

Abstract
Photothermal therapy (PTT) and neoantigen cancer vaccine each offers minimally invasive and highly specific cancer therapy; however, they are not effective against large established tumors due to physical and biological barriers that attenuate thermal ablation and abolish anti-tumor immunity. Here, we designed and performed comparative study using small (~ 50 mm3) and large (> 100 mm3) tumors to examine how tumor size affects the therapeutic efficiency of PTT and neoantigen cancer vaccine. We show that spiky gold nanoparticle (SGNP)-based PTT and synergistic dual adjuvant-based neoantigen cancer vaccine can efficiently regress small tumors as a single agent, but not large tumors due to limited internal heating and immunosuppressive tumor microenvironment (TME). We report that PTT sensitizes tumors to neoantigen cancer vaccination by destroying and compromising the TME via thermally induced cellular and molecular damage, while neoantigen cancer vaccine reverts local immune suppression induced by PTT and shapes residual TME in favor of anti-tumor immunity. The combination therapy efficiently eradicated large local tumors and also exerted strong abscopal effect against pre-established distant tumors with robust systemic anti-tumor immunity. Thus, PTT combined with neoantigen cancer vaccine is a promising nano-immunotherapy for personalized therapy of advanced cancer.
Keywords: Photothermal therapy, neoantigen, cancer vaccine, nano-immunotherapy
Graphical Abstract
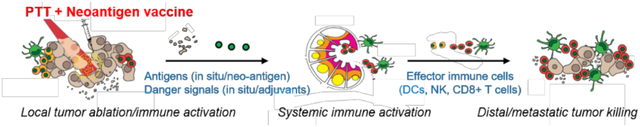
Photothermal therapy and neoantigen cancer vaccine together synergize and improve local tumor ablation, local and systemic immune activation, and tumor trafficking of effector immune cells, leading to efficient control of large local tumors as well as distant metastatic tumors. Photothermal therapy combined with neoantigen cancer vaccination offers a promising strategy for personalized therapy of advanced cancer.
1. Introduction
Photothermal therapy (PTT) offers minimally invasive and targeted cancer therapy using photosensitizers that can absorb low energy near-infrared (NIR) light and induce local heat transfer for thermal ablation of tumors.[1] PTT can minimize collateral damage to the surrounding normal tissues by region-selective administration of photosensitizer and NIR light into the tumor areas.[2] However, the short range of heat diffusion near photosensitizer and limited light penetration typically lead to incomplete ablation of large tumors and rapid tumor recurrence from residual cancer cells in the PTT-treated tumor margin.[3] In addition, although recent studies demonstrated that PTT can stimulate innate and adaptive immune responses by promoting the release of pro-inflammatory cytokines and chemokines, maturation of dendritic cells (DCs), and activation of effector T cells,[4] the resulting anti-tumor immunity is generally sub-optimal for inhibiting distant tumors. Moreover, PTT has been reported to promote local immune suppression by upregulation of inhibitory enzymes and checkpoint ligands[3, 5] and recruitment of immunosuppressive cells,[4b, 6] resulting in the outgrowth of cancer cells in the treatment margin. PTT has been extensively studied to treat local tumors that are directly accessible for laser irradiation, and it has been shown that residual distant tumors can be treated by PTT combined with other therapies, including immune adjuvants, immune checkpoint blockers, adoptive T cell transfer, and chemotherapies.[3, 4b, 5–7] However, it still remains challenging for PTT-based therapies to effectively eliminate large established tumors and exert robust abscopal effect against disseminated systemic metastasis.
Therapeutic cancer vaccine is a clinically relevant cancer immunotherapy that can generate endogenous tumor-specific immune responses to destroy cancer cells.[8] Neoantigens formed by non-synonymous cancer mutations are highly tumor-specific and immunogenic as they are entirely absent in normal cells and thus can bypass the central tolerance.[9] Neoantigen-specific cytotoxic T cells (CTLs) were implicated not only in the rejection of mouse cancers[10] but also improved prognosis in human cancers,[11] highlighting the promise of neoantigen vaccines for personalized cancer immunotherapy.[12] Recent clinical trials have demonstrated the potential of neoantigen-based cancer vaccination in small cohorts of patients.[13] However, traditional neoantigen vaccines employed in these initial trials generated low levels of neoantigen-specific CTLs, potentially due to the weak immune stimulation capacity of the soluble vaccines.[13] In addition, the immunosuppressive tumor microenvironment (TME) presents a major obstacle to cancer immunotherapy as it supports tumor growth and progression with multiple immunosuppressive mechanisms, including various immunosuppressive cells, ligands, cytokines, enzymes, as well as the stromal cells, and other extracellular matrix components.[14] In particular, advanced stage tumors are characterized by a large fraction of immunosuppressive stromal cells in TME[15] that hampers the in vivo performance and therapeutic efficacy of cancer vaccines.[8b, 16] Thus, inefficient induction of tumor-specific CTLs and immunosuppressive TME are critical hurdles to overcome for the development of successful immunotherapies against advanced stage cancers.[8b]
Here, we report that PTT combined with neoantigen cancer vaccine efficiently eradicates large local tumors as well as pre-established metastatic tumors, whereas individual treatment of PTT or neoantigen vaccine is not effective in this late-stage tumor model. We have previously reported that NIR-PTT using spiky gold nanoparticles (SGNPs) exhibited limited anti-tumor efficacy against large established tumors (~ 100 mm3) due to insufficient internal heating.[7b] In this study, we performed a comparative study using small (~ 50 mm3) and large (> 100 mm3) tumors and report that SGNP-based NIR-PTT effectively eliminated small tumors but not large tumors. In parallel, we have developed a potent neoantigen cancer vaccine using a dual adjuvant combination of Toll-like receptor (TLR)-3 agonist pIC and TLR-9 agonist CpG.[17] We have shown that pIC and CpG strongly activated DCs in a synergistic manner by stimulating distinct TLR signals and that a simple soluble vaccine composed of dual TLR agonists and neoantigen peptides generated potent neoantigen-specific CD8+ T cell response as high as ~ 30% in the systemic circulation, leading to the eradication of small tumors (~ 50 mm3). However, large tumors (> 100 mm3) significantly diminished the therapeutic efficiency of dual TLR agonist-based neoantigen cancer vaccine, potentially due to the immunosuppressive TME. Importantly, the combination of PTT and neoantigen cancer vaccine led to complete regression of large primary tumors. Furthermore, the combination therapy also exerted robust abscopal effect against pre-established metastatic tumors for which neither PTT nor neoantigen cancer vaccine was effective. Mechanistically, PTT exerted anti-tumor efficacy by direct thermal ablation of tumor tissue, while neoantigen cancer vaccine elicited anti-tumor immunity in the local tumors as well as in the peripheral tissues. PTT debulked large established tumors with cellular and molecular damage, augmented anti-tumor immunity, and sensitized the tumors to anti-tumor immune cells elicited by neoantigen cancer vaccine, leading to robust local and abscopal effect. Thus, PTT combined with neoantigen cancer vaccine represents a promising cancer therapy for the treatment of advanced cancer.
2. Results
2.1. Photothermal therapy against small and large tumors
We synthesized and utilized SGNPs as a NIR photosensitizer for photothermal cancer therapy as previously described.[7b] SGNPs were tuned to exhibit absorption peak at ~808 nm, the wavelength of NIR laser, for the maximum laser absorption and photothermal conversion (Figure 1A). SGNPs were further surface-passivated with polyethyleneglycol (PEG) for improving colloidal stability and reducing systemic toxicity and immunogenicity.[18] Transmission electron microscope (TEM) image confirmed uniform nano-spiky structures (Figure 1B), with the effective diameter of 44 ± 8.1 nm calculated from the projected areas of individual particles (Figure 1C). For PTT in vivo, we established MC38 colon carcinoma model by inoculating 5 × 105 MC38 cells subcutaneously on the right flank of C57BL/6 mice. On day 9 when the tumor volume reached 47 ± 17 mm3, we performed intratumoral injection of PBS or SGNPs (100 fmol), followed by continuous-wave laser irradiation (808 nm, 0.7 W/cm2 for 5 min) one day later (Figure 1D). SGNP treatment increased the tumor temperature by +12 °C (Figure 1E) and efficiently inhibited tumor growth (Figure 1F), with 63% of animals eliminating tumors (P < 0.001 compared with PBS, Figure 1G). In contrast, animals treated with PBS exhibited a significantly lower temperature increase of +6 °C and failed to inhibit tumor growth with all animals succumbing to tumors by day 35 (Figure 1E–G). In order to examine the impact of PTT on the induction of systemic anti-tumor T cell response, we analyzed peripheral blood mononuclear cells (PBMCs) on day 7 after laser irradiation for the frequency of CD8+ T cells specific to Adpgk peptide, a neoantigen exclusively expressed in MC38 cells.[12c] Flow cytometric analysis using Adpgk-major histocompatibility complex class I (H-2Db) tetramer indicated that SGNP-PTT induced minimal Adpgk-specific CD8+ T cell response in the systemic circulation (Figure 1H).
Figure 1.
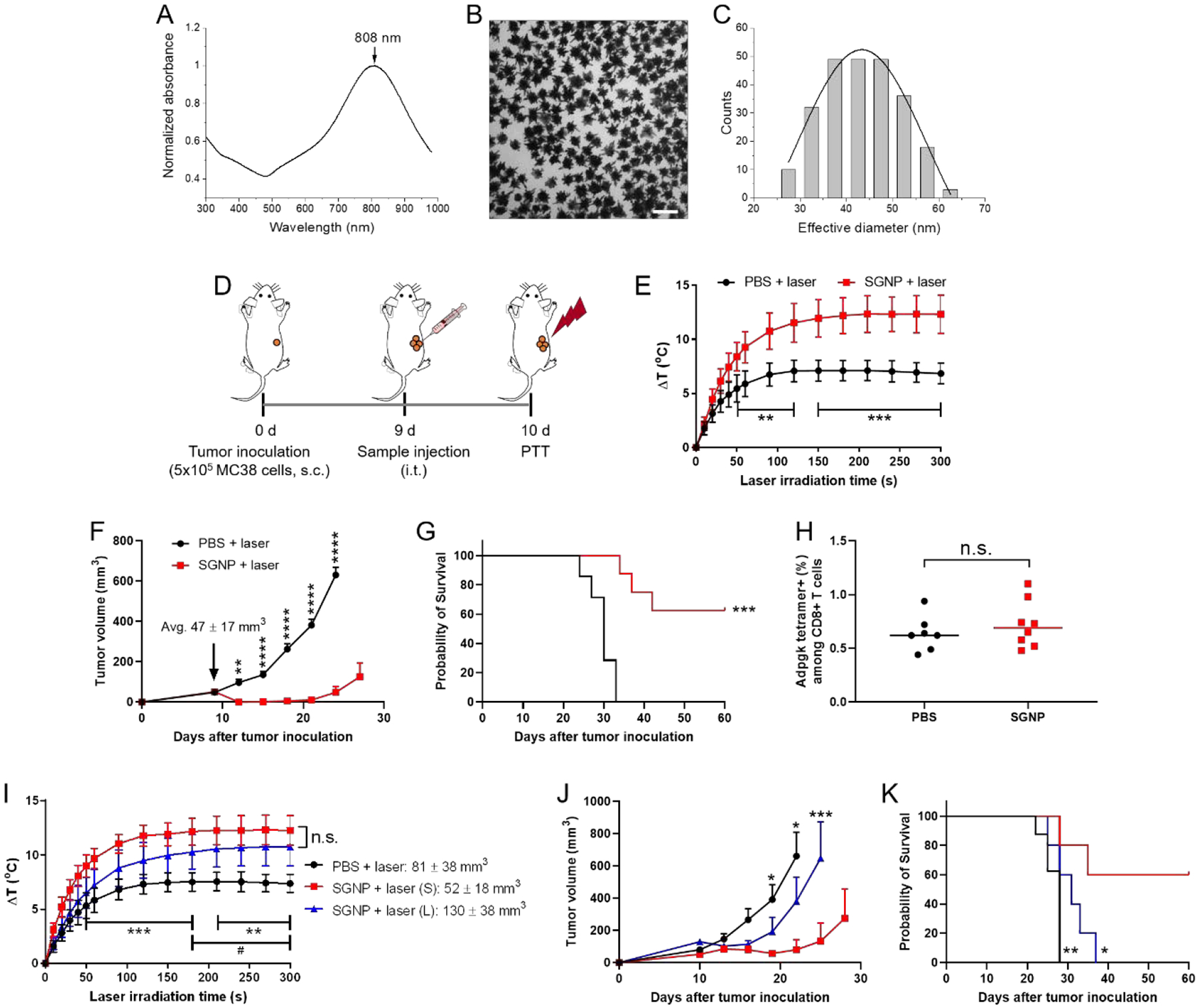
Tumor ablation by SGNP-mediated NIR PTT. (A-C) Absorption spectrum (A), TEM image (B) and the corresponding size distribution (C) of SGNPs. Scale bar = 100 nm. (D) Schematic of PTT regimen. (E) Increase in the local temperature in tumors during laser irradiation. (F,G) Tumor growth (F), and Kaplan–Meier survival curve (G) of MC38 tumor-bearing mice after administration of PBS or SGNP, followed by laser irradiation. (H) Frequency of Adpgk-specific CD8+ T cells in PBMCs, measured by flow cytometry after 7 days post laser irradiation. (I-K) Local temperature increase in the tumors during laser irradiation (I), tumor growth (J), and Kaplan–Meier survival curve (K) of MC38 tumor-bearing mice depending on the tumor size. Small and large tumors are denoted by (S) and (L), respectively. The data show mean ± s.d. (E,I) or s.e.m. (F,J), with n = 7 (PBS + laser) or n = 8 (SGNP + laser) for (E-H) and n = 8 (PBS) or n = 5 (other groups) for (I-K). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, analyzed by unpaired, two-tailed t-test with Welch’s correction (H), two-way ANOVA with Bonferroni multiple comparisons post-test (E,F,I,J), or by log-rank (Mantel–Cox) test (G,K), for comparison with SGNP + laser (S) (*), or PBS + laser vs. SGNP + laser (L) (#).
Next, we examined the therapeutic efficacy of PTT against large local tumors. We controlled the tumor size by adjusting the number of MC38 cells inoculated on the mice. Small or large tumors were established with 5 × 105 (same as above) or 2 × 106 MC38 cells, respectively. Mice were treated with SGNP on day 9 and laser irradiation on day 10 as above. Administration of PBS, followed by laser irradiation, served as the control group. Upon SGNP-PTT, small tumors (52 ± 18 mm3) exhibited higher local tumor temperature than large tumors (130 ± 38 mm3) although they were not statistically different (Figure 1I). SGNP-PTT exhibited reproducible therapeutic efficacy against small tumors as in Figure 1F and G, leading to efficient tumor inhibition (Figure 1J) and long-term survival for 60% mice (Figure 1K). However, despite elevated local tumor temperature (Figure 1I), SGNP-PTT was not effective against large tumors, showing minimal benefit in tumor growth inhibition or survival (Figure 1J,K), compared with the PBS control. Regardless of the tumor size, SGNP-PTT failed to induce systemic anti-tumor T cells, as shown by the basal level of Adpgk-specific CD8+ T cells among PBMCs (Supporting information Figure S1). Overall, these results demonstrated that SGNP-PTT eliminated small, local tumors by direct thermal ablation without eliciting measurable systemic anti-tumor T cells; however, SGNP-PTT was not effective against large tumors.
2.2. Neoantigen cancer vaccination against small and large tumors
It has been well-documented that immunostimulatory pIC and CpG elicit immune responses and promote cytokine production via Toll-interleukin 1 receptor domain–containing adapter inducing interferon-β (TRIF) and myeloid differentiation factor 88 (MyD88) pathways, respectively.[19] We used bone marrow-derived dendritic cells (BMDCs) to treat with the dual adjuvants and examined their synergistic activity. We measured pro-inflammatory cytokines MyD88-dependent IL-12p70 and TRIF-dependent interferon-β (IFN-β) secreted from BMDCs with a pairwise combination of pIC and CpG.[20] These cytokines are independently produced via distinct signaling pathways.[21] In particular, IL-12p70 is known to be critical for the generation of T helper 1 (Th1)-polarized response and priming of anti-tumor CD8+ T cells,[22] and IFN-β is crucial for anti-tumor efficacy.[23] When used as a single agent, the dose of pIC employed in our study (1 – 100 μg/ml) was sub-optimal for the induction of IL-12p70 and IFN-β, whereas CpG at the dose of > 100 ng/ml promoted IL-12p70 production, but not IFN-β (Figure 2A,C). Notably, the combination of pIC and CpG triggered markedly increased secretion of IL-12p70 and IFN-β by BMDCs at the doses of pIC > 1 μg/ml and CpG > 100 ng/ml. To reveal the synergy in a quantitative manner, synergistic fold-increase was calculated by dividing the observed cytokine concentration by the predictive additive concentration from the individual adjuvant treatment,[24] which clearly showed the synergistic effect of pIC and CpG combination, with the maximum 9.4-fold and 21-fold increase in the production of IL-12p70 and IFN-β, respectively (Figure 2B,D). The highest fold-increase was induced by the intermediate concentrations of pIC and CpG, suggesting that the balanced contribution of pIC and CpG is crucial for the synergy. We also examined the dual TLR agonist combination using monophosphoryl lipid A (MPLA), a widely used TLR-4 agonist.[25] We observed similar synergistic IL12-p70 production for CpG + MPLA (Supporting information Figure S2), whereas no cytokine was detected for pIC + MPLA (data not shown).
Figure 2.
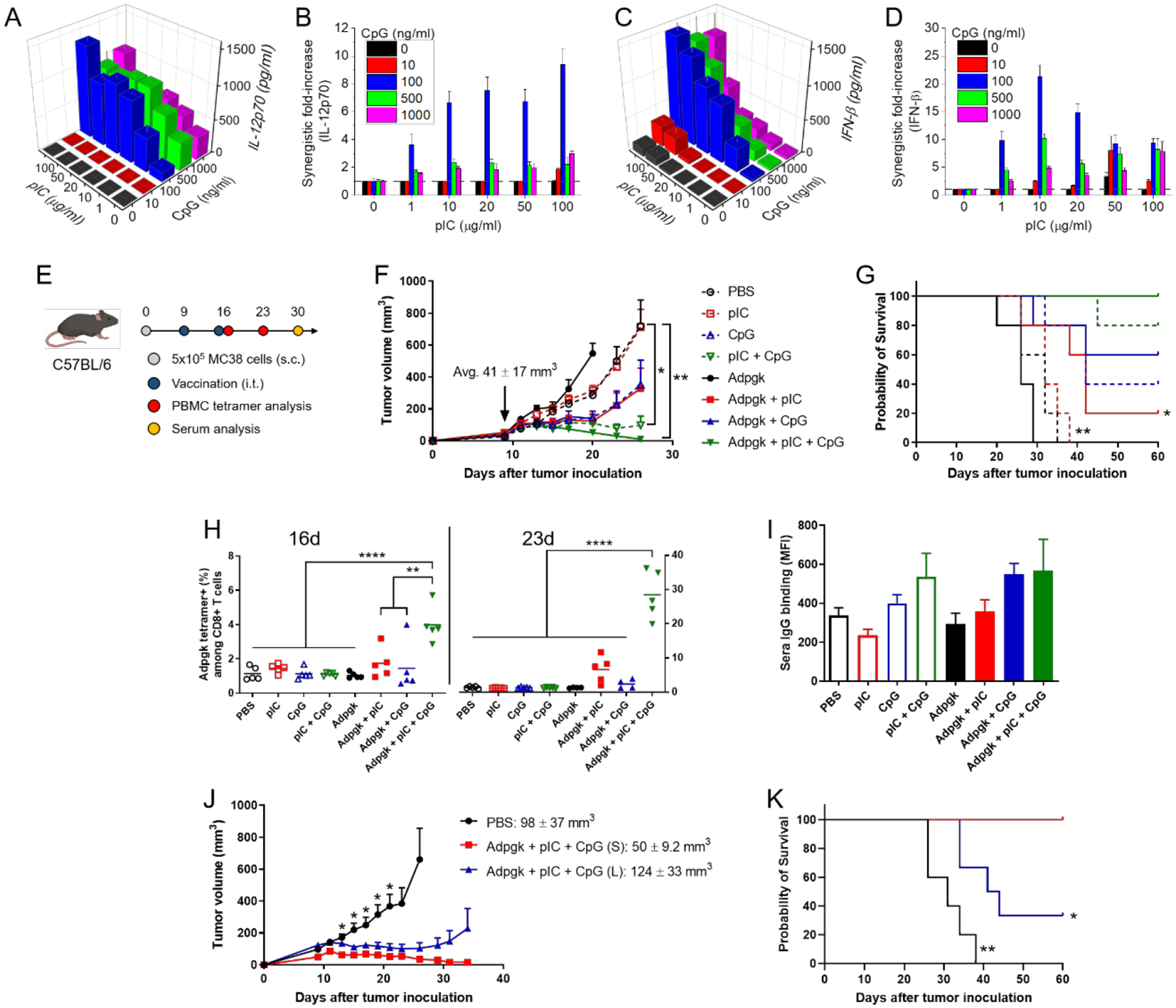
Dual pIC and CpG adjuvants for neoantigen cancer vaccination. (A-D) Secretion of IL-12p70 (A) and IFN-β (C) from BMDCs measured after the combinational treatment of pIC (0 – 100 μg/ml) and CpG (0 – 1000 ng/ml) for 24 h. The synergistic fold increases in the IL-12p70 (B) and IFN-β (D) were calculated over the expected additive value. (E) Schematic of vaccination regimen. (F,G) Tumor growth (F), and Kaplan–Meier survival curve (G) of MC38 tumor-bearing mice after administration of various combination of pIC, CpG, and Adpgk peptide. (H) Frequency of Adpgk-specific CD8+ T cells in PBMCs measured by flow cytometry after 7 days of prime (d16) and boost (d23) vaccination. (I) Mean fluorescence intensity (MFI) of MC38 cell-binding sera IgG collected and analyzed after 2 weeks of boost vaccination. (J-K) Tumor growth (J), and Kaplan–Meier survival curve (K) of MC38 tumor-bearing mice depending on the tumor size. Small and large tumors are denoted by (S) and (L), respectively. The data show mean ± s.d. (A-D) or s.e.m. (F,I,J), with n = 5 for (A-D and F-I), and n = 5 (PBS), n = 4 (Adpgk + pIC + CpG (S)), or n = 6 (Adpgk + pIC + CpG (L)) for (J-K). *P < 0.05, **P < 0.01, and ****P < 0.0001, analyzed by one-way (F,H) or two-way (J) ANOVA with Bonferroni multiple comparisons post-test, or by log-rank (Mantel–Cox) test (G,K), for comparison with Adpgk + pIC + CpG (S).
Having shown the potency of pIC and CpG combination in vitro, we examined the dual adjuvants combined with Adpgk neoantigen peptide as a personalized cancer vaccine in the MC38 tumor model. C57BL/6 mice were inoculated subcutaneously with 5 × 105 MC38 cells on the right flank on day 0. When the tumor volume reached 41 ± 17 mm3 on day 9, vaccines were administered directly into tumors, followed by boost injection on day 16 (Figure 2E). We chose the intratumoral route of vaccination in our study as preclinical and clinical studies have shown that intratumoral delivery of immunotherapy could elicit immunity without overt systemic exposure.[26] To assess synergistic immune stimulation by the dual adjuvants, we compared the therapeutic efficacy of single vs. dual adjuvants formulated with or without Adpgk peptide. Without Adpgk peptide, injection of pIC (50 μg) alone failed to inhibit tumor growth, whereas CpG (15 μg) exhibited moderate inhibition (Figure 2F). Notably, pIC + CpG showed greater tumor suppression than CpG, which indicates that synergistic immune stimulation is linked to strong anti-tumor effect (Figure 2F). Moreover, addition of Adpgk (15 μg) to either pIC or CpG improved their anti-tumor efficacy (Figure 2F,G). Importantly, Adpgk + pIC + CpG triple combination exerted a remarkable anti-tumor effect, leading to complete tumor eradication in 100% mice (Figure 2F,G). On the other hand, Adpgk peptide alone promoted faster tumor growth than PBS, potentially by inducing immunological tolerance,[27] which demonstrates the indispensable role of strong immune adjuvants in cancer vaccination. Tetramer staining performed on 7 days after prime (16d) and boost (23d) showed robust elicitation of Adpgk-specific CD8+ T cells by Adpgk + pIC + CpG, achieving 4.0 ± 1.1% tetramer+ among CD8+ T cells in PBMCs after the first injection (2.3- and 2.7-fold higher than Adpgk + pIC and Adpgk + CpG, respectively, P < 0.01) and 28 ± 7.0% after the boost injection (4.3- and 12-fold higher than Adpgk + pIC and Adpgk + CpG, respectively, P < 0.0001) (Figure 2H). Omitting any single component from Adpgk + pIC + CpG vaccine resulted in significantly decreased Adpgk-specific CD8+ T cell response (Figure 2H), demonstrating that all three components of Adpgk, pIC, and CpG are required for induction of robust tumor-specific CD8+ T cells. In addition, the dual adjuvant treatment induced serum IgG that bound live MC38 cells, showing effective elicitation of humoral immunity (Figure 2I).[28]
Having demonstrated the therapeutic potency of Adpgk + pIC + CpG vaccine against small tumors, we next investigated its anti-tumor efficacy against large tumors. Tumor size was controlled by the number of MC38 cells inoculated, as described above. Whereas Adpgk + pIC + CpG reproducibly induced strong anti-tumor effect and eliminated small tumors (50 ± 9.2 mm3), it was not effective against large tumors (124 ± 33 mm3), with only 33% complete response rate (Figure 2J,K). Increase in the tumor size had no impact on the priming of Adpgk-specific CD8+ T cells or the production of MC38 cell-binding sera IgG in the systemic circulation (Supporting information Figure S3). Taken together, Adpgk + pIC + CpG generates strong anti-tumor cellular and humoral immune responses and efficiently eradicates small tumors; however, its efficacy is significantly diminished against large tumors, likely due to the immunosuppressive TME associated with fully established tumors.[15]
2.3. Treatment of large primary tumors and distant metastasis with photothermal therapy combined with neoantigen cancer vaccine
We next asked whether PTT and neoantigen cancer vaccine could complement each other and provide a potent combination therapy against large established tumors. In parallel, we sought to examine an abscopal effect of the combination therapy against metastatic tumors, which are characteristic features of advanced cancer that contribute to poor patient survival.[29] To test the hypothesis, we employed a bilateral tumor model that presents large primary tumors in one side and pre-established experimental metastasis in the other side. C57BL/6 mice were inoculated subcutaneously with 2×106 and 2×105 MC38 cells on the right and left flank for establishing primary and contralateral tumors, respectively. On day 9 when the primary tumors reached 113 ± 30 mm3, the primary tumors were directly administered with Adpgk + pIC + CpG (dose of 15, 50, and 15 μg, respectively), SGNP (100 fmol), or their combination (denoted as combo), followed by laser irradiation on the next day (Figure 3A). The contralateral tumors were monitored without any treatment in order to examine the systemic anti-tumor effects induced by the local treatment of primary tumors. Intratumoral injection of PBS, followed by laser irradiation, was used as a control group. As expected, SGNP or combo treatment significantly increased the temperature of tumor tissues, compared with PBS or Adpgk + pIC + CpG (Figure 3B), confirming SGNP-mediated photothermal heating of tumors. This also indicated that pre-mixing and co-administration of Adpgk + pIC + CpG did not alter SGNPs for their photothermal effect. We confirmed that SGNP exhibited negligible non-specific binding of Adpgk, pIC, and CpG (Supporting information Figure S4), possibly due to anti-fouling PEG surface coating on SGNP. Importantly, the combo-PTT completely ablated large primary tumors in 100% mice (Figure 3C,F). On the other hand, mice treated with either SGNP-PTT or Adpgk + pIC + CpG vaccine exhibited 0% and 43% rate of complete tumor regression, respectively (Figure 3C,F). Importantly, the combo-PTT also exerted robust abscopal effect against the untreated contralateral tumors (Figure 3D,E,F), significantly delaying the tumor growth and extending the animal survival. In contrast, untreated contralateral tumors in SGNP-PTT or Adpgk + pIC + CpG vaccine group exhibited the similar growth rates to that of PBS control group (Figure 3D,E,F). Overall, these results indicate that PTT combined with neoantigen cancer vaccination exert robust anti-tumor efficacy against local primary tumors as well as untreated distant tumors.
Figure 3.
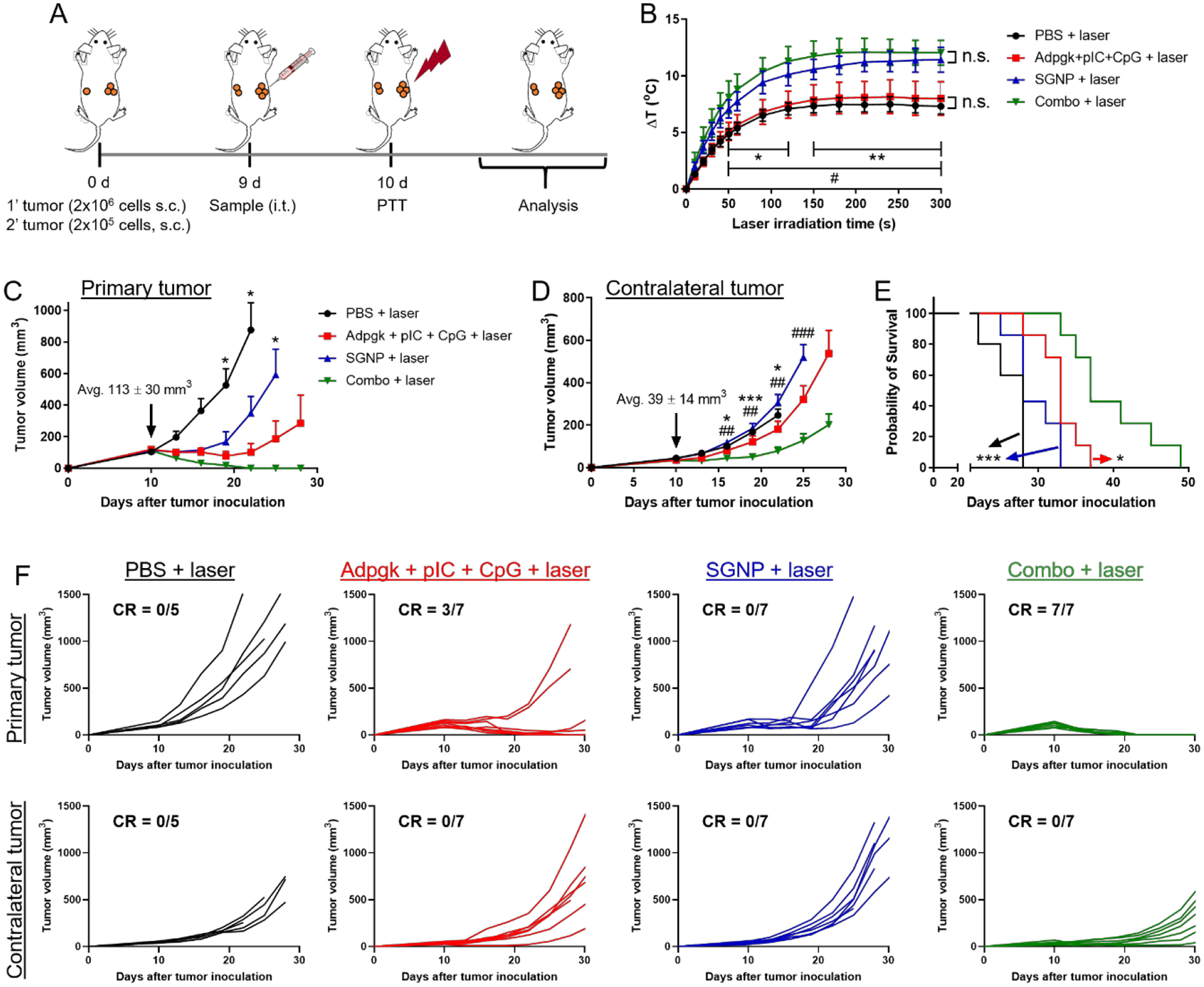
PTT-vaccine combination for the treatment of large primary tumors and pre-established experimental metastasis. (A) Schematic of treatment regimen. (B) Local temperature increase in the tumors during laser irradiation. (C-F) Average tumor growth of the treated primary tumors (C) and untreated contralateral tumors (D), and the overall Kaplan–Meier survival curves (E). (F) Individual tumor growth with fraction of complete tumor regression (CR). The data show mean ± s.d. (B) or s.e.m. (C,D), with n = 5 (PBS + laser) or n = 7 (other groups) for (B-F). *P < 0.05, **P < 0.01, and ***P < 0.001, analyzed by two-way ANOVA with Bonferroni multiple comparisons post-test (B-D), or by log-rank (Mantel–Cox) test (E). *, # in (B) indicate statistically significant differences between PBS + laser vs. SGNP or Combo + laser (*); and Adpgk + pIC + CpG + laser vs. SGNP or Combo + laser (#). * in (C and E) indicate statistically significant differences compared with Combo + laser. *, # in (D) indicate statistically significant differences between PBS + laser vs. Combo + laser (*); and SGNP + laser vs. Combo + laser (#).
2.4. Immune response in the directly treated local tumors
Since the combination therapy led to strong anti-tumor immunity as evidenced by the inhibition of untreated distant tumors, we sought to delineate the immune response triggered by the individual or the combination therapy. We analyzed the primary tumors on day 17 (7 days after laser irradiation) in the bilateral tumor model as in Figure 3A. Thermal ablation of tumor tissue by SGNP-PTT or combo-PTT significantly decreased the frequency of immune cells within the local tumors, including DCs, macrophage, CD8+ T cells, CD4+ T cells, and NK cells (Supporting information Figure S5), probably due to heat-mediated depletion of immune cells non-specifically within the TME. Nevertheless, the frequency of Adpgk-specific CD8+ T cells in the treated tumors was significantly increased by PTT and vaccine (Figure 4A). PTT also promoted cytotoxic activity of NK cells as shown by the upregulation of CD107a,[30] which was associated with the expression of MULT-1 on tumor cells, a stress-induced ligand for NKG2D receptor [31] (Figure 4B and Supporting information Figure S6). In addition, the combo-PTT treatment promoted DC activation and maturation as shown by the upregulation of a co-stimulatory marker CD40 (Figure 4C). Although the combo-PTT treatment also increased the expression of CD86 on DCs, the difference was not statistically significant (Figure 4D). Furthermore, tumors treated with SGNP-PTT or combo-PTT had significantly increased the frequencies of neutrophils (Figure 4E), compared with the PBS control. As neutrophils migrate to the sites of inflammation as a part of the wound healing process,[32] we speculate that PTT-mediated tissue damage induced acute inflammation and intratumoral infiltration of neutrophils.[33]
Figure 4.
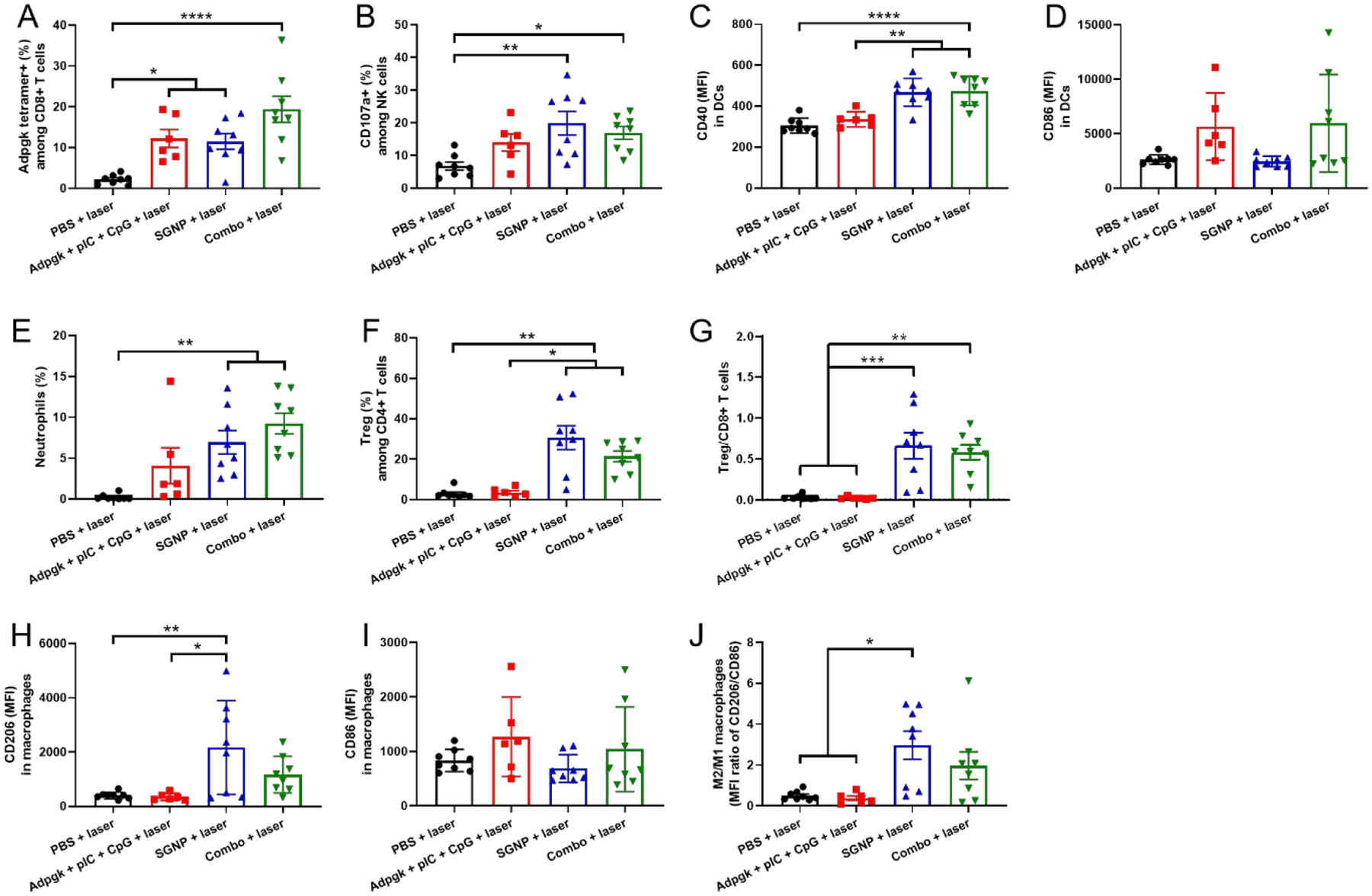
Immune cell analysis in the directly treated primary tumors after 7 days of laser irradiation. (A,B) Frequency of Adpgk-specific CD8+ T cells (A) and CD107a+ NK cells (B). (C,D) MFI of CD40 (C) and CD86 (D) in DCs. (E-G) Frequency of neutrophils (E) and Tregs (F), and the ratio of Treg/CD8+ T cells (G). (H-J) MFI of CD206 (H) and CD86 (I) in macrophages and the corresponding ratio of M2/M1-like macrophages (J). The data show mean ± s.e.m., with n = 6 (Adpgk + pIC + CpG + laser) or n = 8 (other groups). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, analyzed by one-way ANOVA with Bonferroni multiple comparisons post-test.
On the other hand, tumors treated with SGNP-PTT or combo-PTT exhibited significantly increased frequencies of CD4+CD25+Foxp3+ Tregs, resulting in a higher ratio of Treg/CD8+ T cells, compared with the PBS control or Adpgk + pIC + CpG vaccine groups (Figure 4F,G). SGNP-PTT induced upregulation of CD206 on macrophages within the tumors (Figure 4H). Accordingly, SGNP-PTT increased the ratio of CD206-expressing M2-like macrophage to CD86-expressing M1-like macrophage,[34] compared with the PBS or Adpgk + pIC + CpG treatments (Figure 4I,J). In contrast, the combo-PTT treatment did not exhibit statistically significant difference in the ratio of M2/M1-like macrophages, compared with the PBS control. Taken all together, these results indicate that PTT elicited both anti-inflammatory response by M2 macrophages/Tregs and pro-inflammatory response by DCs, T cells, and NK cells in the locally treated tumors, whereas vaccination mostly promoted pro-inflammatory response by DCs and T cells. The combination of PTT with vaccination promoted activation of DCs and tumor-specific T cell response to the greater extents than individual treatments, while maintaining robust NK cell activity mediated by PTT, thus leading to strong anti-tumor immune response for complete tumor regression after local treatment.
2.5. Immune response in the untreated distant tumors
Lastly, we also analyzed immune cells in the untreated contralateral tumors to reveal key immune cells potentially implicated in the inhibition of distant metastasis. The combo-PTT therapy significantly increased the frequency of CD8+ T cells and NK cells in the contralateral tumors, compared with the PBS control (Figure 5A,C). In addition, the high frequency of tumor infiltrating CD8+ T cells and NK cells was associated with their activation as shown by Adpgk tetramer staining and upregulation of CD107a, respectively (Figure 5B,D). Adpgk + pIC + CpG also elicited Adpgk-specific CD8+ T cells to the similar extent as the combo-PTT therapy. These results indicate that systemic activation of CD8+ T cells and their homing to metastatic tumors were mainly driven by vaccination rather than PTT. On the other hand, there was no significant change in the population of DCs, macrophages, CD4+ T cells, and neutrophils within the contralateral tumors by any treatment (Supporting information Figure S7). Nonetheless, the functional analysis indicated that the combo-PTT therapy led to upregulation of CD40 and CD86 among tumor-infiltrating DCs (Figure 5E,F). Mice that received the combo-PTT treatment exhibited an increased frequency of Tregs in the contralateral tumors (Figure 5G). However, the ratio of Treg/CD8+ T cells for the combo-PTT group remained similar to other groups due to the concurrent increase in the frequency of CD8+ T cells (Figure 5H). In addition, the combo-PTT treatment promoted CD86-expressing M1-like macrophages whereas CD206-expressing M2-like macrophages remained unchanged, leading to a significantly decreased ratio of M2/M1-like macrophages, compared with the PBS or SGNP-PTT groups (Figure 5I–K). Overall, the comb-PTT treatment promoted systemic activation of DCs, macrophages, CD8+ T cells, and NK cells and their trafficking into distant tumors, which could foster TME in favor of anti-tumor immunity for exerting robust abscopal effect against untreated metastatic tumors.
Figure 5.
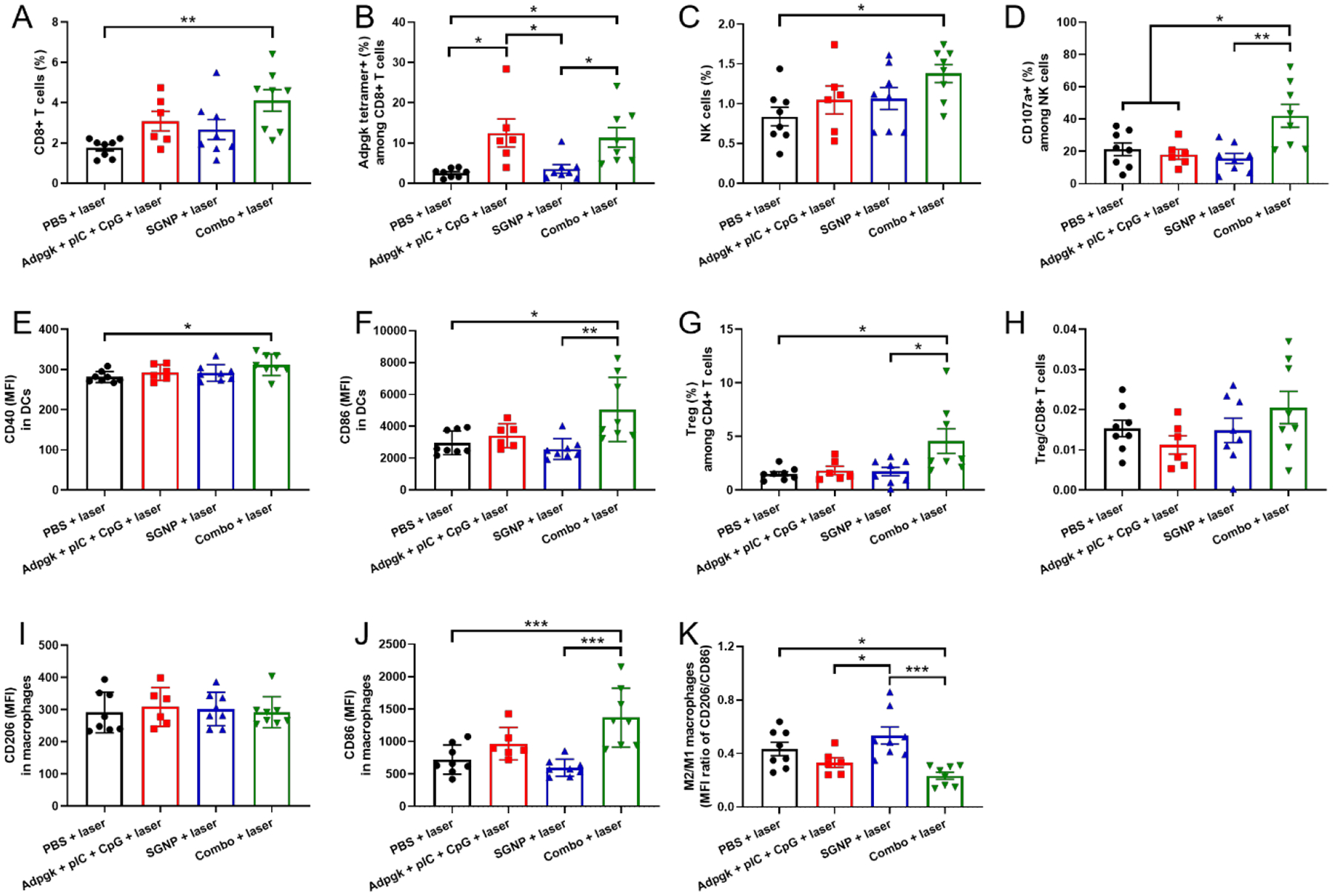
Immune cell analysis in the untreated contralateral tumors. (A-D) Frequency of CD8+ T cells (A) and NK cells (C), and their activation analyzed by Adpgk tetramer staining (B) and CD107a expression (D). (E,F) MFI of CD40 (E) and CD86 (F) in DCs. (G,H) Frequency of Tregs (G) and the ratio of Treg/CD8+ T cells (H). (I-K) MFI of CD206 (I) and CD86 (J) in macrophages and the corresponding ratio of M2/M1-like macrophages (K). The data show mean ± s.e.m., with n = 6 (Adpgk + pIC + CpG + laser) or n = 8 (other groups). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001, analyzed by one-way ANOVA with Bonferroni multiple comparisons post-test.
3. Discussion
In this study, we performed comparative study by categorizing tumors by their size (small vs. large) and demonstrated markedly reduced therapeutic efficiency of NIR-PTT and neoantigen cancer vaccination against large tumors. PTT exhibited inefficient eradiation of large tumors by low internal heating and heterogeneous heat distribution that caused mild hyperthermia with substantial margin beyond effective PTT.[3, 6c, 35] On the other hand, large tumors develop complex and dynamic TME for supporting tumor growth, which endows immune evasion and limits the efficacy of cancer immunotherapy.[14a] We hypothesized that PTT and cancer vaccine together can synergistically improve anti-tumor efficiency against large tumors with their complementary therapeutic effects. PTT can ablate tumor cells and other TME components by thermally-induced cellular and molecular damage, leading to debulking of tumor mass and altering the TME. In parallel, cancer vaccine can eliminate residual tumor burden that becomes susceptible to immunotherapy with compromised TME. Indeed, we demonstrated that the combination of PTT and cancer vaccine can efficiently eradicate large primary tumors, achieving complete tumor regression in 100% of the treated mice. Moreover, the combination therapy also exerted a robust abscopal effect against distant metastatic tumors, suggesting its therapeutic potential against advanced stage tumors featured by large primary tumor and systemic metastasis.
PTT non-specially killed cells in the tumors, including not only cancer cells but also immune cells pre-existing in the tumors. On the other hand, PTT-treated tumors were enriched with neutrophils that infiltrated tumors post PTT. Neutrophils are the most abundant leukocytes in the blood that quickly migrate to the sites of infection or tissue damage and resolve the potentially harmful inflammatory response.[32] Thus, our data indicates that PTT promotes acute inflammation in the locally treated tumors, possibly by causing tissue injury and necrotic cell death,[33] followed by recruitment of neutrophils into inflamed tumors.[36] Neutrophils were more abundant in the tumors treated with SGNP-PTT than the tumors treated with Adpgk + pIC + CpG, suggesting that PTT-induced inflammation can attract neutrophils more efficiently than adjuvant-mediated inflammation.[37] PTT also resulted in the high frequencies of immunosuppressive M2-like macrophages and Tregs in residual tumors. Although PTT could also trigger the activation of local immune cells, including DCs, CD8+ T cells, and NK cells, they were not sufficient to overcome local immune suppression and prevent tumor growth. Notably, the enrichment of neutrophils, Tregs, and M2-like macrophages in the PTT-treated tumors are reminiscent of those observed during tumor relapse after surgical tumor resection.[38] It has been demonstrated that PTT-mediated inflammation accelerated tumor regrowth and therapy resistance although the exact mechanism of action has not been elucidated.[39] Our results in this report indicate that PTT-mediated tissue injury generates acute inflammatory response potentially implicated for tissue regeneration and wound healing,[40] which is likely to induce immunosuppressive milieu in residual tumors to support tumor relapse.
The synergy between pIC and CpG is associated with simultaneous stimulation of TRIF and MyD88 pathways that not only amplifies the individual signals but also activates unique signals and gene expressions.[20–21, 41] The synergistic cytokine production by BMDCs was abolished when MyD88-dependent CpG,[25] not TRIF-dependent pIC, was replaced by TRIF-biased MPLA, suggesting that dual adjuvant synergy stemmed from co-stimulation of MyD88 and TRIF pathways. Surprisingly, our study has shown that neoantigen vaccination with dual TLR agonists of pIC plus CpG resulted in complete regression of MC38 tumors (~ 50 mm3) with strong induction of neoantigen-specific CD8+ T cells in the systemic circulation, as high as ~30% after the boost vaccination. Soluble vaccines consisting free form of antigens and adjuvants are known to elicit weak tumor-specific T cells because of rapid clearance and poor co-delivery of antigens and adjuvants by their unfavorable pharmacological properties, which results in poor vaccine performance and anti-tumor efficacy.[42] This also applies for recent clinical trials of personalized cancer vaccine in which soluble neoantigen vaccines generated rather low frequency of neoantigen-specific CD8+ T cells (generally less than 1% in blood).[13a–c, 13e] Although molecular modifications of antigens/adjuvants and the utilization of sophisticated delivery platforms have been shown to improve vaccine performance,[43] complicated synthesis and post-modification processes for vaccine delivery systems pose technical and manufacturing challenges for clinical translation.[9b] Our study not only confirms the previous finding of strong immune stimulation by the combination of pIC and CpG but also extends their utility toward enhancing the immunogenicity of neoantigen in the form of simple soluble vaccine. Thus, our study offers a promising strategy for improving clinical neoantigen cancer vaccine by employing dual adjuvants for synergistic combinatory effects.
Nevertheless, the anti-tumor efficacy of dual adjuvant-based neoantigen cancer vaccine was limited against large tumors (>100 mm3). This was observed despite the fact that tumor size did not alter the induction of neoantigen-specific CD8+ T cells in the systemic circulation. These results suggest that the tumor-intrinsic factor, likely immunosuppressive TME, plays a detrimental role in vaccine performance. This is consistent with the notion that therapeutic cancer vaccine is most effective for small tumors in early stage before immunosuppressive TME is fully established, as cancer cells program and exploit TME for resisting cancer immunotherapy with multiple immune evasion mechanisms.[8b, 16, 28] Importantly, we have demonstrated that PTT can synergize with cancer vaccine to effectively eliminate large established tumors. While we have not identified the exact mechanisms of action for PTT and vaccine combo treatment responsible for their efficacy against large tumors, we speculate that PTT sensitizes tumors to vaccination by debulking large tumors and destroying the physical barriers of TME, thus turning TME susceptible to systemically activated anti-tumor immune cells. In line with this, tumors treated with neoantigen cancer vaccine plus PTT exhibited increased neoantigen-specific CD8+ T cells, compared with PTT or vaccine alone. In addition, vaccination alleviated immunosuppression in PTT-treated tumors by reducing M2-polarization of macrophages, while PTT activated NK cells and DCs together with vaccination. These features are associated with favorable anti-tumor immune responses that promote regression of large primary tumors by the combo treatment.
The combination therapy also exerted robust abscopal effect against pre-established metastatic tumors. PTT alone barely affected the composition and activation status of immune cells, whereas neoantigen cancer vaccine significantly increased neoantigen-specific CD8+ T cells in the contralateral tumors. These results suggest that PTT only modulates local immunity in the directly treated tumors whereas vaccination promotes both local and systemic immunity. Moreover, neoantigen cancer vaccine combined with PTT exhibited enhanced activation and/or tumor infiltration of CD8+ T cells, NK cells, DCs, and macrophages, compared with either treatment alone. We speculate that PTT-triggered release of tumor antigens and danger signals synergize with cancer vaccination, leading to augmentation of systemic immune activation. Overall, these results show that combo-PTT treatment promotes systemic activation and tumor trafficking of key effector cells for cancer immunotherapy, including CD8+ T cells and NK cells,[7b] resulting in robust anti-tumor activity against distant metastatic tumors.
4. Conclusion
In summary, we have demonstrated that PTT combined with neoantigen cancer vaccine can efficiently eradicate large primary tumors as well as pre-established metastatic tumors by complementary therapeutic actions and synergistic anti-tumor immune activation. The combination of PTT and neoantigen cancer vaccine presents a new nano-immunotherapy that offers a promising approach for personalized therapy of advanced cancer.
5. Experimental Section
Materials and equipment:
L-ascorbic acid was obtained from Fisher Chemical. pIC (high molecular weight, 1.5–8 kb) was purchased from InvivoGen, and CpG1826 was obtained from Integrated DNA Technology. Adpgk peptide (ASMTNMELM) was synthesized by Genemed Synthesis. RPMI 1640, penicillin-streptomycin (PS), beta-mercaptoethanol (β-ME), and ACK lysis buffer were purchased from Gibco. Fetal bovine serum (FBS) was obtained from Corning. Granulocyte-macrophage colony-stimulating factor (GM-CSF) was received from Genscript. Other chemicals were obtained from Sigma-Aldrich unless otherwise stated. UV-Vis absorption spectra were obtained using BioTek synergy neo microplate reader. Transmission electron microscope images were acquired using JEOL 1400-plus and analyzed using the ImageJ software (NIH, Bethesda, MD). Laser irradiation was performed using a 808 nm continuous wave diode laser (China Daheng Group Inc., Beijing, China). Tumor temperature was measured using a mini-hypodermic thermocouple probe coupled with digital thermometer (OMEGA engineering, Inc.). Flow cytometry was performed using ZE5 Cell Analyzer (Bio-Rad) and the data were analyzed using FlowJo 10.5 software.
Synthesis of SGNPs:
SGNPs were synthesized using seed-mediated, surfactant-free method as described previously.[7b] Briefly, seed GNPs were prepared by boiling 1.5 mmol of HAuCl4 and 4.5 mmol of sodium citrate tribasic dehydrate in 300 ml deionized (DI) water for 10 min. Then, the resulting citrate-stabilized seed GNPs were cooled at room temperature and stored at 4 °C before use. For the synthesis of SGNP, 10 ml of citrate-stabilized seed GNPs were diluted in 300 ml DI water and sequentially mixed with 60 μmol of HAuCl4, 300 μl of 1 M HCl, 9 μmol of AgNO3, and 120 μmol of L-ascorbic acid with vigorous stirring, followed by 200 nmol of poly(ethylene glycol) methyl ether thiol (MW 6,000). After stirring for 2 h at room temperature, the mixture was centrifuged at 3,000x g for 1 h and the SGNP pellets were further purified by passing through illustra NAP-10 column (GE healthcare life sciences), followed by storage at 4 °C until further use.
In vitro BMDC study:
BMDCs were collected from C57BL/6 mice and maintained in RPMI 1640 supplemented 10% FBS, 1% PS, 20 ng/ml GM-CSF, and 50 μM β-ME according to the literature.[44] Immature BMDCs were plated at a density of 1 × 105 cells/well in 96 well plates and incubated overnight at 37 °C under 5% CO2. Cells were then incubated with a pairwise combination of pIC and CpG at the concentrations of 0, 1, 10, 20, 50, and 100 μg/ml pIC and 0, 10, 100, 500, and 1000 ng/ml CpG. After 24 h, cells were centrifuged, and cell culture media were collected for the analysis of cytokine secretion. The concentrations of IL12-p70 and IFN-β were measured using ELISA kit by following the manufacture’s instruction (R&D system).
In vivo cancer therapy:
Animals were cared for following the federal, state, and local guidelines. University of Michigan, Ann Arbor is an AAALAC international accredited institution, and all work conducted on animals was approved by the Institutional Animal Care and Use Committee (IACUC) with the protocol # PRO00008587. Female C57BL/6 mice (5–6 weeks) were purchased from Jackson Laboratory (USA). C57BL/6 mice were subcutaneously inoculated with 5 × 105 (small tumors) or 2 × 106 (large tumors) MC38 cells on the right flank and randomly sorted on day 9 for matching similar average tumor volume per group. For PTT, SGNP (50 μl in PBS, 100 fmol) or blank PBS (50 μl) were directly injected into tumors, followed by laser irradiation (0.7 W/cm2, 5 min) on the next day. The local tumor temperature was measured during laser irradiation by inserting a thermocouple probe into the center of tumor region. For vaccination, mice were intratumorally administered with various combination of Adpgk peptide (15 μg), pIC (50 μg), and CpG (15 μg) on days 9 and 16. Blood was collected after 7 days of laser irradiation or vaccination for PBMC analysis. Serum was collected on day 30 for the analysis of IgG responses against tumor cells. For the bilateral tumor model, C57BL/6 mice were subcutaneously inoculated with 2 × 106 and 2 × 105 MC38 cells on the right (primary tumor) and left (contralateral tumor) flank, respectively, and randomly sorted on day 9 when the average primary tumor volume reached > 100 mm3. Then, primary tumors were directly injected with SGNP, Adpgk + pIC + CpG, or their combination at the dose of 100 fmol SGNP and 15/50/15 μg Adpgk/pIC/CpG in 50 μl PBS. Blank PBS was used as a control group. PTT was performed on the next day by exposing primary tumors to the laser irradiation (0.7 W/cm2, 5 min), while contralateral tumors were left untreated. Tumor microenvironment analysis was performed by sacrificing mice and excising tumors after 7 days of laser irradiation. The sizes of tumors were measured twice a week using a digital caliper, and the tumor volume was calculated as V = (width)2 × length × 1/2. The mice were euthanized when the tumors reached the maximum permitted size (1.5 cm in any dimension) or when tumor developed ulceration.
Flow cytometric analysis:
For analysis of Adpgk-specific CD8+ T cells in the systemic circulation, PBMCs were collected by submandibular bleeding, followed by the removal of red blood cells using ACK lysis buffer. PBMCs were stained with Adpgk peptide-MHC tetramer tagged with PE (H-2Db-restricted ASMTNMELM, NIH Tetramer Core Facility) and anti-CD8α-APC (BD Biosciences, No. 553035). Serum IgG specific to MC38 cells was analyzed by incubating MC38 cells in vitro with immune serum, followed by staining with secondary anti-IgG-PE antibody (Invitrogen, No. 12401082). For the tumor microenvironment analysis, tumor tissues were harvested on day 7 after laser irradiation, cut into small pieces, and treated with 1 mg/ml of collagenase type IV and 0.1 mg/ml of DNase I in RPMI for 30 min at 37 °C. Single cells were obtained by passing the cell suspension through a 70-μm strainer, and then stained with the following antibody-fluorophore conjugates; CD40-BV605 (BD Biosciences, No. 745218), CD11c-PE (Invitrogen, No. 12011482), CD86-PE/Cy7 (BD Biosciences, No. 560582) for CD11c+ DCs, CD11b-BV605 (Biolegend, No. 101237), F4/80-FITC (Biolegend, No. 123107), CD86-PE/Cy7 (BD Biosciences, No. 560582), CD206-APC (Biolegend, No. 141708) for CD11b+F4/80+ macrophages, CD3-FITC (Biolegend, No. 100204), CD8-BV605 (BD Biosciences, No. 563152), Adpgk peptide-MHC tetramer-PE (NIH Tetramer Core Facility) for CD3+CD8+ T cells, CD3-FITC, CD4-BV605 (BD Biosciences, No. 743156), Foxp3-PE (Invitrogen, No. 12577382), CD25-PE/Cy7 (BD Biosciences, No. 561780) for CD3+CD4+ T cells and CD3+CD4+CD25+Foxp3+ Tregs, NK1.1-FITC (Biolegend, No. 108706), CD3-PE (BD Biosciences, No. 561799), CD107a-APC (BD Biosciences, No. 560646) for CD3-NK1.1+ NK cells, and Ly6c-FITC (BD Biosciences, No. 553104), CD11b-PE (Invitrogen, No. 12011282), and Ly6G-APC (BD Biosciences, No. 560599) for CD11b+Ly6c-Ly6G+ neutrophils. For the analysis of surface expression of NKG2D ligand, tumor cells were stained with CD45-FITC (BD Biosciences, No. 553080) and hamster anti-mouse MULT-1 antibody (Invitrogen, No. 14586382), followed by secondary antibody staining with goat anti-hamster IgG-PE (Invitrogen, No. 12411283). Flow cytometric analysis was performed with live and intact cells by suspending cells in DAPI solution and gating out DAPI-positive populations.
Statistical analysis:
For the animal studies, after tumors were established, we excluded mice with excessively large or small tumors (10–15% of animals), and the remaining mice were sorted randomly to match similar average tumor volume. Statistical analysis was performed using Prism 8.4.2 software (GraphPad Software) by one-way or two-way ANOVA with Bonferroni multiple comparisons post-test for multiple groups or two-tailed, unpaired t-test with Welch’s correction between two groups. Statistical significance for survival curve was calculated by the log-rank (Mantel–Cox) test. Data were approximately normally distributed, and variance was similar between the groups. Statistical significance is indicated as *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Supplementary Material
Acknowledgements
This work was supported in part by NIH (R01CA210273, R01AI127070, R01DK125087, and U01CA210152) and the University of Michigan Rogel Cancer Center Support Grant (P30CA46592). J.J.M. is supported by NSF CAREER Award (1553831). K.S.P. acknowledges financial support from the UM TEAM Training Program (DE007057 from NIDCR). We acknowledge the NIH Tetramer Core Facility (contract HHSN272201300006C) for the provision of MHC-I tetramers.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Kyung Soo Park, Department of Biomedical Engineering, Biointerfaces Institute, University of Michigan.
James J. Moon, Department of Pharmaceutical Sciences, Department of Biomedical Engineering, Biointerfaces Institute, University of Michigan, Ann Arbor, MI 48108, USA.
References
- [1].Hwang S, Nam J, Jung S, Song J, Doh H, Kim S, Nanomedicine 2014, 9, 2003. [DOI] [PubMed] [Google Scholar]
- [2].Dickerson EB, Dreaden EC, Huang X, El-Sayed IH, Chu H, Pushpanketh S, McDonald JF, El-Sayed MA, Cancer Letters 2008, 269, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Peng J, Xiao Y, Li W, Yang Q, Tan L, Jia Y, Qu Y, Qian Z, Advanced Science 2018, 5, 1700891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Toraya-Brown S, Sheen MR, Zhang P, Chen L, Baird JR, Demidenko E, Turk MJ, Hoopes PJ, Conejo-Garcia JR, Fiering S, Nanomedicine: Nanotechnology, Biology and Medicine 2014, 10, 1273; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bear AS, Kennedy LC, Young JK, Perna SK, Mattos Almeida JP, Lin AY, Eckels PC, Drezek RA, Foster AE, PLOS ONE 2013, 8, e69073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Huang L, Li Y, Du Y, Zhang Y, Wang X, Ding Y, Yang X, Meng F, Tu J, Luo L, Sun C, Nature Communications 2019, 10, 4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Chen Q, Xu L, Liang C, Wang C, Peng R, Liu Z, Nature Communications 2016, 7, 13193; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang C, Xu L, Liang C, Xiang J, Peng R, Liu Z, Advanced Materials 2014, 26, 8154; [DOI] [PubMed] [Google Scholar]; c) Liu Y, Maccarini P, Palmer GM, Etienne W, Zhao Y, Lee C-T, Ma X, Inman BA, Vo-Dinh T, Scientific Reports 2017, 7, 8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a) Guo L, Yan DD, Yang D, Li Y, Wang X, Zalewski O, Yan B, Lu W, ACS Nano 2014, 8, 5670; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nam J, Son S, Ochyl LJ, Kuai R, Schwendeman A, Moon JJ, Nature Communications 2018, 9, 1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Tang J, Shalabi A, Hubbard-Lucey VM, Annals of Oncology 2018, 29, 84; [DOI] [PubMed] [Google Scholar]; b) Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, Mellstedt H, Nature Reviews Clinical Oncology 2014, 11, 509. [DOI] [PubMed] [Google Scholar]
- [9].a) Schumacher TN, Schreiber RD, Science 2015, 348, 69; [DOI] [PubMed] [Google Scholar]; b) Sahin U, Türeci Ö, Science 2018, 359, 1355. [DOI] [PubMed] [Google Scholar]
- [10].a) Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber W-J, Mulder GE, Toebes M, Vesely MD, Lam SSK, Korman AJ, Allison JP, Freeman GJ, Sharpe AH, Pearce EL, Schumacher TN, Aebersold R, Rammensee H-G, Melief CJM, Mardis ER, Gillanders WE, Artyomov MN, Schreiber RD, Nature 2014, 515, 577; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA, Science 2015, 348, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA, Genome Research 2014, 24, 743; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rooney Michael S., Shukla Sachet A., Wu Catherine J., Getz G, Hacohen N, Cell 2015, 160, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, Boegel S, Schrörs B, Vascotto F, Castle JC, Tadmor AD, Schoenberger SP, Huber C, Türeci Ö, Sahin U, Nature 2015, 520, 692; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie W-R, Hildebrand WH, Mardis ER, Linette GP, Science 2015, 348, 803; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, Modrusan Z, Mellman I, Lill JR, Delamarre L, Nature 2014, 515, 572. [DOI] [PubMed] [Google Scholar]
- [13].a) Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, Li S, Lieb DJ, Eisenhaure T, Gjini E, Stevens J, Lane WJ, Javeri I, Nellaiappan K, Salazar AM, Daley H, Seaman M, Buchbinder EI, Yoon CH, Harden M, Lennon N, Gabriel S, Rodig SJ, Barouch DH, Aster JC, Getz G, Wucherpfennig K, Neuberg D, Ritz J, Lander ES, Fritsch EF, Hacohen N, Wu CJ, Nature 2017, 547, 217; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, Shukla SA, Hu Z, Li L, Le PM, Allesøe RL, Richman AR, Kowalczyk MS, Abdelrahman S, Geduldig JE, Charbonneau S, Pelton K, Iorgulescu JB, Elagina L, Zhang W, Olive O, McCluskey C, Olsen LR, Stevens J, Lane WJ, Salazar AM, Daley H, Wen PY, Chiocca EA, Harden M, Lennon NJ, Gabriel S, Getz G, Lander ES, Regev A, Ritz J, Neuberg D, Rodig SJ, Ligon KL, Suvà ML, Wucherpfennig KW, Hacohen N, Fritsch EF, Livak KJ, Ott PA, Wu CJ, Reardon DA, Nature 2019, 565, 234; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, Omokoko T, Vormehr M, Albrecht C, Paruzynski A, Kuhn AN, Buck J, Heesch S, Schreeb KH, Müller F, Ortseifer I, Vogler I, Godehardt E, Attig S, Rae R, Breitkreuz A, Tolliver C, Suchan M, Martic G, Hohberger A, Sorn P, Diekmann J, Ciesla J, Waksmann O, Brück A-K, Witt M, Zillgen M, Rothermel A, Kasemann B, Langer D, Bolte S, Diken M, Kreiter S, Nemecek R, Gebhardt C, Grabbe S, Höller C, Utikal J, Huber C, Loquai C, Türeci Ö, Nature 2017, 547, 222; [DOI] [PubMed] [Google Scholar]; d) Hu Z, Leet DE, Allesøe RL, Oliveira G, Li S, Luoma AM, Liu J, Forman J, Huang T, Iorgulescu JB, Holden R, Sarkizova S, Gohil SH, Redd RA, Sun J, Elagina L, Giobbie-Hurder A, Zhang W, Peter L, Ciantra Z, Rodig S, Olive O, Shetty K, Pyrdol J, Uduman M, Lee PC, Bachireddy P, Buchbinder EI, Yoon CH, Neuberg D, Pentelute BL, Hacohen N, Livak KJ, Shukla SA, Olsen LR, Barouch DH, Wucherpfennig KW, Fritsch EF, Keskin DB, Wu CJ, Ott PA, Nature Medicine 2021; [Google Scholar]; e) Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanović S, Gouttefangeas C, Platten M, Tabatabai G, Dutoit V, van der Burg SH, thor Straten P, Martínez-Ricarte F, Ponsati B, Okada H, Lassen U, Admon A, Ottensmeier CH, Ulges A, Kreiter S, von Deimling A, Skardelly M, Migliorini D, Kroep JR, Idorn M, Rodon J, Piró J, Poulsen HS, Shraibman B, McCann K, Mendrzyk R, Löwer M, Stieglbauer M, Britten CM, Capper D, Welters MJP, Sahuquillo J, Kiesel K, Derhovanessian E, Rusch E, Bunse L, Song C, Heesch S, Wagner C, Kemmer-Brück A, Ludwig J, Castle JC, Schoor O, Tadmor AD, Green E, Fritsche J, Meyer M, Pawlowski N, Dorner S, Hoffgaard F, Rössler B, Maurer D, Weinschenk T, Reinhardt C, Huber C, Rammensee H-G, Singh-Jasuja H, Sahin U, Dietrich P-Y, Wick W, Nature 2019, 565, 240. [DOI] [PubMed] [Google Scholar]
- [14].a) Munn DH, Bronte V, Current Opinion in Immunology 2016, 39, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Anderson KG, Stromnes IM, Greenberg PD, Cancer Cell 2017, 31, 311; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Motz Greg T., Coukos G, Immunity 2013, 39, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Zou W, Nature Reviews Cancer 2005, 5, 263; [DOI] [PubMed] [Google Scholar]; b) Quail DF, Joyce JA, Nature Medicine 2013, 19, 1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJM, Nature Reviews Cancer 2016, 16, 219. [DOI] [PubMed] [Google Scholar]
- [17].Akira S, Takeda K, Nature Reviews Immunology 2004, 4, 499. [DOI] [PubMed] [Google Scholar]
- [18].Suk JS, Xu Q, Kim N, Hanes J, Ensign LM, Advanced Drug Delivery Reviews 2016, 99, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].a) Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S, Science 2003, 301, 640; [DOI] [PubMed] [Google Scholar]; b) Häcker H, Vabulas RM, Takeuchi O, Hoshino K, Akira S, Wagner H, Journal of Experimental Medicine 2000, 192, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhu Q, Egelston C, Vivekanandhan A, Uematsu S, Akira S, Klinman DM, Belyakov IM, Berzofsky JA, Proceedings of the National Academy of Sciences 2008, 105, 16260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A, Nature Immunology 2005, 6, 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Schmidt CS, Mescher MF, The Journal of Immunology 2002, 168, 5521; [DOI] [PubMed] [Google Scholar]; b) Colombo MP, Trinchieri G, Cytokine & Growth Factor Reviews 2002, 13, 155. [DOI] [PubMed] [Google Scholar]
- [23].Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G, Nature Reviews Immunology 2015, 15, 405. [DOI] [PubMed] [Google Scholar]
- [24].Nam J, La W-G, Hwang S, Ha YS, Park N, Won N, Jung S, Bhang SH, Ma Y-J, Cho Y-M, Jin M, Han J, Shin J-Y, Wang EK, Kim SG, Cho S-H, Yoo J, Kim B-S, Kim S, ACS Nano 2013, 7, 3388. [DOI] [PubMed] [Google Scholar]
- [25].Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC, Science 2007, 316, 1628. [DOI] [PubMed] [Google Scholar]
- [26].a) Marabelle A, Tselikas L, de Baere T, Houot R, Annals of Oncology 2017, 28, xii33; [DOI] [PubMed] [Google Scholar]; b) Aznar MA, Tinari N, Rullán AJ, Sánchez-Paulete AR, Rodriguez-Ruiz ME, Melero I, The Journal of Immunology 2017, 198, 31. [DOI] [PubMed] [Google Scholar]
- [27].a) Melief CJM, van der Burg SH, Nature Reviews Cancer 2008, 8, 351; [DOI] [PubMed] [Google Scholar]; b) Toes RE, Blom RJ, Offringa R, Kast WM, Melief CJ, The Journal of Immunology 1996, 156, 3911. [PubMed] [Google Scholar]
- [28].Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds AM, Kumari S, Kelly RL, Kwan BH, Abraham W, Hu K, Mehta NK, Kauke MJ, Suh H, Cochran JR, Lauffenburger DA, Wittrup KD, Irvine DJ, Nature Medicine 2016, 22, 1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Maringe C, Walters S, Butler J, Coleman MP, Hacker N, Hanna L, Mosgaard BJ, Nordin A, Rosen B, Engholm G, Gjerstorff ML, Hatcher J, Johannesen TB, McGahan CE, Meechan D, Middleton R, Tracey E, Turner D, Richards MA, Rachet B, Gynecologic Oncology 2012, 127, 75; [DOI] [PubMed] [Google Scholar]; b) Maringe C, Walters S, Rachet B, Butler J, Fields T, Finan P, Maxwell R, Nedrebø B, Påhlman L, Sjövall A, Spigelman A, Engholm G, Gavin A, Gjerstorff ML, Hatcher J, Johannesen TB, Morris E, McGahan CE, Tracey E, Turner D, Richards MA, Coleman MP, Acta Oncologica 2013, 52, 919; [DOI] [PubMed] [Google Scholar]; c) Walters S, Maringe C, Butler J, Rachet B, Barrett-Lee P, Bergh J, Boyages J, Christiansen P, Lee M, Wärnberg F, Allemani C, Engholm G, Fornander T, Gjerstorff ML, Johannesen TB, Lawrence G, McGahan CE, Middleton R, Steward J, Tracey E, Turner D, Richards MA, Coleman MP, The IMWG, British Journal of Cancer 2013, 108, 1195; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Walters S, Maringe C, Coleman MP, Peake MD, Butler J, Young N, Bergström S, Hanna L, Jakobsen E, Kölbeck K, Sundstrøm S, Engholm G, Gavin A, Gjerstorff ML, Hatcher J, Johannesen TB, Linklater KM, McGahan CE, Steward J, Tracey E, Turner D, Richards MA, Rachet B, Thorax 2013, 68, 551. [DOI] [PubMed] [Google Scholar]
- [30].Alter G, Malenfant JM, Altfeld M, Journal of Immunological Methods 2004, 294, 15. [DOI] [PubMed] [Google Scholar]
- [31].Nice TJ, Coscoy L, Raulet DH, Journal of Experimental Medicine 2009, 206, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LAJ, Perretti M, Rossi AG, Wallace JL, The FASEB Journal 2007, 21, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].a) McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CCM, Beck PL, Muruve DA, Kubes P, Science 2010, 330, 362; [DOI] [PubMed] [Google Scholar]; b) Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T, Nature Immunology 2008, 9, 1179. [DOI] [PubMed] [Google Scholar]
- [34].a) Raimondo TM, Mooney DJ, Proceedings of the National Academy of Sciences 2018, 115, 10648; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jaynes JM, Sable R, Ronzetti M, Bautista W, Knotts Z, Abisoye-Ogunniyan A, Li D, Calvo R, Dashnyam M, Singh A, Guerin T, White J, Ravichandran S, Kumar P, Talsania K, Chen V, Ghebremedhin A, Karanam B, Bin Salam A, Amin R, Odzorig T, Aiken T, Nguyen V, Bian Y, Zarif JC, de Groot AE, Mehta M, Fan L, Hu X, Simeonov A, Pate N, Abu-Asab M, Ferrer M, Southall N, Ock C-Y, Zhao Y, Lopez H, Kozlov S, de Val N, Yates CC, Baljinnyam B, Marugan J, Rudloff U, Science Translational Medicine 2020, 12, eaax6337; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li Q, Shen A, Wang Z, RSC Advances 2020, 10, 16537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Li Y, He L, Dong H, Liu Y, Wang K, Li A, Ren T, Shi D, Li Y, Advanced Science 2018, 5, 1700805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].a) Zhang L, Zhang Y, Xue Y, Wu Y, Wang Q, Xue L, Su Z, Zhang C, Advanced Materials 2019, 31, 1805936; [DOI] [PubMed] [Google Scholar]; b) Qiu Q, Li C, Yan X, Zhang H, Luo X, Gao X, Liu X, Song Y, Deng Y, Biomaterials 2021, 269, 120652. [DOI] [PubMed] [Google Scholar]
- [37].a) Creagh EM, O’Neill LAJ, Trends in Immunology 2006, 27, 352; [DOI] [PubMed] [Google Scholar]; b) Chen GY, Nuñez G, Nature Reviews Immunology 2010, 10, 826; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kolaczkowska E, Kubes P, Nature Reviews Immunology 2013, 13, 159. [DOI] [PubMed] [Google Scholar]
- [38].a) Xue J, Zhao Z, Zhang L, Xue L, Shen S, Wen Y, Wei Z, Wang L, Kong L, Sun H, Ping Q, Mo R, Zhang C, Nature Nanotechnology 2017, 12, 692; [DOI] [PubMed] [Google Scholar]; b) Wu M, Zhang H, Tie C, Yan C, Deng Z, Wan Q, Liu X, Yan F, Zheng H, Nature Communications 2018, 9, 4777; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Predina J, Eruslanov E, Judy B, Kapoor V, Cheng G, Wang L-C, Sun J, Moon EK, Fridlender ZG, Albelda S, Singhal S, Proceedings of the National Academy of Sciences 2013, 110, E415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dong Q, Wang X, Hu X, Xiao L, Zhang L, Song L, Xu M, Zou Y, Chen L, Chen Z, Tan W, Angewandte Chemie International Edition 2018, 57, 177. [DOI] [PubMed] [Google Scholar]
- [40].a) Wynn TA, Vannella KM, Immunity 2016, 44, 450; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) de Oliveira S, Rosowski EE, Huttenlocher A, Nature Reviews Immunology 2016, 16, 378; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Boothby IC, Cohen JN, Rosenblum MD, Science Immunology 2020, 5, eaaz9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].a) Tross D, Petrenko L, Klaschik S, Zhu Q, Klinman DM, Molecular Immunology 2009, 46, 2557; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Arsenault RJ, Kogut MH, He H, Cellular Signalling 2013, 25, 2246. [DOI] [PubMed] [Google Scholar]
- [42].Nam J, Son S, Park KS, Moon JJ, Advanced Science, n/a, 2002577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].a) Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, Van Egeren DS, Park C, Irvine DJ, Nature 2014, 507, 519; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhu G, Lynn GM, Jacobson O, Chen K, Liu Y, Zhang H, Ma Y, Zhang F, Tian R, Ni Q, Cheng S, Wang Z, Lu N, Yung BC, Wang Z, Lang L, Fu X, Jin A, Weiss ID, Vishwasrao H, Niu G, Shroff H, Klinman DM, Seder RA, Chen X, Nature Communications 2017, 8, 1954; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lynn GM, Sedlik C, Baharom F, Zhu Y, Ramirez-Valdez RA, Coble VL, Tobin K, Nichols SR, Itzkowitz Y, Zaidi N, Gammon JM, Blobel NJ, Denizeau J, de la Rochere P, Francica BJ, Decker B, Maciejewski M, Cheung J, Yamane H, Smelkinson MG, Francica JR, Laga R, Bernstock JD, Seymour LW, Drake CG, Jewell CM, Lantz O, Piaggio E, Ishizuka AS, Seder RA, Nature Biotechnology 2020, 38, 320; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ, Nature Materials 2017, 16, 489; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu H, Huang B, Sohail M, Luo S, Um SH, Khant H, Goodwin JT, Ramos J, Chiu W, Irvine DJ, Nature materials 2011, 10, 243; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Ni Q, Zhang F, Liu Y, Wang Z, Yu G, Liang B, Niu G, Su T, Zhu G, Lu G, Zhang L, Chen X, Science Advances 2020, 6, eaaw6071; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Cheng K, Zhao R, Li Y, Qi Y, Wang Y, Zhang Y, Qin H, Qin Y, Chen L, Li C, Liang J, Li Y, Xu J, Han X, Anderson GJ, Shi J, Ren L, Zhao X, Nie G, Nature Communications 2021, 12, 2041; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Zhang J, Chen X, Xue T, Cheng Q, Ye X, Wang C, Yu Y, Ji X, Wu M, Zhang X, Zheng Y, Wu B, Liang X, Mei L, Journal of biomedical nanotechnology 2020, 16, 1394. [DOI] [PubMed] [Google Scholar]
- [44].Lutz MB, Kukutsch N, Ogilvie ALJ, Rößner S, Koch F, Romani N, Schuler G, Journal of Immunological Methods 1999, 223, 77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.