

Abstract
TMEM106B has been recently implicated in multiple neurodegenerative diseases. Here, Rademakers et al. report a late‐onset cerebellar Purkinje cell loss and progressive decline in motor function and gait deficits in a conventional Tmem106b−/− mouse model. By using high‐power microscopy and bulk RNA sequencing, the authors further identify lysosomal and immune dysfunction as potential underlying mechanisms of the Purkinje cell loss.
![]()
Dear Sir,
With great interest, we read the work of Stroobants et al., published online October 5th 2020 in Brain Pathology, in which the authors reported late‐onset cerebellar Purkinje cell loss and progressive decline in motor function and gait deficits in their CRISPR Tmem106b−/− mouse model (1). Here, we report similar age‐dependent Purkinje cell death and motor deficits in a conventional Tmem106b−/− mouse model. By high‐power microscopy and bulk RNA sequencing, we further identify lysosomal and immune dysfunction as potential underlying mechanisms of the Purkinje cell loss.
TMEM106B was first identified as a genetic risk factor for a specific pathological subtype of frontotemporal lobar degeneration (FTLD) characterized by abnormal inclusions of TAR DNA‐binding protein 43 (TDP‐43) (FTLD‐TDP) (2). Subsequent studies established TMEM106B as a major modifier of disease risk, with the strongest association in FLTD patients with mutations in the progranulin gene (FTLD‐GRN) (3, 4). Since then, TMEM106B has been further implicated in other neurodegenerative diseases such as Alzheimer’s disease (AD) (5) and hypomyelinating leukodystrophy (6) as well as in normal aging (7). However, the precise function of TMEM106B remains unknown.
In previous work, we showed that the loss of Tmem106b leads to myelination deficits, likely due to developmental deficits (8). To further characterize the function of Tmem106b in vivo, we performed a thorough histological analysis of the brains from wild‐type (WT) and Tmem106b‐deficient (Tmem106b−/−) mice. Notably, hematoxylin and eosin stains revealed increased gaps between Purkinje cells in Tmem106b−/− mice as compared with WT mice at 15 months of age suggesting the loss of Purkinje cells in the Tmem106b−/− cerebellum (Figure 1A). In contrast, no obvious difference in the thickness of the molecular and granular cell layers of the cerebellum was observed between WT and Tmem106b−/− mice (Figure 1A). The increased gap between Purkinje cells in Tmem106b−/− mouse cerebellum was further validated by the immunofluorescence staining of the Purkinje cell markers calbindin and inositol 1,4,5‐trisphosphate receptor (IP3R) (Figure 1A). In line with these findings, quantification in these 15 month‐old animals revealed a nearly 50% decrease of Purkinje cell number in the Tmem106b−/− mice (p < 0.0001) when compared to WT mice (Figure 1D). With aging (23 months), we found that the number of surviving Purkinje cells was even further reduced in Tmem106b−/− as compared to WT mice (p < 0.0001) (Figure 1E). We next examined Purkinje cell numbers in younger mice; however, no significant difference in terms of Purkinje cell number was observed between Tmem106b−/− and WT mice at 3 and 8 months of age (Figure 1B, C). These findings suggest that the loss of the Purkinje cells observed at an older age is unlikely to be a consequence of developmental deficits. Importantly, regardless of the age of the animals, Tmem106b+/− mice showed no signs of Purkinje cell loss (Tmem106b+/− vs WT, p > 0.05 at all ages) (Figure 1B–E). Notably, in the study from Stroobants et al. (1), prominent loss of Purkinje cells was demonstrated at 18 months of age whereas 9‐months was described as an age preceding prominent neuronal loss. While it is difficult to compare the results from independent studies, the timing of the age‐dependent Purkinje cell loss observed in our Tmem106b−/− mice appears potentially similar to this model. This consistency between models is interesting given that our mice were previously shown to have limited residual expression of Tmem106b. This residual expression was sufficient to significantly delay motor deficits and death in double knock‐out Tmem106b−/−; Grn−/− mice; however, does not appear to result in an obvious rescue or delay of the Purkinje cell loss phenotypes reported here (9). Together these data suggest that the loss of Tmem106b leads to age‐dependent Purkinje cell death.
FIGURE 1.

Purkinje cell loss and motor deficits in Tmem106b−/− mice. (A) Representative images of Hematoxylin and eosin (H&E) stain and Calbindin and IP3R immunostaining in cerebellums of 15‐month‐old Tmem106b+/+ (WT) and Tmem106b−/− (KO) mouse brains. (B–E) Quantification of Purkinje cells in the cerebellums of WT, Tmem106b+/− (Het), and KO mouse brains at indicated ages (n = 4–17 per group). Graphs represent the mean ± SD. Data were analyzed by One‐way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. NS, not significant, ****p < 0.0001. (F) Representative images of Gfap and Iba‐1 immunostaining in cerebellums of 15‐month‐old Tmem106b WT and KO mouse brains. (G–H) Quantification of Gfap (G) and Iba‐1(H) immunointensities in the cerebellums of WT and KO mouse brains in F (n = 3–4 per group). Graphs represent the mean ± SD. Data were analyzed by Student’s t‐test. *p < 0.05, **p < 0.01. (I) Dowel test for Tmem106b WT, Het, and KO mice (n = 14–16 per group). Graphs represent the mean ± SD. Data were analyzed by One‐way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. NS, not significant, **p < 0.01
Previously, we also showed that the loss of Tmem106b leads to mild microglia and astroglia activation across different brain regions at 11–12 months of age (9). To study the glial activation in aged Tmem106b−/− mice, we examined the cerebellums of 15‐month‐old mice by immunohistochemistry (IHC) staining. This analysis showed that the loss of Tmem106b results in an approximately 2‐fold increase of immunoreactivity of Gfap (p < 0.01) and Iba‐1 (p < 0.05) in the cerebellum as compared to WT mice (Figure 1F–H). Similar to the observation from Stroobants et al. (1), gliosis was pronounced in the molecular layer and white matter tract. We also observed that there were significantly more lysosomal enlarged Purkinje neurons and microglia cells in Tmem106b−/− as compared to WT mice as evidenced by Lamp1 immunofluorescence staining (Figure S1). The enlarged lysosomes in Purkinje cells were mainly located in the axon hillock (Figure S1A). Instead of single enlarged lysosomes as observed in Stroobants et al. (1), we often observed multiple clustered enlarged lysosomes. It is possible that, with time, these clustered enlarged lysosomes might fuse to form single enlarged lysosomes. Regardless, these results demonstrated the involvement of gliosis and further suggest lysosomal dysfunction contributes to cerebellar Purkinje cell loss in Tmem106b‐deficient mice.
Since cerebellar Purkinje cells are known to play an important role in controlling balance, we next investigated motor function in aged Tmem106b−/−, Tmem106b+/−, and WT mice (15 months of age) using a dowel test. In line with the Purkinje cell numbers (Figure 1D), the latency of time on the dowel rod was significantly reduced in Tmem106b−/− mice as compared to WT mice (p < 0.01), whereas no difference between Tmem106b+/− and WT mice was observed (p > 0.05) (Figure 1I). These findings were in line with impaired treadmill performance and gait defects reported in the study from Stroobants et al. (1) and together, they suggest that the loss of Purkinje cell in the cerebellum of Tmem106b−/− mice is sufficient to induce motor deficits.
To better understand the molecular mechanism underlying Tmem106b deficiency‐induced Purkinje cell loss, we performed bulk RNA sequencing on mouse cerebellar samples. We included three Tmem106b+/+ (WT) and three Tmem106b−/− mice, all at 15 months of age. After gene normalization and filtering (log2 RPKM > 1), we identified a total of 14,250 genes expressed in both genotypes for inclusion in the differential gene expression study. We identified 1149 differentially expressed genes (DEGs) at the nominal significance level (572 down‐regulated and 577 up‐regulated; p‐value < 0.05) comparing Tmem106b−/− to WT mice (Figure 2A, Table S1). Hierarchical cluster analysis of the top 100 most up‐regulated and down‐regulated DEGs showed a consistent gene expression pattern within each genotype (Figure 2B). Finally, we validated several of the most down (Tnc, Ebf2, Alpk2, and Cck)‐ and up (Serpina3n, Fcrls, Lyz2, and Lpl)‐ regulated DEGs among top fold changed genes by quantitative PCR (qPCR) in 15‐month‐old mouse cerebellums, which confirmed the RNAseq data and further revealed that the expression changes were specific to Tmem106b−/− mice, since no significant difference in gene expression was observed between Tmem106b+/− and WT mice, with the exception of one gene (Figure 2C–J). To further understand the functional relevance of the identified DEGs, we performed a pathway analysis. Immune and inflammation‐related cellular processes including complement system, phagosome formation, and triggering receptor expressed on myeloid cells‐1 (TREM1) signaling were identified as significantly enriched pathways among the observed DEGs (Figure 2K), with key genes being upregulated in Tmem106b−/− as compared to WT cerebellar tissues. To validate the expression of the complement system, we performed immunofluorescence staining for C3b, a cleavage product of C3. The immunofluorescence staining showed increased C3b expression in microglia but not astroglia and neurons, suggesting that up‐regulated complement pathway is mainly due to changes in microglia (Figure S2). Notably, loss of progranulin has also been shown to cause complement system activation (10). Next, we sought to identify highly correlated gene clusters/modules that were either up‐ or down‐regulated in a genotype‐specific manner through a weighted gene co‐expression network analysis (WGCNA). Through this analysis, we identified 20 modules of which two were significantly correlated with Tmem106b genotype: one up‐regulated and one down‐regulated module (Figure S3A). Enrichment analyses revealed that the significantly upregulated module (yellow module) was enriched for immune and inflammatory‐related processes [defense response FDR = 1.73E‐04; inflammatory response FDR = 4.84E‐04 (Figure S3B)] further confirming the consequence of Tmem106b loss on the immune response and inflammation. The downregulated module (brown) was enriched for neuronal components including neuron part (FDR = 3.40E‐08), neuron projection (FDR = 4.2E‐08), and synapse part (FDR = 1.43E‐07) (Figure S3C) likely reflecting the neuronal loss we observed in Tmem106b−/− cerebellums. Interestingly, Purkinje cell‐enriched components including dendritic tree (FDR = 1.47E‐06) and dendrite (FDR = 1.23E‐06) (Figure S3C) were also enriched in the downregulated module further supporting that the neuronal loss is likely due to Purkinje cell loss. Taken together, these results are in line with Purkinje cell loss in Tmem106b−/− cerebellum and further implicate inflammation in Tmem106b deficiency‐induced Purkinje cell loss.
FIGURE 2.
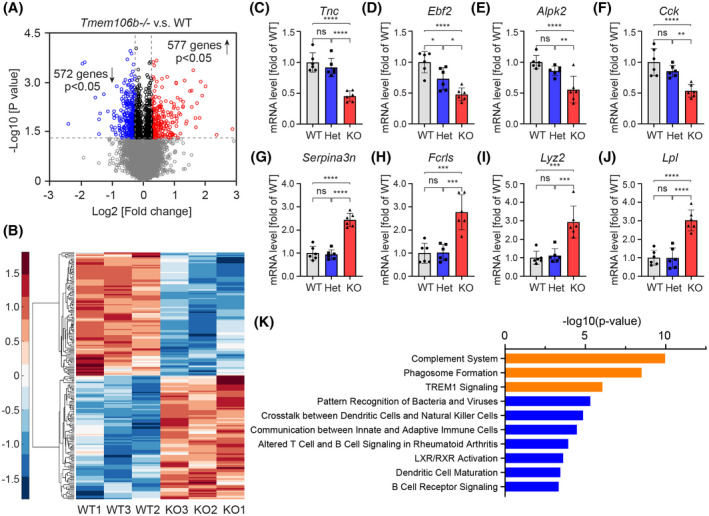
Transcriptomic analysis of cerebellar tissue of Tmem106b−/− and WT mice. (A) Volcano plots show differentially expressed genes (DEGs) between Tmem106b−/− and WT mice using nominal significant p‐value < 0.05. Red and blue colors represent up‐regulated and down‐regulated genes in Tmem106b−/− mice, respectively. (B) Hierarchical clustering of top 100 up‐regulated and down‐regulated DEGs between Tmem106b−/− and WT mice. Red and blue colors represent upregulated and downregulated genes in Tmem106b−/− mice, respectively. (C–J) q‐PCR validation of selected down‐ (D–G) and up‐ (H–K) regulated DEGs using 15‐month‐old cerebellar tissue from WT, Tmem106b+/− (Het) and Tmem106b−/− (KO) mice. Graphs represent the mean ± SD. Data were analyzed by One‐way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test (n = 6 per group). NS, no significant, **p < 0.01, ***p < 0.001, ****p < 0.0001. (K) Top 10 cellular processes identified by the ingenuity pathway analysis from the DEGs between Tmem106b−/− and WT mice. The top three most significant cellular processes are highlighted in orange
In summary, we reported here that the deficiency of Tmem106b results in an age‐dependent loss of cerebellar Purkinje cells accompanied by motor function deficits. We also demonstrated that Tmem106b deficiency results in lysosomal enlargement in both Purkinje cells and microglia, and increased neuro‐inflammation including complement system activation. Together with the independent findings from Stroobants et al. (1), these data suggest that, in addition to myelination (8), Tmem106b also plays important role in maintaining the health and survival of cerebellar Purkinje cells during aging.
CONFLICT OF INTEREST
All authors declare that they have no conflicts of interest.
AUTHOR CONTRIBUTIONS
X. Zhou, R. Rademakers, and A. Nicholson designed and performed experiments and analyzed the data. Y. Ren, S. Koga, H. Phuoc, M. Brooks, W. Qiao, Z. Quicksall, B. Matchett, R. Perkerson, A. Kurti, M. Castanedes‐Casey, V. Phillips, A. Librero, C. Fernandez De Castro, M. Baker, S. Roemer, and M. Murray performed experiments and analyzed the data. Y. Asmann, J. Fryer, G. Bu, and D. Dickson designed experiments and analyzed the data. X. Zhou and R. Rademakers wrote the manuscript and supervised the work; all other authors edited the manuscript.
Supporting information
Table S1
TABLE S1 Differentially expressed genes (DEGs) between Tmem106b−/− and Tmem106b+/+ mouse cerebellums with individual animal log2RPKM
Supplementary Material
ACKNOWLEDGMENTS
RR received funding from the National Institutes of Health (NIH) (grant R35 NS097261) and the Bluefield Project to Cure FTD. XZ is supported by a research fellowship from The Bluefield Project to Cure FTD and a Developmental grant from the Mayo Clinic ADRC (NIH P30 AG062677).
Contributor Information
Rosa Rademakers, Email: rosa.rademakers@uantwerpen.vib.be.
Xiaolai Zhou, Email: zhouxiaolai@gzzoc.com.
DATA AVAILABILITY STATEMENT
The authors confirm that the data supporting the findings of this study are available within the article and/or its Supporting Information. Raw RNAseq data can be accessed at Gene expression Omnibus (GEO) repository (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE165105).
REFERENCES
- 1.Stroobants S, D'Hooge R, Damme M. Aged Tmem106b knockout mice display gait deficits in coincidence with Purkinje cell loss and only limited signs of non‐motor dysfunction. Brain Pathol. 2020. 10.1111/bpa.12903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van Deerlin VM, Sleiman PM, Martinez‐Lage M, Chen‐Plotkin A, Wang LS, Graff‐Radford NR, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP‐43 inclusions. Nat Genet. 2010;42(3):234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus‐Hernandez M, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76(5):467–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pottier C, Zhou X, Perkerson RB 3rd, Baker M, Jenkins GD, Serie DJ, et al. Potential genetic modifiers of disease risk and age at onset in patients with frontotemporal lobar degeneration and GRN mutations: a genome‐wide association study. Lancet Neurol. 2018;17(6):548–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murray ME, Cannon A, Graff‐Radford NR, Liesinger AM, Rutherford NJ, Ross OA, et al. Differential clinicopathologic and genetic features of late‐onset amnestic dementias. Acta Neuropathol. 2014;128(3):411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simons C, Dyment D, Bent SJ, Crawford J, D'Hooghe M, Kohlschutter A, et al. A recurrent de novo mutation in TMEM106B causes hypomyelinating leukodystrophy. Brain. 2017;140(12):3105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhinn H, Abeliovich A. Differential aging analysis in human cerebral cortex identifies variants in TMEM106B and GRN that regulate aging phenotypes. Cell Syst. 2017;4(4):404–15.e5. [DOI] [PubMed] [Google Scholar]
- 8.Zhou X, Nicholson AM, Ren Y, Brooks M, Jiang P, Zuberi A, et al. Loss of TMEM106B leads to myelination deficits: implications for frontotemporal dementia treatment strategies. Brain. 2020;143(6):1905–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou X, Brooks M, Jiang P, Koga S, Zuberi AR, Baker MC, et al. Loss of Tmem106b exacerbates FTLD pathologies and causes motor deficits in progranulin‐deficient mice. EMBO Rep. 2020;21(10):e50197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, et al. Progranulin deficiency promotes circuit‐specific synaptic pruning by microglia via complement activation. Cell. 2016;165(4):921–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
TABLE S1 Differentially expressed genes (DEGs) between Tmem106b−/− and Tmem106b+/+ mouse cerebellums with individual animal log2RPKM
Supplementary Material
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and/or its Supporting Information. Raw RNAseq data can be accessed at Gene expression Omnibus (GEO) repository (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE165105).