

ABSTRACT
Sporadic late‐onset nemaline myopathy (SLONM) is a rare adult‐onset non‐hereditary disease with subacute proximal muscle and often axial muscle weakness, characterized by the presence of nemaline bodies in skeletal muscle biopsies. Considering its association with concurrent monoclonal gammopathy of undetermined significance (MGUS), the disease is classified into two major subtypes (1) SLONM without MGUS (SLONM‐noMGUS) and (2) with MGUS (SLONM‐MGUS) association. SLONM associated with HIV infection (SLONM‐HIV) is also reported. SLONM‐MGUS has been shown to be associated with poorer prognosis and required aggressive treatment including high‐dose melphalan and autologous stem cell transplantation. The approach is currently debatable as recent reports suggested effectiveness of intravenous immunoglobulin as initial treatment with indifference of overall survival despite the presence of MGUS. Our study aimed to find an underlying basis by review of pathological features in 49 muscle biopsy proven‐SLONM from two large tertiary centers in Japan and Germany (n = 49: SLONM‐noMGUS = 34, SLONM‐MGUS = 13, SLONM‐HIV = 2). We compared pathological findings in SLONM‐noMGUS and SLONM‐MGUS and focused on the presence of any detectable inflammatory features by immunohistochemistry. The clinical and histological features in SLONM‐noMGUS and SLONM‐MGUS were not distinctively different except for more common regenerating fibers (>5% of myofibers) present in SLONM‐MGUS (p < 0.01). HLA‐ABC expression and fine granular p62 were observed in 66.7% and 78.3% of SLONM, respectively. The predominant inflammatory cells were CD68+ cells. The inflammatory cells showed positive correlations with the percentage of nemaline‐containing fibers (p < 0.001). In conclusion, inflammatory features are present although rather mild in SLONM. This finding contributes to the hypothesis of an acquired inflammatory disease pathogenesis and opens the possibility to offer immunotherapy in SLONM with inflammatory features regardless of the monoclonal gammopathy status.
Keywords: monoclonal gammopathy, muscle biopsy, nemaline myopathy, SLONM, sporadic
Retroactive myopathological study of 49 SLONM highlights common immunopathological elements among SLONM‐MGUS and SLONM‐noMGUS. CD68+ cells were the most common inflammatory cells observed in SLONM.
![]()
1. INTRODUCTION
Sporadic late‐onset nemaline myopathy (SLONM) is a rare, subacute adult‐onset non‐hereditary myopathy pathologically characterized by the presence of nemaline bodies in muscle fibers (1). Since the first two reports in 1966 (2, 3), to our best knowledge, less than 200 cases have been reported in the literature. The common clinical presentations in SLONM include bilateral proximal muscle weakness and muscle atrophy with normal or slightly elevated levels of creatine kinase (CK) (4). Presence of axial muscle weakness, bulbar muscle weakness, and distal muscle weakness are typical but less frequent (4). Respiratory symptoms have been associated with unfavorable outcome (5, 6). Presence of nemaline bodies in a substantial number of myofibers compatible with the diagnosis of SLONM are also reported to be present in HIV‐associated myopathy (SLONM‐HIV) (7, 8, 9, 10, 11, 12, 13, 14, 15), one case with concurrent HIV and HTLV‐2 infection (16), and rarely in association with other diseases that may be associated with dysimmune conditions including but not limited to vitiligo (2), Hodgkin disease (17), hypothyroidism (18, 19) systemic lupus erythematosus (20, 21), Sjogren syndrome (22), myasthenia gravis (23, 24), amyloid myopathy,(25) multiple myeloma (26), and POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes) syndrome (Crow–Fukase syndrome) (27). Concurrent monoclonal gammopathy of undetermined significance (MGUS) is present in approximately half of the reported cases of SLONM (4, 5, 6, 25, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48). Of note, three cases of SLONM associated with biclonal gammopathy are reported (6, 49). SLONM patients without MGUS (SLONM‐noMGUS) were heterogenous but reported to be associated with favorable response after immunotherapy (e.g., corticosteroids, mycophenolate, azathioprine, cyclophosphamide, plasma exchange, and intravenous immunoglobulin [IVIg]) with or without the combination of chemotherapy (e.g., lenalidomide and thalidomide) (1, 4, 5). Moreover, SLONM patients with concurrent MGUS (SLONM‐MGUS) were reported to have variable response to immunotherapy and chemotherapy (1, 4, 5, 6, 25, 28, 29, 30, 31, 32, 33, 34, 36, 38, 40, 41, 43, 44, 46, 47, 48) and were associated with a more aggressive clinical course mainly caused by respiratory involvement (1, 5, 6). Notably, long‐term follow‐up in two SLONM‐noMGUS patients showed clinical improvement or stable disease without any treatment (5, 6) while one patient died because of heart disease at the age of 85 years (5). Untreated SLONM‐MGUS patients had grim prognosis (5). Although the pathomechanism and role of MGUS in SLONM are unclear, the hematologic treatment regimen including high‐dose melphalan (HDM) followed by autologous stem cell transplantation (ASCT) has been applied to SLONM‐MGUS (33, 35). The reason for this approach was its efficacy in plasma cell dyscrasias associated with MGUS and other severe neuromuscular disorders (i.e., POEMS syndrome and amyloid light chain amyloidosis) (33, 35). The SLONM‐MGUS patients treated with HDM‐ASCT or other regimen that include ASCT generally achieved positive response even in long‐term follow‐up (4, 6, 33, 34, 35, 37, 40, 42, 45, 47). Because SLONM‐MGUS is regarded as a rapidly progressive condition, the HDM‐ASCT regimen has been recommended to be used early in the disease course while the patient is still be able to tolerate such aggressive regimen. However, the recent monocentric study by Naddaf et al. challenges this recommendation by showing the effectiveness of IVIg‐based immunotherapy as the first line treatment in SLONM‐MGUS (6); the findings are supported by several similar observations (25, 36, 41, 44, 48). Interestingly, Naddaf et al. also demonstrated indifference of overall survival rates in SLONM with and without MGUS (6). These recent observations prompted us to re‐investigate the pathological features in SLONM with and without MGUS for possible explanations.
In this study, we performed an exhaustive myopathological retrospective study of muscle biopsies of patients diagnosed with SLONM. We aimed to describe the pathological features of SLONM with and without MGUS and focused on inflammatory feature(s) that may differ with respect to the treatment options of the two conditions.
2. MATERIALS AND METHODS
2.1. Standard protocol approvals, registrations, and patient consent
We included all muscle biopsy from patients pathologically diagnosed as having SLONM at the Charité—Universitätsmedizin Berlin, Germany and the National Center of Neurology and Psychiatry (NCNP), Japan during January 2011–December 2019. Informed consent was obtained from all patients at each institution involved. The official ethical standards committee at the Charité ‐ Universitätsmedizin Berlin (EA2/163/17) and the institutional review board of NCNP approved the study.
2.2. Clinical information
We collected relevant clinical information from pathology requisition forms and associated letters from the attending physicians (Table 1). Respiratory weakness was determined by the presence of dyspnea, decreased vital capacity, or required respiratory support. Cardiomyopathy was determined by the presence of abnormal electrocardiogram or decreased ejection fraction (<55%).
TABLE 1.
Clinical features of patients in this study
| SLONM (total = 49) | SLONM‐noMGUS (n = 34) | SLONM‐MGUS (n = 13) | SLONM‐HIV (n = 2) | |
|---|---|---|---|---|
| Age of onset (M ± SD, years) | 60.4 ± 11.9a | 61.8 ± 10.8b | 57.4 ± 14.8c | 53c |
| Age at biopsy (M ± SD, years) | 63.1 ± 11.7 | 64.6 ± 10.3 | 61.3 ± 14.8 | 51 ± 2.8 |
| Sex, female | 32 [65.3] | 25 [73.5] | 7 [53.8] | 0 |
| CK (M ± SD, U/L) | 304.3 ± 615.8d | 322.6 ± 702.7a | 227.4 ± 326.2b | 600c |
| Lower than normal | 6 [13.0]e | 4 [12.1]c | 2 [18.2]b | 0 |
| Normal | 22 [47.8]e | 15 [45.5]c | 6 [54.5]b | 1 [50.0] |
| >1‐ to ≤5‐fold | 17 [37.0]e | 13 [39.4]c | 3 [27.3]b | 1 [50.0] |
| >5‐fold | 1 [2.2]e | 1 [3.0]c | 0b | 0 |
| κ‐light chain MGUS | 6 [50]c | 0 | 6 [50]c | 0 |
| λ‐light chain MGUS | 6 [50]c | 0 | 6 [50]c | 0 |
| Signs and symptoms | ||||
| Myalgia | 20 [40.8] | 15 [44.1] | 4 [30.8] | 1 [50.0] |
| Weight loss | 13 [26.5] | 7 [20.6] | 4 [30.8] | 2 [100.0] |
| Camptocormia/scoliosis | 3 [6.1] | 2 [5.9] | 1 [7.7] | 0 |
| Proximal muscle weakness | 43 [87.8] | 28 [82.4] | 13 [100.0] | 2 [100.0] |
| Distal muscle weakness | 17 [34.7] | 9 [26.5] | 6 [46.2] | 2 [100.0] |
| Neck muscle weakness | 32 [65.3] | 23 [67.6] | 9 [69.2] | 0 |
| Winged scapula | 8 [16.3] | 6 [17.6] | 2 [15.4] | 0 |
| Facial muscle weakness | 9 [18.4] | 5 [14.7] | 3 [23.1] | 1 [50.0] |
| Bulbar muscle weakness | 17 [34.7] | 12 [35.3] | 4 [30.8] | 1 [50.0] |
| Respiratory weaknessf | 20 [40.8] | 13 [38.2] | 7 [53.8] | 0 |
| Required respiratory support | 8 [16.3] | 6 [17.6] | 2 [15.4] | 0 |
| Cardiomyopathyg | 10 [20.4] | 8 [23.5] | 1 [7.7] | 1 [50.0] |
| Treatment(s) before biopsy | 11 [22.4] | 9 [26.5] | 2 [15.4] | 0 |
Abbreviations: CK, creatine kinase; HIV, human immunodeficiency virus; MGUS, monoclonal gammopathy of undetermined significance; NA, not available; SLONM, sporadic late‐onset nemaline myopathy; SLONM‐HIV, SLONM associated with HIV; SLONM‐MGUS, SLONM with MGUS association; SLONM‐noMGUS, SLONM without MGUS association.
Continuous data are shown as mean (M) ± standard deviation (SD) while categorical data are shown as number and [percentage].
Information not available in four cases.
Information not available in two cases.
Information not available in one case.
Information not available in seven cases.
Information not available in three cases.
Determined by dyspnea, decreased vital capacity, or required respiratory support.
Determined by abnormal electrocardiogram or decreased ejection fraction.
2.3. Histochemical and immunohistochemical evaluation
We performed the following routine histochemical and immunohistochemical (IHC) staining methods; hematoxylin and eosin (H&E), modified Gomori trichrome (mGT), nicotinamide adenine dinucleotide tetrazolium reductase (NADH‐TR), acid phosphatase (ACP), alkaline phosphatase (ALP), serialized ATPases, class I human major histocompatibility complex (MHC) (HLA‐ABC; clone W6/32, Thermo Fisher Scientific), MHC class II (HLA‐DR; clone B308, Affinity BioReagents), membrane attack complex (MAC; C5b‐9, clone aE11, Dako), neonatal myosin heavy chain (MHCn; clone WB‐MHCn, Leica), CD3 (polyclonal, Abcam), CD8 (clone DK25, Dako), CD20 (clone L26, eBioscience), CD68 (clone KP1, Dako), p62 (SQSTM1 D‐3, Santa Cruz Biotechnology and rabbit polyclonal 91526, Abcam), and MxA (Mx 1/2/3, Santa Cruz Biotechnology). ISG15 (ab14374, Abcam) and Siglec1 (HSn7D2, Novus Biological) were additionally performed in 11 cases. LAMP‐2 (clone 5H2, Santa Cruz Biotechnology), HSP‐70 (ab6535, Abcam), BAG3 (ab47124, Abcam), LC3 (0260‐100/LC3‐2G6, nanoTools Art), and alpha‐B‐crystallin (CRYAB) (ab13496, Abcam) were additionally performed in nine cases. JT, AU, IN, and WS performed the pathological evaluation.
We used mGT stain to identify nemaline bodies characterized as deep red or greenish red thread‐or rod‐like structures (Figure 1A,C,D) or as sand‐like small granules (Figure 1B). H&E stains were routinely performed in all cases (Figure 1E–H). We manually counted and calculated the percentage of muscle fibers‐containing nemaline bodies in 10 random areas of mGT stain at 400x magnification (high power fields, HPF; 0.575 mm2). The quantitative evaluations were performed on the entire tissue section/available material if the specimen was not sufficient for 10 HPFs evaluation. We categorized muscle fibers‐containing nemaline bodies into round type (Figure 1B,C), angular type (Figure 1A), and cap type (Figure 1D). We qualitatively classified muscle biopsies based on the predominant type (> 50%) of nemaline bodies and nemaline‐containing fibers. We categorized and documented intermyofibrillary network changes in nemaline‐containing fibers using NADH‐TR as follows: dark atrophic fibers (Figure 1I), non‐atrophic fibers with dark areas (Figure 1J), moth‐eaten fibers (Figure 1K), and lobulated fibers (Figure 1L). If present, intranuclear rods (not shown), cytoplasmic vacuolization (Figure 1A,B), ACP‐positive fibers, nemaline‐containing fibers highlighted by ACP (Figure 1M–P), increased ALP activity in perimysium, and type 1 fiber predominance were documented. We counted and calculated regenerating fibers in 10 random HPFs using sarcoplasmic expression of MHCn as a surrogate marker. We recorded the number of CD3, CD8, CD20, and CD68‐positive cells in 10 random HPFs. We determined the positivity of HLA‐ABC and HLA‐DR in non‐necrotic and non‐regenerating fibers by sarcolemmal and sarcoplasmic expression, respectively. The degree of positivity was qualitatively categorized by the number of positive myofibers in the section: score 0 negative, score 1: <25%, score 2: 25%–75%, and score 3: >75%. We determined MAC expression by the presence of granular deposition on sarcolemma or capillaries. Expression of p62 was considered as positive either by punctate/dot‐like or small granular sarcoplasmic expression patterns. The degree of involvement by vacuolar fibers, ACP‐positive fibers, MAC, MxA, ISG15, Siglec1, p62, LAMP‐2, HSP‐70, BAG3, LC3, and CRYAB was qualitatively categorized: score 0 negative, score 1 positive on single fiber/capillary (focally), score 2 positive on many fibers/capillaries (focally), and score 3 positive on many fibers/capillaries (diffusely).
FIGURE 1.
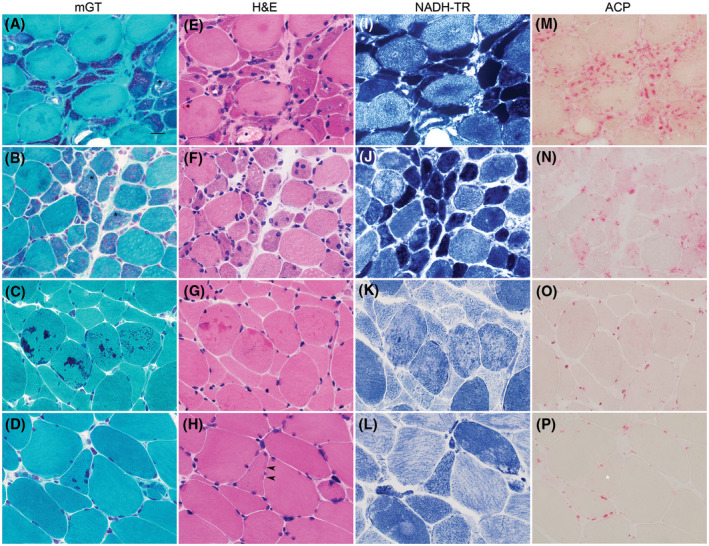
Histological findings in SLONM. Nemaline bodies are best demonstrated by mGT (A–D) as deep red or greenish red thread‐or rod‐like structures (A, C, D) or as sand‐like small granules (B) in myofibers. The regions within myofibers containing nemaline bodies can be categorized into 3 major types: angular type (A), round type (B and C), and cap type (D). Vacuolated fibers are occasionally observed (A and B, black asterisks). In H&E (E–H), parts of the myofibers containing nemaline bodies stain darker than the adjacent areas. The change is subtle with small number of nemaline bodies (D and H, black arrowheads). Intermyofibrillar changes in nemaline containing myofibers demonstrated by NADH‐TR include dark atrophic fibers (I), dark areas, non‐atrophic fibers (J), moth‐eaten fibers (K) and lobulated fibers (L). ACP often highlights areas within fibers containing nemaline bodies (M‐O). ACP staining can be subtle or negative in fiber with small numbers of nemaline bodies (P, white asterisk). SLONM: sporadic late‐onset nemaline myopathy. A‐P 60x magnification, bar = 20 microns; A‐D: mGT, modified Gomori Trichrome; E‐H: H&E, hematoxylin and eosin; I‐L: NADH‐TR, nicotinamide adenine dinucleotide dehydrogenase‐tetrazolium reductase; M‐P: ACP, acid phosphatase)
2.4. Statistics
We conducted statistical analyses using Excel for Mac version 16.4.2 and GraphPad Prism version 9.0.0 for macOS (GraphPad Software, San Diego, California USA, www.graphpad.com). We performed the independent two‐tailed t test for the continuous variables and the two‐tailed Fisher exact test for the categorical variables. Friedman test followed by Dunn's multiple comparisons was used to compare paired groups. Spearman correlation was calculated for the association between continuous variables and between continuous and categorical variables. The p value of <0.05 was considered statistically significant.
3. RESULTS
We identified 49 muscle biopsies previously diagnosed as having SLONM including 34 SLONM‐noMGUS, 13 SLONM‐MGUS, and 2 SLONM‐HIV. We compared the clinical and histological features of SLONM‐noMGUS and SLONM‐MGUS. Because the number of SLONM‐HIV was limited in this study, we did not include them into the comparison.
3.1. Clinical features
The relevant clinical features of SLONM are summarized in Table1. The age of onset in SLONM ranged from 34 to 80 years old (60.4 ± 11.9 years old). Thirty‐six patients (36/44, 81.8%) had the disease onset during their 5th–7th decades (Figure 2). SLONM was more common in women (32/49, 65.3%). The CK level ranged from 16 to 3870 U/L (304.3 ± 615.8 U/L). Thirty‐nine patients (39/46, 84.8%) were associated with normal to mildly elevated CK level (≤5 times of normal value). One patient with markedly elevated CK level (3870 U/L, 23.5 times of normal value) had vital capacity decreased to 72% and ejection fraction to 25%. The three most common signs and symptoms in this study were proximal muscle weakness (43/49, 87.8%), neck muscle weakness (32/49, 65.3%), and respiratory weakness (20/49, 40.8%). Respiratory supports including non‐invasive and tracheostomy invasive ventilation were documented in eight patients (8/49, 16.3%). Ten patients (10/49, 20.4%) had cardiac abnormalities detected by abnormal electrocardiogram or echocardiogram. Eleven patients (11/49, 22.4%) had corticosteroid treatment before muscle biopsy; two patients had a combined regimen of corticosteroid with IVIg and corticosteroid with IVIg and chemotherapy. The percentages of kappa and lambda‐light chain proteinemia present in SLONM‐MGUS were not different (6/12, 50%); the information was not available in one SLONM‐MGUS patient. The clinical features of SLONM‐noMGUS and SLONM‐MGUS patients were not distinctively different (p > 0.05).
FIGURE 2.
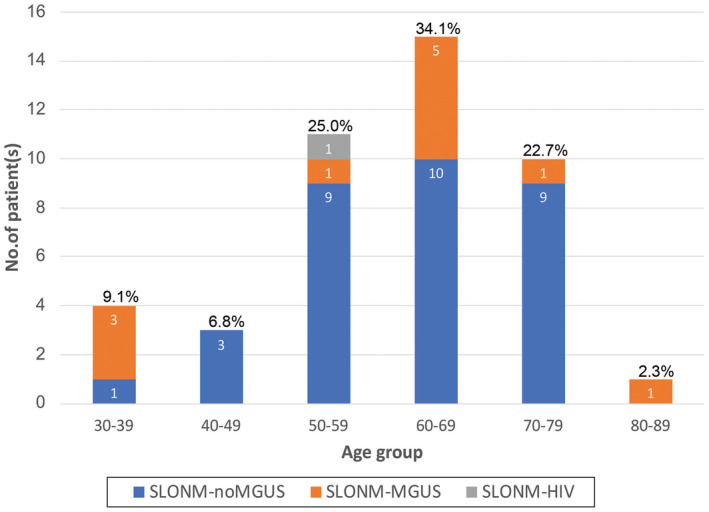
Distribution of the age of onset in SLONM. The majority of patients (81.8%) have the age of onset during their 5th and 7th decades
3.2. Pathological features
The histological features are summarized in Table 2. The most common biopsy site in this study was the biceps brachii (26/43, 60.5%). The percentages of myofibers‐containing nemaline bodies varied from 1.1% to 93.7%. Twenty‐three of SLONM (23/49, 46.9%) had nemaline‐containing fibers less than or equal to 10% of myofibers in a biopsy. Notably, 15 muscle biopsies (15/49, 30.6%) had nemaline‐containing myofibers less than or equal to 5% (Figure 3). Muscle biopsies were mainly composed of round‐type nemaline fibers (23/49, 46.9%) and rod‐shaped nemaline bodies (27/49, 55.1%). Seven biopsies contained intranuclear rods (7/49, 14.3%). Twenty‐five biopsies showed sarcoplasmic vacuolization (25/49, 51.0%); most of which was observed as single fiber, focally (12/49, 24.5%). Intermyofibrillary sarcoplasmic alterations detected by NADH‐TR were observed in most cases (48/49, 98.0%); the most frequent change were dark atrophic fibers (30/49, 61.2%). There was no ALP activity in perimysial connective tissue in any biopsy. ACP‐positive fibers were observed in most cases (48/49, 98.0%); mainly present as focal distribution of many positive fibers (25/49, 51.0%). Presence of ACP positivity in nemaline‐containing fibers was observed in 40 biopsies (40/49, 81.6%). The results of the Spearman correlation indicated a strong association between ACP score and the percentage of nemaline‐containing fibers (r = 0.69, p = 3.6e‐8) (Table 3). There was predominance of type 1 fibers in 12 biopsies (12/49, 25%). The histological features of SLONM‐noMGUS and SLONM‐MGUS muscle biopsy were not distinctively different (p > 0.05).
TABLE 2.
Biopsy site and Pathological features of SLONM patients
| SLONM (total = 49) | SLONM‐noMGUS (n = 34) | SLONM‐MGUS (n = 13) | SLONM‐HIV (n = 2) | |
|---|---|---|---|---|
| Biopsy site | ||||
| Upper extremity, proximal muscle | ||||
| Biceps | 26 [60.5]a | 19 [63.3]b | 7 [53.8] | NAc |
| Deltoid | 2 [4.7]a | 1 [3.3]b | 1 [7.7] | NAc |
| Lower extremity, proximal muscle | ||||
| Quadriceps, NOS | 5 [11.6]a | 4 [13.3]b | 1 [7.7] | NAc |
| Rectus femoris | 3 [7.0]a | 1 [3.3]b | 2 [15.4] | NAc |
| Vastus lateralis | 3 [7.0]a | 1 [3.3]b | 2 [15.4] | NAc |
| Semimembranosus | 1 [2.3]a | 1 [3.3]b | 0 | NAc |
| Lower extremity, distal muscle | ||||
| Gastrocnemius | 1 [2.3]a | 1 [3.3]b | 0 | NAc |
| Othersd | 2 [4.7]a | 2 [6.7]b | 0 | NAc |
| Histology | ||||
| Major type of muscle fiber‐containing nemaline bodies | ||||
| Round | 23 [46.9] | 16 [47.1] | 7 [53.8] | 0 |
| Angular | 12 [20.4] | 7 [20.6] | 3 [23.1] | 2 [100.0] |
| Cap | 6 [12.2] | 5 [14.7] | 1 [7.7] | 0 |
| Mixed | 8 [16.3] | 6 [17.6] | 2 [15.4] | 0 |
| Major type of nemaline bodies | ||||
| Rod‐like | 27 [55.1] | 18 [52.9] | 8 [61.5] | 1 [50.0] |
| Sand‐like | 15 [30.6] | 12 [35.3] | 2 [15.4] | 1 [50.0] |
| Mixed | 7 [14.3] | 4 [11.8] | 3 [23.1] | 0 |
| Intranuclear rod | 7 [14.3] | 3 [8.8] | 3 [23.1] | 1 [50.0] |
| Vacuolar change | 25 [51.0] | 18 [52.9] | 5 [38.5] | 2 [100.0] |
| Score 1 | 12 [24.5] | 8 [23.5] | 3 [23.1] | 1 [50.0] |
| Score 2 | 8 [16.3] | 5 [14.7] | 2 [15.4] | 1 [50.0] |
| Score 3 | 5 [10.2] | 5 [14.7] | 0 | 0 |
| Intermyofibrillary changes observed in NADH‐TR | 48 [98.0] | 33 [97.1] | 13 [100.0] | 2 [100.0] |
| Dark atrophic | 30 [61.2] | 22 [64.7] | 6 [46.2] | 2 [100.0] |
| Focal dark, not atrophic | 12 [24.5] | 8 [23.5] | 4 [30.8] | 0 |
| Lobulated | 16 [32.7] | 10 [29.4] | 5 [38.5] | 1 [50.0] |
| Moth‐eaten | 17 [34.7] | 14 [41.2] | 3 [23.1] | 0 |
| Other | 1 [2.0] | 1 [2.9] | 0 | 0 |
| ACP‐positive fibers | 48 [98.0] | 33 [97.1] | 13 [100.0] | 2 [100.0] |
| Score 1 | 19 [38.8] | 14 [41.2] | 4 [30.8] | 1 [50.0] |
| Score 2 | 25 [51.0] | 16 [47.1] | 8 [61.5] | 1 [50.0] |
| Score 3 | 4 [8.2] | 3 [8.8] | 1 [7.7] | 0 |
| ACP positive on myofibers‐containing nemaline bodies | 40 [81.6] | 27 [79.4] | 11 [84.6] | 2 [100.0] |
| Type 1 fiber predominance | 12 [24.5] | 9 [26.5] | 1 [7.7] | 2 [100.0] |
Abbreviations: ACP, acid phosphatase; SLONM‐HIV, SLONM associated with HIV; SLONM‐MGUS, SLONM with MGUS association; SLONM‐noMGUS, SLONM without MGUS association.
Information not available in six cases.
Information not available in four cases.
Information not available in two cases.
Sternocleidomastoid, splenius.
FIGURE 3.
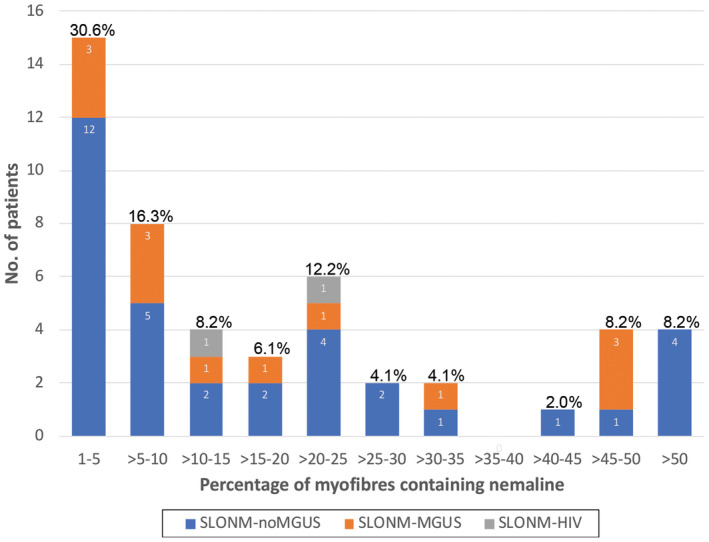
Percentage of nemaline‐containing fibers in SLONM. The diagnosis of SLONM can be challenging due to the limited number of nemaline‐containing fibres present in muscle biopsies. In our study, fifteen muscle biopsies (15/49, 30.6%) had nemaline‐containing myofibres below 5%
TABLE 3.
Spearman correlation between the percentage of nemaline‐containing fibers and the degree of ACP positivity, HLA‐ABC, and p62 expression
| ACP | HLA‐ABC | p62 | |
|---|---|---|---|
| Spearman r | 0.69 | 0.49 | 0.39 |
| 95% confidence interval | 0.50–0.82 | 0.23–0.68 | 0.10–0.61 |
| p value (two‐tailed) | 3.6e‐8 | 4.2e‐4 | 8.0e‐3 |
Abbreviations: ACP, acid phosphatase; HLA, human leukocyte antigen; p62 (SQSTM1).
SLONM‐noMGUS contained smaller numbers of regenerating fibers comparing to those in SLONM‐MGUS. Most SLONM‐noMGUS had regenerating fibers below 5% (27/34, 81.8% p = 7.8e‐3) (Table 4); there was a weak correlation between the percentage of regenerating fibers and myofiber‐containing nemaline bodies (r = 0.35, p = 1.9e‐2) . The most common infiltrating inflammatory cells in SLONM were CD68+ macrophages (99.9 ± 94.3) (Figure 4). There was moderate to strong association between the percentage of nemaline‐positive fibers and the number of infiltrating inflammatory cells (Table 5). Thirty‐two muscle biopsies (32/48, 66.7%) showed HLA‐ABC expression (Figure 4). One muscle biopsy (1/48, 2.1%) showed HLA‐DR positivity in several perifascicular fibers. MAC expression on capillaries (29/48, 60.4%) was more common than the expression on sarcolemma (6/48, 12.5%) but most of the expression was evaluated as score 1 (19/48, 39.6%). Thirty‐six (36/46, 78.3%) muscle biopsy had fine granular p62 expression pattern. Punctate/dot‐like p62 expression pattern was observed in two SLONM‐MGUS biopsies (2/11, 18.2%), and one SLONM‐noMGUS biopsy (1/33, 3.0%) (Table 4). We compared staining pattern and total staining score of LAMP2, CRYAB, BAG3, HSP70, and LC3 in sequential sections from nine muscle biopsies to those of p62. There was no significant difference between p62 score and LAMP2, CRYAB, BAG3 to HSP70 scores. In contrast, the p62 score (2.7 ± 0.7) and LC3 score (0.1 ± 0.3) were significantly different (p = 2.7e‐3). The association between the percentage of nemaline‐containing fibers and the score of HLA‐ABC and p62 expression was moderate (r = 0.49, p = 4.2e‐4) and weak (r = 0.39, p = 8.0e‐3), respectively (Table 3). Notably, in available sequential sections, overlapping HLA‐ABC and p62 expression was observed in 4/9 biopsies (44.4%); HLA‐ABC was negative in 2/9 biopsies. There was no MxA expression observed in SLONM. Staining for ISG15 showed score1 in 3 of 11 cases. Staining for Siglec1 showed score 2 positivity in two biopsies (one of them also showed ISG15 positivity) and score 1 positivity in three non‐ISG15‐positive biopsies. Except for the smaller number of MHCn‐defined regenerating fibers, the histological features of SLONM‐noMGUS and SLONM‐MGUS muscle biopsy were not distinctively different (p > 0.05).
TABLE 4.
Inflammatory features in SLONM
| SLONM | SLONM‐noMGUS | SLONM‐MGUS | SLONM‐HIV | |
|---|---|---|---|---|
| (total = 49) | (n = 34) | (n = 13) | (n = 2) | |
| Immunohistochemistry | ||||
| MHCn, regenerating fibers (%) | 5.8 ± 9.3a | 5.1 ± 9.9b | 9.0 ± 7.9c | 0.65 ± 0.5 |
| ≤1% | 16 [36.4]a | 13 [39.4]b | 3 [27.3]c | 2 [100.0] |
| >1 to ≤5% | 15 [34.1]a | 14 [42.4]b | 1 [9.1]c | 0 |
| >5 to ≤10% | 5 [11.4]a | 2 [6.1]b | 3 [27.3]c | 0 |
| >10% | 8 [18.2]a | 4 [12.1]b | 4 [36.4]c | 0 |
| CD3 in 10 HPF | 19.4 ± 27.7b | 19.0 ± 30.6 | 19.3 ± 20.5b | 28.0 ± 22.6 |
| CD8 in 10 HPF | 17.9 ± 26.4b | 18.0 ± 29.8 | 17.8 ± 17.0b | 18.0 ± 17.0 |
| CD20 in 10HPF | 2.1 ± 4.6c | 1.7 ± 3.7 | 3.2 ± 6.8c | 0 |
| CD68 in 10HPF | 99.9 ± 94.3b | 105.1 ± 104.8 | 89.0 ± 63.4b | 78.0 ± 87.7 |
| HLA‐ABC | 32 [66.7]b | 23 [67.6] | 8 [66.7]b | 1 [50.0] |
| Score 1 | 10 [20.8]b | 5 [14.7] | 5 [41.7]b | 0 |
| Score 2 | 13 [27.1]b | 9 [26.5] | 3 [25.0]b | 1 [50.0] |
| Score 3 | 9 [18.8]b | 9 [26.5] | 0b | 0 |
| HLA‐DR | 1 [2.1]b | 0 | 1 [8.3]b | 0 |
| MAC capillary deposition | 29 [60.4]b | 21 [61.8] | 6 [50.0]b | 2 [100] |
| Score 1 | 19 [39.6]b | 11 [32.4] | 6 [50.0] b | 2 [100] |
| Score 2 | 8 [16.7]b | 8 [23.5] | 0b | 0 |
| Score 3 | 2 [4.2]b | 2 [5.9] | 0b | 0 |
| MAC sarcolemmal deposition | 6 [12.5]b | 5 [14.7] | 1 [8.3]b | 0 |
| Score 1 | 2 [4.2]b | 2 [5.9] | 0b | 0 |
| Score 2 | 3 [6.3]b | 2 [5.9] | 1 [8.3]b | 0 |
| Score 3 | 1 [2.1]b | 1 [2.9] | 0b | 0 |
| MAC capillary/sarcolemmal deposition, mixed | 9 [18.8]b | 7 [20.6] | 2 [16.7]b | 0 |
| p62 (fine granular pattern) | 36 [78.3]a | 26 [78.8]b | 8 [72.7]c | 2 [100] |
| Score 1 | 6 [13.0]a | 4 [12.1]b | 2 [18.2]c | 0 |
| Score 2 | 12 [26.1]a | 10 [30.3]b | 2 [18.2]c | 0 |
| Score 3 | 18 [39.1]a | 12 [36.4]b | 4 [36.4]c | 2 [100] |
Abbreviations: CD, cluster of differentiation; HLA‐ABC, human leukocyte antigen‐ABC; HLA‐DR, human leukocyte antigen‐DR; HPF, high power field (40x objective); MAC, membrane attack complex (C5b‐9); MHCn, neonatal myosin; p62 (STQM1); SLONM‐HIV, SLONM associated with HIV; SLONM‐MGUS, SLONM with MGUS association; SLONM‐noMGUS, SLONM without MGUS association.
Information not available in three cases.
Information not available in one case.
Information not available in two cases.
FIGURE 4.
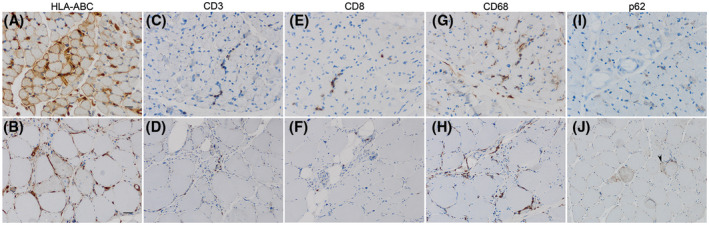
Representative immunopathological findings. Two representative biopsies showing different degrees of sarcolemmal HLA‐ABC positivity (A and B) CD3 (C and D), CD8 (E and F), CD68 (G and H), and p62 (I and J). There is small number of CD3+ and CD8+ cells infiltration while CD68+cells infiltration is more prominent. Pattern of p62 positivity is heterogenous. Fine granular pattern is demonstrated in J (black arrowhead) (A, C, E, G, I 40x magnification bar = 20 microns; B, D, F, H, J 20x magnification bar = 50 microns)
TABLE 5.
Spearman correlation between the percentage of nemaline‐containing fibers and the number of inflammatory cell infiltration in 10 high power fields
| CD3 | CD8 | CD20 | CD68 | |
|---|---|---|---|---|
| Spearman r | 0.60 | 0.61 | 0.36 | 0.55 |
| 95% confidence interval | 0.37–0.76 | 0.38–0.76 | 0.07–0.59 | 0.30–0.73 |
| p value (two‐tailed) | 7.7e‐6 | 6.5e‐6 | 1.3e‐3 | 6.9e‐5 |
SLONM‐HIV showed a predominant angular shape of nemaline‐containing myofibers but overall myopathological changes observed in SLONM‐HIV were comparable to those in the other two entities (SLONM‐MGUS and SLONM‐noMGUS).
4. DISCUSSION
We describe a large cohort (n = 49) of SLONM patients of whom 13 also suffered from MGUS. The most common clinical features of patients in our study, which included adult‐onset proximal muscle weakness, neck muscle weakness, and respiratory weakness were suggestive of the diagnosis of SLONM. The pathological confirmation, however, may sometimes be problematic caused by the limited number of nemaline‐containing fibers present in muscle biopsies and requires expert knowledge of this myopathological feature. Based on the seldom reported increased ACP activity in single case reports and small case series of SLONM (5, 17, 50, 51), we explored if ACP staining would be useful for the diagnostic process. In our study, 81.6% of SLONM showed ACP positivity in nemaline‐containing fibers and the percentage of nemaline‐containing fibers correlated well with ACP‐staining score. Thus, we suggest that identification of ACP‐positive fibers could be helpful to identify nemaline‐containing fibers. ACP‐staining is a commonly used practice in myopathology, giving diagnostic information in myopathies that are characterized by pathologic activity such as Pompe disease (GSD II) (52), Danon disease (53), X‐linked myopathy with excessive autophagy (XMEA) (54), inflammatory myopathies (55), and many more. The most common histological findings included round‐type nemaline‐containing fibers, rod‐like nemaline bodies, and NADH‐TR dark atrophic fibers.
Thirty‐two SLONM patients (66.7%) had variable sarcolemmal HLA‐ABC expression as a core feature of myositis. CD68+ macrophages were the most common inflammatory cells observed in SLONM. There were smaller numbers of CD3+ and CD8+ cells present in all biopsies. CD20+ cells were rarely present in SLONM. Moderate to strong association between the number of inflammatory cells and the percentage of nemaline‐containing fibers suggested that inflammation may be involved in pathogenesis of the disease. Direct infiltration of myofibers by T cells was not observed in any of the biopsies. Although presence of CD68+ macrophages in necrotic myofibers or myophagocytoses may occur as a feature independent of any primary inflammation, presence of T cells (and B cells) in SLONM overall exceeded what is generally found in “pure” resorptive processes.
It is unlikely that the complement cascade is involved in SLONM pathogenesis considering the low degree of capillary and sarcolemmal MAC expression. We did not find strong evidence suggestive of type 1 interferon‐related pathway activation in SLONM by virtual absence of any MxA, and only very mild ISG15, or mild Siglec1 expression in single biopsies. Notably, 36 patients (78.3%) had fine granular p62 expression pattern similar to those observed in immune‐mediated necrotizing myopathy (IMNM) (56), while only three cases showed a coarse p62 staining pattern in some fibers. Unlike in IMNM, the p62 staining pattern correlated well with LAMP2, CRYAB, BAG3, and HSP70 but not with LC3. Expression of p62 within fibers lined by HLA‐ABC was identified in our SLONM cohort (irrespective of the MGUS status). These findings, however, could suggest insufficient function of the autophagosomal pathway putatively involved in dysfunctional macroautophagy of nemaline fragments thereby reflecting the pathogenesis of nemaline formation in SLONM. Overall histological and immunohistochemical features in SLONM‐noMGUS and SLONM‐MGUS were not distinctively different.
The challenge in this study was its retrospective nature including that the clinical information and tissue repository were limited in some cases. For that reason, we could not correlate pathology features with clinical severity or treatment outcome. The small number of SLONM‐HIV patients limited further studies and comparison of this entity with the other two defined here.
Inflammatory features including endomysial or perivascular inflammatory cell infiltration and HLA‐ABC expression were documented as separate single case report or small case series of SLONM‐noMGUS (2, 4, 5, 57), SLONM‐MGUS (4, 5, 39, 41, 44), and SLONM‐HIV (4, 8, 15). Our study adds a collective in‐depth analysis of the inflammatory features of SLONM. Although this study did not identify the differences with respect to the features studied here, between SLONM‐noMGUS and SLONM‐MGUS, it highlights common immunopathological elements among both entities that could be fundamental for further studying their pathogenesis.
Relatively mild sarcolemmal HLA‐ABC expression and some minor CD3 and CD8 immunoreactive T cells may occur in the context of several neuromuscular diseases that are not primarily considered inflammatory myopathies. The significance of such changes has been mentioned in the past at several occasions and is beginning to be understood and put in relevant contexts.(61, 62, 63) This phenomenon, often times mentioned as “accompanying inflammatory changes,” if correctly diagnosed individually may open (additional) therapeutic approaches even in primarily genetic diseases such as Duchenne muscular dystrophy (DMD) (64) or dysferlinopathy (LGMDR2) and anoctaminopathy (LGMDR12) (65), and many others. Also in some acquired for example, toxic conditions, mild inflammatory changes have been described (66). This is the case for certain drugs for example, hydroxychloroquine, colchicine, amiodarone, or immune checkpoint‐inhibitors such as PD1‐ or PDL1‐targeting anticancer drugs (67).
SLONM‐MGUS was previously suggested to have autoimmune etiology by the presence of the same type of immunoglobulin deposits on sarcolemma (30) and the responsiveness to immunotherapy including IVIg (6, 30, 36). Although patients with SLONM‐MGUS were shown to achieve long‐term benefit from hematologic treatment, it is currently a matter of debate whether the responsiveness is caused by the result of immunomodulation or ablation of any abnormal plasma cell clone. IVIg‐based treatment, moreover, was reported to have variable outcomes in patients suffering from SLONM‐MGUS (5, 6, 25, 36, 38, 41, 43, 44, 48). Thus, the treatment of choice in SLONM‐MGUS has to be considered on a case‐by‐case basis. Interestingly, according to the available information, three SLONM‐MGUS patients without inflammatory features had a good long‐term response (>24 months) to IVIg (36, 44). The other patients reported in the literature so far, were either stable (5, 44) or becoming worse (38, 43). One SLONM‐MGUS patient with perivascular inflammation showed disease progression after IVIg treatment (44). In one SLONM‐noMGUS patient without inflammatory features, a combined treatment including IVIg, corticosteroids, and methotrexate did not improve but stabilized the disease conditions over a 6‐year period (5). It is our great interest whether inflammatory features identified at the time of muscle biopsy in SLONM would influence the treatment option and predict responsiveness to these regimens. Answering this question would certainly require a prospective study and an international effort since the disease is exceedingly rare. Hence, we believe that our study has raised awareness not only for the disease and their subtypes but also for immune‐inflammatory features which may become relevant for individualized treatment options in the future.
ACKNOWLEDGMENTS
The study was supported partly by Intramural Research Grant (2–5) for the Neurological and Psychiatric Disorders of NCNP. This study was supported by a grant of the Deutsche Gesellschaft für Muskelkranke e.V. (DGM) to Dr. Uruha, Dr. Dittmayer, and Dr. Stenzel. The authors appreciate the technical assistance by Ms. Kaoru Tatezawa, Kazuko Iwasawa, Naho Fushimi, Miyuki Matsuda, Sylvia Stefaniak, and Petra Matylewski.
Jantima Tanboon, Akinori Uruha, Ichizo Nishino and Werner Stenzel are contributed equally to this work.
DATA AVAILABILITY STATEMENT
Data are not provided in the article and additional information on methods and materials may be shared upon request.
REFERENCES
- 1.Uruha A, Benveniste O. Sporadic late‐onset nemaline myopathy with monoclonal gammopathy of undetermined significance. Curr Opin Neurol. 2017;30:457–63. [DOI] [PubMed] [Google Scholar]
- 2.Engel AG. Late‐onset rod myopathy (a new syndrome?): light and electron microscopic observations in two cases. Mayo Clin Proc. 1966;41:713–41. [PubMed] [Google Scholar]
- 3.Engel WK, Resnick JS. Late‐onset rod myopathy: a newly recognized acquired and progressive disease. Neurology. 1966;16:308–9. [Google Scholar]
- 4.Schnitzler LJ, Schreckenbach T, Nadaj‐Pakleza A, Stenzel W, Rushing EJ, Van Damme P, et al. Sporadic late‐onset nemaline myopathy: clinico‐pathological characteristics and review of 76 cases. Orphanet J Rare Dis. 2017;12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chahin N, Selcen D, Engel AG. Sporadic late onset nemaline myopathy. Neurology. 2005;65:1158–64. [DOI] [PubMed] [Google Scholar]
- 6.Naddaf E, Milone M, Kansagra A, Buadi F, Kourelis T. Sporadic late‐onset nemaline myopathy: clinical spectrum, survival, and treatment outcomes. Neurology. 2019;93:e298–e305. [DOI] [PubMed] [Google Scholar]
- 7.Dalakas MC, Pezeshkpour GH, Flaherty M. Progressive nemaline (rod) myopathy associated with HIV infection. N Engl J Med. 1987;317:1602–3. [DOI] [PubMed] [Google Scholar]
- 8.Simpson DM, Bender AN. Human immunodeficiency virus‐associated myopathy: analysis of 11 patients. Ann Neurol. 1988;24:79–84. [DOI] [PubMed] [Google Scholar]
- 9.Cabello A, Martínez‐Martín P, Gutiérrez‐Rivas E, Madero S. Myopathy with nemaline structures associated with HIV infection. J Neurol. 1990;237:64–5. [DOI] [PubMed] [Google Scholar]
- 10.Dwyer BA, Mayer RF, Lee SC. Progressive nemaline (rod) myopathy as a presentation of human immunodeficiency virus infection. Arch Neurol. 1992;49:440. [DOI] [PubMed] [Google Scholar]
- 11.Miró O, Masanés F, Pedrol E, García‐Carrasco M, Mallolas J, Casademont J, et al. A comparative study of the clinical and histological characteristics between classic nemaline myopathy and that associated with the human immunodeficiency virus. Med Clin (Barc). 1995;105:500–3. [PubMed] [Google Scholar]
- 12.Feinberg DM, Spiro AJ, Weidenheim KM. Distinct light microscopic changes in human immunodeficiency virus‐associated nemaline myopathy. Neurology. 1998;50:529–31. [DOI] [PubMed] [Google Scholar]
- 13.Madonia P, Wilson J, Bican O, Willis M, Bass P 3rd. HIV, rods, and the muscles‐a discussion about HIV‐associated nemaline rod myopathy. J La State Med Soc. 2012;164:320–3. [PubMed] [Google Scholar]
- 14.Silva AMS, Mendonça RH, Moreno CAM, Estephan EP, Helito PVP, Carvalho MS, et al. Clinical, histological and radiological responses to methylprednisolone in HIV‐associated rod myopathy. Neuromuscul Disord. 2017;27:756–9. [DOI] [PubMed] [Google Scholar]
- 15.Gonzales MF, Olney RK, So YT, Greco CM, McQuinn BA, Miller RG, et al. Subacute structural myopathy associated with human immunodeficiency virus infection. Arch Neurol. 1988;45:585–7. [DOI] [PubMed] [Google Scholar]
- 16.Maytal J, Horowitz S, Lipper S, Poiesz B, Wang CY, Siegal FP. Progressive nemaline rod myopathy in a woman coinfected with HIV‐1 and HTLV‐2. Mt Sinai J Med. 1993;60:242–6. [PubMed] [Google Scholar]
- 17.Portlock CS, Boland P, Hays AP, Antonescu CR, Rosenblum MK. Nemaline myopathy: a possible late complication of Hodgkin's disease therapy. Hum Pathol. 2003;34:816–8. [DOI] [PubMed] [Google Scholar]
- 18.Reyes MG, Tal A, Abrahamson D, Schwartz M. Nemaline myopathy in an adult with primary hypothyroidism. Can J Neurol Sci. 1986;13:117–9. [DOI] [PubMed] [Google Scholar]
- 19.Pavlu J, Carey MP, Winer JB. Hypothyroidism and nemaline myopathy in an adult. J Neurol Neurosurg Psychiatry. 2006;77:708–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dimitri D, Dubourg O. Sporadic late‐onset nemaline myopathy in a patient with systemic lupus erythematosus. J Neurol. 2013;260:3171–3. [DOI] [PubMed] [Google Scholar]
- 21.Hindocha A, Klimiuk P, Roberts M, Pal P, Evangelista T, Lochmüller H, et al. Co‐presentation of adult‐onset systemic lupus erythematosus and nemaline myopathy. Rheumatology (Oxford). 2017;56:2034–5. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki M, Shimizu Y, Takeuchi M, Kobayashi M, Iwata M, Uchiyama S. Sporadic late‐onset nemaline myopathy in a patient with primary Sjögren's syndrome. J Neurol. 2012;259:358–60. [DOI] [PubMed] [Google Scholar]
- 23.Cao L, Wang Y, Liu X, Hu Y, Li N, Qiu G, et al. Adult‐onset nemaline myopathy coexisting with myasthenia gravis: a case report. Medicine (Baltimore). 2016;95:e2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanatani M, Adachi T, Sakata R, Watanabe Y, Hanajima R. [A case of sporadic late‐onset nemaline myopathy associated with myasthenia gravis positive for anti‐titin antibody and anti‐Kv1.4 antibody]. Rinsho Shinkeigaku. 2020;60:489–94. [DOI] [PubMed] [Google Scholar]
- 25.Wang M, Lei L, Chen H, Di L, Pang M, Lu Y, et al. Monoclonal gammopathy with both nemaline myopathy and amyloid myopathy. Neuromuscul Disord. 2017;27:942–6. [DOI] [PubMed] [Google Scholar]
- 26.Montagnese F, Portaro S, Musumeci O, Migliorato A, Moggio M, Fagiolari G, et al. Sporadic late‐onset nemaline myopathy in a woman with multiple myeloma successfully treated with lenalidomide/dexamethasone. Muscle Nerve. 2015;51:934–5. [DOI] [PubMed] [Google Scholar]
- 27.Kanamori T, Kusumoto S, Okita K, Hagiwara S, Kato C, Nakashima T, et al. Sporadic late‐onset nemaline myopathy with monoclonal gammopathy of undetermined significance mimicking POEMS syndrome. Rinsho Ketsueki. 2018;59:161–6. [DOI] [PubMed] [Google Scholar]
- 28.Engel WK, Oberc MA. Abundant nuclear rods in adult‐onset rod disease. J Neuropathol Exp Neurol. 1975;34:119–32. [DOI] [PubMed] [Google Scholar]
- 29.Seitz RJ, Toyka KV, Wechsler W. Adult‐onset mixed myopathy with nemaline rods, minicores, and central cores: a muscle disorder mimicking polymyositis. J Neurol. 1984;231:103–8. [DOI] [PubMed] [Google Scholar]
- 30.Eymard B, Brouet JC, Collin H, Chevallay M, Bussel A, Fardeau M. Late‐onset rod myopathy associated with monoclonal gammopathy. Neuromuscul Disord. 1993;3:557–60. [DOI] [PubMed] [Google Scholar]
- 31.Deconinck N, Laterre EC, Van den Bergh PY. Adult‐onset nemaline myopathy and monoclonal gammopathy: a case report. Acta Neurol Belg. 2000;100:34–40. [PubMed] [Google Scholar]
- 32.Keller CE, Hays AP, Rowland LP, Moghadaszadeh B, Beggs AH, Bhagat G. Adult‐onset nemaline myopathy and monoclonal gammopathy. Arch Neurol. 2006;63:132–4. [DOI] [PubMed] [Google Scholar]
- 33.Voermans NC, Minnema M, Lammens M, Schelhaas HJ, Kooi AV, Lokhorst HM, et al. Sporadic late‐onset nemaline myopathy effectively treated by melphalan and stem cell transplant. Neurology. 2008;71:532–4. [DOI] [PubMed] [Google Scholar]
- 34.Voermans NC, Benveniste O, Minnema MC, Lokhorst H, Lammens M, Meersseman W, et al. Sporadic late‐onset nemaline myopathy with MGUS: long‐term follow‐up after melphalan and SCT. Neurology. 2014;83:2133–9. [DOI] [PubMed] [Google Scholar]
- 35.Benveniste O, Laforet P, Dubourg O, Solly S, Musset L, Choquet S, et al. Stem cell transplantation in a patient with late‐onset nemaline myopathy and gammopathy. Neurology. 2008;71:531–2. [DOI] [PubMed] [Google Scholar]
- 36.Milone M, Katz A, Amato AA, Soderland CA, Segarceanu M, Young NP, et al. Sporadic late onset nemaline myopathy responsive to IVIg and immunotherapy. Muscle Nerve. 2010;41:272–6. [DOI] [PubMed] [Google Scholar]
- 37.Novy J, Rosselet A, Spertini O, Lobrinus JA, Pabst T, Kuntzer T. Chemotherapy is successful in sporadic late onset nemaline myopathy (SLONM) with monoclonal gammopathy. Muscle Nerve. 2010;41:286–7. [DOI] [PubMed] [Google Scholar]
- 38.Hanisch F, Schneider I, Müller T, Romeike BF, Stoltenburg G, Holzhausen HJ, et al. Treatability of sporadic late onset nemaline myopathy. Nervenarzt. 2013;84:955–61. [DOI] [PubMed] [Google Scholar]
- 39.Doppler K, Knop S, Einsele H, Sommer C, Wessig C. Sporadic late onset nemaline myopathy and immunoglobulin deposition disease. Muscle Nerve. 2013;48:983–8. [DOI] [PubMed] [Google Scholar]
- 40.Maeda MH, Ohta H, Izutsu K, Shimizu J, Uesaka Y. Sporadic late‐onset nemaline myopathy as a rare cause of slowly progressive muscle weakness with young adult onset. Muscle Nerve. 2015;51:772–4. [DOI] [PubMed] [Google Scholar]
- 41.Mizuno Y, Mori‐Yoshimura M, Okamoto T, Oya Y, Nishino I, Murata M. [Two cases of sporadic late onset nemaline myopathy effectively treated with immunotherapy]. Rinsho Shinkeigaku. 2016;56:605–11. [DOI] [PubMed] [Google Scholar]
- 42.Belkhribchia MR, Tazi I, Louhab N, Kissani N, Mahmal L, Pereon Y. Autologous stem cell transplantation in a patient with sporadic late‐onset nemaline myopathy and monoclonal gammopathy: first Moroccan experience. Presse Med. 2017;46:122–5. [DOI] [PubMed] [Google Scholar]
- 43.Belhomme N, Maamar A, Le Gallou T, Minot‐Myhié MC, Larralde A, Champtiaux N, et al. Rare myopathy associated to MGUS, causing heart failure and responding to chemotherapy. Ann Hematol. 2017;96:695–6. [DOI] [PubMed] [Google Scholar]
- 44.Monforte M, Primiano G, Silvestri G, Mirabella M, Luigetti M, Cuccagna C, et al. Sporadic late‐onset nemaline myopathy: clinical, pathology and imaging findings in a single center cohort. J Neurol. 2018;265:542–51. [DOI] [PubMed] [Google Scholar]
- 45.Kotchetkov R, Dyszkiewicz‐Korpanty A, Kukreti V. Chemotherapy with stem cell transplantation is more effective than immunotherapy in sporadic late onset nemaline myopathy with monoclonal gammopathy. Bone Marrow Transplant. 2018;53:895–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumutpongpanich T, Owattanapanich W, Tanboon J, Nishino I, Boonyapisit K. Sporadic late‐onset nemaline myopathy with monoclonal gammopathy of undetermined significance (SLONM‐MGUS): an alternative treatment using cyclophosphamide‐thalidomide‐dexamethasone (CTD) regimen. Neuromuscul Disord. 2018;28:610–3. [DOI] [PubMed] [Google Scholar]
- 47.Truffert A, Iancu Ferfoglia R, Lobrinus JA, Samii K, Kohler A. Sporadic late onset nemaline myopathy with monoclonal gammopathy of undetermined significance: two cases with long term stability. Eur J Transl Myol. 2020;30:9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okhovat AA, Nilipour Y, Boostani R, Vahabizad F, Najmi S, Nafissi S, et al. Sporadic late‐onset nemaline myopathy with monoclonal gammopathy of undetermined significance: report of four patients. Neuromuscul Disord. 2021;31:29–34. [DOI] [PubMed] [Google Scholar]
- 49.Irodenko VS, Lee HS, de Armond SJ, Layzer RB. Adult nemaline myopathy with trabecular muscle fibers. Muscle Nerve. 2009;39:871–5. [DOI] [PubMed] [Google Scholar]
- 50.Paulus W, Peiffer J, Becker I, Roggendorf W, Schumm F. Adult‐onset rod disease with abundant intranuclear rods. J Neurol. 1988;235:343–7. [DOI] [PubMed] [Google Scholar]
- 51.Nonaka I, Ishiura S, Arahata K, Ishibashi‐Ueda H, Maruyama T, Ii K. Progression in nemaline myopathy. Acta Neuropathol. 1989;78:484–91. [DOI] [PubMed] [Google Scholar]
- 52.Tsuburaya RS, Monma K, Oya Y, Nakayama T, Fukuda T, Sugie H, et al. Acid phosphatase‐positive globular inclusions is a good diagnostic marker for two patients with adult‐onset Pompe disease lacking disease specific pathology. Neuromuscul Disord. 2012;22:389–93. [DOI] [PubMed] [Google Scholar]
- 53.Sugie K, Yamamoto A, Murayama K, Oh SJ, Takahashi M, Mora M, et al. Clinicopathological features of genetically confirmed Danon disease. Neurology. 2002;58:1773–8. [DOI] [PubMed] [Google Scholar]
- 54.Yan C, Tanaka M, Sugie K, Nobutoki T, Woo M, Murase N, et al. X‐linked myopathy with excessive autophagy (XMEA) A new congenital form of X‐linked autophagic vacuolar myopathy. Neurology. 2005;65:1132–4. [DOI] [PubMed] [Google Scholar]
- 55.De Bleecker JL, De Paepe B, Aronica E, de Visser M, ENMC Myositis Muscle Biopsy Study Group , Amato A, et al. 205th ENMC International Workshop: pathology diagnosis of idiopathic inflammatory myopathies Part II 28–30 March 2014, Naarden, The Netherlands. Neuromuscul Disord. 2015;25(3):268–72. [DOI] [PubMed] [Google Scholar]
- 56.Fischer N, Preuße C, Radke J, Pehl D, Allenbach Y, Schneider U, et al. Sequestosome‐1 (p62) expression reveals chaperone‐assisted selective autophagy in immune‐mediated necrotizing myopathies. Brain Pathol. 2020;30:261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gyure KA, Prayson RA, Estes ML. Adult‐onset nemaline myopathy: a case report and review of the literature. Arch Pathol Lab Med. 1997;121:1210–3. [PubMed] [Google Scholar]
- 58.Kamieniecka Z. Late onset myopathy with rod‐like particles. Acta Neurol Scand. 1973;49:547–51. [DOI] [PubMed] [Google Scholar]
- 59.Nakamura K, Shibuya K, Nishino I, Kuwabara S. Dropped head in sporadic late‐onset nemaline myopathy. Intern Med. 2019;58:1967–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.De Stefano L, Volpi N, De Stefano P, Ginanneschi F, Frati E, Rossi A. Sporadic late‐onset nemaline myopathy in a patient with silicone breast implants. Clin Neurol Neurosurg. 2020;196:105999. [DOI] [PubMed] [Google Scholar]
- 61.Flanigan KM, Campbell K, Viollet L, Wang W, Gomez AM, Walker CM, et al. Anti‐dystrophin T cell responses in Duchenne muscular dystrophy: prevalence and a glucocorticoid treatment effect. Hum Gene Ther. 2013;24:797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Confalonieri P, Oliva L, Andreetta F, Lorenzoni R, Dassi P, Mariani E, et al. Muscle inflammation and MHC class I up‐regulation in muscular dystrophy with lack of dysferlin: an immunopathological study. J Neuroimmunol. 2003;142:130–6. [DOI] [PubMed] [Google Scholar]
- 63.Nikolaus M, Tietze A, Schweizer L, Kaindl AM, Stenzel W, Schuelke M, et al. Fulminant cerebral venous thrombosis associated with the m.3243A>G MELAS mutation: a new guise for an old disease. Brain Dev. 2019;41:901–4. [DOI] [PubMed] [Google Scholar]
- 64.Mendell JR, Campbell K, Rodino‐Klapac L, Sahenk Z, Shilling C, Lewis S, et al. Dystrophin immunity in Duchenne's muscular dystrophy. N Engl J Med. 2010;363:1429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Claeys KG, Gorodinskaya O, Handt S, Reimann J, Kress W, Kornblum C, et al. Diagnostic challenge and therapeutic dilemma in necrotizing myopathy. Neurology. 2013;81:932–5. [DOI] [PubMed] [Google Scholar]
- 66.Doughty CT, Amato AA. Toxic myopathies. Continuum (Minneap Minn). 2019;25:1712–31. [DOI] [PubMed] [Google Scholar]
- 67.Touat M, Maisonobe T, Knauss S, Ben Hadj Salem O, Hervier B, Auré K, et al. Immune checkpoint inhibitor‐related myositis and myocarditis in patients with cancer. Neurology. 2018;91:e985–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are not provided in the article and additional information on methods and materials may be shared upon request.