitak SA, Torkenczy KA, Rosenkrantz JL, Fields AJ, Christiansen L, Wong MH, et al. Sequencing thousands of single‐cell genomes with combinatorial indexing. Nat Methods. 2017;14:302–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yin YI, Jiang Y, Lam K‐W, Berletch JB, Disteche CM, Noble WS, et al. High‐throughput single‐cell sequencing with linear amplification. Mol Cell. 2019;76:676–690.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Macaulay IC, Haerty W, Kumar P, Li YI, Hu TX, Teng MJ, et al. G&T‐seq: parallel sequencing of single‐cell genomes and transcriptomes. Nat Methods. 2015;12:519–22. [DOI] [PubMed] [Google Scholar]
- 172.Dey SS, Kester L, Spanjaard B, Bienko M, van Oudenaarden A. Integrated genome and transcriptome sequencing from the same cell. Nat Biotechnol. 2015;33:285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sarnat HB, Flores‐Sarnat L. Infantile tauopathies: hemimegalencephaly; tuberous sclerosis complex; focal cortical dysplasia 2; ganglioglioma. Brain Dev. 2015;37:553–62. [DOI] [PubMed] [Google Scholar]
- 174.Sarnat H, Flores‐Sarnat L, Crino P, Hader W, Bello‐Espinosa L. Original article Hemimegalencephaly: foetal tauopathy with mTOR hyperactivation and neuronal lipidosis. Folia Neuropathol. 2012;4:330–45. [DOI] [PubMed] [Google Scholar]
- 175.Lee VM‐Y, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–59. [DOI] [PubMed] [Google Scholar]
- 176.Scarmeas N, Honig LS, Choi H, Cantero J, Brandt J, Blacker D, et al. Seizures in alzheimer disease. Arch Neurol. 2009;66(8):992–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien‐Ly N, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tai XY, Koepp M, Duncan JS, Fox N, Thompson P, Baxendale S, et al. Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: a study of temporal lobe resections. Brain. 2016;139:2441–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Stefanidou M, Beiser AS, Himali JJ, Peng TJ, Devinsky O, Seshadri S, et al. The bi‐directional association between epilepsy and dementia. The Framingham Heart Study. Neurology. 2020;95:e3241–e3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Boldrini M, Fulmore CA, Tartt AN, Simeon LR, Pavlova I, Poposka V, et al. Human hippocampal neurogenesis persists throughout aging. Cell Stem Cell. 2018;22:589–599.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sorrells SF, Paredes MF, Cebrian‐Silla A, Sandoval K, Qi D, Kelley KW, et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature. 2018;555:377–81. [DOI] [PMC free article] [PubMed] [Google Scholar]