

Abstract
We report on a case of EWSR1‐PATZ1 rearranged brain tumor occurring in a 17 month‐old child, originally interpreted as an infantile glioblastoma. Our case shows important analogies with the 2 previously reported cases, including the intraventricular location, the histologic appearance (pushing borders, oligodendrocyte‐like morphology, rich vascular network) and the glioneural immunophenotype, supporting the role of these features as relevant clues to the diagnosis. On the other hand, our case displays unique characteristics, i.e. the onset in an infant, the presence of a focal high‐grade component and the leptomeningeal dissemination, pointing to the importance of considering this entity in the differential diagnosis of an infantile glial/glioneural tumor.

Keywords: EWSR1‐PATZ1 rearranged CNS tumor, glioneural tumor, high‐grade, infantile, leptomeningeal dissemination
EWSR1‐PATZ1 fusions, first described in small round or spindle cell sarcomas (1) have recently been reported in rare low‐ and high‐grade CNS tumors (1, 2, 3, 4, 5, 6). Among the cerebral cases, only three have been provided with a complete clinico‐pathological description (4, 6). We report a case of EWSR1‐PATZ1 brain tumor displaying unique clinico‐pathological and molecular features.
A 17‐month old child was brought to the emergency room with drowsiness thought to be related to a tricycle bike trauma. MRI scan showed a large right intraventricular mass (10 × 6 × 7 cm), without evidence of perilesional edema, associated with midline shift (Figure 1A–C). The mass, hyperintense on T2 and FLAIR sequences, showed heterogeneous postcontrast enhancement with cystic, necrotic, and hemorrhagic areas (Figure 1A–C). Intra and peritumoral ectatic vessels were present (Figure 1A). Linear leptomeningeal enhancement involved the brain stem, the cranial nerves, and focally the spinal cord, consistent with leptomeningeal dissemination (Figure 1A). CSF cytology was negative for malignant cells. A partial tumor resection was achieved at surgery with residual tumor involving the body and atrium of the lateral ventricle. Based on a histological diagnosis of infantile glioblastoma (WHO grade IV), the patient was treated with chemotherapy according to national guidelines (7), with primary tumor and leptomeningeal involvement showing a partial response. After four courses of high‐dose chemotherapy, a second surgery was performed with gross total resection of the residual tumor. After surgical recovery, chemotherapy was started again. The patient has been currently receiving the fifth cycle of high‐dose chemotherapy, with last MRI showing a residual leptomeningeal involvement limited to the brain stem. The first resection specimen showed a well demarcated, moderately cellular tumor composed of cells with round monomorphous nuclei and oligodendroglial‐like morphology (Figure 1D,E). Focally cells showed abundant clear cytoplasm. A striking feature was the presence of a rich vascular network consisting of delicate branching capillaries (Figure 1E). Microvascular proliferation was focally present. Isolated microcalcifications and rare cysts were also seen (Figure 1F). While most of the tumor lacked mitotic activity, a small component (<5% of the tumor volume) showed high cellularity, high mitotic activity (7/10 HPF) and pseudo‐palisading necrosis (Figure 1G,H). As for the immunophenotype, GFAP and synaptophysin expression was focal in both the low and the high‐grade components (Figure 1I–L); OLIG2 was focally expressed in the low‐grade component, whereas it was diffuse in the high‐grade component (Figure 1M,N). Furthermore, only the high‐grade area lacked H3K27me3 expression, which was instead retained in the dominant low grade component, likely representing a secondary event associated with clonal evolution (Figure 1O,P); p53, IDH1R132H, BRAFV600E, H3K27M, L1CAM, and p65/RELA were negative and ATRX expression was maintained throughout the neoplasm. Ki67 was approximately 20% in the high‐grade compared to 2% in the low‐grade component. The second tumor specimen did not show high‐grade areas. The morphological features with high mitotic index and necrosis suggested the diagnosis of “infantile glioblastoma, grade IV”, a recently defined tumor category with a characteristic methylation profile, driven by kinase fusions in half of the cases (8). Methylation analysis of both tumor samples yielded a no match result (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162537). CGH‐array, SNP‐array, and CNV analysis from methylation profiling exclusively showed an overly complex rearrangement of chromosome 22, with multiple gains and losses, suggesting the occurrence of an intrachromosomal gene fusion (Figure 1Q). No additional chromosomal abnormalities were observed. RNA sequencing analysis revealed a EWSR1‐PATZ1 fusion (breakpoints: GRCh38 chr22:29285885‐chr22:31345600) (https://www.ncbi.nlm.nih.gov/sra/PRJNA681741), which was confirmed by RT‐PCR and Sanger sequencing (Supporting Information) in keeping with the complex chromosome 22 alterations, as both EWSR1 and PATZ1 map on this region (Figure 1Q,R). Additionally, a heterozygous mutation p.R393W (c.1177C > T) in ERBB4 receptor, involving a highly conserved motif of the L2 domain was found. This variant, reported in dbSNP (rs55671017) and COSMIC (COSV53532907) database, may sensitize the tumor to tyrosine kinase inhibitors (9). Secondary molecular alterations affecting cell cycle genes, for example, CDKN2A, and possibly contributing to tumor aggressiveness, have been reported in EWSR1‐PATZ1 rearranged tumors, with a higher frequency in sarcomas compared to CNS tumors (1). To our knowledge, this is the first EWSR1‐PATZ1 rearranged tumor bearing an activating tyrosine kinase mutation.
FIGURE 1.
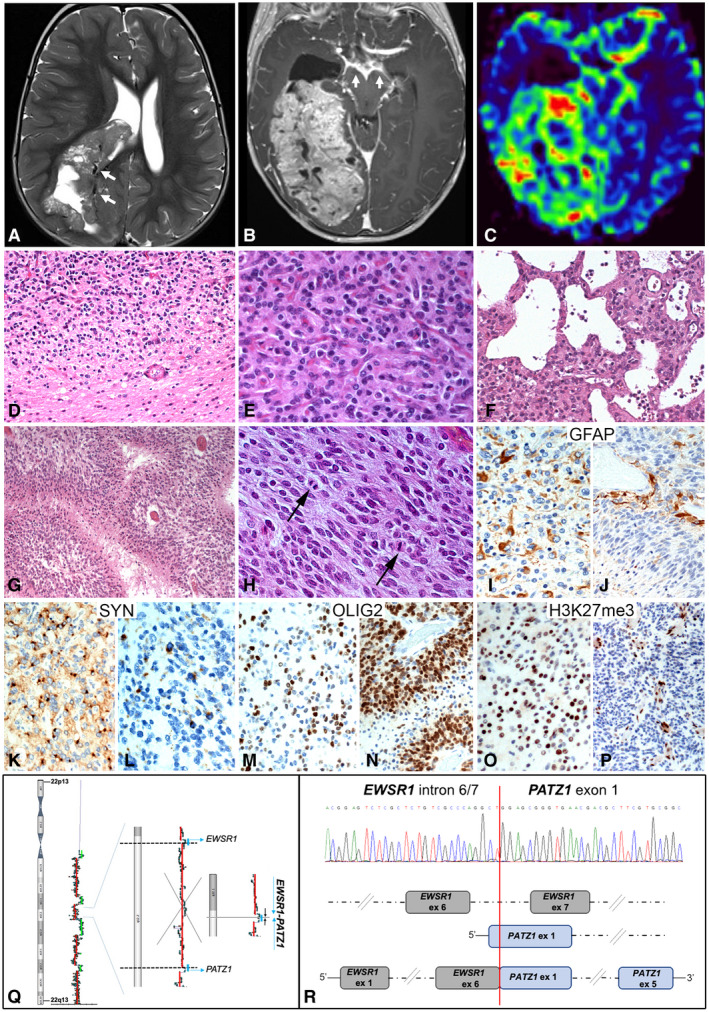
T2w (A) and Gd T1w (B) axial MRI images showed a hyperintense right intraventricular mass with cystic and necrotic areas (A), intra and peritumoral ectasic vessels (arrows, A), heterogeneous postcontrast enhancement (B) and leptomeningeal involvement (arrows, B). Perfusion‐weighted images showed increased relative cerebral blood flow (C). The tumor featured pushing borders (D), a rich vascular network (E), and rare cysts (F). A high‐grade area with hypercellularity, high mitotic index, and pseudo‐palisading necrosis was present (G‐H). GFAP and Synaptophysin were focally expressed in both low‐grade (I, K) and high‐grade (J, L) components. OLIG2 and H3K27me3 expression was respectively focal and retained in the low‐grade component (M, O), whereas was diffuse and lost in the high‐grade component (N, P). SNP‐array analysis showed chromosome 22 chromothripsis. Parts of EWSR1 and PATZ1 genes were retained within a large lost region of chromosome 22 (Q). Nucleotide sequence of EWSR1‐PATZ1 fusion transcript around the breakpoint including EWSR1 intron 6/7 and PATZ1 exon 1 (R, upper part). Cartoon of EWSR1‐PATZ1 fusion protein with splicing of EWSR1 intron 6/7 (R, lower part)
Our case shows analogies with two previously reported cases (6). Remarkably, they were all endoventricular, involving either the lateral ventricles or the fourth ventricle. From a histological standpoint, they shared sharp borders, a monotonous cellularity with oligodendrocyte‐like or clear cell morphology, and, most strikingly, a rich vascular network. The immunophenotype was also very similar, as all three cases displayed a glioneural phenotype with co‐expression of OLIG2, GFAP, and synaptophysin (6). Beside such similarities, some characteristics of our case are unique, that is the onset in an infant, the occurrence of a high‐grade area and the leptomeningeal involvement. No previous EWSR1‐PATZ1 cerebral tumors have been reported in infants and the median age in this group of patients, in the current literature, is 21 years (range 7‐50) (1, 2, 3, 4, 5, 6).
This case confirms the previously reported characteristic clinico‐pathological features of EWSR1‐PATZ1 glioneuronal tumors, for example, endoventricular location, oligodendrocyte‐like morphology and rich vascular network. At the same time, the current case expands the clinico‐pathological spectrum of CNS tumors harboring this fusion to include infants. If EWSR1‐PATZ1 glioneuronal tumors truly represent a distinct tumor entity remains to be determined.
AUTHOR CONTRIBUTIONS
Study conception: Rossi, Barresi, Colafati, Giangaspero, Alaggio. Acquisition of data: Rossi, Barresi, Giovannoni, Alesi, Ciolfi, Colafati, Diomedi‐Camassei, Miele, Cacchione, Quacquarini, Carai, Tartaglia, Mastronuzzi. Analysis and interpretation of data: Rossi, Alesi, Miele, Ciolfi, Tartaglia, Giannini, Giangaspero, Alaggio. Drafting of manuscript: Rossi, Barresi, Giangaspero, Giannini, Mastronuzzi. Critical revision: Rossi, Barresi, Giovannoni, Alesi, Ciolfi, Colafati, Diomedi‐Camassei, Miele, Cacchione, Quacquarini, Carai, Tartaglia, Giannini, Giangaspero, Mastronuzzi, Alaggio. All authors approved the final version of the manuscript to be published.
CONFLICT OF INTEREST
None of the authors declare conflict of interest.
ETHICS
Patient’s parents have given written consent to the publication of the case details.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
Ministry of Health, 5 × 1000 funds 2020 to R.A (202005_ONCO_ALAGGIO). We wish to thank Mr. Gabriele Bacile for iconography preparation.
DATA AVAILABILITY STATEMENT
Methylation data are openly available in [GEO] at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162537. RNASeq data are openly available in [SRA] at https://www.ncbi.nlm.nih.gov/sra/PRJNA681741.
REFERENCES
- 1.Bridge JA, Sumegi J, Druta M, Bui MM, Henderson‐Jackson E, Linos K, et al. Clinical, pathological, and genomic features of EWSR1‐PATZ1 fusion sarcoma. Mod Pathol. 2019;32:1593–604. [DOI] [PubMed] [Google Scholar]
- 2.Guadagno E, Vitiello M, Francesca P, Cali G, Caponnetto F, Cesselli D, et al. PATZ1 is a new prognostic marker of glioblastoma associated with the stem‐like phenotype and enriched in the proneural subtype. Oncotarget. 2017;8:59282–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson A, Severson E, Gay L, Vergilio JA, Elvin J, Suh J, et al. Comprehensive genomic profiling of 282 pediatric low‐ and high‐grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist. 2017;22:1478–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez‐Nunez O, Cafferata B, Santi M, Ranganathan S, Pearce TM, Kulich SM, et al. The spectrum of rare central nervous system (CNS) tumors with EWSR1‐non‐ETS fusions: experience from three pediatric institutions with review of the literature. Brain Pathol. 2020. 10.1111/bpa.12900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD, et al. Genetic alterations in uncommon low‐grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol. 2016;131:833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siegfried A, Rousseau A, Maurage CA, Pericart S, Nicaise Y, Escudie F, et al. EWSR1‐PATZ1 gene fusion may define a new glioneuronal tumor entity. Brain Pathol. 2019;29:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antonelli M, Korshunov A, Mastronuzzi A, Diomedi‐Camassei F, Andrea Carai A, Colafati GS, et al. Long‐term survival in a case of ETANTR with histological features of neuronal maturation after therapy. Virchows Arch. 2015;466:603–7. [DOI] [PubMed] [Google Scholar]
- 8.Clarke M, Mackay A, Ismer B, Pickles JC, Tatevossian RG, Newman S, et al. Infant high‐grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov. 2020;10:942–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lau C, Killian KJ, Samuels Y, Rudloff U. ERBB4 mutation analysis: emerging molecular target for melanoma treatment. Methods Mol Biol. 2014;1102:461–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Methylation data are openly available in [GEO] at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162537. RNASeq data are openly available in [SRA] at https://www.ncbi.nlm.nih.gov/sra/PRJNA681741.