

Abstract
Background:
In the PACIFIC trial, durvalumab significantly improved progression-free and overall survival (PFS/OS) versus placebo, with manageable safety, in unresectable, stage III non-small-cell lung cancer (NSCLC) patients without progression after chemoradiotherapy (CRT). We report exploratory analyses of outcomes by tumour cell (TC) programmed death-ligand 1 (PD-L1) expression.
Patients and methods:
Patients were randomly assigned (2:1) to intravenous durvalumab 10 mg/kg every 2 weeks or placebo ≤12 months, stratified by age, sex, and smoking history, but not PD-L1 status. Where available, pre-CRT samples were tested for PD-L1 expression (immunohistochemistry) and scored at pre-specified (25%) and post hoc (1%) TC cut-offs. Treatment-effect hazard ratios (HRs) were estimated from unstratified Cox proportional hazards models (Kaplan–Meier-estimated medians).
Results:
In total, 713 patients were randomly assigned, 709 of whom received at least 1 dose of study treatment durvalumab (n = 473) or placebo (n = 236). Some 451 (63%) were PD-L1-assessable: 35%, 65%, 67%, 33%, and 32% had TC ≥25%, <25%, ≥1%, <1%, and 1%–24%, respectively. As of 31 January 2019, median follow-up was 33.3 months. Durvalumab improved PFS versus placebo (primary-analysis data cut-off, 13 February 2017) across all subgroups [HR, 95% confidence interval (CI); medians]: TC ≥25% (0.41, 0.26–0.65; 17.8 versus 3.7 months), <25% (0.59, 0.43–0.82; 16.9 versus 6.9 months), ≥1% (0.46, 0.33–0.64; 17.8 versus 5.6 months), <1% (0.73, 0.48–1.11; 10.7 versus 5.6 months), 1%–24% [0.49, 0.30–0.80; not reached (NR) versus 9.0 months], and unknown (0.59, 0.42–0.83; 14.0 versus 6.4 months). Durvalumab improved OS across most subgroups (31 January 2019 data cut-off; HR, 95% CI; medians): TC ≥ 25% (0.50, 0.30–0.83; NR versus 21.1 months), <25% (0.89, 0.63–1.25; 39.7 versus 37.4 months), ≥1% (0.59, 0.41–0.83; NR versus 29.6 months), 1%–24% (0.67, 0.41–1.10; 43.3 versus 30.5 months), and unknown (0.60, 0.43–0.84; 44.2 versus 23.5 months), but not <1% (1.14, 0.71–1.84; 33.1 versus 45.6 months). Safety was similar across subgroups.
Conclusions:
PFS benefit with durvalumab was observed across all subgroups, and OS benefit across all but TC <1%, for which limitations and wide HR CI preclude robust conclusions.
Keywords: durvalumab, immunotherapy, non-small-cell lung cancer, PACIFIC, PD-L1 expression, stage III
INTRODUCTION
Immune checkpoint blockade (ICB) of the programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1) pathway has shown promise in several advanced tumours.1–3 Durvalumab is a selective, high-affinity, human IgG1 monoclonal antibody that targets PD-L1 and occludes its binding to PD-1 and CD80 [B7–1].4 In the PACIFIC trial of unresectable, stage III non-small-cell lung cancer (NSCLC) patients whose disease had responded or stabilised after concurrent chemoradiotherapy (cCRT),5,6 durvalumab significantly improved progression-free survival (PFS) [hazard ratio (HR), 0.52; 95% confidence interval (CI), 0.42–0.65; P < 0.0001; median 16.8 versus 5.6 months] and overall survival (OS) [HR, 0.68; 95% CI, 0.53–0.87; P = 0.00251; median not reached (NR) versus 28.7 months] versus placebo, with a manageable safety profile and without compromising patient-reported outcomes.5–8
These results have led to the growing recognition of the ‘PACIFIC regimen’ (durvalumab after cCRT) as the standard of care in this setting, and to global approvals of durvalumab for treatment of patients with unresectable, stage III NSCLC in the absence of disease progression following platinum-based cCRT.7,9,10 However, in Europe, based on the results of post hoc analyses requested by the European Medicines Agency (EMA), patients must also have tumours that express PD-L1 on ≥1% of tumour cells (TCs).7
PD-L1 expression is up-regulated in several tumour types, including NSCLC, and preclinical evidence suggests that tumour PD-L1 expression increases following radiotherapy or chemotherapy.11–17
PD-L1 expression alone is not an absolute differentiator of those who benefit and those who do not13,18; however, its value as a predictive biomarker for PD-1/PD-L1 ICB has been recognised in clinical guidelines for the stage IV/metastatic NSCLC setting, with several therapies approved with companion or complementary diagnostic immunohistochemistry assays to assess PD-L1 expression on malignant tumour and/or immune cells.19–21
In the PACIFIC trial, patient provision of archived, pre-cCRT tumour tissue samples was optional and enrolment was not restricted based on PD-L1 expression.5,6 Nonetheless, PFS and OS benefit with durvalumab versus placebo was demonstrated irrespective of pre-cCRT, PD-L1 TC expression, based on tumour tissue (where available) tested and scored at pre-specified cut-offs.5,6 Herein, we report exploratory analyses of efficacy and safety from the PACIFIC trial based on tumour PD-L1 expression, using pre-specified and post hoc PD-L1 cut-offs, which includes updated post hoc OS outcomes, approximately 3 years after the last patient was randomly allocated to treatment.
METHODS
Patients
PACIFIC (NCT02125461), a randomised, double-blind, international, multicentre, phase III trial, has been described elsewhere.5,6 Briefly, eligible patients had documented unresectable, stage III NSCLC according to the Staging Manual in Thoracic Oncology version 7 of the International Association for the Study of Lung Cancer. Patients must have received two or more cycles of platinum-based cCRT, with no evidence of disease progression after cCRT, and completed radiotherapy within 1–42 days of randomisation.
All patients provided written informed consent for participation, which was approved by relevant ethics committees and carried out in accordance with the International Conference on Harmonisation Guidelines on Good Clinical Practice and the Declaration of Helsinki.
Study design and treatment
Patients were randomised 2:1 to receive intravenous durvalumab 10 mg/kg, or placebo, every 2 weeks for up to 12 months or until confirmed progression, alternative anticancer therapy initiation, unacceptable toxicity, or consent withdrawal. Randomisation was stratified by age (<65 versus ≥65 years), sex (male versus female), and smoking history (current/former smoker versus never smoked), but not PD-L1 status. Patients were followed for survival and permitted retreatment with the assigned trial regimen after initial completion of 12 months treatment, if (i) disease control was achieved at the end of 12 months treatment, (ii) the disease progressed during follow-up after the initial 12 months of treatment, and (iii) the patient had not received another subsequent systemic anticancer therapy.
End points and assessments
The primary end points were PFS [per RECIST version 1.1 by blinded, independent, central review (BICR)] and OS. Secondary end points included: objective response rate (ORR), duration of response (DoR), and time to death or distant metastasis (TTDM), all assessed per BICR; 24-month OS; and safety (graded using the Common Terminology Criteria for Adverse Events version 4.03).
Optional, pre-cCRT archival tumour samples were tested retrospectively for PD-L1 expression using the fully validated VENTANA PD-L1 (SP263) immunohistochemistry assay (Ventana Medical Systems, Tucson, AZ). Testing was carried out at a central laboratory by pathologists trained and qualified by Ventana to score the samples at validated pre-specified (TC 25%) and post hoc (TC 1%) cut-offs.
Statistical analyses
The PD-L1 subgroup analyses reported here were based on the following data cut-off (DCO) dates: 13 February 2017 (DCO for the primary analysis of PFS) for PFS and related secondary efficacy end points (ORR, DoR, ongoing response, and TTDM); 22 March 2018 for OS and safety (DCO for the primary analysis of OS and an updated analysis of safety for patients completing the initial 12 months of treatment); and 31 January 2019 for updated OS.22
Pre-specified analyses of PFS and ORR were carried out for the PD-L1 TC ≥25% and <25% patient subgroups (and for patients with unknown PD-L1 status); exploratory, post hoc analyses of OS, DoR, and TTDM were also carried out for these subgroups. Additional analyses were carried out for the exploratory, post hoc TC ≥1% and <1% subgroups (PFS, OS, ORR, DoR, and TTDM) and a TC 1%–24% subgroup (PFS and OS only). Adverse event (AE) data was summarised for all subgroups.
For time-to-event end points, the treatment effect of durvalumab versus placebo within each subgroup was estimated by an HR (and corresponding 95% CI) using unstratified Cox proportional hazards; no adjustment for multiple comparisons was planned. The Kaplan–Meier method was used to estimate medians and associated 95% CIs. Response rate CIs were estimated using the Clopper-Pearson method. AEs and post-discontinuation, disease-related, anticancer therapy were descriptively summarised.
SAS® version 9.2 was used for all aforementioned analyses.
An exploratory, multiple-imputation model (described in the supplementary Methods, available at Annals of Oncology online) was used to impute missing data (using SAS® version 9.4) and estimate the OS treatment effect (HR and 95% CI) for the TC ≥1% and <1% subgroups, based on the DCO for the primary analysis.
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at: https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
RESULTS
Patients and treatment
In total, 713 patients underwent randomisation of whom 709 received durvalumab (n/N = 473/476) or placebo (n/N = 236/237). Of the randomized patients, 451 (63%) had archived, pre-cCRT samples suitable for determination of PD-L1 expression; 262 patients (37%) either did not provide a sample (n = 168; 24%) or provided one that was inadequate for testing (n = 94; 13%), resulting in unknown PD-L1 status. Among patients with known PD-L1 status, 159 (35%) had TC ≥25%, 303 (67%) had TC ≥1%, and 144 (32%) had TC 1%–24% (Figure 1).
Figure 1. Summary of patient distribution by tumour PD-L1 expression status (intention-to-treat population).
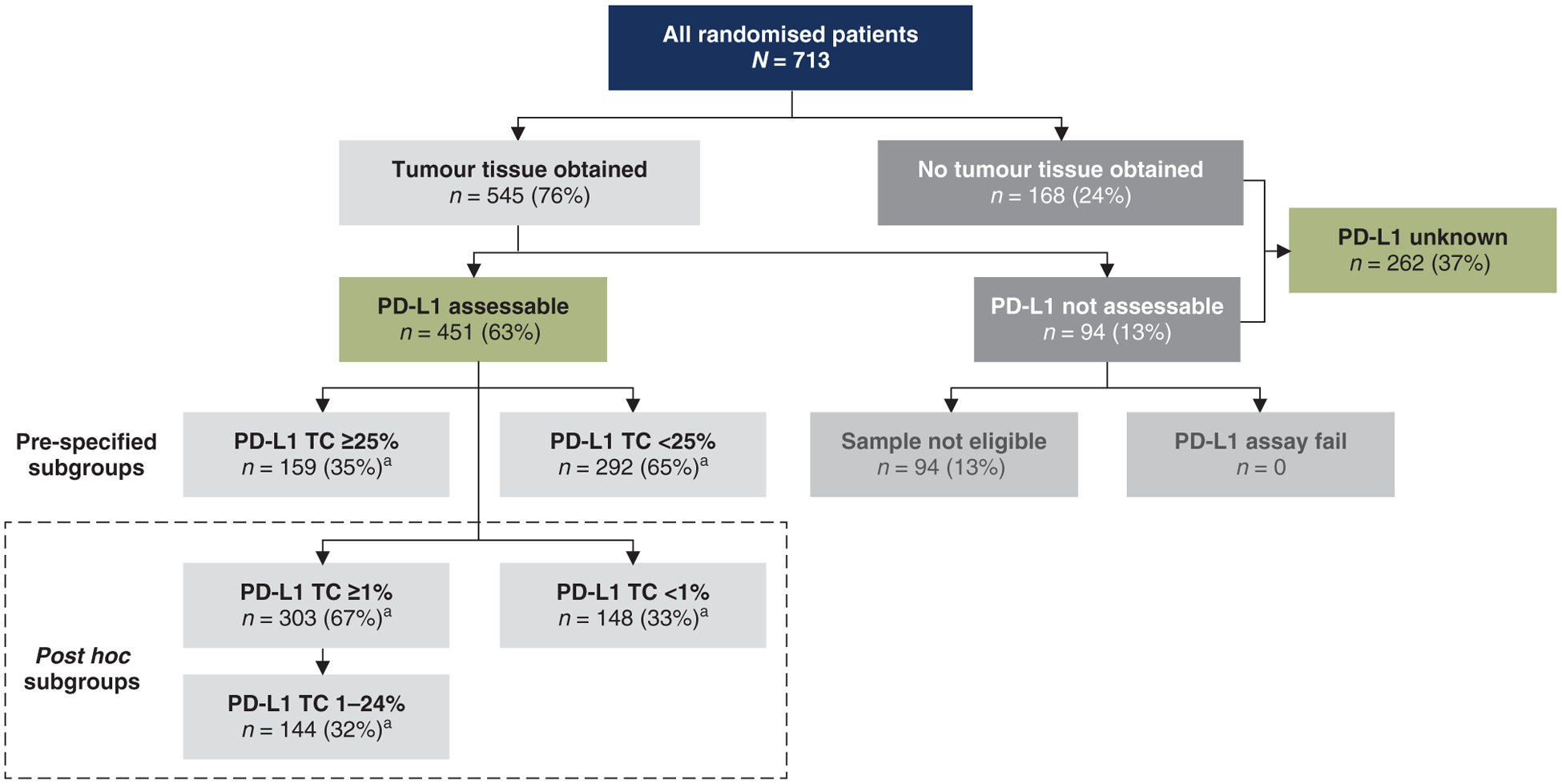
PD-L1, programmed death-ligand 1; TC, tumour cell.
a Percentages based on patients with known PD-L1 status (n = 451).
Post hoc evaluation (described in the supplementary Methods, available at Annals of Oncology online) identified clinically meaningful differences in baseline prognostic factors between PD-L1 assessable and PD-L1 unknown patients. Overall, 51% of PD-L1 assessable patients and 36% of PD-L1 unknown patients (i.e. patients with missing PD-L1 status) had squamous histology; this corresponded to an odds ratio of 0.54 (95% CI, 0.39–0.75) that samples were PD-L1 evaluable among patients with non-squamous histology versus squamous histology (i.e. patients with squamous histology were more likely to be PD-L1 assessable).
Baseline patient and disease characteristics and prior therapy, including best response to prior cCRT, were broadly well-balanced between the treatment arms within the PD-L1 subgroups (supplementary Tables S1 and S2, available at Annals of Oncology online). However, there were several notable differences within the TC <1% subgroup: proportionally more patients in the durvalumab arm, versus the placebo arm, were aged ≥65 years (48% versus 36%), Asian (33% versus 22%), male (79% versus 69%), had squamous tumour histology (59% versus 48%), and had stage IIIB disease (48% versus 41%).
A higher proportion of patients completed the protocol-defined, 12 months of treatment in the durvalumab arm compared with the placebo arm across all PD-L1 subgroups. Within the durvalumab arm, more patients in the PD-L1-enriched subgroups completed 12 months of treatment [TC ≥25% (55%) versus <25% (44%) and TC ≥1% (51%) versus <1% (41%)], which was seemingly driven by a lower incidence of disease progression in the PD-L1-enriched subgroups (supplementary Table S3, available at Annals of Oncology online). Fewer patients in the durvalumab arm received immunotherapy as anticancer therapy, after (any-cause) discontinuation of study treatment across all subgroups (supplementary Table S4, available at Annals of Oncology online); for example, in the TC <1% subgroup 11% and 34% of patients in the durvalumab and placebo arms, respectively, received subsequent immunotherapy.
Efficacy
At the DCO for its primary analysis, PFS favoured durvalumab, versus placebo, across all PD-L1 subgroups (Figure 2A–F). For example, among patients with TC ≥25%, HR for PFS with durvalumab versus placebo was 0.41 (95% CI, 0.26–0.65), corresponding to a median PFS of 17.8 months with durvalumab versus 3.7 months with placebo. Notably, PFS favoured durvalumab, versus placebo, in patients with TC < 1% [HR, 0.73 (95% CI, 0.48–1.11); median: 10.7 versus 5.6 months, respectively].
Figure 2. PFS by tumour PD-L1 expression status (BICR; intention-to-treat population).a.
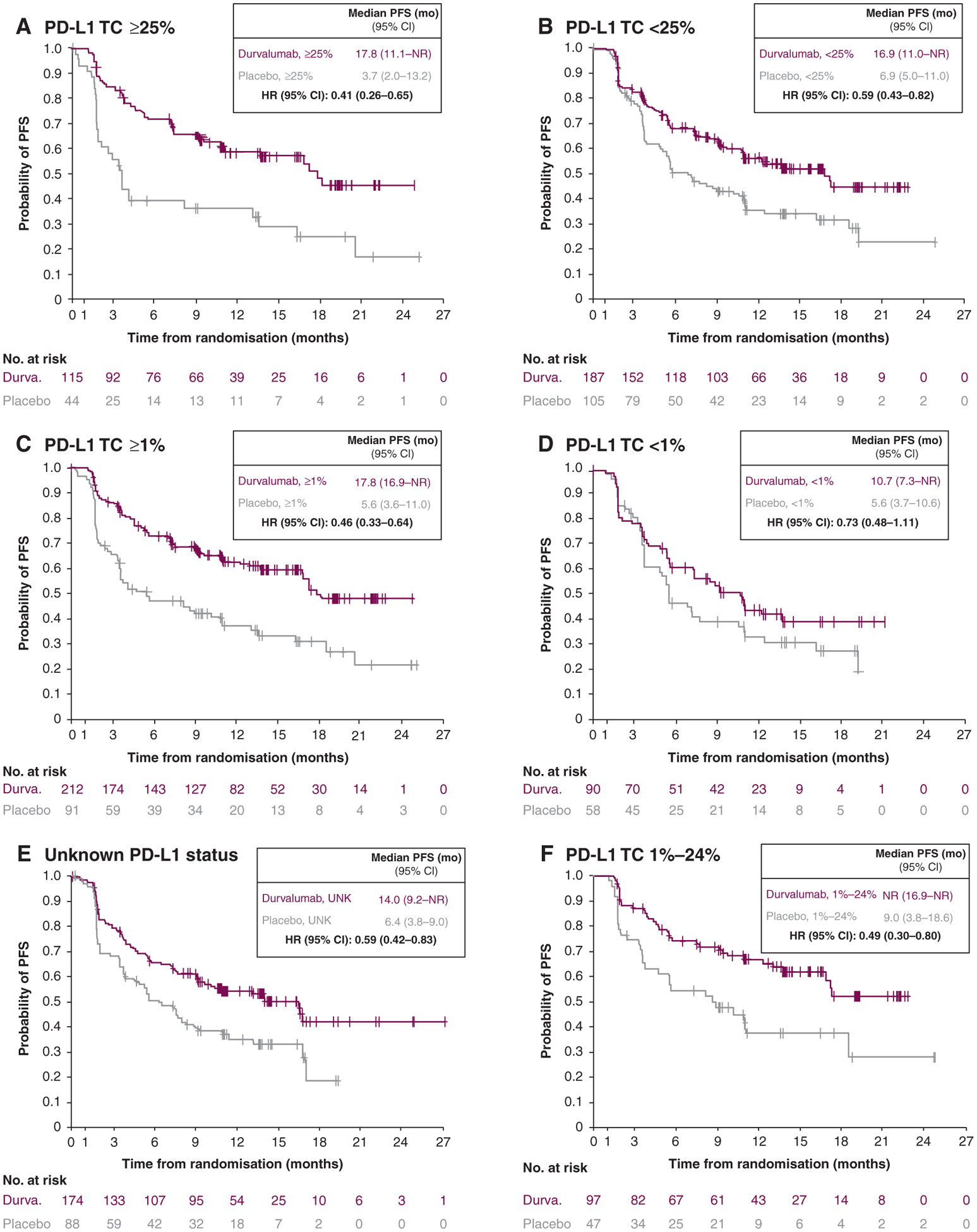
BICR, blinded independent central review; CI, confidence interval; DCO, data cutoff; Durva., durvalumab; HR, hazard ratio; mo, months; NR, not reached; PD-L1, programmed death-ligand 1; PFS, progression-free survival; TC, tumour cell; UNK, unknown.
a DCO was 13 February 2017 (DCO for the primary analysis of PFS): median duration of follow-up of 14.5 months (range, 0.2–29.9).
At the DCO for its primary analysis, OS favoured durvalumab, versus placebo, across all PD-L1 subgroups but one, patients with TC <1% (HR, 1.36; 95% CI, 0.79–2.34) (supplementary Figure S1, available at Annals of Oncology online).6,7
At the DCO for its updated analysis (approximately 3 years after the last patient was randomly allocated to the study), the observed OS results were similar (Figure 3A–F). The updated OS benefit with durvalumab, versus placebo, was observed across all subgroups except patients with TC <1% [HR, 1.14 (95% CI, 0.71–1.84)], although HR had shifted closer to 1 since the primary analysis.
Figure 3. Updated OS by tumour PD-L1 expression status (Intention-to-treat population).a.
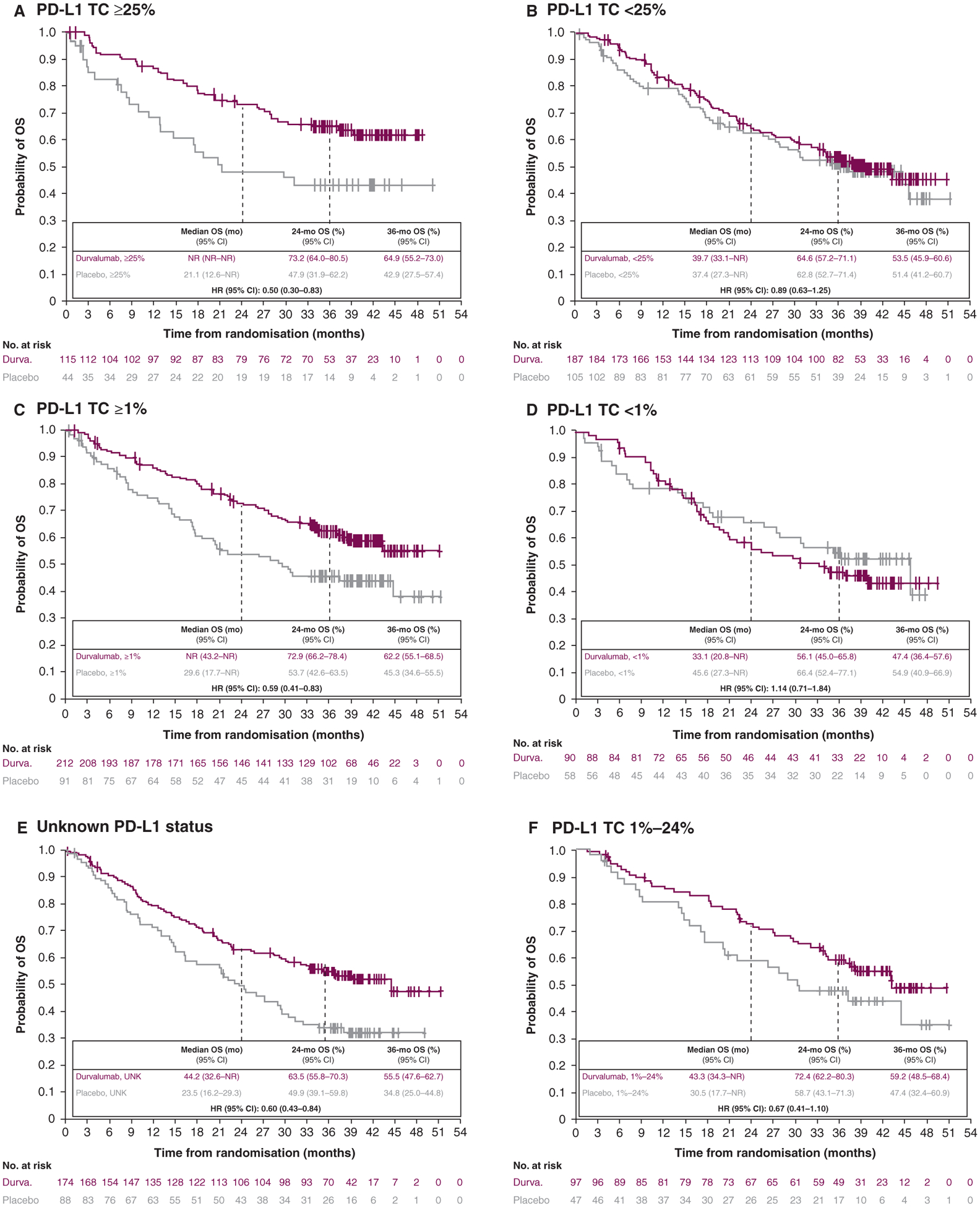
CI, confidence interval; DCO, data cutoff; Durva., durvalumab; HR, hazard ratio; mo, month; NR, not reached; OS, overall survival; PD-L1, programmed-death ligand 1; TC, tumour cell; UNK, unknown.
a DCO was 31 January 2019: median duration of follow-up of 33.3 months (range, 0.2–51.3).
The results of the exploratory multiple imputation model for OS (using the DCO for its primary analysis), which imputed missing data for the TC ≥1% and <1% subgroups, were similar: durvalumab improved OS versus placebo in the TC ≥1% subgroup [HR, 0.52 (95% CI, 0.38–0.70)] but not in the TC <1% subgroup [HR, 1.18 (95% CI, 0.73–1.89)].
Of note, based on a post hoc restricted mean survival time (RMST) analysis (using the DCO for the primary analysis of OS, as described in the supplementary Methods, available at Annals of Oncology online), there was no evidence for detriment in OS with durvalumab compared with placebo in the TC <1% subgroup [difference in RMST (95% CI), −0.6 months (−3.4 to 2.3)].
At the DCO for the primary analysis of PFS, TTDM favoured durvalumab, versus placebo, across all PD-L1 subgroups but one, the TC <1% subgroup, in which the evidence was inconclusive [HR, 0.93 (95% CI, 0.52–1.67)] (supplementary Table S5, available at Annals of Oncology online).
At the DCO for the primary analysis of PFS, ORR was greater with durvalumab, versus placebo, across all PD-L1 subgroups (supplementary Table S6, available at Annals of Oncology online), ranging from 24.7% to 31.0% with durvalumab versus 11.7% to 21.6% with placebo. Median DoR was numerically longer with durvalumab compared with placebo in the TC <1% and <25% and unknown subgroups, but NR in either treatment arm in the TC ≥1% and ≥25% subgroups.
Safety
The safety profile of durvalumab across PD-L1 subgroups was broadly consistent with that reported for durvalumab in the full analysis set,6 with similar incidences of all-cause, any-grade AEs between durvalumab- and placebo-treated patients (Table 1). AEs leading to discontinuation were more common with durvalumab, versus placebo, across all but one of the subgroups; for patients with TC <1%, a higher proportion experienced AEs leading to discontinuation with placebo (17.5%) compared with durvalumab (11.0%). The incidences of serious AEs were similar across subgroups. Any-grade pneumonitis/radiation pneumonitis was more common with durvalumab across all PD-L1 subgroups, ranging from 30.8% to 35.7% with durvalumab and 17.4% to 29.9% with placebo (supplementary Table S7, available at Annals of Oncology online); however, the incidences of grade 3 and grade 5 pneumonitis/radiation pneumonitis in the treatment arms were low (no grade 4 events were reported) and similar across all subgroups (e.g. within the TC <1% subgroup, 2.2% versus 3.5% were grade 3 and 1.1% versus 1.8% were grade 5 with durvalumab and placebo, respectively).
Table 1.
Safety summary by tumour PD-L1 expression status (as-treated population).a
| Number of patients (%) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PD-L1 TC <1% | PD-L1 TC ≥1% | PD-L1 TC <25% | PD-L1 TC ≥25% | PD-L1 TC 1%–24% | PD-L1 TC unknown | |||||||
| Durvalumab (n = 91) |
Placebo (n = 57) |
Durvalumab (n = 213) |
Placebo (n = 90) |
Durvalumab (n = 189) |
Placebo (n = 103) |
Durvalumab (n = 115) |
Placebo (n = 44) |
Durvalumab (n = 98) |
Placebo (n = 46) |
Durvalumab (n = 171) |
Placebo (n = 87) |
|
| Any-grade all-causality AEs | 88 (96.7) | 54 (94.7) | 205 (96.2) | 83 (92.2) | 184 (97.4) | 100 (97.1) | 109 (94.8) | 37 (84.1) | 96 (98.0) | 46 (100) | 167 (97.7) | 85 (97.7) |
| Grade 3/4 | 28 (30.8) | 14 (24.6) | 72 (33.8) | 21 (23.3) | 57 (30.2) | 22 (21.4) | 43 (37.4) | 13 (29.5) | 29 (29.6) | 8 (17.4) | 55 (32.2) | 31 (35.6) |
| Outcome of death | 3 (3.3) | 4 (7.0) | 8 (3.8) | 4 (4.4) | 6 (3.2) | 5 (4.9) | 5 (4.3) | 3 (6.8) | 3 (3.1) | 1 (2.2) | 10 (5.8) | 7 (8.0) |
| Leading to discontinuation | 10 (11.0) | 10 (17.5) | 36 (16.9) | 5 (5.6) | 31 (16.4) | 12 (11.7) | 15 (13.0) | 3 (6.8) | 21 (21.4) | 2 (4.3) | 27 (15.8) | 8 (9.2) |
| Serious AEs | 20 (22.0) | 11 (19.3) | 64 (30.0) | 18 (20.0) | 52 (27.5) | 17 (16.5) | 32 (27.8) | 12 (27.3) | 32 (32.7) | 6 (13.0) | 54 (31.6) | 25 (28.7) |
Includes AEs with an onset date on or after the date of the first dose, or pre-treatment AEs that increase in severity on or after the date of first dose, up to and including 90 days following the date of last dose of study medication or up to and including the date of initiation of the first subsequent therapy (whichever occurs first).
AE, adverse event; DCO, data cut off; OS, overall survival; PD-L1, programmed death-ligand 1; TC, tumour cell.
DCO was 22 March 2018 (DCO for the primary analysis of OS and an updated analysis of safety for patients completing the initial 12 months of treatment): median duration of follow-up of 25.2 months (range, 0.2–43.1).
DISCUSSION
Although the PACIFIC trial was not designed to evaluate durvalumab based on archival tumour PD-L1 expression, the results of these exploratory analyses support treatment benefit with durvalumab versus placebo irrespective of archival, pre-specified tumour PD-L1 expression status. Durvalumab treatment was associated with improved PFS and ORR, versus placebo, across all PD-L1 subgroups, including patients with unknown PD-L1 status. Additionally, durvalumab improved OS, and TTDM, versus placebo, across all subgroups but one, namely the post hoc TC <1% subgroup. Safety outcomes were comparable across PD-L1 subgroups and consistent with the full analysis set.5,6
PD-1/PD-L1 ICB has improved outcomes for patients with advanced-stage NSCLC. However, not all patients benefit from PD-1/PD-L1 ICB as monotherapy for first-line treatment of metastatic NSCLC. This observation has driven identification of predictive biomarkers of response to enhance patient selection, leading to regulatory restrictions on the use of some therapies, based on minimum threshold levels of PD-L1 expression. Based on this experience in metastatic NSCLC, durvalumab was approved in the EU for patients with locally advanced, unresectable NSCLC whose disease has not progressed following receipt of platinum-based chemotherapy and radiotherapy; however, due to the post hoc analyses, its approval was restricted to those whose tumours express PD-L1 on ≥1% of TCs.7 However, there are a number of limitations to the results underlying this restriction. For example, in the analysis of OS for the post hoc TC <1% subgroup, in which the sample size was relatively small (n = 148), the estimated CI included 1 and, therefore, no definitive conclusions can be drawn. Further limitations include the exploratory, post hoc nature of these results and the lack of assayable tumour samples for approximately 40% of randomised patients in this trial, as recently cited by a panel of international lung cancer experts who disagreed with the EMA’s decision.23 In addition, the panel noted the following: PD-L1 assessment was analysed in pre-cCRT samples, which, based on the hypothesis that cCRT may alter PD-L1 expression, may have left them inaccurate predictors of response; with only 63% of patients PD-L1 assessable and 148 patients with TC <1%, there is no guarantee that samples were missing at random, indicating potential bias; and, since randomisation was not stratified by PD-L1 expression status, there may have been prognostic imbalances in baseline characteristics across subgroups, leading to unreliability and bias in the results, thereby confounding OS data. Indeed, patients in the placebo arm within the TC <1% subgroup were more likely to be younger (aged <65 years), white, and female, and to have non-squamous histology and stage IIIA disease; such differences may have accounted for the over-performance among these patients (with respect to OS) relative to the placebo arm of the full analysis set. Additional limitations include the unplanned nature of this analysis and the small sample size of the TC <1% subgroup. The number of OS events (n = 60) in the TC <1% subgroup was inadequate to sufficiently power this analysis, which, based on the trial’s pre-specified statistical analysis plan, would have required a high benefit target (HR = 0.43) to demonstrate meaningful results. Finally, although PD-L1 expression is, at best, an imperfect predictor of response for PD-1/PD-L1 inhibitors given as monotherapy, its role is uncertain when such agents are given in sequence or in combination with other therapies (e.g. CRT or other immunotherapies).
Besides demonstrating efficacy across the PD-L1 subgroups, durvalumab exhibited a manageable safety profile irrespective of tumour PD-L1 expression status. Moreover, exploratory analyses of patients from the PACIFIC trial found that PD-L1 expression had no clinically meaningful impact on patient-reported outcomes (symptoms, functioning, and global health status/quality of life).24 Importantly, durvalumab was effective and well tolerated in patients with unknown PD-L1 status (for whom durvalumab was associated with a greater than 20-month benefit in OS). This addresses an unmet clinical need, as up to 40% of patients do not have tumour biopsies suitable for histological PD-L1 assessment (e.g. due to inadequate tissue collection using fine needle aspiration), and cytological assessment of PD-L1 expression, while feasible, is not yet widely standardised in routine clinical practice.25
Conclusion
In conclusion, these findings demonstrated that treatment benefit with durvalumab versus placebo was evident in patients with unresectable, stage III NSCLC from the PACIFIC trial, with consistently manageable safety. PFS benefit with durvalumab was observed irrespective of tumour PD-L1 expression, and OS benefit, in post hoc analyses, was consistently observed in patients with TC PD-L1 expression ≥1%. However, a small sample size with too few events, overlapping CIs, and inadvertent prognostic imbalances favouring the placebo group preclude robust conclusions regarding OS in patients with TC PD-L1 expression <1%. Consequently, prospectively planned studies to assess outcomes with immunotherapies in patients with different levels of tumour PD-L1 expression are warranted (e.g. the phase III PACIFIC-5 trial of durvalumab after concurrent or sequential CRT in patients with stage III NSCLC26), since the PACIFIC trial was not designed to do so.
Supplementary Material
{kind=link}
ACKNOWLEDGEMENTS
The authors would like to thank the patients, their families and caregivers, and all investigators involved in this study. Medical writing support, which was in accordance with Good Publication Practice (GPP3) guidelines, was provided by Aaron Korpal, PhD, and Andrew Gannon, MS, MA, of Cirrus Communications (Manchester, UK), an Ashfield company, and was funded by AstraZeneca. Professor Corinne Faivre-Finn is supported by a grant from the National Institute for Health Research Manchester Biomedical Research Centre.
FUNDING
This study was funded by AstraZeneca (ClinicalTrials.gov: NCT02125461).
DISCLOSURE
LP-A is a board member of Genomica and has received honoraria from Roche/Genentech, Eli Lilly, Pfizer, Boehringer Ingelheim, Bristol-Myers Squibb, Merck Sharp and Dohme, AstraZeneca, Merck Serono, Pharmamar, Novartis, Celgene, Sysmex, Amgen, and Incyte, and travel, accommodations or expenses from Roche, AstraZeneca, AstraZeneca Spain, Merck Sharp and Dohme, Bristol-Myers Squibb, Eli Lilly, and Pfizer; AS has received advisory fees from Array BioPharma and Incyte, honoraria from CytomX Therapeutics, AstraZeneca/MedImmune and Merck, research funding from LAM Therapeutics, and institutional research support from Roche, AstraZeneca/MedImmune, Boehringer Ingelheim, Astellas Pharma, Novartis, NewLink Genetics, Incyte, AbbVie, Ignyta, LAM Therapeutics, TrovaGene, Takeda, MacroGenics, CytomX Therapeutics, Astex Pharmaceuticals, Bristol-Myers Squibb, Loxo, and Arch Therapeutics; DR has received honoraria from Merck and Nanobiotix, consultant fees from AstraZeneca and Suvica, and advisory board fees from AstraZeneca, Merck, Genentech, and Nanobiotix; DP has received advisory or lecture fees from AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Celgene, Daiichi Sankyo, Eli Lilly, Merck, MedImmune, Novartis, Pfizer, prIME Oncology, Peer CME, and Roche, honoraria from AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Celgene, Eli Lilly, Merck, Novartis, Pfizer, prIME Oncology, Peer CME, and Roche, institutional research funding from AstraZeneca, Bristol-Myers Squibb, AbbVie, Boehringer Ingelheim, Eli Lilly, Merck, Novartis, Pfizer, Roche, Medimmune, Sanofi-Aventis, Taiho Pharma, Novocure, and Daiichi Sankyo, and travel, accommodations or expenses from AstraZeneca, Bristol-Myers Squibb, Boehringer Ingelheim, Roche, Merck, Novartis, prIME Oncology, and Pfizer; DD has received institutional research funding from E.R. Squibb and Sons, AstraZeneca, Boehringer Ingelheim, Genentech, Eli Lilly and Company, Novartis Pharmaceuticals, Pfizer, Celgene, and Roche; AV has received honoraria from AstraZeneca, Gilead, and Seattle Genetics; RH has received advisory fees and honoraria from AstraZeneca, Merck Sharp and Dohme, Novartis, Roche, Bristol-Myers Squibb, and Eli Lilly; SS has received research grants from Varian Medical Systems and ViewRay Inc., and honoraria from AstraZeneca, Eli Lilly, Merck Sharp and Dohme, and Celgene; CJL has received honoraria from Roche/Genentech, Eli Lilly, Pfizer, Boehringer Ingelheim, Bristol-Myers Squibb, Merck Sharp and Dohme, AstraZeneca, Novartis, Celgene, Takeda, and Gilead; and travel, accommodations or expenses from Roche, AstraZeneca, Merck Sharp and Dohme, Bristol-Myers Squibb, Eli Lilly and Pfizer; BAP has received advisory fees from AstraZeneca and Bristol-Myers Squibb; and institutional research support from Bristol-Myers Squibb. A-MB and PAD are full-time employees of AstraZeneca with stock ownership; HB is an independent contractor, funded by AstraZeneca; CW was a full-time employee of AstraZeneca when the work was completed; SJA has received advisory fees from Bristol-Myers Squibb, Novartis, Merck, Boehringer Ingelheim, AstraZeneca/MedImmune, Cellular Biomedicine Group, and Memgen; CF-F has received research funding and travel support from Merck, AstraZeneca, and Elekta, and travel support from Pfizer; the remaining authors declare no potential conflicts of interest.
REFERENCES
- 1.Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27(4):450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herzberg B, Campo MJ, Gainor JF. Immune checkpoint inhibitors in non-small cell lung cancer. Oncologist. 2017;22(1):81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–1086. [DOI] [PubMed] [Google Scholar]
- 4.Stewart R, Morrow M, Hammond SA, et al. Identification and characterization of MEDI4736, an antagonistic anti-PD-L1 monoclonal antibody. Cancer Immunol Res. 2015;3(9):1052–1062. [DOI] [PubMed] [Google Scholar]
- 5.Antonia SJ, Villegas A, Daniel D, et al. Durvalumab after chemoradiotherapy in stage III non-small-cell lung cancer. N Engl J Med. 2017;377(20):1919–1929. [DOI] [PubMed] [Google Scholar]
- 6.Antonia SJ, Villegas A, Daniel D, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med. 2018;379(24):2342–2350. [DOI] [PubMed] [Google Scholar]
- 7.European Medicines Agency. Durvalumab (Imfinzi). Summary of product characteristics 2018. Available at https://www.ema.europa.eu/en/documents/product-information/imfizi-epar-product-information_en.pdf.AccessedJanuary 13, 2020.
- 8.Hui R, Ozguroglu M, Villegas A, et al. Patient-reported outcomes with durvalumab after chemoradiotherapy in stage III, unresectable, non-small-cell lung cancer (PACIFIC): a randomised, controlled, phase 3 study. Lancet Oncol. 2019;20(12):1670–1680. [DOI] [PubMed] [Google Scholar]
- 9.US Food and Drug Safety Administration. IMFINZI (Durvalumab) Label 2018. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761069s002lbl.pdf.AccessedOctober 15, 2019. [Google Scholar]
- 10.Pharmaceuticals and Medical Devices Agency (PMDA). List of Approved Products Financial Year 2018. Available at https://www.pmda.go.jp/english/review-services/reviews/approved-information/drugs/0002.html.AccessedOctober 15, 2019.
- 11.Chen DS, Irving BA, Hodi FS. Molecular pathways: next-generation immunotherapy–inhibiting programmed death-ligand 1 and programmed death-1. Clin Cancer Res. 2012;18(24):6580–6587. [DOI] [PubMed] [Google Scholar]
- 12.Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8(6):467–477. [DOI] [PubMed] [Google Scholar]
- 13.Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48(3):434–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu CT, Chen WC, Chang YH, et al. The role of PD-L1 in the radiation response and clinical outcome for bladder cancer. Sci Rep. 2016;6: 19740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dovedi SJ, Cheadle EJ, Popple AL, et al. Fractionated radiation therapy stimulates antitumor immunity mediated by both resident and infiltrating polyclonal T-cell populations when combined with PD-1 blockade. Clin Cancer Res. 2017;23(18):5514–5526. [DOI] [PubMed] [Google Scholar]
- 16.Peng J, Hamanishi J, Matsumura N, et al. Chemotherapy induces programmed cell death-ligand 1 overexpression via the nuclear factor-kB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res. 2015;75(23):5034–5045. [DOI] [PubMed] [Google Scholar]
- 17.McCall NS, Dicker AP, Lu B. Beyond concurrent chemoradiation: the emerging role of PD-1/PD-L1 inhibitors in stage III lung cancer. Clin Cancer Res. 2018;24(6):1271–1276. [DOI] [PubMed] [Google Scholar]
- 18.Kordbacheh T, Honeychurch J, Blackhall F, et al. Radiotherapy and anti-PD-1/PD-L1 combinations in lung cancer: building better translational research platforms. Ann Oncol. 2018;29(2):301–310. [DOI] [PubMed] [Google Scholar]
- 19.Tsao MS, Le Teuff G, Shepherd FA, et al. PD-L1 protein expression assessed by immunohistochemistry is neither prognostic nor predictive of benefit from adjuvant chemotherapy in resected non-small cell lung cancer. Ann Oncol. 2017;28(4):882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.US Food and Drug Safety Administration. KEYTRUDA (pembrolizumab) Label 2018. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125514s034lbl.pdf.AccessedOctober 15, 2019.
- 21.US Food and Drug Safety Administration. OPDIVO (nivolumab) Label 2019. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125554s070lbl.pdf.AccessedOctober 15, 2019.
- 22.Gray JE, Villegas A, Daniel D, et al. Brief report: three-year overall survival with durvalumab after chemoradiotherapy in stage III NSCLC - update from PACIFIC. J Thorac Oncol. 2020;15(2):288–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peters S, Dafni U, Boyer M, et al. Position of a panel of international lung cancer experts on the approval decision for use of durvalumab in stage III non-small-cell lung cancer (NSCLC) by the Committee for Medicinal Products for Human Use (CHMP). Ann Oncol. 2019;30(2): 161–165. [DOI] [PubMed] [Google Scholar]
- 24.Garassino MC, Paz-Ares L, Hui R, et al. Patient-reported outcomes with durvalumab by PD-L1 expression in unresectable, stage III NSCLC (PACIFIC). Oral presentation at the European Lung Cancer Congress. April10–13, 2019; Geneva, Switzerland (LBA2). [Google Scholar]
- 25.Dietel M, Bubendorf L, Dingemans AM, et al. Diagnostic procedures for non-small-cell lung cancer (NSCLC): recommendations of the European Expert Group. Thorax. 2016;71(2):177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu Y-L, Wang L, Sendur MAN, et al. PACIFIC-5: phase 3 study of durvalumab after either concurrent or sequential chemoradiotherapy (CRT) in patients with stage III NSCLC. Ann Oncol. 2019;30(9):339TiP. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.