

Abstract
Muscle pathology in inclusion body myositis (IBM) typically includes inflammatory cell infiltration, muscle fibers with rimmed vacuoles and cytochrome c oxidase (COX)‐deficient fibers. Previous studies have revealed clonal expansion of large mitochondrial DNA (mtDNA) deletions in the COX‐deficient muscle fibers. Technical limitations have prevented complete investigations of the mtDNA deletions and other mtDNA variants. Detailed characterization by deep sequencing of mtDNA in muscle samples from 21 IBM patients and 10 age‐matched controls was performed after whole genome sequencing with a mean depth of mtDNA coverage of 46,000x. Multiple large mtDNA deletions and duplications were identified in all IBM and control muscle samples. In general, the IBM muscles demonstrated a larger number of deletions and duplications with a mean heteroplasmy level of 10% (range 1%‐35%) compared to controls (1%, range 0.2%‐3%). There was also a small increase in the number of somatic single nucleotide variants in IBM muscle. More than 200 rearrangements were recurrent in at least two or more IBM muscles while 26 were found in both IBM and control muscles. The deletions and duplications, with a high recurrence rate, were mainly observed in three mtDNA regions, m.534‐4429, m.6330‐13993, and m.8636‐16072, where some were flanked by repetitive sequences. The mtDNA copy number in IBM muscle was reduced to 42% of controls. Immunohistochemical and western blot analyses of IBM muscle revealed combined complex I and complex IV deficiency affecting the COX‐deficient fibers. In conclusion, deep sequencing and quantitation of mtDNA variants revealed that IBM muscles had markedly increased levels of large deletions and duplications, and there were also indications of increased somatic single nucleotide variants and reduced mtDNA copy numbers compared to age‐matched controls. The distribution and type of variants were similar in IBM muscle and controls indicating an accelerated aging process in IBM muscle, possibly associated with chronic inflammation.
Keywords: inclusion body myositis, muscle disease, mitochondrial DNA, mtDNA deletions, mtDNA duplications, mtDNA rearrangements, mtDNA point mutations
Muscle pathology in inclusion body myositis (IBM) typically includes cytochrome c oxidase‐deficient muscle fibers indicating mitochondrial DNA defects. Deep sequencing and quantification of mtDNA variants revealed that IBM muscles had markedly increased levels of large deletions and duplications and reduced mtDNA copy numbers compared to age‐matched controls. The distribution and type of variants were similar in IBM muscle and controls indicating an accelerated aging process in IBM muscle.
1. INTRODUCTION
Inclusion body myositis (IBM), a chronic inflammatory myopathy affecting mainly elderly people, is the most common acquired myopathy in individuals over 50 years of age (1). Clinically, the disease has a slowly progressive course over many years, primarily affecting the thigh and long finger flexor muscles, and dysphagia is common (2, 3, 4, 5). The myopathy leads to severe muscle weakness and many individuals become wheelchair dependent for ambulation. The muscle pathology is characterized by endomysial inflammatory cell infiltration, muscle fibers with rimmed vacuoles and protein aggregates with typical ultrastructural and immunohistochemical characteristics (6). Besides, there are regenerating muscle fibers and mitochondrial alterations with cytochrome c oxidase (COX)‐deficient muscle fibers and eventually the muscle tissue is replaced by connective and fat tissues (7, 8).
The mitochondrial pathology presents as ragged red fibers and frequent muscle fibers with deficiency of COX (8, 9). The mitochondrial alterations are usually prominent in IBM and were described as characteristic findings in 1975 (10), a few years after Yunis and Samaha identified IBM as a separate entity among the idiopathic inflammatory myopathies in 1971 (11). In some patients, the mitochondrial pathology is the predominant histopathological change and IBM may be regarded as a mitochondrial disease (12).
The molecular basis for the mitochondrial respiratory chain deficiency in IBM muscle was first described in 1993 by investigations of mitochondrial DNA (mtDNA) and its transcripts using Southern blot and in situ hybridization analyses (13). Multiple mtDNA deletions are clonally expanded in muscle fibers, concomitant with reduced levels of normal mtDNA in the same fibers. The COX‐deficient muscle fibers correspond to deficiency of COX in segments of the muscle fibers, which range in length from 75 µm to more than 1 mm along the fibers (14). The COX‐deficient segments rarely display invasion of inflammatory cells or rimmed vacuoles (15, 16). Still, an association between the number of inflammatory cells in the muscle and the abundance of respiratory chain deficient fibers has been demonstrated, implying a possible direct link between inflammation and mtDNA damage (17). The respiratory chain‐deficient fibers have also been shown to be more atrophic than other fibers, indicating that the respiratory chain deficiency may lead to muscle fiber atrophy (17).
The clinical importance of the mtDNA alterations is difficult to evaluate on the background of other present changes. However, the number of COX‐deficient fibers is similar or even higher in some IBM patients than in patients with mitochondrial myopathies who suffer from muscle fatigue and weakness due to a primary mtDNA variant such as a single mtDNA deletion.
In addition to the direct effects of somatic mtDNA variants on the respiratory chain synthesis, assembly and function, there are several studies pointing to other mitochondrial defects in IBM such as dysfunctional mitophagy and mitokine dysregulation (for review see (12)).
The collective research results demonstrate that mitochondrial dysfunction caused by somatic mtDNA variants leads to muscle tissue impairment in IBM. Therefore, the mitochondrial changes are not only a biomarker but also part of the pathobiology of IBM. To better understand the pathobiology of mtDNA alterations in IBM, attempts to map deletion breakpoints and identify single nucleotide variants (SNVs) have been made (18). However, the methods did not allow for complete identification of deletion breakpoints and SNVs. In this study we investigated in detail the somatic mtDNA variants (deletions, duplications, and SNVs) and mtDNA copy numbers in muscle samples from IBM patients and age‐matched controls by deep sequencing of mtDNA applying whole genome sequencing (WGS). We found reduced mtDNA copy numbers, higher levels of large mtDNA deletions and duplications, and more frequent SNVs in IBM muscle than in age‐matched controls. Since normal aging is known to be associated with reduced mtDNA copy numbers and accumulation of mtDNA deletions and SNVs, our results indicate an accelerated mitochondrial muscle aging process in IBM.
2. MATERIAL AND METHODS
2.1. Patients and controls
Muscle samples from 21 IBM patients (age range 50‐74 years) with mitochondrial alterations in their muscle biopsies were selected for investigation. Age, gender, disease duration at the time of biopsy and basic histopathological data are summarized in Table 1. The European Neuromuscular Centre (ENMC) criteria for clinico‐pathologically defined IBM were applied (6). Control skeletal muscle included anonymized muscle biopsy specimens from ten individuals (age range 56‐80 years) who had been investigated for a possible muscle disorder, but in whom the clinical and pathological investigations excluded muscle disease (Table 1).
TABLE 1.
IBM and control muscle samples
| Sample | Sex | Age at biopsy (years) | IBM disease duration at biopsy (years) | COX deficiency | Endomysial fibrosis | Inflammation | |
|---|---|---|---|---|---|---|---|
| IBM | 584 | M | 67 | 10 | +++ | +++ | +++ |
| 585 | F | 61 | 1 | + | + | +++ | |
| 586 | F | 67 | 7 | +++ | ++ | ++ | |
| 587 | M | 63 | 1 | +++ | ++ | ++ | |
| 588 | M | 63 | 3 | ++ | ++ | +++ | |
| 589 | M | 65 | 6 | ++ | ++ | ++ | |
| 590 | F | 69 | 11 | + | + | ++ | |
| 591 | F | 72 | 9 | ++ | +++ | +++ | |
| 632 | M | 69 | 8 | +++ | +++ | +++ | |
| 633 | M | 54 | 5 | ++ | +++ | +++ | |
| 634 | M | 75 | 5 | +++ | ++ | +++ | |
| 635 | M | 73 | 2 | +++ | ++ | ++ | |
| 636 | F | 69 | 6 | + | + | + | |
| 637 | M | 50 | 2 | ++ | +++ | +++ | |
| 638 | M | 58 | 2 | ++ | + | + | |
| 639 | M | 65 | 4 | +++ | +++ | +++ | |
| 640 | M | 61 | 4 | +++ | + | +++ | |
| 641 | F | 69 | 3 | +++ | +++ | +++ | |
| 642 | F | 69 | 2 | ++ | ++ | ++ | |
| 643 | F | 59 | 9 | ++ | +++ | +++ | |
| 644 | M | 74 | 3 | + | ++ | ++ | |
| Controls | 592 | F | 61 | a | (−) | − | − |
| 593 | F | 66 | a | − | − | − | |
| 594 | M | 72 | a | (−) | − | − | |
| 595 | M | 72 | a | (−) | − | − | |
| 596 | M | 69 | a | (−) | − | − | |
| 645 | F | 73 | a | (−) | − | − | |
| 646 | M | 56 | a | − | − | − | |
| 647 | F | 60 | a | (−) | − | − | |
| 648 | M | 80 | a | (−) | − | − | |
| 649 | F | 70 | a | − | − | − |
Cytochrome c oxidase (COX) deficient fibers: −, none; (−), occasional; +, few; ++, many; +++, large proportion (>10%).
Endomysial fibrosis: −, none; +, slight; ++, moderate; +++, marked.
Inflammation (endomysial inflammatory cell infiltration): −, none; +, slight; ++, moderate; +++, intense.
Abbreviations: F, female; IBM, Inclusion body myositis; M, male.
No IBM disease.
2.2. Morphological analysis
Open skeletal muscle biopsy was performed in all individuals (Table 1). Specimens were snap‐frozen in isopentane cooled by liquid nitrogen for cryostat sectioning and histochemistry. Standard techniques were applied for enzyme histochemistry (19). For immunohistochemistry (IHC), which was performed in selected patients’ muscle biopsies, sections were fixed in 4% formaldehyde at 4°C for 10 min, washed in TBS‐T for 10 min, permeabilized in a graded methanol series (70% 10 min, 95% 10 min, 100% 20 min, 95% 10 min, and 70% 10 min), washed in TBS‐T for 5 min, and further processed in a Dako Autostainer using the Dako EnVision FLEX High pH kit. The primary antibodies, all from Abcam, were applied for 1 hr: anti‐NDUFB8 (complex I, ab110242, dilution 1:100); anti‐SDHB (complex II, ab14714, dilution 1:500); anti‐MTCO1 (complex IV, ab14705, dilution 1:2000); anti‐VDAC1 (a mitochondrial membrane protein and mitochondrial marker in IHC, ab14734, dilution 1:2000).
2.3. Molecular genetic analyses
Total genomic DNA was isolated from the muscle biopsy specimens using standard protocols. DNA was subjected to WGS using the TruSeq™ PCR free library preparation kit (Illumina, San Diego, CA, USA). Illumina's HiSeq X platform was used for sequencing (Illumina, San Diego, CA, USA).
The paired‐end reads from the WGS were aligned to the reference genome (hg19) using the CLC Biomedical Genomics workbench (Qiagen). Data were analyzed using Ingenuity Variant Analysis (IVA) (www.ingenuity.com/products/variant‐analysis) (Qiagen).
To identify small somatic mtDNA variants, such as SNVs and small deletions or insertions, which potentially could affect respiratory chain activity, variants were searched for in regions encoding tRNAs and polypeptides. A level of at least 1% heteroplasmy was set to exclude sequencing errors and below 20% to not include most of the inherited variants, which are frequently homoplasmic or show a high heteroplasmy level.
To identify variants in nuclear genes, which are associated with multiple mtDNA deletions, we searched for single nucleotide polymorphisms (SNPs), insertions and deletions in the coding regions of 12 such nuclear genes: SPG7, DNA2, RNASEH1, SLC25A4, OPA1, TWNK, POLG, POLG2, TYMP, RRM2B, TK2, and MPV17 (19).
To identify large mtDNA deletions and duplications, alignment to chrM (rCRS assembly, NC_012920.1) was performed (20, 21). After realignment, a mean chrM coverage depth of 46,000x was observed. The mitochondrial DNA coverage was normalized to account for shifts due to deletions and duplications by considering the coverage at positions m.4000‐6000 in the mitochondrial genome, a region with low amounts of deletions/duplications, for all the samples. Gapped alignments, indicative of deletions/duplications, were clustered and visualized as described (20, 21, 22). A deletion or duplication was considered only if it was supported by five sequencing reads with a minimum heteroplasmy level of 0.01%. Deletions were reclassified as likely duplications in case of overlap with replication origins, as previously described (20, 21). Heteroplasmy levels for individual deletions and duplications were estimated by comparing the number of reads supporting the corresponding breakpoints to the total number of reads overlapping the breakpoints (including wild type).
Analysis of mtDNA copy number in relation to nuclear DNA was estimated as previously described: mtDNA copy number = mitochondrial genome coverage x 2/ nuclear genome coverage (23).
2.4. Western blot analysis of respiratory chain subunits
Western blot was performed on protein extracted from fresh‐frozen skeletal muscle biopsy specimens from seven IBM patients and three controls. An enhanced chemiluminescent detection western blotting system (Fujifilm LAS‐4000 system) was used for detection. The primary antibodies, all from Abcam, used in the analyses were: anti‐NDUFB8 (complex I, ab110242, dilution 1:1000), anti‐SDHB (complex II, ab14714, dilution 1:1000), anti‐UQCRC2 (complex III, ab14745, dilution 1:2000), anti‐MTCO1 (complex IV, ab 14705, dilution 1:1000), and anti‐ATPB (complex V, ab14730, dilution 1:2000).
3. RESULTS
3.1. Morphological analysis
All IBM patient biopsies revealed inflammatory myopathy with presence of rimmed vacuoles and mitochondrial changes including numerous COX‐deficient fibers typical for IBM. The age of the patients at biopsy, estimated disease duration, degree of inflammation, endomysial fibrosis, and number of COX deficient fibers are presented in Table 1. COX‐deficient fibers in the IBM muscles varied from approximately 2% to 15 %. Many COX‐deficient fibers were atrophic and serial sections, including immunohistochemical staining of respiratory chain subunits, revealed deficiency of complex I and IV in the COX‐deficient fibers (Figure 1). The control muscle biopsies did not show any inflammatory cell infiltration or fibrosis but occasional COX‐deficient fibers as expected in relation to the age of the controls.
FIGURE 1.
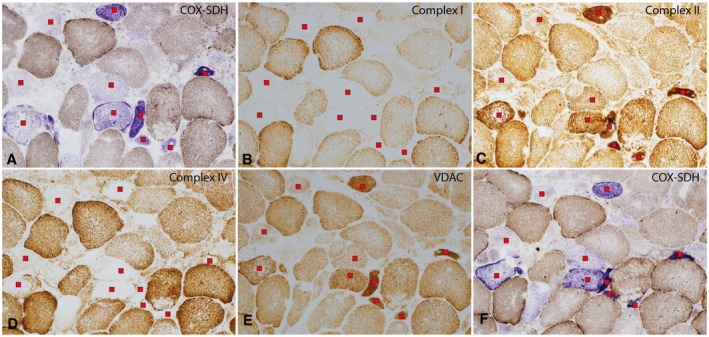
Enzyme‐histochemical (A and F) and immuno‐histochemical (B‐E) analysis of serial sections of an IBM muscle biopsy (sample 588). (A and F) Several muscle fibers with reduced cyotochrome c oxidase (COX) activity (marked with a red square) at two different levels. The fibers appear blue in the combined COX/succinate dehydrogenase (SDH) enzyme histochemical staining. Some of the COX‐deficient fibers show mitochondrial proliferation and several are atrophic. (B) The NDUFB8 subunit in complex I is reduced in the COX‐deficient fibers. (C) The SDHB subunit in complex II shows normal presence in relation to the distribution of mitochondria. (D) The COX1 subunit in complex IV is reduced in the COX‐deficient fibers. (E) VDAC shows the distribution of mitochondria. (A‐E) Red squares indicate COX deficient fibers
3.2. Analysis of mtDNA variants and copy number
By deep sequencing of skeletal muscle mtDNA, with a mean depth of coverage of 46,000x, followed by bioinformatic analysis, detailed characterization of mtDNA in muscle samples from 21 patients with IBM and ten age‐matched controls was performed. Multiple large mtDNA deletions and duplications were identified in all IBM muscles and controls at a stringent clustering threshold of minimum five reads supporting a breakpoint (Figure 2, Figure S1). The IBM patients demonstrated, in general, a higher level of large‐scale rearrangements with a mean level of heteroplasmy of 10% (range 1%‐35%) compared to 1% in controls (range 0.2%‐3%) (Figure 3A,B). There was a clear correlation between the number of different deletions/duplications and the heteroplasmy level (Figure 3C). More than 200 rearrangements were recurrent in at least 2 or more IBM patients while 26 were found in both patients and controls and only six were unique to and recurrent in only controls (Figure 4). The deletions/duplications with a high recurrence rate were mainly observed in three regions, m.534‐4429, m.6330‐13993, and m.8636‐16072, where some were flanked by repetitive sequences.
FIGURE 2.
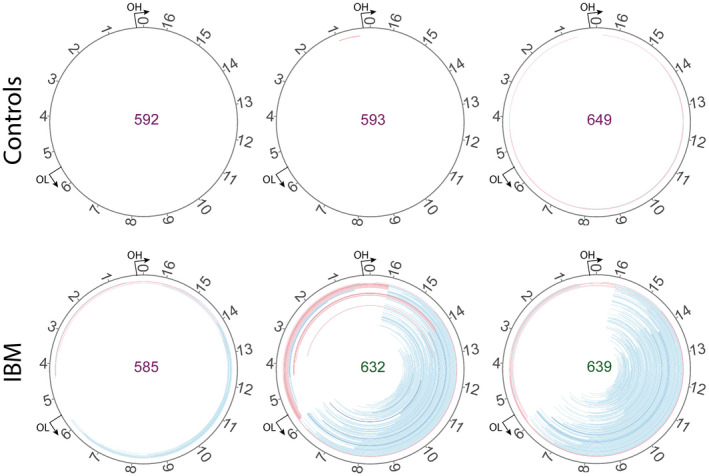
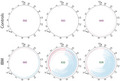
Circular map of the deletions (blue) and duplications (red) identified by the pipeline in three IBM and three control samples (all 31 samples are illustrated in Figure S1). The color intensity is related to the abundance of each rearrangement. A heteroplasmy cut‐off of 0.05% is used for visualization. The deletions are localized mainly to the major arc of mtDNA between the origins of heavy (OH) and light (OL) chain replication
FIGURE 3.
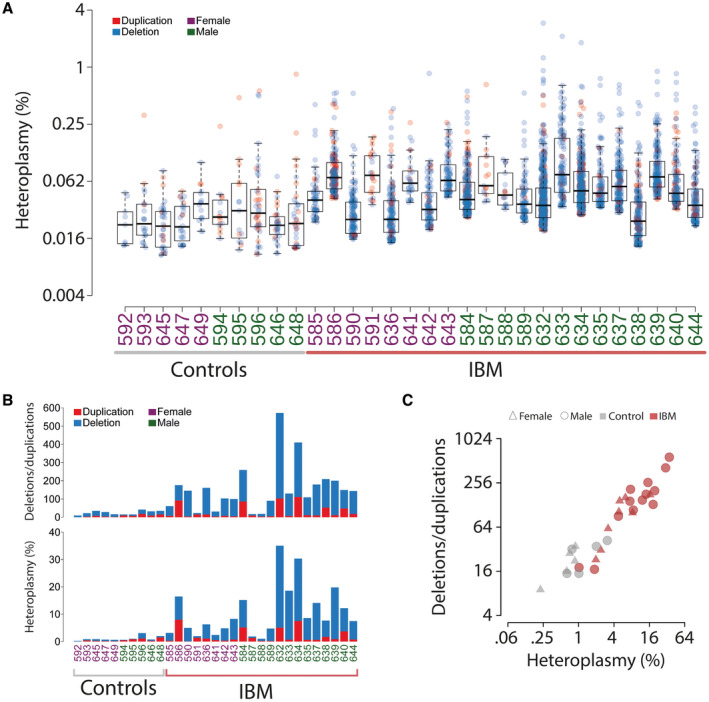
Overview of structural mtDNA alterations in muscle samples from IBM patients and age‐matched controls without muscle disease. (A) Sample‐wise distribution of heteroplasmy of the identified deletions and duplications. The color intensity is related to the abundance of each rearrangement. (B) The number of deletions and duplications in each sample and their respective contribution to sum % heteroplasmy. (C) The IBM muscle samples show a higher amount of deletions and duplications in the mitochondrial genome than the controls and the number of structural alterations correlates with the level of heteroplasmy
FIGURE 4.
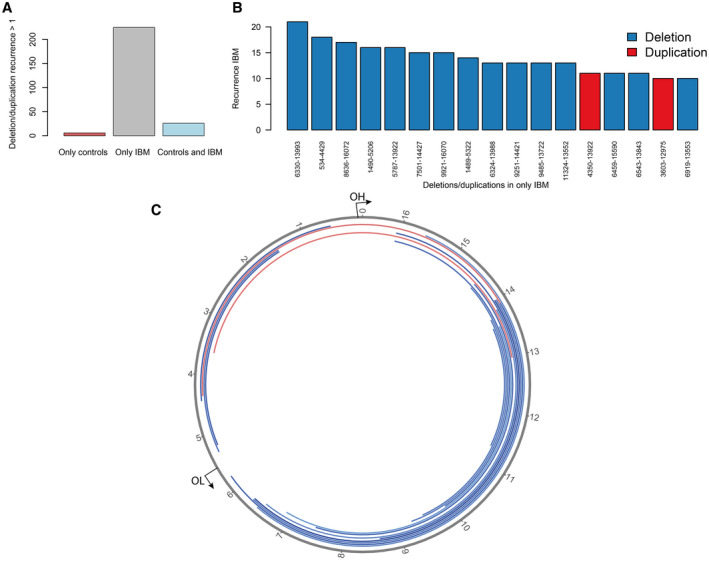
Recurrent mtDNA rearrangements. (A) The number of recurrent deletions and duplications in IBM and control muscles. (B) The most frequently recurrent deletions and duplications among IBM samples. One specific deletion was identified in all 21 IBM muscle samples. (C) Circular map of the most frequently recurrent deletions (blue) and duplications (red) illustrated in (B). The origins of heavy (OH) and light (OL) strand replication are indicated
Analysis of small somatic mtDNA variants such as SNVs and small insertions or deletions in coding regions showed that 10 of the 21 IBM muscles had altogether 16 such variants (Table 2). Two of the 10 control muscles were identified with one such variant each, indicating a slight increase in the occurrence of such somatic variants in IBM muscle.
TABLE 2.
Identified variants in tRNA and protein coding genes in mtDNA at a heteroplasmy level between 1 and 20% in muscle samples from IBM patients and controls
| Gene symbol | Transcript variant | Protein variant | Heteroplasmy level (% (sample)) | |
|---|---|---|---|---|
| IBM (n = 21) | Controls (n = 10) | |||
| MT‐TL1 | m.3243A > T | 5.05 (637) | ||
| MT‐ND2 | m.4553T > C | p.F28F | 1.06 (596) | |
| MT‐TW | m.5522G > A | 1.31 (649) | ||
| MT‐CO1 | m.6698delA | p.E266fs*6 | 1.11 (584) | |
| 1.19 (586) | ||||
| 1.08 (591) | ||||
| MT‐CO2 | m.7741T > C | p.N52N | 5.53 (643) | |
| MT‐ATP6; MT‐ATP8 | m.8533G > A | p.T56T; p.E3K | 5.06 (586) | |
| MT‐ATP6 | m.8764G > A | p.A80T | 4.24 (635) | |
| MT‐ND4 | m.11954A > T | p.N399Y | 1.01 (632) | |
| m.11957A > C | p.M400L | 1.08 (632) | ||
| m.11959_11960insa | p.L401_V402insb | 1.14 (632) | ||
| m.11964T > G | p.V402G | 1.88 (632) | ||
| 1.31 (633) | ||||
| MT‐CYB | m.15148G > A | p.P134P | 5.30 (638) | |
| m.15553G > A | p.K269K | 3.81 (642) | ||
| m.15607A > G | p.K287K | 1.89 (641) | ||
| m.15734G > A | p.A330T | 1.85 (635) | ||
Abbreviations: IBM, inclusion body myositis; n, number of investigated muscle samples.
TTATTCGGCGCATGAGCTGGAGTC.
FGAWAGVL.
Estimations of mtDNA copy number revealed a reduced amount of mtDNA in the IBM muscles with a mean of 1660 mtDNA copies per 1 nuclear DNA copy (range 809‐3535) compared to controls (mean 3936; range 2115‐6196) (Figure 5). No clear correlation between mtDNA copy number and levels of heteroplasmy was observed.
FIGURE 5.
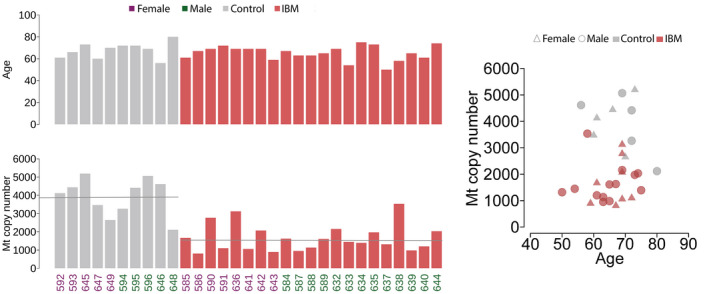
Age (years) of the patients and controls and estimated mtDNA (Mt) copy number relative to the nuclear DNA of each sample. The IBM patient samples show reduced mtDNA copy number on average. The mean level is indicated
3.3. Analysis of nuclear genes associated with multiple mtDNA deletions
In order to investigate the occurrence of nuclear gene variants that could possibly contribute to a mild dysfunction of the replication machinery of mtDNA we investigated 12 genes, which are known to be associated with multiple mtDNA deletions in muscle. By WGS the presence of variants in exons or splice sites were identified in these genes. Very few, mostly synonymous variants and/or common SNVs, were identified and there were no differences between the patients and controls (Table S1). The controls harbored on average 6.4 variants (2.3 missense) and the IBM patients harbored on average 6.3 variants (2.2 missense) in coding regions in these 12 genes. Very high heteroplasmy levels of mtDNA deletions and duplications (>30%) were detected in 2 IBM muscles (sample 632 and 634). However, no likely pathogenic variants were identified in any of the 12 genes associated with multiple mtDNA deletions in these two cases (Table S1).
3.4. Western blot analysis of respiratory chain subunits
To investigate the overall effect of the mtDNA alterations, we examined the respiratory chain subunits at the protein level in muscle homogenate in seven IBM muscles and three controls. Subunits of complex I, III, IV, and V in the respiratory chain are partly encoded by mtDNA, whereas complex II subunits are all encoded by nuclear DNA. In IBM muscle a consistent reduction in complex I and III was observed, whereas the reduction of complex IV was more variable (Figure 6). The expression of complex II was also variable but did not show any reduction in IBM muscle. For complex V no apparent difference between the patients and controls was observed.
FIGURE 6.
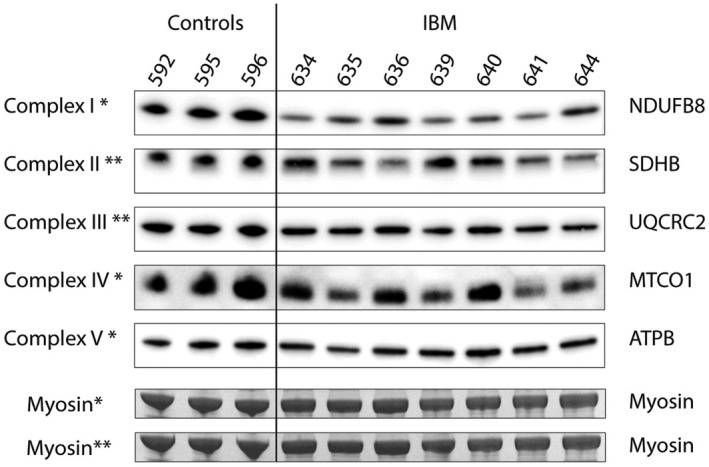
Western blot analyses using antibodies to subunits of the respiratory complexes I to V. There is a consistent decrease of NDUFB8 (Complex I) protein in the muscle samples from seven IBM patients compared to controls. Complex II is increased in some IBM samples, whereas complex III and IV are variably reduced, and complex V appears similar to controls. The analysis was performed in two different gels. Results from complex I, IV, and V analyzed in one gel are indicated with *, and complex II and III analyzed in the other gel are indicated with **. The bands corresponding to myosin heavy chain in the lower part of the panel were used as loading control for the corresponding gels
4. DISCUSSION
The muscle pathology in IBM is characterized by inflammatory cell infiltration and autophagic vacuoles in addition to frequent COX deficient muscle fibers and mitochondrial proliferation (8). The mitochondrial pathology in IBM is similar to what is found in several inherited mitochondrial myopathies caused by primary or secondary mtDNA variants.
Several previous studies have described multiple large‐scale mtDNA deletions in IBM muscle, but in this study, we have characterized all the breakpoints in detail and quantified the mtDNA rearrangements for the first time. We also, for the first time, demonstrate the presence of mtDNA duplications in IBM muscle and identified SNVs and small deletions or insertions. Our results show that there is an increase of all these mtDNA alterations in IBM muscle compared to age‐matched controls. We also found a mild decrease of mtDNA copy number in agreement with previous studies (24).
A group of age‐related inherited muscle diseases where the primary genetic defect resides in the nuclear DNA is characterized by somatic multiple different mtDNA deletions (25). This is caused by defects in proteins involved in mtDNA maintenance and results in COX deficiency in muscle fiber segments (19, 26). One of the nuclear genes in this group is POLG that encodes mtDNA polymerase gamma A (PolγA) (27) and defective PolγA induces mtDNA deletions during mtDNA replication (21). Defective PolγA may also cause mtDNA duplications and, to a minor extent, mtDNA SNVs (22, 28). Detailed mapping of the mtDNA rearrangements in POLG‐associated diseases using WGS has only been performed in a limited number of cases, but these results indicate that the distribution of deletions and duplications within the mitochondrial genome are to some extent overlapping with those in IBM muscle as described in this study (21, 22). Patients with multiple mtDNA deletions in muscle, due to mutations in POLG, differ in the clinical expression from IBM patients in that external ophthalmoplegia, which is very frequent in such PolγA patients, is not a feature of IBM (see also below). Unlike IBM, patients with PolγA‐associated disease frequently show a multiorgan disorder with encephalopathy since PolγA is expressed in all tissues. In addition, the typical distribution of muscle weakness seen in IBM patients is not present in PolγA‐associated myopathy.
It has been speculated that in some IBM patients, pathogenic missense variants in POLG and other nuclear genes involved in mtDNA maintenance could contribute to the development of a high number of mtDNA deletions. An earlier study identified POLG variants in some IBM patients, but no conclusions regarding their contribution to the mtDNA deletions could be drawn (9). In the present study, we investigated 12 nuclear genes known to be associated with multiple mtDNA deletion disorders. However, the results did not indicate that there were any variants in these genes, contributing to high levels of mtDNA deletions in IBM. In most hereditary diseases associated with multiple mtDNA deletions, extraocular muscles are involved, causing progressive external ophthalmoplegia and ptosis. Such extraocular muscle involvement is not seen in IBM, supporting the concept that genetic variants in mtDNA maintenance proteins are not engaged in the multiple mtDNA deletions seen in IBM.
The deficiency of respiratory chain enzymes in IBM involves both complex IV (COX) and complex I, but not complex II (as shown in Figure 1), indicating defects in mtDNA (17). The western blot analysis of the respiratory chain subunits in muscle homogenate in the present study indicated that complex I is the most vulnerable complex in the respiratory chain in IBM, as also found in inherited mitochondrial diseases with multiple mtDNA deletions. The results show that the deficiency observed at the single cell level by immunohistochemistry can also be seen in muscle homogenate by western blot analysis, indicating an overall perturbed function of the oxidative phosphorylation in IBM muscle.
Aging is associated with a reduction of mtDNA copy number and accumulation of somatic mtDNA rearrangements and SNVs in various tissues including muscle (23, 26, 29, 30). In the present study, the mean heteroplasmy level, that is, the amount of deletions and duplications in relation to normal mtDNA in IBM muscle, was 10% and ranged from 1% to 35%. This may be compared to the age‐matched controls without muscle disease, where the level of heteroplasmy was 1% (range 0.2%‐3%). However, the distribution of deletions and duplications showed overlap between the IBM muscle and controls, indicating that the somatic mtDNA rearrangements in IBM muscle may be regarded as an acceleration of the normal aging process. Besides, reduced mtDNA copy number and increased SNVs may be part of such an accelerated muscle mitochondrial aging.
Age‐related progressive decline in muscle function and muscle mass is referred to as sarcopenia. The cause of sarcopenia is manifold but chronic inflammation as part of aging, so called “inflamm‐aging” or “senoinflammation” (31, 32, 33), is considered an essential factor leading to sarcopenia (34, 35). Mitochondrial dysfunction is another hallmark of sarcopenia (26, 36), and it has been proposed that dysfunctional mitochondria and damaged mtDNA are parts of the pathogenesis of sarcopenia (37). Proximal muscle weakness associated with COX deficiency and multiple mtDNA deletions in the elderly has been described as a disease entity, “Late‐onset mitochondrial myopathy,” and considered a type of accelerated aging (38). In these patients, the mean number of COX‐deficient fibers was 1.6%, which is less than what is usually found in inclusion body myositis (9, 17). In another disease entity named “Polymyositis with cytochrome c oxidase negative muscle fibres” (39), the patients had a high percentage (range 4%‐27%) of COX‐deficient fibers and were on average 9 years older, at the onset of disease, than polymyositis patients lacking COX‐deficient fibers. These findings indicate that aging in combination with inflammation, which is also a hallmark of IBM, may drive the generation of multiple mtDNA deletions and duplications.
Several facts thus point to an association between chronic inflammation and multiple mtDNA deletions and duplications and reduced mtDNA copy number, especially in aging. It may be hypothesized that there is a link between chronic inflammation and dysfunction of the machinery involved in mtDNA maintenance, resulting in the accumulation of mtDNA with various rearrangements, especially in postmitotic tissues such as muscle. It is unclear what the mechanisms may be, but a link between inflammation and mtDNA depletion has been proposed in a murine model of inflammation‐induced colon tumorigenesis (40). The suggested mechanism resulting in reduced mtDNA copy number was inflammation‐induced DNA methylation with secondary silencing of PolγA expression (40).
In conclusion, we have performed a detailed mtDNA characterization regarding somatic deletions, duplications, SNVs and also mtDNA copy number in IBM muscle and compared it with normal aging. Our results indicate a similar pattern of mtDNA defects but much more pronounced in IBM than in normal aging muscle. There are also similarities with the pattern of mtDNA alterations seen in PolγA‐associated disease with multiple mtDNA deletions. These findings point to a defective mtDNA replication machinery in IBM muscle leading to an accelerated aging process, which may be related to the chronic inflammation that is a hallmark of IBM and also a part of normal aging.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICAL STATEMENT
The study was approved by the Regional Ethics Committee at the University of Gothenburg, Sweden and conducted according to the Declaration of Helsinki of 1975.
AUTHOR CONTRIBUTIONS
Study conception: Carola Hedberg‐Oldfors, Erik Larsson Lekholm, Maria Falkenberg, Anders Oldfors; Acquisition of data: Swaraj Basu, Carola Hedberg‐Oldfors, Ulrika Lindgren, Christopher Lindberg, Ulrika Lindgren, Kittichate Visuttijai, Anders Oldfors; Analysis and interpretation of data: Swaraj Basu, Carola Hedberg‐Oldfors, Erik Larsson Lekholm, Maria Falkenberg, Anders Oldfors; Drafting of manuscript: Swaraj Basu, Carola Hedberg‐Oldfors, Ulrika Lindgren, Kittichate Visuttijai, Anders Oldfors; Critical revision: Erik Larsson Lekholm, Maria Falkenberg, Anders Oldfors.
Supporting information
ACKNOWLEDGMENTS
The authors acknowledge the Clinical Genomics Stockholm facility at Science for Life Laboratory for providing assistance in next generation sequencing.
Funding information
This work was supported by grants from the Swedish Research Council (No 2018‐02821 to AO) and the ALF agreement (ALFGBG‐716821 to AO and ALFGBG‐727491 to MF) and the Western Sweden Muscle Foundation (to AO and UL)
DATA AVAILABILITY STATEMENT
The data are available upon request.
REFERENCES
- 1.Tawil R, Griggs RC. Inclusion body myositis. Curr Opin Rheumatol. 2002;14(6):653–7. [DOI] [PubMed] [Google Scholar]
- 2.Benveniste O, Guiguet M, Freebody J, Dubourg O, Squier W, Maisonobe T, et al. Long‐term observational study of sporadic inclusion body myositis. Brain. 2011;134(Pt 11):3176–84. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg SA. Inclusion body myositis: clinical features and pathogenesis. Nat Rev Rheumatol. 2019;15(5):257–72. [DOI] [PubMed] [Google Scholar]
- 4.Lindberg C, Persson L, Björkander J, Oldfors A. Inclusion body myositis ‐ clinical, morphological, physiological and laboratory findings in 18 cases. Acta Neurol Scand. 1994;89(2):123–31. [DOI] [PubMed] [Google Scholar]
- 5.Mastaglia FL, Needham M. Inclusion body myositis: a review of clinical and genetic aspects, diagnostic criteria and therapeutic approaches. J Clin Neurosci. 2015;22(1):6–13. [DOI] [PubMed] [Google Scholar]
- 6.Rose MR. 188th ENMC International Workshop: Inclusion Body Myositis, 2–4 December 2011, Naarden, The Netherlands. Neuromuscul Disord. 2013;23(12):1044–55. [DOI] [PubMed] [Google Scholar]
- 7.Dahlbom K, Geijer M, Oldfors A, Lindberg C. Association between muscle strength, histopathology, and magnetic resonance imaging in sporadic inclusion body myositis. Acta Neurol Scand. 2019;139(2):177–82. [DOI] [PubMed] [Google Scholar]
- 8.Dahlbom K, Lindberg C, Oldfors A. Inclusion body myositis: morphological clues to correct diagnosis. Neuromuscul Disord. 2002;12(9):853–7. [DOI] [PubMed] [Google Scholar]
- 9.Lindgren U, Roos S, Hedberg Oldfors C, Moslemi AR, Lindberg C, Oldfors A. Mitochondrial pathology in inclusion body myositis. Neuromuscul Disord. 2015;25(4):281–8. [DOI] [PubMed] [Google Scholar]
- 10.Carpenter S, Karpati G, Eisen A. A morphologic study in polymyositis: clues to pathogenesis of different types. In: Bradley W, editor. Recent advances in myology. Amsterdam: Excerpta Medica; 1975. p. 374–9. [Google Scholar]
- 11.Yunis EJ, Samaha FJ. Inclusion body myositis. Lab Invest. 1971;25(3):240–8. [PubMed] [Google Scholar]
- 12.De Paepe B. Sporadic inclusion body myositis: an acquired mitochondrial disease with extras. Biomolecules. 2019;9(1):15. 10.3390/biom9010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oldfors A, Larsson NG, Lindberg C, Holme E. Mitochondrial DNA deletions in inclusion body myositis. Brain. 1993;116(2):325–36. [DOI] [PubMed] [Google Scholar]
- 14.Oldfors A, Moslemi AR, Jonasson L, Ohlsson M, Kollberg G, Lindberg C. Mitochondrial abnormalities in inclusion‐body myositis. Neurology. 2006;66:S49–55. [DOI] [PubMed] [Google Scholar]
- 15.Horvath R, Fu K, Johns T, Genge A, Karpati G, Shoubridge EA. Characterization of the mitochondrial DNA abnormalities in the skeletal muscle of patients with inclusion body myositis. J Neuropathol Exp Neurol. 1998;57(5):396–403. [DOI] [PubMed] [Google Scholar]
- 16.Molnar M, Schroder JM. Pleomorphic mitochondrial and different filamentous inclusions in inflammatory myopathies associated with mtDNA deletions. Acta Neuropathol. 1998;96(1):41–51. [DOI] [PubMed] [Google Scholar]
- 17.Rygiel KA, Miller J, Grady JP, Rocha MC, Taylor RW, Turnbull DM. Mitochondrial and inflammatory changes in sporadic inclusion body myositis. Neuropathol Appl Neurobiol. 2015;41(3):288–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moslemi AR, Lindberg C, Oldfors A. Analysis of multiple mitochondrial DNA deletions in inclusion body myositis. Hum Mutat. 1997;10(5):381–6. [DOI] [PubMed] [Google Scholar]
- 19.Dubowitz V, Sewry C, Oldfors A. Muscle biopsy. A practical approach, 5th ed. Amsterdam: Elsevier; 2021. ISBN: 978‐0‐7020‐7471‐4. [Google Scholar]
- 20.Basu S, Xie X, Uhler JP, Hedberg‐Oldfors C, Milenkovic D, Baris O, et al. Accurate mapping of mitochondrial DNA deletions and duplications using deep sequencing. PLOS Genet. 2020;16(12):e1009242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Persson O, Muthukumar Y, Basu S, Jenninger L, Uhler JP, Berglund AK, et al. Copy‐choice recombination during mitochondrial L‐strand synthesis causes DNA deletions. Nat Commun. 2019;10(1):759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hedberg‐Oldfors C, Macao B, Basu S, Lindberg C, Peter B, Erdinc D, et al. Deep sequencing of mitochondrial DNA and characterization of a novel POLG mutation in a patient with arPEO. Neurol Genet. 2020;6(1):e391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding J, Sidore C, Butler TJ, Wing MK, Qian Y, Meirelles O, et al. Assessing mitochondrial DNA variation and copy number in lymphocytes of ~2,000 Sardinians using tailored sequencing analysis tools. PLoS Genet. 2015;11(7):e1005306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Catalan‐Garcia M, Garrabou G, Moren C, Guitart‐Mampel M, Hernando A, Diaz‐Ramos A, et al. Mitochondrial DNA disturbances and deregulated expression of oxidative phosphorylation and mitochondrial fusion proteins in sporadic inclusion body myositis. Clin Sci. 2016;130(19):1741–51. [DOI] [PubMed] [Google Scholar]
- 25.Young MJ, Copeland WC. Human mitochondrial DNA replication machinery and disease. Curr Opin Genet Dev. 2016;38:52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeBalsi KL, Hoff KE, Copeland WC. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age‐related diseases. Ageing Res Rev. 2017;33:89–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rahman S, Copeland WC. POLG‐related disorders and their neurological manifestations. Nat Rev Neurol. 2019;15(1):40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kollberg G, Jansson M, Perez‐Bercoff A, Melberg A, Lindberg C, Holme E, et al. Low frequency of mtDNA point mutations in patients with PEO associated with POLG1 mutations. Eur J Hum Genet. 2005;13(4):463–9. [DOI] [PubMed] [Google Scholar]
- 29.Fayet G, Jansson M, Sternberg D, Moslemi AR, Blondy P, Lombes A, et al. Ageing muscle: clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul Disord. 2002;12(5):484–93. [DOI] [PubMed] [Google Scholar]
- 30.Herbst A, Widjaja K, Nguy B, Lushaj EB, Moore TM, Hevener AL, et al. Digital PCR quantitation of muscle mitochondrial DNA: age, fiber type, and mutation‐induced changes. J Gerontol A Biol Sci Med Sci. 2017;72(10):1327–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung HY, Kim DH, Lee EK, Chung KW, Chung S, Lee B, et al. Redefining chronic inflammation in aging and age‐related diseases: proposal of the senoinflammation concept. Aging Dis. 2019;10(2):367–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fulop T, Larbi A, Witkowski JM. Human Inflammaging. Gerontology. 2019;65(5):495–504. [DOI] [PubMed] [Google Scholar]
- 33.Piber D, Olmstead R, Cho JH, Witarama T, Perez C, Dietz N, et al. Inflammaging: Age and systemic, cellular, and nuclear inflammatory biology in older adults. J Gerontol A Biol Sci Med Sci. 2019;74(11):1716–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta S, Dhillon RJS, Hasni S. Sarcopenia: a rheumatic disease? Rheum Dis Clin North Am. 2018;44(3):393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Riuzzi F, Sorci G, Arcuri C, Giambanco I, Bellezza I, Minelli A, et al. Cellular and molecular mechanisms of sarcopenia: the S100B perspective. J Cachexia Sarcopenia Muscle. 2018;9(7):1255–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Picca A, Calvani R, Bossola M, Allocca E, Menghi A, Pesce V, et al. Update on mitochondria and muscle aging: all wrong roads lead to sarcopenia. Biol Chem. 2018;399(5):421–36. [DOI] [PubMed] [Google Scholar]
- 37.Pinti M, Cevenini E, Nasi M, De Biasi S, Salvioli S, Monti D, et al. Circulating mitochondrial DNA increases with age and is a familiar trait: Implications for “inflamm‐aging”. Eur J Immunol. 2014;44(5):1552–62. [DOI] [PubMed] [Google Scholar]
- 38.Johnston W, Karpati G, Carpenter S, Arnold D, Shoubridge EA. Late‐onset mitochondrial myopathy. Ann Neurol. 1995;37(1):16–23. [DOI] [PubMed] [Google Scholar]
- 39.Blume G, Pestronk A, Frank B, Johns DR. Polymyositis with cytochrome oxidase negative muscle fibres. Early quadriceps weakness and poor response to immunosuppressive therapy. Brain. 1997;120(Pt 1):39–45. [DOI] [PubMed] [Google Scholar]
- 40.Maiuri AR, Li H, Stein BD, Tennessen JM, O'Hagan HM. Inflammation‐induced DNA methylation of DNA polymerase gamma alters the metabolic profile of colon tumors. Cancer Metab. 2018;6:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data are available upon request.