

Abstract
Imbalance of oxidants is a universal contributor to the failure of implanted devices and tissues. A sustained oxidative environment leads to cytotoxicity, prolonged inflammation, and ultimately host rejection of implanted devices/grafts. The incorporation of antioxidant materials can inhibit this redox/inflammatory cycle and enhance implant efficacy. Cerium oxide nanoparticles (CONP) is a highly promising agent that exhibits potent, ubiquitous, and self-renewable antioxidant properties. Integrating CONP as surface coatings provides ease in translating antioxidant properties to various implants/grafts. Herein, we describe the formation of CONP coatings, generated via the sequential deposition of CONP and alginate, and the impact of coating properties, pH, and polymer molecular weight, on their resulting redox profile. Investigation of CONP deposition, layer formation, and coating uniformity/thickness on their resulting oxidant scavenging activity identified key parameters for customizing global antioxidant properties. Results found lower molecular weight alginates and physiological pH shift CONP activity to a higher H2O2 to O2−-scavenging capability. The antioxidant properties measured for these various coatings translated to distinct antioxidant protection to the underlying encapsulated cells. Information gained from this work can be leveraged to tailor coatings towards specific oxidant-scavenging applications and prolong the function of medical devices and cellular implants.
Keywords: biomaterials, coatings, cerium, diabetes, antioxidants
Graphical Abstract
Functionalization of biomaterials with a nanoscale antioxidant coating can be achieved using LbL technique which incorporates cerium oxide nanoparticles (CONP) and alginate. The redox activity of the coatings can be manipulated by altering factors such as pH of CONP and the molecular weight of alginate. This work presents an array of coating formulations with variable H2O2/O2−-scavenging capabilities.
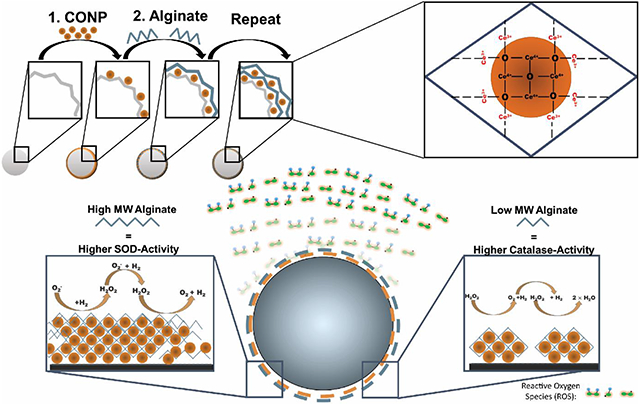
1. Introduction
A promising antioxidant for local oxidative stress scavenging is cerium oxide nanoparticles. While initially used for nonmedical applications (e.g., corrosion prevention, optical devices, UV absorbers), cerium oxide nanoparticles have been recently explored for biomedical applications as a therapy for redox imbalances in inflammatory diseases, such as cancer, diabetes, wound healing, neurodegenerative, and cardiovascular conditions. [1, 2] Cerium oxide exists in two states: cerium sesquioxide (Ce2O3) in the trivalent (Ce3+) state; and cerium dioxide (CeO2) in the tetravalent (Ce4+) state.[3] Cerium oxide uniquely undergoes continuous valence state cycling, permitting self-renewal after interacting with different reactive species. The capacity to rapidly cycle between valence states provides multi-enzymatic antioxidant activity. Specifically, a shift from Ce3+ to Ce4+ results in superoxide dismutase (SOD)-mimetic activity, which scavenges superoxide (O2−), while a shift from Ce4+ to Ce3+ provides catalase-mimetic activity, which deactivates H2O2.[4] This theoretically inexhaustible redox capability sets cerium oxide apart other metal oxides such as transition metals (e.g. iron, zinc, and titanium) and other lanthanides (yttrium).[5, 6] In the nanoparticle form, cerium oxide nanoparticles (CONP) exhibit elevated catalytic capabilities, as its high surface-area-to-volume leads to increased oxygen vacancies and a higher ratio of Ce3+/Ce4+.[7] The CONP valence ratio can be further manipulated by intrinsic factors, such as the synthesis method, aging, surface stabilizers, and size.[8, 9, 10, 11] However, extrinsic factors such as temperature, pH, and interactions with specific molecules can also influence the Ce3+/ Ce4+ ratio.[8, 12, 13]
CONP have been explored with these appealing properties as a treatment for a wide range of inflammatory diseases. These antioxidant nanoparticles can be engineered to target specific diseased tissues and cells and modulate the redox activity according to its microenvironment.[2] For biomedical implant applications, however, the infusion of nanoparticles into the implant site presents challenges in retention and potential clinical toxicity.[14] To bypass these issues, several groups have immobilized nanoparticles within various biomaterials to incorporate superior redox properties of CONP and circumvent toxicity concerns.[15] Leveraging established methods for integrating nanoparticles into nanoscale coatings via layer-by-layer (LbL)[16], CONP was recently immobilized into a conformal coating on the implant surface (Figure 1A&B). Functional LbL antioxidant coatings were generated by alternating layers of alginate and CONP onto metallic, ceramic, and polymeric biomaterial surfaces of varying size and shape (Figure 1A&B).[17] Alginate was used as the interconnecting layer due to its antifouling properties and its ease of layer formation with CONP.[17, 18] Resulting CONP-alginate layers exhibited antioxidant capabilities and downstream cytoprotection, demonstrating this approach's potential.
Figure 1. Formation of CONP-based coatings.

A) Ultrathin coatings generated from sequential incubation in CONP and alginate can be formed onto various biomaterial surfaces. B) Investigation of coating glass, hydrogels, and metal alloys via LbL coating with CONP and alginate.
To further investigate the utility and tunability of this LbL approach, we sought to investigate critical material parameters that may direct CONP deposition, layer uniformity, and overall antioxidant activity. Herein, we investigated two primary coating parameters: alginate molecular weight and CONP pH (Figure 2A). Different molecular weight alginates were utilized to investigate the potential role of coordination complexes between Ce3+ and carboxylate anions (COO−) on CONP-alginate layer formation and uniformity. We also explored the impact of Ce3+/COO− interactions on subsequent layer redox activity, as modulation of Ce3+ availability may shift this antioxidant particle towards a catalase-mimetic activity.[12, 19] CONP pH was also adjusted from a standard acidic to a net neutral suspension. The pH of the CONP solution can serve multiple roles in coating properties, with the potential to alter not only alginate interactions but also CONP-redox activity.[20]
Figure 2. Layer-by-layer coatings of CONP and alginate onto idealized silicon wafers was dependent on material properties.

A) Schematic of the six distinct groups tested, screening acidic and neutral pH of CONP in suspension and three alginate molecular weights. The effect of these factors on B) coating thickness and C-E) rate of layering onto planar silica wafers was evaluated via ellipsometry. Two-way and one-way ANOVA with Tukey's post-hoc test: ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05, where * represents differences between groups and γ compares groups with same alginate MW but different CONP pH.
Herein, we conducted a comprehensive study on the effect of CONP pH and alginate molecular weight on layer formation and resulting antioxidant activity. First, the development and resulting thickness and uniformity of coatings were evaluated. Next, the CONP concentration, Ce4+/Ce3+ ratio, and activity of resulting layers were uniquely characterized and contextualized. Finally, the implications of these features in the generation of antioxidant coatings capable of altering deleterious host responses to implants were discussed.
2. Results
2.1. Characterization of the Impact of pH and Alginate Molecular Weight on CONP/Alginate Coatings on Biomaterial Platforms
To investigate the role of CONP and alginate interactions on the layer properties, we manipulated two primary parameters: alginate molecular weight and CONP pH. To modulate alginate molecular weight (MW), high guluronic alginate sources, specifically MVG (MW > 200 kDa), LVG (MW = 200-75 kDa), and vLVG, (MW < 75 kDa), were used. CONP pH was adjusted from its classic acidic suspension (pH = 4, designated CONP4) to a net neutral suspension (pH = 7.4; designated as CONP7.4) using sucrose to stabilize the nanoparticles and prevent immediate pH-driven precipitation.[13, 21] Other agents, such as poly(acrylic acid), dextran, sucrose, and citric acid, were screened as potential stabilizers, but sucrose provided simplicity in generation and did not impact CONP activity (unlike other methods). DLS characterize of both CONP4 and CONP7.4 measured minimal changes in particle size; 6.8 ± 2.5 and 8 ± 4.5 nm, respectively. Although the use of saccharides for CONP stabilization has been previously reported (e.g., glucose and dextran), this is the first report of the use of sucrose, to the best of our knowledge.[13, 21]
The thickness of CONP/alginate layers formed on an idealized planar silica surface was tracked to characterize the impact of CONP pH and alginate MW on layer formation. Six groups were tested: A) CONP4 & MVG; B) CONP4 & LVG; C) CONP4 & vLVG; D) CONP7.4 & MVG; E) CONP7.4 & LVG; and F) CONP7.4 & vLVG (Figure 2A). Each layer was distinctly deposited onto a dry silicon wafer, with excess material rinsed away before the next layer's addition. Figure 2B shows that coating thickness was measured after every CONP/Alginate bilayer via ellipsometry. Examination of total coating thickness after 12-layers found alginate molecular weight, but not CONP pH, to impact overall layer thickness (p < 0.0001 & 0.121, respectively; 2-way ANOVA). A positive correlation was found between alginate molecular weight and coating thickness (r = 0.98 and 0.99 for CONP4 and CONP7.4, respectively). Results implicate alginate molecular weight as the dominant parameter in layering thickness when coating planar silica surfaces.
After characterizing the total coating thickness at 12 layers, the rate of layer formation at different coating stages was evaluated. Globally, CONP pH impacted the efficiency of layer deposition. Specifically, for the first layers, acidic CONP formed thicker layers more efficiently than neutral CONP (p<0.0001) with a 40, 37.7, and 20.2% increase in coating thickness for alginate MVG, LVG, and vLVG, respectively, when compared to their CONP7.4 analog (Figure 2C). The phenomenon was reversed for the next four layers for MVG and LVG combinations, where the CONP7.4 accumulated 42.9 (p=0.002) and 90% (p=0.0002) faster than CONP4, respectively. No significant difference in material accumulation between acidic and physiological pH CONP was measured for alginate vLVG for layers four to eight (Figure 2D). For the final four layers, while alginate MVG exhibited no impact in CONP pH, the rate of layer thickness was higher at CONP7.4 than CONP4 when coating with alginate LVG (108.67%) and vLVG (73.1%), Figure 2E. Ellipsometry results indicate that on a planar, idealized surface, acidic CONP accelerated material deposition during early stages and plateaued at later stages. In contrast, the material deposition was more discrete and linear at physiological CONP. Moreover, the higher MW alginate showed accelerated material deposition at earlier stages.
While ellipsometry is widely used to measure coating thickness at the nanometer scale accurately, measurements must be done under anhydrous conditions. In addition, LbL coatings can be impacted by the material surface charge, hydrophobicity, topography, and even geometry. To examine the versatility of these coatings to be applied to various surfaces and examine coatings under hydrated conditions, coatings were applied to alginate hydrogel beads. Bright-field microscopy images of alginate microbeads coated with alginate and CONP illustrated cerium within each coating formulation (Figure 3A). Interestingly, coatings showed alginate precipitates at the surface of beads coated when the higher molecular weight alginate (i.e., MVG) was used. In contrast, coatings formed using alginate vLVG exhibited a decreased presence of precipitates.
Figure 3. CONP/alginate LbL coatings onto hydrogels were dependent on coating material properties.

A) Bright-field images of alginate microbeads allowed for visual evaluation of overall coating morphology via optical changes in bead transparency. B) 3D confocal projection images of alginate (labeled via carboxyfluorescein) deposited onto alginate microbeads following CONP/alginate coating permitted image quantification of % area coverage of alginate-CF (C) to investigate the effect of MW and CONP pH on overall coating uniformity. D) Cross-sectional confocal images of alginate-CF within CONP/alginate coatings at the center of the microbead permitted image quantification of overall coating thickness (E) for each formulation. Scale= 200 μm. Two-way and one-way ANOVA with Tukey's post-hoc test:****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05, where * represents differences between groups, Δ compares six-and twelve-layer coatings, and γ compares groups with same alginate MW but varying CONP pH.
To further investigate coating morphology, the alginate used in the CONP/alginate coatings was fluorescently tagged and resulting 6- and 12-layer coated hydrogel beads were visualized by confocal imaging (Figure 3B & D). Image analysis of 3-D image projections of coated beads characterized alginate coating uniformity via percent area covered (Figure 3C), while alginate coating thickness was measured from cross-sectional images (Figure 3E).
Visualization of the alginate-carboxyfluorescein (CF) coatings formed using the CONP-alginate layers validated the previously observed differences in alginate precipitation and highlighted differences in alginate deposition. Analysis of alginate coverage implicated differences in coating uniformity associated with alginate molecular weight. The longer-chain alginate coatings (MVG) resulted in complete coverage of the hydrogel bead after only 6-layers, with no difference in % area covered between 6 and 12-layers (CONP4: p >0.9999, CONP7.4: p=0.61); this indicates that the higher molecular weight alginate results in more efficient microbead coverage. For the lower molecular weights, a more dramatic change between 6 and 12 layers was observed. For example, coatings formed using alginate vLVG-CF exhibited a 60% increase in coverage between layers 6 and 12, regardless of the pH. For the lower molecular weight alginates (LVG and vLVG), the neutral CONP enhanced coating uniformity. Specifically, coatings formed using the intermediate alginate molecular weight (i.e., LVG) and CONP7.4 exhibited no significant change in coating uniformity between 6 and 12-layers. In contrast, LVG + CONP4 coatings exhibited a significant 39.1% increase in percent coverage between 6 and 12 layers (p < 0.001). Further investigation of this data showed that the MW of alginate influences the surface coverage (6-layers, p<0.0001; 12-layer coatings, p=0.018). However, the pH during coating fabrication can also influence the uniformity of the coatings (6-layers, p=0.0204; 12-layer coatings, p=0.0066), and the effect of these two factors are dependent on each other (MW x pH: 6-layers, p<0.0001; 12-layer coatings, p=0.0002). Investigation of individual comparisons show that at acidic pH, surface coverage increases as MW of alginate increases (6-layers, p<0.0001; 12-layer coatings, p=0.031). At physiological pH the initial six layers follow this trend (p<0.0001), however 12-layer coatings show higher surface coverage with the lowest MW alginate (vLVG) compared to LVG (p=0.0001) and statistical equivalent to alginate MVG (p=0.306). Individual comparisons of these results are illustrated in Figure 3B&C and statistical differences are further described in Table 1.
Table 1.
Effect of MW of Alginate on Percent Surface Coverage: Mean Difference
| CONP4 | CONP7.4 | |||||
|---|---|---|---|---|---|---|
| MVG vs. LVG |
MVG vs. vLVG |
LVG vs. vLVG |
MVG vs. LVG |
MVG vs. vLVG |
LVG vs. vLVG |
|
| 6-layers | 35.79% (p<0.0001) |
51.82% (p<0.0001) |
16.04% (p=0.0137) |
−3.747% (p=0.7516) |
30.09% (p<0.0001) |
33.84% (p<0.0001) |
| 12-layers | 7.020% (p=0.429) |
13.29% (p=0.0240) |
6.265% (p=0.4286) |
15.16% (p=0.0234) |
−7.185% (p=0.3065) |
−22.34% (p=0.0001) |
To further understand the differences in alginate deposition via CONP/alginate coatings onto hydrogels, a cross-sectional image, approximately halfway through the microbead, was captured to calculate each coating's ring thickness (Figure 3D&E). After 6-layers, the effect of alginate molecular weight on layer thickness was only noted for coatings formed using acidic pH. Specifically, while MVG, LVG, and vLVG coatings formed using CONP7.4 exhibited equivalent thickness after 6-layers, replacement with CONP4 resulted in a positive correlation between coating thickness and alginate molecular weight, with CONP4/MVG resulting in 40 and 52.2% thicker coatings than alginate CONP4/LVG and CONP4/vLVG, respectively. After 12-layers, the effect of CONP pH on the ring thickness followed a similar trend, with the acidic CONP decreasing coating thickness with decreasing MW (p<0.001). Specifically, coatings formed using vLVG exhibited significantly thinner coatings than those formed using MVG and LVG (45.9 and 32.5% decrease, respectively). Furthermore, only coatings formed using alginate vLVG showed a significant change in coating thickness when the CONP pH was adjusted to 7.4, with a 67% increase in thickness (p < 0.0001). Overall, evaluating alginate deposition in CONP/alginate coatings onto hydrogel microbeads implicates that acidic CONP results in thinner coatings for the lower molecular weight alginates (LVG and vLVG). The assessment method, however, cannot conclude if these thinner coatings result from tighter CONP-alginate interactions or decreased alginate deposition.
Understanding the effect of these coating formulations on alginate uniformity and thickness can be useful; however, these results can be independent of the amount of cerium captured within each coating formulation, as the amount of alginate deposited is not a direct indicator of cerium concentration with the coatings. To capture this information, coated beads were processed and analyzed via Induced Coupled Plasma- Mass Spectrometry (ICP-MS). Interestingly, the concentration of cerium was not directly correlated with alginate coating thickness and uniformity. While alginate deposition analysis showed minimal differences between 6 and 12-layer coatings formed using neutral CONP, the cerium concentration increased two-fold for 12-layer versus 6-layer beads for coatings formed using MVG and LVG (Figure 4A). The impact of layer number on cerium content was more pronounced with acidic CONP, with all alginate types exhibiting a two to three-fold increase in cerium content when the layer number was increased from six to twelve. The difference in fold-change between acidic and physiological pH (Figure 4B) correlated with ellipsometry results, which showed that the rate of layer formation at acidic pH is higher at early coating stages. In contrast, at physiological pH, there was less variation in rate between layers.
Figure 4. Deposition of CONP during CONP/alginate layer-by-layer formation was dependent on coating parameters.

A) Cerium concentration per 50 beads was quantified via ICP-MS. B) The fold change of cerium accumulation between twelve-layer from six-layer beads was quantified to the rate of CONP deposition between groups. C) The amount of cerium per micrometer of alginate-CF was calculated to understand the efficiency of cerium accumulation among groups. Two-way and one-way ANOVA with Tukey's post-hoc test: ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05, where * represents differences between groups, Δ compares six-and twelve-layer coatings, and γ compares groups with same alginate MW but varying pH of CONP.
To further understand these results, the cerium concentration per coating was normalized to the coating thickness, as assessed via fluorescent alginate imaging. Results show that the ratio of cerium to coating thickness increased as the number of CONP/Alginate bilayers was doubled (Figure 4C). Globally, these results identify the CONP4 and MVG or LVG coatings as superior in CONP deposition after 12 layers, when compared to the other tested combinations. Alternatively, the use of CONP7.4 and vLVG results in the smallest amount of CONP deposition.
2.2. Effect of alginate MW and CONP pH multi-enzymatic activity of coatings:
The goal of developing CONP-based coatings was to provide antioxidant protection at the biomaterial interface. CONP has the potential to impart a spectrum of antioxidant activity, as it can cycle continuously between its tetra- (Ce4+) and trivalent (Ce3+) states (Figure 1C). The Ce4+ state exhibits a more catalase-like activity, while the Ce3+ state mimics superoxide dismutase (SOD). Since the molecular weight of alginate and the pH of CONP during fabrication affected the uniformity and thickness of the coatings, it was hypothesized that these parameters could also impact global oxidant activity. If changing these parameters alter the ratio of available Ce4+/Ce3+ in the coatings, different coatings could be then be used to target diverse oxidative stress conditions in which either H2O2 or SO is predominant.
To understand the effect of alginate MW and CONP pH in the Ce4+/Ce3+ ratio, cyclic voltammetry (CV) was used. First, stainless steel (SS) electrodes were coated with the different formulations of CONP and alginate. These coated working electrodes were then analyzed via CV, which permits evaluation of the reduction and oxidation potential of the coating (see example traces in (Figure 5A&B). The evaluation of the maximum anodic peak on a CV graph provides insight into the electrode's reductive capacity, while the maximum cathodic CV peak characterizes its oxidative capacity (Figure S1). Translating this to CONP, an increase in the reductive capacity indicates an elevated Ce4+/Ce3+ ratio and an elevated catalase-mimetic activity. Alternatively, an increase in oxidative capacity translates to a decreased Ce4+/Ce3+ ratio and an elevated SOD-mimetic activity. Thus, evaluating alterations in the coated wire's anodic and cathodic properties provides insight into the CONP/alginate coating's preferential redox activity.
Figure 5. The redox activity of different formulations on coated stainless-steel wires establishes variance of activity with CONP/alginate coating properties.

Representative traces of reduction/oxidation curves for wires coated using A) acidic and B) neutral pH CONP assessed via cyclic voltammetry. Dashed lines: Max peak cathodic and anodic currents (as indicated) from uncoated wire. C) Since variations in cerium concentration can affect the peak current, the ratio of reduction peak potential (Ce4+) to oxidation peak potential (Ce3+) was calculated. Two-way and one-way ANOVA with Tukey's post-hoc test:****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05, where * represents differences between groups, Δ compares six-and twelve-layer coatings, and γ compares groups with same alginate MW but varying pH of CONP.
All CONP/alginate coating formulations resulted in an increased peak anodic current, when compared to uncoated control electrodes (see example 12-layer traces in Figure 5A&B). Increasing layers from six to twelve further elevated peak anodic levels for all coating combinations, except those formed using MVG alginate. Examination of the peak cathode levels revealed a differing trend, where the presence of the CONP/alginate coating either decreased or unaltered this value compared to uncoated control wires. The quantification of the max anodic and cathodic currents of 12-layer coatings measured via CV is summarized in Figure S1. The measured anodic max current was significantly impacted by both alginate type and CONP pH (p < 0.0001 for both parameters; 2-way ANOVA), with coatings formed using LVG alginate, imparting the most consistent and significant effect. Coatings formed using LVG were not impacted by pH (p = 0.81), while coatings formed using vLVG were dramatically affected (56.1 versus 15.5 μAmps for CONP4 versus CONP7.4; p < 0.0001). Despite these differences, results collectively indicate that CONP/Alginate coatings increase the metal wire's reductive capacity, thereby exhibiting elevated catalase-mimetic activity. Examination of the cathodic properties shows that CONP/alginate imparted minimal effects on oxidative capacity. Only the CONP4/MVG and CONP7.4/vLVG coatings significantly decreased the maximum cathodic current compared to uncoated wire. This decrease in oxidative activity indicates that these specific coatings may exhibit decreased SOD-mimetic activity. A caveat of this analysis was that these max peak current changes were dependent on several factors, including the cerium concentration deposited onto the wires.
Since a higher cerium concentration can skew the max peak current, a ratio of the max anodic current to the max cathodic current was calculated (Figure S1).[22, 23] For MVG-based coatings, the Ce4+/Ce3+ ratio was unaltered when the number of layers was increased from six to twelve (p = 0.0619), regardless of the CONP pH used. In contrast, for the lower molecular weight alginates of LVG and vLVG, the addition of six new layers increased the Ce4+/Ce3+ ratio, independent of the CONP pH (p < 0.0001 for all). A focused analysis of the twelve-layer alginate coatings found both CONP pH and alginate molecular weight significantly impacting the Ce4+/Ce3+ ratio (p = 0.005 and < 0.0001, respectively), with MVG exhibiting the lowest Ce4+/Ce3+ ratio when compared to coatings formed using the lower MW alginates. Coatings formed using vLVG were most impacted by CONP pH, with a significant decrease in catalase-mimetic activity when CONP pH was increased from 4 to 7.4 (p = 0.0037). Overall, the normalization of the anodic to the cathodic peaks showed a positive correlation between decreasing alginate molecular weight and the Ce4+/Ce3+ ratio (p <0.0001). There was also a positive correlation between the acidity of CONP during coating fabrication and the Ce4+/Ce3+ ratio (p<0.0054). However, pH only impacted the Ce4+/Ce3+ ratio for lower MW alginate, due to the interaction between these two factors (p < 0.0008). Overall, analysis of the synergistic effect of CONP pH and alginate MW on the coatings’ multi-enzymatic activity indicates that an array of coating formulations can induce in a wide range of antioxidant properties.
2.3. Characterization of Scavenging Capabilities in Hydrogels:
CV indications of skewed multi-enzymatic activity of CONP/Alginate coatings was validated by incubating coatings with distinct environmental oxidants. For evaluation of SOD-mimetic activity, superoxide (SO) scavenging was measured. For catalase-mimetic activity, TMB and H2O2 oxidation, was measured. Tests were conducted for CONP/Alginate coatings formed onto alginate microbeads.
For SO generation, xanthine and xanthine oxidase was added to the incubation media. All CONP coating formulations demonstrated SOD-mimetic activity with significant SO scavenging, when compared to uncoated controls (p < 0.0001); however, there was no difference in SO consumption between any of the coating groups (p = 0.097) (Figure 6A&B).
Figure 6. Scavenging of exogenous SO was not impacted by CONP/alginate coating parameters onto alginate microbeads.

A) 6-layer and B) 12-layer beads coated with the variable CONP/alginate coatings indicating scavenging of superoxide, but no significant differences between groups.
The catalase-mimetic activity was assessed in coated alginate beads via oxidation of TMB substrate, as TMB exhibits a color change when CONP is reduced from Ce4+ to Ce3+. For all CONP coating formulations, TMB absorbance values in the incubating solution were elevated, compared to uncoated controls, indicating increased TMB oxidation (Figure S2). Additionally, increasing the layers from six to twelve further increased ambient TMB oxidation for all groups, except for CONP7.4/MVG. Coatings, however, also exhibited a blue color following TMB exposure, indicating binding of the oxidized TMB within the CONP/alginate layers (Figure S2). Since the assay only measured color changes in the incubating solution, the full catalase-like activity of the coatings was not fully captured by this method.
As catalase activity accelerates H2O2 oxidation into oxygen, the CONP coated microbeads' capacity to deactivate H2O2 was quantified.[24] In addition, to explore the full potential of CONP scavenging capabilities, the same coated beads were exposed to a total of three distinct and sequential challenges of 100 μM H2O2. On the first H2O2 challenge, 12-layer beads coated with CONP4 showed similar activity, although LVG coatings were slightly (14.9%) more active than vLVG (p = 0.0002). At physiological pH, alginate LVG contained the moderately higher scavenging capability (11-14%) compared to both MVG (p = 0.003) and vLVG (p = 0.0006). After two additional H2O2 bolus challenges, the scavenging capabilities between alginate LVG was still superior to vLVG, regardless of pH (CONP4 and CONP7.4 p = 0.04 and 0.017, respectively). There was no difference between MVG and the lower MW alginate coatings at either or physiological pH. Globally, acidic conditions did not impact the H2O2-scavenging capabilities of 12-layer coatings (p = 0.198). Examination of 6-layer coatings indicated exhaustion of H2O2-scavenging capabilities for coatings formulated using physiological pH, with the greatest decline in activity measured for CONP7.4/vLVG coatings (see Figure 7A&B).
Figure 7. The H2O2 scavenging capability of CONP/alginate coatings on hydrogels was dependent on coating parameters, after repeated exposure to the exogenous H2O2.

Change in H2O2 levels following repeated exposure of 6-layer (A) and 12-layer (B) coated microbeads. Three incubation periods of distinct 100 μM H2O2 challenges are shown in three compounded bars 1st (bottom), 2nd (middle), and 3rd (top). The statistics for the sum of total H2O2 consumed after three H2O2 challenges are shown above the bars. Differences in scavenging capabilities between H2O2 challenges are represented as tau (τ), where τ τ τ τ p < 0.0001, τ τ τ p < 0.001, τ τ p < 0.01, and τ p < 0.05. The amount of H2O2 scavenged per total cerium content is shown for 6-layer (C) and 12-layer (D) beads. For all graphs, statistical differences are represented as two-way and one-way ANOVA with Tukey's post-hoc test: ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05, where * represents differences between groups, τ compares H2O2 scavenged between challenges, Δ compares six-and twelve-layer coatings, and γ compares groups with same alginate MW but varying pH of CONP.
As reported earlier, the CONP content among coating formulations varied, and the scavenging capability is dependent on the amount of cerium. To incorporate this parameter into assessment of enzymatic activity, the H2O2 scavenging capacity for each coating formulation was normalized to its measured cerium content (Figure 7C&D). As shown, 12-layer coatings formed using the lower MW alginate (vLVG) were superior in catalase-mimetic antioxidant activity at both acidic (p = 0.0007 and < 0.0001 for LVG and MVG, respectively) and at physiological pH (p<0.0001 for both LVG and MVG). Furthermore, an increase in CONP pH resulted in increased H2O2-scavenging per cerium content for both 6- and 12-layer beads (p<0.0001). Specifically, 12-layer coatings showed that CONP7.4/MVG and CONP7.4/vLVG formulations have a higher H2O2-scavenging capability than when coated with acidic pH CONP (p=0.0177 and p=0.0002, respectively). These results confirm that an increase in catalase-mimetic activity can be achieved by decreasing the MW of alginate and increasing the pH of CONP during fabrication.
Overall, the H2O2-scavenging studies validate the CV results that a decrease in MW of alginate and an increase in CONP pH increases the coatings' catalase-/SOD-activity ratio. The disparity in MW between MVG and vLVG allowed us to understand the effect of MW on CONP redox orientation. Based on these results, alginate LVG formulations were removed for the following studies to focus on groups with the most evident changes in redox activity. In the following studies, the coatings' ability to protect encapsulated cells from H2O2 and SO was assessed to understand the applications of these coatings.
2.4. Examination of H2O2/SO Protection of Coatings on Encapsulated MIN6 cells
Potent antioxidant coatings can protect tissue grafts from local oxidative stress generated by the host in response to the implantation procedure. In one approach, antioxidant coatings can serve to transplanted insulin-producing β-cells from the deleterious effects of extracellular reactive oxygen species generated at the implant site. To assess the ability of CONP/alginate coatings to protect β-cells from oxidative stress, MIN6 β-cells were encapsulated within alginate microbeads coated with or different formulations of CONP and alginate.
To understand the baseline impact of CONP/Alginate coatings on beta-cell metabolism, metabolic activity was assessed post-coating. For this work, coatings generated using alginate MVG and vLVG were studied, given that they exhibited the most distinct differences in coating properties and activity. For all formulations tested, 6-layer coatings did not impact cellular metabolic activity (p = 0.3037; Figure S3A). However, at the 12-layer dosage, selected formulations decreased cellular responses. Specifically, CONP4/MVG coatings imparted the most impact, with a decrease of 18.9% in metabolic activity when compared to the uncoated control (p < 0.001; Figure 8A). Decreasing the MW of alginate mitigated the effect, while replacing the CONP to net neutral reduced the negative impact of the MVG coating to ~5%. Combining both a neutral CONP and a lower molecular weight alginate (vLVG) did not synergize to decrease the coating’s effect in cell metabolism further, as the results were statistically equivalent to CONP7.4/MVG and CONP4/vLVG coatings (p > 0.999 and 0.271, respectively). Globally, while alternations could decrease the cell impact to only ~5%, both parameters of CONP pH and alginate molecular weight had a significant impact on the base metabolic activity of the encapsulated cells (p < 0.0001 and 0.0002, respectively; 2-way ANOVA).
Figure 8. The impact of CONP/alginate coatings on the baseline beta cell metabolic activity was dependent on coating material properties.

The baseline metabolic activity (A) and insulin secretory response to a glucose challenge (B) of encapsulated cells following coating with 12-layers of the designated coating formulations. The effect of 12-L coatings on oxidative cell health was measured via reduced:oxidized glutathione (GSH:GSSG) ratio (C), total glutathione (D), and oxidized glutathione (E). Grey dashed line: untreated controls. Two-way and one-way ANOVA with Tukey's post-hoc test: ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05, where * represents differences between groups, Δ compares 12-L coatings to 0-L beads, and γ compares groups with same alginate MW and varying pH of CONP.
To further investigate the coating effect on beta cell health and function, insulin secretion in response to a high glucose challenge (i.e., 16.7 mM) was quantified. There was no significant difference between 0-L controls and 12-L coated beads, regardless of formulation (Figure 8B, p = 0.0619). Results implicate that coatings did not impact insulin secretion, both in terms of cell function and protein diffusion out of the coated microbeads.
Additional investigation into the antioxidant profile of the encapsulated cells was also conducted via measurement of both total intracellular glutathione, which includes both reduced glutathione (GSH) and oxidized glutathione (GSSG)[25]. In addition, GSSG levels were quantified. Globally, the GSH:GSSG ratio provides a comparable index of oxidative cell health, with reduced ratios implicating increased oxidative stress[26]. As summarized in Figure 8C, coatings formed using acidic CONP either imparted no change (p = 0.999 and 0.468 for coatings formed using MVG or vLVG respectively). Coatings using CONP stabilized at physiological pH exhibited increased oxidative cell health, as indicated by an increased GSH:GSSG ratio (MVG, p < 0.001; vLVG, p = 0.0015). The beneficial impacts of coatings formed using vLVG appear to be driven by elevated total glutathione levels, while coatings formed using CONP4 coatings caused a decrease in total glutathione levels (Figure 8D). These impacts correlate with metabolic activity results. Of interest, all CONP-based coatings conveyed a significant decrease in the presence of oxidative glutathione when compared to uncoated controls (Figure 8E).
Despite the modest decrease in baseline metabolic activity for selected coatings, a robust antioxidant protection would impart a net positive impact on prolonging the survival of encapsulated cell therapies. To examine the potency of the various coating formulations on protecting the underlying cells from oxidative injury, coated microbeads were incubated with either H2O2 or SO. Following the oxidative challenge, downstream impacts on metabolic activity were assessed and compared to unchallenged controls. The exposure of uncoated microbeads to H2O2 or SO conveyed significant impacts on the metabolic activity of the encapsulated cells, with a 33.3 or 34.2% decrease in cellular metabolic activity, respectively. Coating encapsulated cell microbeads with 6-layers of CONP and alginate did not impart substantial protection, regardless of the formulation (Figure S3B). Increasing the layer number to 12-layers showed enhanced protection, depending on the coating formulation (Figure 9A). Specifically, beads coated with CONP4/MVG fully protected against both H2O2- and SO-mediated loss in metabolic activity, with values statistically equivalent to the non-challenged controls (p = 0.635 and 0.704, respectively). Alternatively, coatings with CONP4/vLVG were only able to fully protect against an H2O2 insult (p = 0.814 and p < 0.0001 versus untreated controls and H2O2-treated controls, respectively), with no measurable impact on superoxide protection (p < 0.0001 and p = 0.845 versus untreated controls and SO-treated controls, respectively).
Figure 9. Protection of encapsulated beta cells from exogenous ROS insults was dependent on coating material properties.

The impact of oxidative stress from either H2O2 or SO on the underlying cells was evaluated and summarized as fold change from untreated controls (grey dashed line). Metabolic activity (A) and insulin secretory response to a glucose challenge (B) of encapsulated cells following treatment with H2O2 or Xa/XO (SO generating). Uncoated or microbeads coated with 12-layers of the designated coating formulations were tested. The effect of 12-L coatings on protecting cells was also measured via reduced:oxidized glutathione (GSH:GSSG) ratio (C), total glutathione (D), and oxidized glutathione (E). Grey dashed line: untreated controls. Two-way and one-way ANOVA with Tukey's post-hoc test: ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05, where * represents differences between groups, Δ compares 12-L coatings to 0-L beads, and γ compares groups with same alginate MW and varying pH of CONP.
Examination of the effect of CONP pH on cytoprotection from external oxidants, coated formed using alternating layers of CONP7.4 and alginate MVG or vLVG were examined. Even though the CONP7.4/MVG exhibited a ~2-fold increase in CONP content over the CONP7.4/vLVG formulation, their protective impact was generally similar. When exposed to an exogenous H2O2 challenge, both MVG and vLVG coatings formed using CONP7.4 imparted a protective effect, mitigating declines in metabolic activity from 33% to only 7.34 and 6.76%, respectively (p < 0.001 for both when compared to treated controls). Resulting metabolic activities for CONP7.4/MVG and CONP7.4/vLVG coated cells were statistically identical (p = 0.999) although they technically imparted different statistical benefits when compared to untreated controls (p = 0.06 and 0.03, respectively). For Xa/XO challenged microbeads, the protective nature of the coatings was less pronounced with a modest, but significant, shift from 34.1% (untreated controls) to 25.3 and 21.2% (p < 0.0001) for MVG and vLVG coated beads, respectively; this level of protection was not comparable to untreated controls (p < 0.0001 for both coating formulations). Even though different coating formulations had varying protective effects when assessed via metabolic activity, insulin secretion in response to glucose was minimally altered (Figure 9B). It should be noted, however, that characterizing beta cell functional health under acute oxidative stress via insulin release is complicated, as oxidative insults can stimulate insulin secretion[27].
To investigate coating impacts on oxidative cell health, intracellular glutathione levels (total and oxidative) was quantified for cells within coated or uncoated beads exposed to an ROS challenge (Figure 9C-E). In concordance with metabolic activity results, uncoated beads showed a significant decrease in GSH:GSSG ratio following H2O2 (p=0.0032) and Xa/XO (p=0.0017) challenge, implicating a decrease in intracellular antioxidant protection. Interestingly, this altered ration was not due to changes in total glutathione concentrations, (p = 0.455 and 0.957 for H2O2 and Xa/XO when compared to untreated controls). Instead, there was a significant elevation in the presence of intracellular oxidized glutathione in both H2O2 and Xa/XO treated uncoated controls (p < 0.0001 for both).
For encapsulated cells coated with CONP-based layers, changes in their antioxidant potential was dependent on the coating composition. CONP4/MVG coated cells treated to H2O2 exhibited an unaltered GSH:GSSG ratio, when compared to unchallenged coated controls (p = 0.538); however, this formulation did not prevent a drop in GSH:GSSG ratio when challenged with Xa/XO (p = 0.0011). This alteration was associated with a decrease in total glutathione (p = 0.0035). Coatings formed using CONP4/vLVG exhibited declines in GSH:GSSG ratio following exposure to either H2O2 and Xa/XO (p < 0.0001). For H2O2 exposed CONP4/vLVG coatings, these intracellular antioxidant alterations were conveyed by elevated levels of oxidized glutathione (p = 0.042 compared to untreated), as total glutathione concentrations remained unchanged (p = 0.206 compared to untreated). For Xa/XO exposed CONP4/vLVG coatings, changes were caused by lower levels in both total and oxidized glutathione (p = 0.014 and < 0.0001, respectively). Finally, none of the coating formulations using CONP7.4 resulted in preservation of their untreated intracellular antioxidant levels, regardless of the type of ROS exposure (p<0.0001).
3. Discussion
An imbalance in oxidants can initiate an endless loop between inflammation and oxidative stress. Research groups have addressed this challenge by engineering methods to supplement biomaterial implants with antioxidant properties. These approaches include redox-sensitive biomaterials such as poly(thioketal) urethane (PTK-UR), that can serve as an oxidant sponge and deliver therapeutic agents as the material degrades.[28, 29] Alternative approaches are based on forming hydrogen-bonded polyphenols onto biomaterials and tissue grafts to protect from oxidative stress-mediated cytotoxicity.[30] In addition to protecting tissue grafts from oxidative damage, these materials can further mitigate inflammation and create a balance between inflammatory and tolerogenic phenotypes.12-14 Although these antioxidant approaches show potential, the materials used in these studies exhibit limited redox capabilities and self-renewing potential.
For longer-term studies, metal oxide nanoparticles such as cerium oxide, manganese, zinc, titanium, and yttrium are more appropriate, as their catalytic activity per unit volume is substantially higher and more durable than that of polyphenols and redox-sensitive polymers.[6, 31] Of these catalytic nanoparticles, cerium oxide exhibits superior self-renewability potential and multi-enzymatic activity.[11, 19, 32] While CONP is a promising antioxidant/anti-inflammatory therapeutic for a wide range of biomedical applications, systemic administration of nanoparticles still raises concerns regarding toxicity and clearance.[14] Therefore, our group has engineered an LbL technique to immobilize redox-active nanoparticles by alternatively depositing CONP and alginate onto the surface of biomaterials. In previous work, resulting LbL coatings exhibited redox activity that protected encapsulated murine β-cells from H2O2-/SO-mediated decreases in metabolism, oxidative stress, and functionality.[17]
In the current work, CONP/Alginate coating combination was further investigated to exploit its full potential. Based on previous literature, it was hypothesized that factors such as molecular weight (MW) of the intermediate anionic layer and the net pH of the CONP could impact the thickness, uniformity, cerium content, and redox activity of the coatings through CONP/alginate interactions.[10, 33, 34, 35-37] Potential interactions between CONP and alginate that can drive the formation of specific nano- and micro-complexes include: i) charge, ii) acidic precipitation of alginate, iii) CONP precipitation onto biomaterials surfaces, iv) hydrogen bonding, v) chelating ligands, and vi) covalent complexes.[38] Manipulation of CONP pH and alginate MW can dictate the primary type of interactions driving CONP/alginate complexes, which can play key roles in controlling coating thickness, uniformity, Ce4+/Ce3+ ratio, and, subsequently, overall enzymatic activity.
Layer-by-layer deposition for different formulations of CONP and alginate resulted in varying thickness and layering rates. As reported in previous LbL studies, polyelectrolyte complexes formed using higher MW polymers typically leads to thicker coatings.[36] For coatings formed using acidic CONP, a positive correlation between high molecular weight alginate and coating thickness was measured via both ellipsometry and fluorescent imaging quantification. The investigation into the efficacy of layer formation was another parameter of interest. Although many LbL coatings grow linearly, several material combinations, such as poly(L-lysine) (PLL)/alginate and PLL/hyaluronic acid, deposit layers in more of an exponential-like manner.[39] Similarly, CONP/alginate coatings formed using acidic CONP exhibited a more exponential growth behavior, followed by linear growth. A steep increase in thickness at the early stages, followed by stabilization at later stages of layering, is a characteristic pattern of several polyelectrolytes and nanoparticle coatings.[40] This is postulated to be due to surface aggregation of the nanoparticles[41]. Furthermore, higher MW polymers yield a steeper exponential growth phase than lower MW polymers, which explains the higher growth rate of alginate MVG and LVG compared to vLVG at early stages.[33]
Previous reports have also shown that the coating thickness of weak polyelectrolyte complexes can be controlled by changing the pH during fabrication.[35, 36] A comparison of coatings formed using a net neutral CONP validated these correlations, as these layers exhibited a more discreet and linear layer deposition that was not significantly impacted by alginate MW. These trends correlate with previous studies showing that polycations exhibit the highest increase in layer thickness at pH 4-6.[35]. Even though the exponential growth behavior of CONP/alginate complexes was accelerated at acidic pH, the self-precipitation of alginate may be another contributing factor to an early increase in thickness.[20] Overall, under both dry or hydrated conditions, the lower MW alginate exhibited more disparate differences in coating thickness and uniformity between early and later stages of coatings.
Independent of coating thickness and coverage, the MW of alginate and CONP pH influenced the amount of CONP entrapped within the coatings. As expected, the concentration of cerium deposited in the coatings increased between six and twelve layers, despite no significant change in coating uniformity on beads. In addition, the concentration of cerium deposited on hydrogel microbeads was positively correlated to alginate MW, with decreasing CONP presence as the alginate MW decreased. CONP pH also played a key role, with the acidic formulation resulting in enhanced CONP presence. Normalization of CONP to coating thickness indicated elevated CONP capture efficiency for the 12-layer group, when compared to the 6-layer coatings. Furthermore, the CONP capture efficiency was the lowest when the neutral CONP formulation and lowest MW alginate was used. Examination of these global trend, indicates that alginate MW and CONP pH impart changes in the interactions between these two materials, resulting in variability in CONP capture efficiency and global layer formation.
Investigation into the established crosslinking behavior of alginate can provide insight into potential CONP-alginate interactions. In alginate cationic crosslinking, cations preferentially align along the longest alginate junctions before creating new junctions.[42, 43] Under this assumption, higher MW alginates should exhibit faster-crosslinking kinetics due to decreased loose-end fraction, which would allow for elevated accommodation of the cationic CONP between adjacent alginate chains.[43] Moreover, high MW chains are more flexible than lower MW alginate, which are more prone to shrink and contract, thereby contributing to intrachain dimerization or precipitation.[43, 44] Acidic conditions can further exacerbate this precipitation since, the alginate chain is composed of mannuronic and guluronic acid monomers, which have pK values of 3.38 and 3.65, respectively.[20] Thus, based on published reports, it is postulated that the higher MW alginates were more prone to precipitation, which was intensified by the presence of the acidic CONP.
To investigate if these distinct CONP-alginate interactions induced variability in antioxidant features, cyclic voltammetry electrochemical analysis was employed.[22] CV analysis of coatings showed anodic and cathodic peaks, indicating that all CONP coatings exhibited fully reversible oxidation and reduction. Examination of differences in the peak magnitude from the cathode and anode regions during electrochemical stimulation indicated differences in the reduction and oxidation potential of the different coating formulations. As the magnitude of these peaks can be influenced by the concentration of cerium deposited onto the SS electrode, the ratio of anodic/cathodic current, and hence the Ce4+/ Ce3+ ratio of the coatings, was investigated and correlated to its overall antioxidant behavior.
The molecular weight of the alginate substantially altered the Ce4+/ Ce3+ ratio of the embedded CONP. Specifically, formulations using alginate vLVG exhibited the highest Ce4+/Ce3+ ratio, while coatings formed using alginate MVG exhibited the lowest Ce4+/Ce3+ ratio. The mechanism driving this observed correlation is not completely clear; however, it is postulated that these shifts are due to the type of interactions between alginate and CONP. Due to the decreased bias of lower MW alginate to self-precipitation, there is enhanced potential for coordinated cerium-alginate interactions, specifically complexes between Ce3+ in the CONP and carboxylate anions (i.e., COO−) in alginate.[12] As the hard and soft acid and base (HSAB) theory explains, Ce3+, a hard acid, can bind to soft bases such as phosphates or hydroxides over sulfates and hydrides.[12] Strong bases, such as phosphate or carboxylate anions, can bind to Ce3+, which can block reversible switching to Ce4+.[45] The inhibition of Ce3+ by carboxylate anions interactions shifts the overall activity of CONP towards Ce4+, which it already favors due to its xenon-like configuration.[19] Therefore, an enhanced presence of coordinated CONP-alginate complex would result in a coating biased towards a catalase-mimetic activity. Contrarily, the highest MW alginate, MVG, likely exhibited the lowest Ce4+/Ce3+ ratio due to a shift from coordinated CONP-alginate complexation to self-participation, which would preserve the baseline Ce4+/Ce3+ ratio of the CONP. This formulation would also result in enhanced CONP deposition, as validated in the ICP-MS measurements. Coatings formed using the alginate LVG appear to follow a more complex pattern, as its MW range is in between the other alginates (MVG > LVG > vLVG) and contains both long and short strands. Due to this characteristic, alginate LVG likely interacts with CONP through both precipitation and coordinated complexes, depending on the MW of each alginate strand. While longer chains will accommodate cations along the length of a chain before it crosslinks with a new chain[42, 43], the short chains will interact with Ce3+ in a discrete manner similar to vLVG, which would shift the redox activity to a SOD-mimetic activity. Resulting coatings formed using LVG would exhibit more CONP deposition, due to enhanced self-precipitation, but also elevated CONP-alginate complexes. Thus, LVG coatings exhibit a global redox potential in between the two other alginate types.
In addition to alginate MW, the effect of pH also plays a role in the Ce4+/Ce3+ ratio of coatings. Several groups have reported changes in redox activity due to pH.[10, 22, 37, 46]. There were no measurable differences in redox activity between CONP4/MVG and CONP7.4/MVG, although there was a 1.6-fold decrease in CONP integration. These results further implicate self-precipitation as the dominant interaction, whereby the pH does not alter CONP activity, but lower pH elevated CONP self-precipitation during coating. For the lowest alginate MW (vLVG), the acidic CONP elevated the Ce4+/Ce3+ ratio of the resulting coatings compared to neutral CONP coatings. This shift has been observed in previous publications, in which an acidic environment leads to an increase in catalase-mimetic (Ce4+) activity.[10] As with MW, the LVG formulations exhibited an intermediate response with only a modest impact of pH on the potential catalytic activity of the CONP.
Once the effects of MW and pH during fabrication were fully characterized, each formulation's properties were assessed in their ability to mitigate different sources of oxidative stress in the microenvironment. Even 6-layer CONP7.4/vLVG, which consumed the least amount of H2O2 (1.043 μM/min), has an equivalent scavenging rate to catalase enzyme (1 μM/min).[47] For the surface area of nanoscale CONP coatings, the H2O2-scavenging rate is comparable to bulk antioxidants such as PTK-UR and to nanoscale antioxidants such as polyphenols.[29, 48]
The catalase-mimetic activity of different coating formulations was tested via TMB oxidation assay, showing equivalent activity between MVG and vLVG. Even though these two coating formulations have different cerium content, the catalase-mimetic activity of vLVG coatings was higher. However, this assay is surface-based, and the change of absorbance in the liquid might not represent the full TMB oxidation. For that reason, catalase-mimetic activity was measured via exposure to three sequential challenges with cytotoxic levels of H2O2 consumption.[48] The H2O2 consumption assay showed similar results to TMB oxidation, where MVG and vLVG had similar scavenging capability. However, coating formulations with alginate vLVG contain less cerium than those of MVG. Normalization of H2O2 to the total cerium content shows that lower MW alginate shifts the activity of the coatings towards a higher catalase/SOD ratio. These results validate the theory explained above that CONP interacts with lower MW alginate primarily via Ce3+-COO− coordination complexes rather than precipitation. These interactions lead to Ce3+ inhibition (responsible for SOD activity), in which case the primary redox modulator is Ce4+ (responsible for catalase activity. Although hydrogen peroxide consumption results concur with the various techniques used to assess the Ce4+/Ce3+ ratio, it is important to point out the caveats of the H2O2 consumption assay: 1) The scavenging capabilities of the coatings were not fully exhausted, and the long-term capacity of these coatings are yet to be investigated. 2) beads were incubated with H2O2 for one hour before measuring scavenging capabilities, which is enough time for these molecules to diffuse through the bead. Even though this data is an accurate indicator of the scavenging rate of H2O2, these results might not result in cell protection if H2O2 diffuses freely through the coatings.
As a final validation of the tunability of the antioxidant coatings in terms of redox activity, the ability of the different coating formulations to protect encapsulated cells was assessed by exposing coated and non-coated encapsulated cells to SO or H2O2. Each formulation showed specificity in protection for either SO or H2O2, depending on coating uniformity, cerium content, and Ce4+/Ce3+ ratio. Beads coated with CONP4/MVG were the most efficient formulation at protecting cells from both SO and H2O2. While CV results found the CONP4/MVG formulation exhibited a preference to the SOD-mimetic side of the redox spectrum, it is suspected that the high concentration of cerium captured in this coating supported the equal protection against H2O2 and SO. Interestingly, CONP4/vLVG exhibited only a distinct preferential protection against only a H2O2 challenge. While coatings formed using alginate vLVG contained almost a 2-fold lower cerium concentration than coatings formed using alginate MVG, the global shift of these vLVG coatings towards a higher Ce4+/Ce3+ ratio allowed the CONP4/vLVG coating to be as effective as CONP4/MVG coatings in catalase-mimetic activity. However, this preferential catalase activity was insufficient in preventing an H2O2-induced decrease in the GSH:GSSG ratio. In addition, coatings formed using CONP7.4 were not as effective in protecting encapsulated cells as their acidic pH counterparts, indicating that their decreased CONP presence reduced their global enzymatic activity.
To expand on coating modularity, manipulations in layer formation and in material selection can be explored. For example, alternating or block layering of alginate and CONP4 and CONP7.4 may result in different layer thicknesses, as well as antioxidant activity, thereby creating coating that excel in broader ROS protection. In addition, other materials besides alginate can be investigated, such as poly(lactic-co-glycolic acid), gelatin, or cellulose acetate[49]. Furthermore, the encapsulation of CONP with polymers (e.g. polystyrene sulfonate) or surface functionalization could expand its capacity to layer or link to numerous other polymers.
Overall, investigation into the effects of CONP and alginate parameters on antioxidant coatings and activity revealed the capacity to modulate antioxidant properties, as well as coating features. The ease in which CONP/alginate coatings were formed on silica, metal, and hydrogels further illustrates the utility of this approach for broad translation to numerous medical implants. For example, this method can potentially mitigate fibrosis and oxidant-mediated electrical/mechanical malfunction of continuous glucose monitors (CGM),[50] cardiovascular stents/valves, and orthopedic prosthesis.[51] The scavenging of oxidants in the local environment could not only suppress foreign body responses, but potentially protect transplanted cells from the deleterious effects of ROS. Future work will seek to translate these coatings to in vivo applications to examine the impact of this redox modularity on host responses that lead to fibrous encapsulation, elevated adaptive immune response, and damage to the implanted cells. Finally, in addition to antioxidant properties, these CONP-coatings can be explored for their capacity to induce angiogenesis or provide anti-bacterial protection[2, 49, 52].
4. Conclusion
Alternating layers of CONP and sodium alginate yielded antioxidant, multi-enzymatic coatings that can be formed on metallic, ceramic, and polymeric surfaces. The uniformity, thickness, and the ratio of SOD/Catalase-mimetic activity can be manipulated through changes in the molecular weight of alginate and the pH of CONP dispersion. In conclusion, the mechanism driving the coating dictates the activity of CONP in coatings. The two major mechanisms driving CONP/alginate interactions are precipitation and coordination complexes. Higher MW alginate is more susceptible to precipitation, and coordination complexes between Ce and carboxylate anions in alginate are the prominent binding mechanism in lower MW coatings. The primary mechanism in each coating formulation dictates its Ce4+/Ce3+ ratio. Coatings with lower MW has a higher catalase-mimetic activity due to Ce3+ inhibition and increased availability of Ce4+. These controllable antioxidant coatings have a broad range of applications, from increasing the electroconductivity of inert surfaces to protecting tissue grafts from oxidative stress and the potential to mitigate FBR to biomaterial implants.
5. Experimental Section/Methods
5.1. Materials
The cerium (IV) oxide nanoparticles dispersion (CONP, 20% wt/wt in H2O, low pH, d < 5 nm) was purchased from Alfa Aesar (Cat# 47232). Three PRONOVA UP grade sodium alginates (Alg) with a G/M ratio of 1.5 were purchased from NovaMatrix and consisted of the following: 1) MVG with MW > 200 kDa, 2) LVG with MW 75-200 kDa, and 3) vLVG with MW < 75 kDa. Sucrose (Cat# S7903) and all other chemicals were reagent grade or higher purity and purchased from Sigma Aldrich. Nanoparticle size was validated via NICOMP ZLS Z3000 (Particle Sizing Systems, Port Richey, FL, USA). A fresh nanoparticle dispersion was prepared by diluting CONP in MOPS/KNO3 buffer (10 × 10–3 m each, pH 7.4) and loaded into a 1 cm rectangular plastic cell cuvette. Dynamic Light Scattering (DLS) spectra measurement consisted of ten runs for 30 seconds, and the size was based on the Gaussian number distribution.
5.2. Preparation of CONP at pH 7.4
The CONP dispersion, as received from the manufacturer, had a pH of 4. To adjust to pH to 7.4, the 20% CONP dispersion was diluted in Sucrose-MOPS buffer (40 mM sucrose, 10 mM MOPS, 125 mM NaCl, 3 mM KCl, pH 7.4) to a final concentration of 3 mg/ml and pH adjusted to 7.4 with 1 M NaOH. Nanoparticle size was acquired following the DLS methods explained in section 5.1. However, nanoparticles were diluted in a 40 mM sucrose-MOPS/KNO3 buffered solution.
5.3. Ellipsometry
A silicon wafer (Si wafer, single side polished, no dopant, 2 inches diameter x 0.5 mm thick) was cut into ~11*13 mm2 pieces. Before coating, the Si wafer was first cleaned with acetone (3x) and plasma treated for 5 min at high-intensity settings using a Plasma Cleaner (Harrick Plasma, PDC-001-HP). The Si wafer was placed in a glass test tube and incubated with a dispersion of CONP (3 mg/mL) for thirty seconds, followed by washes of MOPS-buffer (3x) and de-ionized water (1x). The Si wafer was then incubated with a solution of sodium alginate (3 mg/mL) for thirty seconds, followed by washes of MOPS-buffer (3x), de-ionized water (1x), ethanol (3x), and dried with a stream of argon. The Si wafer thickness was measured every bilayer using a J. A. Woollam Co. alpha-SE spectroscopic ellipsometer, at an angle of 70° and standard mode settings, using the transparent film data model. Three readings at different locations of the Si wafer were collected for each bilayer.
5.4. Cyclic Voltammetry (CV)
Cyclic voltammetry (CV) technique was performed as previously described, with some modifications.[53] Briefly, a two centimeters stainless steel wires (127 um diameter, A-M Systems Inc, WA) were coated with different formulations of CONP and alginate. The coated stainless-steel wire was submerged 1 cm into a MOPS buffer solution at room temperature and connected to an Autolab potentiostat PGSTAT12 (EcoChemie, Utrecht, The Netherlands) using a three-electrode configuration. CV measurements consisted of 0.5 V/s. linear sweeps from −0.6V to 0.8V.
5.5. Alginate Bead Fabrication
A solution of 1.6% Sodium Alg-MVG was prepared by dissolving alginate powder in MOPS buffer. Microbeads were fabricated using an electrostatic encapsulation unit VAR V1 (NISCO Engineering, Zurich, Switzerland) by passing alginate solution through a needle at a constant flow rate of 1 mL/min. Alginate droplets fall into a bath of BaCl2-MOPS solution (10 mM MOPS, 50 mM BaCl2, 3 mM KCl, 50 mM NaCl, 0.2 mM Tween 20, pH 7.4) with a voltage difference of 7.0 kV. Resulting bead diameters were 702.43 ± 86.33 μm (n = 318 beads).
5.6. Alginate Bead Coatings
Alginate beads (~1-1.5 mL alginate beads in MOPS-buffer) were transferred into a 24 well plate, and MOPS-buffer was replaced with a 3 mg/mL dispersion of CONP in MOPS buffer (prepared immediately before use). Beads were incubated at room temperature in CONP dispersion for 30 seconds before removal and multiple washes (3x) with MOPS buffer. Beads were subsequently incubated with a 3 mg/mL solution of Alg-MVG and incubated at RT for 30 seconds, followed by three washes with MOPS-buffer. This procedure was repeated until the desired number of layers is achieved. This procedure was followed for Alg-LVG and Alg-vLVG.
5.7. Microbead Microscopy
Bright-field images of coated alginate beads were collected using a Zeiss AxioObserver Microscope. For fluorescent imaging, fluorescently labeled alginate (VLVG, LVG or MVG) was synthesized by dissolving 25 mg of alginate (VLVG, LVG, or MVG) in 2.5 mL of purified water, followed by the addition of 5 mg of 2-(N-morpholino)ethanesulfonic acid, 5 mg of N-hydroxysuccinimide, and 0.5 mg of 4’-(aminomethyl fluorescein) (CF, dissolved in 100 μL of ethanol). Then, N-(3-dimethylaminopropyl)-N′-ethyl carbodiimide hydrochloride was dissolved in 100 μL of purified water, followed by 20 min stirring. After stirring, 200 μL of 0.25 M NaOH was added at a rate of 7 μL/min, and the solution was stirred for one hour. After one h of stirring, the solution was precipitated with 5 mL of ethanol, resuspended in 2 mL of 50 mM NaCl, and precipitated again in 5 mL of ethanol. The resulting alginate-CF was rinsed two times with a mix of 2 mL of water and 4 mL of ethanol, then 3x with ethanol, and dried under reduced pressure. Beads were coated following the same protocol from section 2.5 but using fluorescent alginate. Fluorescent coatings were quantified via ImageJ by setting a Region of Interest (ROI) around the bead circumference and measuring the percent area covered by Alginate-CF. To measure the coating thickness, a cross-sectional image of the coated beads was exported to ImageJ, ROIs were drawn around the outer and inner diameter of the coating. The area of each circle was obtained using the measurement function in ImageJ. The area of each circle was used to calculate the diameter, and the difference between the inner and outer circle was calculated to obtain the thickness of each coating. The sample size for all fluorescent quantification was ≥ 10.
5.8. Induced Coupled Plasma- Mass Spectrometry (ICP-MS)
A total of fifty alginate microbeads were handpicked (n=4 per group) and digested in 15 mL polypropylene tubes with 0.5 mL nitric acid (Optima grade, Fisher Chemical, Fair Lawn, NJ) at 100°C for 2 hours on a dust-protected hotplate. The digests were subsequently diluted to 15 mL volumes with ultrapure water (EMD Millipore, Burlington, MA). Cerium was quantified using an Agilent 7900 ICP-MS at the University of Florida Analytical Toxicology Core Laboratory, equipped with in-line internal standard addition, and employing He gas mode to minimize polyatomic interferences.
5.9. TMB Oxidation
A total of 50 beads per well (3 well per experiment, 4-6 experiments from different bead coatings) were incubated in a 48 well plate with 500 μL of TMB substrate. The plate was shaken at 500 rpm for 30 min covered from light. After CONP oxidized the TMB substrate, 150 μL of solution (n=3) was transferred to a clear flat bottom 96 well plate for absorbance quantification (650 nm) utilizing a Molecular Devices SpectraMax M5 reader.
5.10. Hydrogen Peroxide Consumption Assay
A total of 50 handpicked microbeads were incubated with 500 μL of a solution containing 320 μM of Xanthine, 93 nM of Xanthine Oxidase from bovine milk (X1875), and 83.33 μM of Cytochrome C. Beads were incubated for 15 min covered from light before transferring 150 μL (n=3) to a 96-well flat-bottom plate to read absorbance at 550 nm. The rate of superoxide consumption was calculated using the following equation .[54] Where ΔA is the difference between reduced and oxidized cytochrome C, V is the total volume per well (150 μL), K is a constant 21*103 cm−1/M, l is the path length, and t is the total time of incubation.
5.11. Protection of Encapsulated β-Cells Assay
Mouse Insulinoma Cells (MIN6) cells were encapsulated in 1.6% (w/v) alginate MVG at a cell loading density of six million cells per mL of alginate. Encapsulation of cells was done under sterile conditions following the same procedure and settings as in section 2.4. After encapsulation, MIN6 cell-containing beads were left to rest for at least one hour before coating with different formulations of CONP and alginate. Coating of encapsulated MIN6 cells followed the same procedure as in section 2.5. However, additional washes before the coating process were needed to ensure cell media was removed entirely to prevent any agents in the cell media from interfering with the coating process. Cell-containing microbeads (n=30 per well) rested in cell media overnight before assessing protection from oxidative stress challenge.
5.12. Insulin Secretion
MIN6 cells response to high glucose (16.67 mM) concentration was assessed following previously published methods.[17] Briefly, control and coated beads (n=30 per well) were exposed to a Krebs buffer solution of with 16.67 mM of high glucose for 90 min at 37 C. Supernatant was then collected for insulin content analysis via ELISA (Mercodia).
5.13. Reduced: Oxidized Glutathione Ratio
A total of 30 beads per well were exposed to either hydrogen peroxide or xanthine/xanthine oxidase system for 2 hrs at 37 C. After ROS challenge, beads were washed to stop oxidative damage and groups were prepared for glutathione quantification. The total glutathione (N=3) and oxidized glutathione (N=3) were measured from coated and control MIN6 cells-containing beads following previous protocols.[17] Briefly, the solution in the wells were replaced with 100 μL of cell-culture-grade water, followed by the addition of 25 μL of either total glutathione lysis reagent or oxidized glutathione lysis reagent. Plates were shaken at 500 rpm for 15 min at RT. After cell lysis, 50 μL of generation reagent was added to each well and plates were incubated for 30 min at RT. Then, 100 μL of detection reagent was added to each well and plates were covered from light and incubated for 15 min at RT. Finally, 100 μL was transferred from each well (n=2) to a 96 well plate for luminescent detection.
5.14. Statistical Analysis
Ellipsometry and CV studies were collected from three scans per group. Alginate bead data was collected from independent replicates (n = 6 to 9) from distinctly separate studies (N = 3) to validate trends. Cell protection data were collected from independent replicates (n = 3) from distinctly separate studies (N ≥ 2) to validate trends. All values were expressed as mean ± SD. Statistical analyses were performed using GraphPad Prism 8.0 software (GraphPad Software, La Jolla, CA, USA). Statistical tests used one- or two-way ANOVA with post-hoc Tukey's multiple comparisons. Statistical significance was considered at p < 0.05 with designations of ****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05.
Supplementary Material
Acknowledgements
This work was supported by NIH grants DK100654 and DK126413. N.J.A. was supported by a T32 Interdisciplinary Graduate Program in Type 1 Diabetes and Biomedical Engineering Predoctoral Training Grant (DK108736).
References
- [1].Castano CE, O’Keefe MJ, Fahrenholtz WG, Current Opinion in Solid State and Materials Science 2015, 19, 69. [Google Scholar]
- [2].Abuid NJ, Gattás-Asfura KM, LaShoto DJ, Poulos AM, Stabler CL, in Nanoparticles for Biomedical Applications, (Eds: Chung EJ, Leon L, Rinaldi C), Elsevier, 2020, 283. [Google Scholar]
- [3].Luches P, Valeri S, Materials 2015, 8, 5818; [DOI] [PMC free article] [PubMed] [Google Scholar]; Conesa J, Surface Science 1995, 339, 337. [Google Scholar]
- [4].Korsvik C, Patil S, Seal S, Self WT, Chemical communications 2007, 1056; [DOI] [PubMed] [Google Scholar]; Heckert EG, Karakoti AS, Seal S, Self WT, Biomaterials 2008, 29, 2705; [DOI] [PMC free article] [PubMed] [Google Scholar]; Pirmohamed T, Dowding JM, Singh S, Wasserman B, Heckert E, Karakoti AS, King JE, Seal S, Self WT, Chemical communications 2010, 46, 2736; [DOI] [PMC free article] [PubMed] [Google Scholar]; Ighodaro OM, Akinloye OA, Alexandria Journal of Medicine 2018, 54, 287. [Google Scholar]
- [5].Hosseini A, Baeeri M, Rahimifard M, Navaei-Nigjeh M, Mohammadirad A, Pourkhalili N, Hassani S, Kamali M, Abdollahi M, Human & experimental toxicology 2013, 32, 544; [DOI] [PubMed] [Google Scholar]; Schubert D, Dargusch R, Raitano J, Chan SW, Biochemical and biophysical research communications 2006, 342, 86. [DOI] [PubMed] [Google Scholar]
- [6].Sims CM, Hanna SK, Heller DA, Horoszko CP, Johnson ME, Montoro Bustos AR, Reipa V, Riley KR, Nelson BC, Nanoscale 2017, 9, 15226; [DOI] [PMC free article] [PubMed] [Google Scholar]; Alkaladi A, Abdelazim AM, Afifi M, International journal of molecular sciences 2014, 15, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Deshpande S, Patil S, Kuchibhatla SVNT, Seal S, Applied Physics Letters 2005, 87, 133113. [Google Scholar]
- [8].Karakoti AS, Munusamy P, Hostetler K, Kodali V, Kuchibhatla S, Orr G, Pounds JG, Teeguarden JG, Thrall BD, Baer DR, Surface and Interface Analysis 2012, 44, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kuchibhatla SVNT, Karakoti AS, Baer DR, Samudrala S, Engelhard MH, Amonette JE, Thevuthasan S, Seal S, The Journal of Physical Chemistry C 2012, 116, 14108; [DOI] [PMC free article] [PubMed] [Google Scholar]; Walkey C, Das S, Seal S, Erlichman J, Heckman K, Ghibelli L, Traversa E, McGinnis JF, Self WT, Environmental Science: Nano 2015, 2, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Asati A, Santra S, Kaittanis C, Nath S, Perez JM, Angewandte Chemie (International ed. in English) 2009, 48, 2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pulido-Reyes G, Rodea-Palomares I, Das S, Sakthivel TS, Leganes F, Rosal R, Seal S, Fernandez-Pinas F, Scientific reports 2015, 5, 15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dahle TJ, Arai Y, International Journal of Environmental Research and Public Health 2015, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Alpaslan E, Yazici H, Golshan NH, Ziemer KS, Webster TJ, ACS Biomaterials Science & Engineering 2015, 1, 1096. [DOI] [PubMed] [Google Scholar]
- [14].Kumari M, Singh SP, Chinde S, Rahman MF, Mahboob M, Grover P, International journal of toxicology 2014, 33, 86; [DOI] [PubMed] [Google Scholar]; Lin W, Huang Y-W, Zhou X-D, Ma Y, International journal of toxicology 2006, 25, 451. [DOI] [PubMed] [Google Scholar]
- [15].Weaver JD, Stabler CL, Acta Biomaterialia 2015, 16, 136; [DOI] [PMC free article] [PubMed] [Google Scholar]; Marino A, Tonda-Turo C, De Pasquale D, Ruini F, Genchi G, Nitti S, Cappello V, Gemmi M, Mattoli V, Ciardelli G, Ciofani G, Biochimica et Biophysica Acta (BBA) - General Subjects 2017, 1861, 386; [DOI] [PubMed] [Google Scholar]; Mandoli C, Pagliari F, Pagliari S, Forte G, Di Nardo P, Licoccia S, Traversa E, Advanced Functional Materials 2010, 20, 1617. [Google Scholar]
- [16].Gil PR, del Mercato LL, del_Pino P, Muñoz_Javier A, Parak WJ, Nano Today 2008, 3, 12; [Google Scholar]; Lengert EV, Koltsov SI, Li J, Ermakov AV, Parakhonskiy BV, Skorb EV, Skirtach AG, Coatings 2020, 10, 1131. [Google Scholar]
- [17].Abuid NJ, Gattás-Asfura KM, Schofield EA, Stabler CL, Advanced Healthcare Materials 2019, 0, 1801493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhou J, Romero G, Rojas E, Ma L, Moya S, Gao C, Journal of Colloid and Interface Science 2010, 345, 241; [DOI] [PubMed] [Google Scholar]; Wang Z, Zhang X, Gu J, Yang H, Nie J, Ma G, Carbohydrate Polymers 2014, 103, 38. [DOI] [PubMed] [Google Scholar]
- [19].Xu C, Qu X, NPG Asia Materials 2014, 6. [Google Scholar]
- [20].Simsek-Ege FA, Bond GM, Stringer J, Journal of Applied Polymer Science 2003, 88, 346. [Google Scholar]
- [21].Li M, Shi P, Xu C, Ren J, Qu X, 2013; [Google Scholar]; Karakoti AS, Kuchibhatla SVNT, Babu KS, Seal S, The Journal of Physical Chemistry C 2007, 111, 17232. [Google Scholar]
- [22].Yang Y, Yang Y, Du X, Chen Y, Zhang Z, Zhang J, Applied Surface Science 2014, 305, 330. [Google Scholar]
- [23].Sarpoushi MR, Nasibi M, Golozar MA, Shishesaz MR, Borhani MR, Noroozi S, Materials Science in Semiconductor Processing 2014, 26, 374. [Google Scholar]
- [24].Sworski TJ, Mahlman HA, Matthews RW, The Journal of Physical Chemistry 1971, 75, 250. [Google Scholar]
- [25].Marí M, Morales A, Colell A, García-Ruiz C, Fernández-Checa JC, Antioxidants & Redox Signaling 2009, 11, 2685; [DOI] [PMC free article] [PubMed] [Google Scholar]; Franco R, Cidlowski JA, Cell Death & Differentiation 2009, 16, 1303. [DOI] [PubMed] [Google Scholar]
- [26].Hao F, Kang J, Cao Y, Fan S, Yang H, An Y, Pan Y, Tie L, Li X, Apoptosis 2015, 20, 1420. [DOI] [PubMed] [Google Scholar]
- [27].Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, Reece JM, Deeney JT, Andersen ME, Corkey BE, Collins S, Diabetes 2007, 56, 1783. [DOI] [PubMed] [Google Scholar]
- [28].Gupta MK, Martin JR, Werfel TA, Shen T, Page JM, Duvall CL, Journal of the American Chemical Society 2014, 136, 14896; [DOI] [PubMed] [Google Scholar]; O’Grady KP, Kavanaugh TE, Cho H, Ye H, Gupta MK, Madonna MC, Lee J, O’Brien CM, Skala MC, Hasty KA, Duvall CL, ACS Biomaterials Science & Engineering 2018, 4, 1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].McEnery MA, Lu S, Gupta MK, Zienkiewicz KJ, Wenke JC, Kalpakci KN, Shimko D, Duvall CL, Guelcher SA, RSC Adv 2016, 6, 109414; [DOI] [PMC free article] [PubMed] [Google Scholar]; Dollinger BR, Gupta MK, Martin JR, Duvall CL, Tissue engineering. Part A 2017, 23, 1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kozlovskaya V, Xue B, Lei W, Padgett LE, Tse HM, Kharlampieva E, Advanced healthcare materials 2015, 4, 686; [DOI] [PMC free article] [PubMed] [Google Scholar]; Pham-Hua D, Padgett LE, Xue B, Anderson B, Zeiger M, Barra JM, Bethea M, Hunter CS, Kozlovskaya V, Kharlampieva E, Tse HM, Biomaterials 2017, 128, 19; [DOI] [PMC free article] [PubMed] [Google Scholar]; Alford A, Kozlovskaya V, Xue B, Gupta N, Higgins W, Pham-Hua D, He L, Urban VS, Tse HM, Kharlampieva E, Chem Mater 2018, 30, 344; [Google Scholar]; Kozlovskaya V, Zavgorodnya O, Chen Y, Ellis K, Tse HM, Cui W, Thompson JA, Kharlampieva E, Adv Funct Mater 2012, 22, 3389; [DOI] [PMC free article] [PubMed] [Google Scholar]; Kozlovskaya V, Kharlampieva E, Drachuk I, Cheng D, Tsukruk VV, Soft Matter 2010, 6, 3596. [Google Scholar]
- [31].Lingaraju K, Raja Naika H, Manjunath K, Basavaraj RB, Nagabhushana H, Nagaraju G, Suresh D, Applied Nanoscience 2016, 6, 703; [Google Scholar]; Mitra RN, Merwin MJ, Han Z, Conley SM, Al-Ubaidi MR, Naash MI, Free radical biology & medicine 2014, 75, 140; [DOI] [PMC free article] [PubMed] [Google Scholar]; Tootoonchi MH, Hashempour M, Blackwelder PL, Fraker CA, Acta Biomater 2017, 59, 327. [DOI] [PubMed] [Google Scholar]
- [32].Das S, Dowding JM, Klump KE, McGinnis JF, Self W, Seal S, Nanomedicine (London, England) 2013, 8, 1483. [DOI] [PubMed] [Google Scholar]
- [33].Porcel C, Lavalle P, Decher G, Senger B, Voegel JC, Schaaf P, Langmuir 2007, 23, 1898. [DOI] [PubMed] [Google Scholar]
- [34].Draget KI, Gåserød O, Aune I, Andersen PO, Storbakken B, Stokke BT, Smidsrød O, Food Hydrocolloids 2001, 15, 485; [Google Scholar]; Wason MS, Colon J, Das S, Seal S, Turkson J, Zhao J, Baker CH, Nanomedicine : nanotechnology, biology, and medicine 2013, 9, 558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Shiratori SS, Rubner MF, Macromolecules 2000, 33, 4213. [Google Scholar]
- [36].Choi I, Suntivich R, Plamper FA, Synatschke CV, Müller AHE, Tsukruk VV, Journal of the American Chemical Society 2011, 133, 9592. [DOI] [PubMed] [Google Scholar]
- [37].Asati A, Kaittanis C, Santra S, Perez JM, 2011;; Grulke E, Reed K, Beck M, Huang X, Cormack A, Seal S, 2014.
- [38].Huang X, Lin S, Shang J, He W, Lan J, Journal of Reinforced Plastics and Composites 2014, 33, 1207. [Google Scholar]
- [39].Li Y, Wang X, Sun J, Chemical Society Reviews 2012, 41, 5998; [DOI] [PubMed] [Google Scholar]; Elbert DL, Herbert CB, Hubbell JA, Langmuir 1999, 15, 5355; [Google Scholar]; Picart C, Lavalle P, Hubert P, Cuisinier FJG, Decher G, Schaaf P, Voegel JC, Langmuir 2001, 17, 7414. [Google Scholar]
- [40].Markarian MZ, Hariri HH, Reisch A, Urban VS, Schlenoff JB, Macromolecules 2012, 45, 1016; [Google Scholar]; Peng C, Thio YS, Gerhardt RA, Ambaye H, Lauter V, Chemistry of Materials 2011, 23, 4548. [Google Scholar]
- [41].Kruk T, Gołda-Cępa M, Szczepanowicz K, Szyk-Warszyńska L, Brzychczy-Włoch M, Kotarba A, Warszyński P, Colloids and Surfaces B: Biointerfaces 2019, 181, 112. [DOI] [PubMed] [Google Scholar]
- [42].Gabriele A, Spyropoulos F, Norton IT, Food Hydrocolloids 2009, 23, 2054. [Google Scholar]
- [43].Farrés IF, Norton IT, Food Hydrocolloids 2014, 40, 76. [Google Scholar]
- [44].Smidsrød O, Faraday discussions of the Chemical Society 1974, 57, 263. [Google Scholar]
- [45].Xue Y, Zhai Y, Zhou K, Wang L, Tan H, Luan Q, Yao X, Chemistry – A European Journal 2012, 18, 11115; [DOI] [PubMed] [Google Scholar]; Singh S, Dosani T, Karakoti AS, Kumar A, Seal S, Self WT, Biomaterials 2011, 32, 6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Alpaslan E, Yazici H, Golshan NH, Ziemer KS, Webster TJ, 2015. [DOI] [PubMed] [Google Scholar]
- [47].Weydert CJ, Cullen JJ, Nat Protoc 2010, 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].de Gracia Lux C, Joshi-Barr S, Nguyen T, Mahmoud E, Schopf E, Fomina N, Almutairi A, Journal of the American Chemical Society 2012, 134, 15758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kalaycıoğlu Z, Kahya N, Adımcılar V, Kaygusuz H, Torlak E, Akın-Evingür G, Erim FB, European Polymer Journal 2020, 133, 109777; [Google Scholar]; Xu Z, Xu Y, Basuthakur P, Patra CR, Ramakrishna S, Liu Y, Thomas V, Nanda HS, Journal of Materials Chemistry B 2020, 8, 9110. [DOI] [PubMed] [Google Scholar]
- [50].Nichols SP, Koh A, Storm WL, Shin JH, Schoenfisch MH, Chem Rev 2013, 113, 2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bekmurzayeva A, Duncanson WJ, Azevedo HS, Kanayeva D, Materials Science and Engineering: C 2018, 93, 1073. [DOI] [PubMed] [Google Scholar]
- [52].Qi M, Li W, Zheng X, Li X, Sun Y, Wang Y, Li C, Wang L, Frontiers in Materials 2020, 7. [Google Scholar]
- [53].Olczak KP, McDermott MD, Otto KJ, ACS Applied Bio Materials 2019, 2, 5597. [DOI] [PubMed] [Google Scholar]
- [54].Hume PS, Anseth KS, J Biomed Mater Res A 2011, 99A, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.