

Abstract
Chimeric antigen receptor (CAR) T-cell therapies have evolved from a research tool to a paradigm-shifting therapy with impressive responses in B cell malignancies. This review summarizes the current state of the CAR T-cell field, focusing on CD19- and B cell maturation antigen-directed CAR T-cells, the most developed of the CAR T-cell therapies. We discuss the many challenges to CAR-T therapeutic success and innovations in CAR design and T-cell engineering aimed at extending this therapeutic platform beyond hematologic malignancies.
Keywords: chimeric, antigen, CAR, T cell, cancer
Introduction
Although anti-tumor immunity by T lymphocytes has been known for decades, translating it into anti-cancer therapies has been challenging. However, biological advances, such as the generation of single chain antibody fragments (scFv) 1, the elucidation of pathways mediating the activation of functional memory T-cells 2,3, and molecular cloning 4 have led to the engineering of chimeric antigen receptor (CAR) T-cells, introducing a new era of cancer immunotherapy 5,6 and permitting the treatment of large groups of patients with genetically-agumented patient-derived T-cells.
The first generation of CAR T-cells fused the scFv antibody fragment to T-cell signaling domains comprising the immunoreceptor tyrosine-based activation motif (ITAM), offering a relatively simple method of endowing T-cells with MHC-independent recognition of antigen7. Over the following two decades, the CAR platform evolved into 2nd generation (2 domain) and 3rd generation (3 domain) CARs that incorporated additional signal transduction domains, including cytoplasmic domains from important T-cell costimulatory receptors such as CD28, CD137 (4–1BB), and CD134 (OX-40) (reviewed in 8,9). These additional signaling domains promote both the persistence and anti-tumor activity of CAR-T-cells following adoptive transfer3,10–14, and were essential to avoid the anergy observed with 1st generation CARs15.
The remarkable ability of a CAR to reprogram T-cell specificity led to attempts at clinical translation. The earliest clinical application used a simple CAR design comprised of a CD4 ectodomain fused to the CD3ζ cytoplasmic domain to treat HIV-infected patients 16 and established both the safety of engineered CAR T-cells, as well as the potential for decade-long persistence of the genetically-modified T-cells 17. Subsequent studies evaluating first and second-generation scFv-based CARs soon followed, leading to the demonstration of robust activity of CD19-specific CAR T-cells and ultimately the regulatory approval of two CAR T-cell therapies for hematologic malignancies in the USA in 2017 and Europe in 2018.
As preclinical models of adoptive T-cell therapy are limited, correlative studies performed during their clinical development to determine the kinetics and quality of the infused CAR T-cells, measure tumor cells dynamics, and assess cytokine levels and repertoires during therapy, have proven pivotal in improving our understanding of these complex therapies and enhancing their clinical application. These correlative studies have highlighted many factors that are essential to safely achieving both deep and durable clinical responses in otherwise treatment-refractory cancers. Here, we discuss the important role of correlative science in developing CAR T-cell therapies, and highlight the challenges still faced during clinical application and the new technologies promising to address these complications to help extend this therapeutic modality beyond B-cell malignancies.
Efficacy and toxicities of CD19-specific CAR T-cell therapies
Normal and malignant B cells uniformly and exclusively expres CD19 18, the dominant signaling moiety of a tetramolecular complex consisting of CD21, CD81, and CD225, which modulates B-cell receptor signaling and mediates immunoglobulin-induced B cell activation19. Given CD19’s broad expression within the B-cell lineage from early pro-B cells to subsets of plasma cells (Fig. 1), and its generally uniform expression on B-cell malignancies 20 this molecule became a prime target of CAR T-cell approaches. The initial encouraging results in relatively small studies in non-Hodgkin’s Lymphoma (NHL) 13,21, Chronic Lymphocytic Leukemia (CLL) 22–24 and Acute Lymphoblastic Leukemia (ALL) 24,25 have since been confirmed in larger cohorts 26–36. So far, the first CLL patients treated with anti-CD19 CAR T-cells have sustained remission beyond 9 years 37, and the first ALL patient to be treated with the same engineered product, has been in remission for more than 7 years 25.
Figure 1.
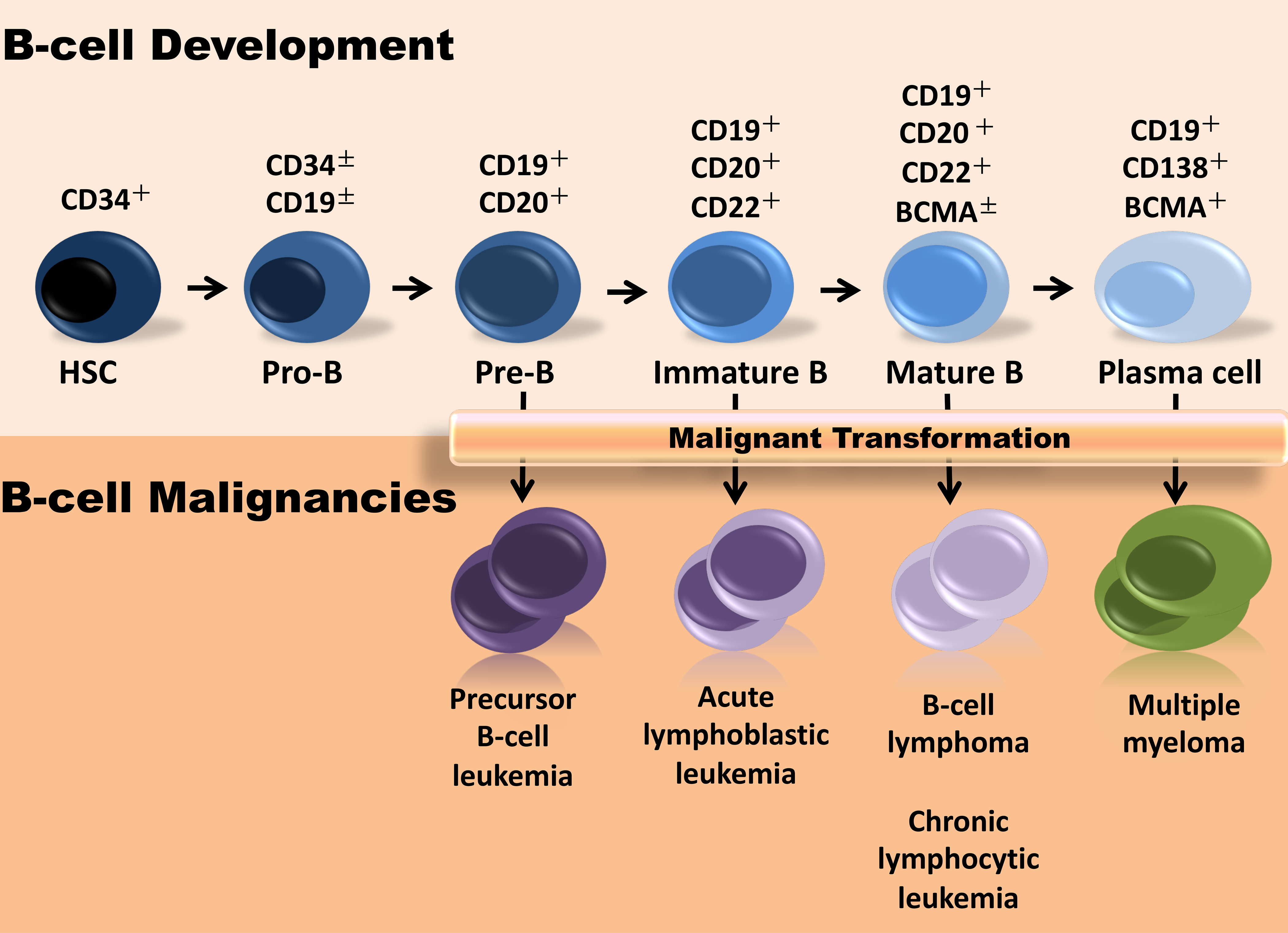
B cell malignancies at the different stages of B cell development. The normal B-cell developmental lymphocytes shown at the top, often share the same immunophenotypic characteristics with the malignant counterparts depicted at the bottom, reflecting the expansion of a dominant clone leading to development of leukemia or lymphoma.
Generally, the overall response rate (ORR) has been highest in B-ALL (>80%), variable in lymphomas (63%~100%) and lower in CLL (50–70%) 28,35,38,39. CLL patients who achieve remission with anti-CD19 CAR T-cell treatment sustained their disease-free state 35,40,41. In ALL, however, only 20–40% of patients sustained remission on this therapy 28,32,33,35,38,39. Loss of CD19 expression is a major mechanism of resistance in ALL, accounting for ~2/3 of relapse cases and is a well-recognized phenomenon in lymphoma as well 42–44. Loss of CAR T-cell engraftment may account for most of the remaining cases of relapse 26. Initial small trials 13,24 followed by larger ones 32,33,45 confirmed the immense potential for this therapy also in NHL. Both the CD28- 33 and 4–1BB cosignaling anti-CD19 CAR T-cells 46 induced complete remission in 40–50% of patients, most of whom remained disease-free. Although clinical responses were generally sustained in NHL, most disease-free patients would display normal B cell recurrence and loss of detectable CAR T-cells, suggesting that other mechanisms were responsible for longterm tumor control in NHL.
Interestingly, although ALL and CLL patients generally achieved their best overall response within the first month following CAR T-cell infusion, lymphoma patients often continued to improve beyond the first month, with some patients not achieving their maximal response until 6 months post-CAR T-cell treatment 32,33,45,47. The reasons for these differences are not understood, and remain an important subject of study in the post-marketing phase.
Although CD19-specific CAR T-cell therapies have shown remarkable clinical activity against B-cell malignancies, these deep and durable responses do come at a cost of some unique adverse effects. Cytokine release syndrome (CRS) is the most frequently observed adverse event in CART19-treated patients. Most CRS is mild or moderate in severity and manageable. However, the frequency of severe CRS across studies, reported in 19.8–38.8% of treated individuals48, has been clouded by the use of diverse grading systems. Fortunately, a new consensus grading system for CRS was recently described, the adoption of which should greatly facilitate comparing its incidence across different CAR T-cell products 49. In addition to CRS, a somewhat unique and unexpected neurotoxicity has also been observed in CD19-specific CAR T-cell-treated patients. This toxicity can range from mild delirium to severe encephalopathy. The incidence of neurotoxicity may depend on the disease and CAR design. Severe neurotoxicity was seldomly reported in CLL patients treated with the BBζ-signaling CAR 24,28,35,50, but observed in every CAR T-cell trial for ALL26,29,33,38,39, more prominently with a CD28ζ signaling CAR 38,39. Myelosuppression has also been observed in anti-CD19 CAR T-cell-treated lymphoma and leukemia patients 33,36,39,51. Additionally, during the first 8 weeks post-infusion, febrile neutropenia, and tumor lysis syndrome is commonly observed in lymphoma patients treated with the BBζ-based CAR T-cells 45. The majority of adverse events have been reversible through supportive care, cytokine inhibitors and glucocorticoid 52.
Generating hypotheses and interventions with correlative studies
Defining the kinetics, homing, and bioactivity of the cell therapy product and the tumor response to treatment in each patient requires diligent monitoring, as these are critical components in the continued translational cycle from the bench to the bed and back again. Furthermore, the US Food and Drug Administration (FDA) mandates that sponsors observe study participants for delayed adverse events for as long as 15 years following the infusion of modified cells (https://www.fda.gov/media/113768/download). To this end, it is desirable to include a correlative studies laboratory in an organization which operates according to Good Clinical Laboratory Practice (https://www.niaid.nih.gov/sites/default/files/gclp.pdf) (Fig. 2) to ensure that biospecimens from patients on cell therapy are handled by qualified personnel following experimental processes specified by Standard Operating Procedure (SOP). Sample analytics and biobanking are two critical activities in such a laboratory, all of which should be carried out using rigorously validated, SOP-defined procedures. As most phase I trials are run in academic centers, some of the analytical methods would have to be developed and validated for novel, innovative therapies such as the CRISPR/Cas9-mediated disruption of endogenous genes in mature T-cells, combined with lentiviral delivery of a tumor-targeting T-cell receptor 53. An example is the frequent monitoring of CAR T-cell bioactivity in terms of changes in cytokine and soluble cytokine receptor levels22,23,25,54–56 in serum early after infusion, given that high-grade toxicities may rapidly develop upon treatment.
Figure 2.
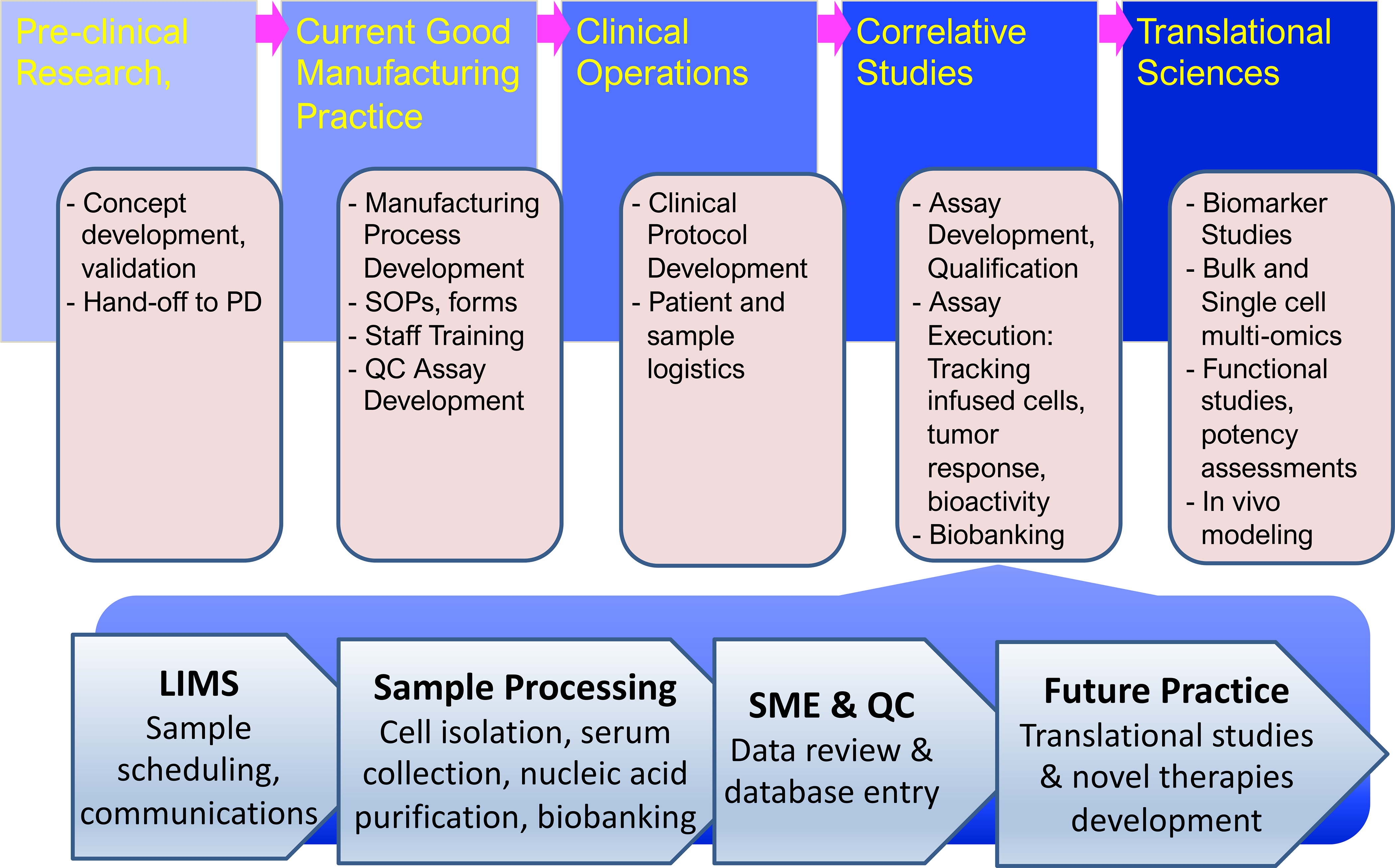
Operational pipeline for integrating correlative studies in translational science laboratories. Novel therapies developed and pre-clinically validated in research laboratories are handed off to the process development (PD) team for scale-up and the development of a current Good Manufacturing Practice (GMP) process. In collaboration with the GMP teams Standard Operating Procedures (SOP) and documentation forms are developed and GMP staff trained in the new procedures. The Correlative Studies Laboratory will, in parallel, ensure that all supportive assays, protocols, and forms are in place, that staff is trained, and that routine, qualified assays are developed and biobanking ensured. This same team is also involved in protocol development, which is lead by the Clinical Operations team with feedback from the study clinicians and the research laboratory that developed the new process. When a new clinical trial begins the Correlative Studies laboratory starts receiving biospecimens from the clinic, manufacturing facility, or collaborating laboratories, and logs these samples into the Laboratory Information Management System (LIMS), to be processed as specified by standard operating procedures and examined using validated assays by qualified personnel. Aliquots are retained from each specimen for future translational studies. The data are reviewed by subject matter experts (SME) before being reviewed by the quality control (QC) manager and entered into a database. A staff statistician cleans and analyzes the data for reporting purposes, e.g. to FDA or for scientific meetings and manuscript preparation.
The value of correlative studies is underscored by the identification of a rise in interleukin-6 (IL-6) levels in association with the onset of CRS in patients, which played a central role in prompting the evaluation of IL-6/IL-6 receptor blockade in severe CRS 25. This insight proved life-saving for many patients, and formed the foundation for co-developing anti-IL-6 and CD19 CAR T-cell therapy, leading to their concurrent FDA approval for severe CRS 57 and B cell ALL, respectively. More extensive analyses of serum from patients in multiple trials have led to the discovery and validation of biomarkers of CRS and neurotoxicity, providing insight into the mechanisms that drive them 58 and potential paths to predicting these complications of CAR T-cell therapy. Although not all studies agree on the precise cytokines 55 or biomarkers 56 to interrogate, they all focus on identifying predictive markers and developing algorithms to distinguish patients at increased risk of developing life-threatening toxicities.
Biobanked cells from patients have played a critical role in identifying mechanisms of resistance to CD19-specific CAR therapy. One of the earliest reports of CART19 in ALL revealed evidence of relapse in the context of loss of CD19 expression, which has been demonstrated to be the dominant resistance mechanism in ALL, occurring through various genetic mechanisms and rare iatrogenic causes 59–61. Early loss of CAR T-cells preceded by normal B cell recovery is another commonly observed event association with relapse 26. Analyses of the T-cells used for manufacturing the CAR T-cells as well as the product itself have revealed a number of associations that link CAR T-cell quality to outcome. In particular, the presence of naive-like CD27+CD45RO- cells in the apheresis product used for CART19 generation was shown to predict engraftment and clinical response in CLL 41. The reinfusion of relapsing leukemia patients with a murine scFv-based CAR has been associated with reduced expansion compared with first infusion 62,30,31, suggesting that an immune-mediated mechanism may underlie resistance to retreatment. Humanizing or developing a fully human scFv fragment might therefore enhance therapeutic success 62. Recently, defects in death receptor signaling have been identified in a subset of ALL that is resistant to CD19-specific CAR T-cell therapy, providing additional resistance mechanisms beyond CD19 loss 63.
Correlative studies also reveal differential kinetics of the CAR T-cells in responding and non-responding patients with CLL 28 and ALL 29,64,65, which led to the development of an in vitro, proliferation-based potency assay 41,66. This correlation between clinical response and in vivo CART19 cell proliferation, was not evident in trials of the same product when used for NHL 32,45,67, in contrast with a CD28-costimulated CAR 33, suggesting that the costimulatory domain dominated the differential expansion kinetics of CART19 for NHL.
Correlative studies sometimes also provide unexpected observations that can lead to new ideas. The rather dramatic expansion of an ultra-low dose of CAR T-cells (1.4×107) followed by the eradication of the leukemic mass in one patient 23 suggested that the proliferative response was key to the anti-tumor response 11,68.
In aggregate, correlative studies followed by mechanistic investigations based on samples and data from patients treated with CD19-specific CAR T-cells in clinical studies continue to improved our understanding of therapy-related toxicities and mechanisms of escape.
The impact of T-cell biology and CAR engineering on clinical responses
Although CART19 therapy has been efficacious in ALL and NHL, many factors contributing to patient response remain poorly understood. As patient-derived T-cells are used to target a tumor-associated cell surface protein, the immune system is repurposed to treat the malignancy. Thus, the therapeutic efficacy still depends on T-cell memory and effector functions. This also includes T-cell fitness, which is affected by the malignancy and prior therapies, and, most importantly, the ability of the CAR-redirected T-cells to sustain the anti-tumor response, because most tumors exist in actively growing and dormant phases which can last from several several years to decades 69–71. By harnessing T-cells, this form of immunotherapy abides by similar target cell quiescence-reactivation principles to induce a cure. Naïve and memory T-cells retain the ability to proliferate vigorously in response to cognate antigen recognition, in contrast to their effector progeny that has lost that ability and instead directly lyse the tumor. Two studies recently confirmed that this therapy depends on a functional, self-renewing T-cell pool, by demonstrating that in CLL the advanced age of the patient population in combination with effector-memory skewing limited CAR T-cell functionality (Figure 3) 41,72. Further, response to therapy in CLL can be predicted based on the presence of a pool of more functional early memory cells 41. CAR T-cells and other therapies that rely on immune system activation may therefore have limited effect in malignancies that terminally skew T-cell differentiation or occur in aged populations where T-cells are less functional at baseline. That baseline functionality of the T-cell pool plays a significant role in dictating response rates was confirmed in a separate study, which revealed that CD8+ T-cell dysfunction at apheresis and the rapid expression of immune checkpoint molecules after infusion marked CAR T-cells from non-responding and partially responding ALL patients 65. Therefore, CAR T-cells are subject to inhibition via endogenous immune checkpoint pathways such as PD-1 73. Inhibitory receptor/ligand interactions normally dampen T-cell functions to prevent an overactive immune response and sustain a memory T-cell pool. In CAR T-cells this can result in failure to eliminate the tumor and loss of T-cell persistence. Whereas checkpoint blockade can improve responses, other immune-suppressive factors in the microenvironment can impair CAR T-cell function. Immune-suppressive cytokines, metabolic competition, and high inhibitory ligand expression levels all serve to modulate the function of cell-based therapies 73–77.
Figure 3.
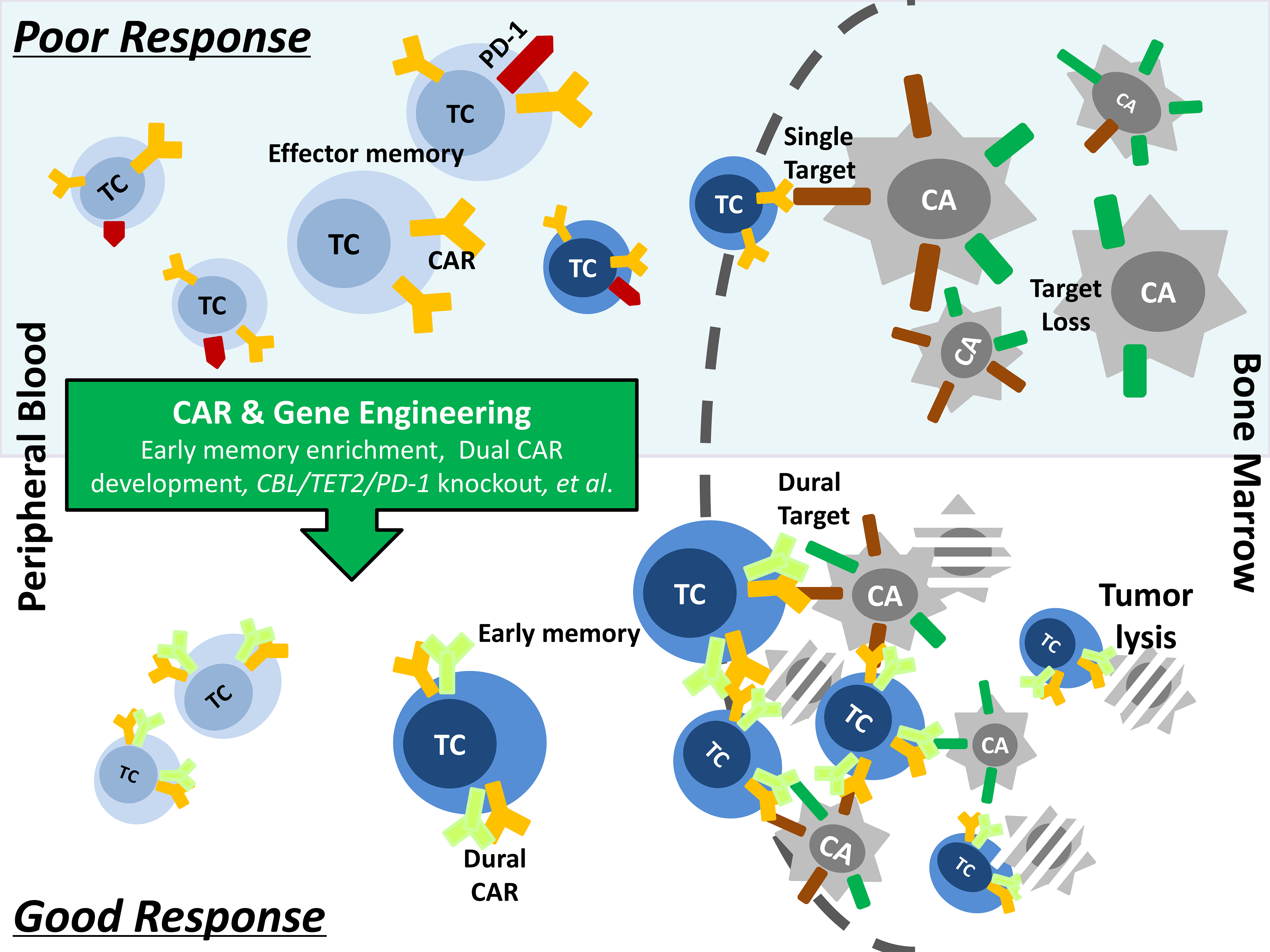
Current strategies to overcome the hurdles of poor response to autologous CAR-T-cell therapy. Several factors, such as low frequencies of early memory CAR-T-cells in the infusion product, over-expression of checkpoint inhibitory molecules on the apheresis T-cells and loss of target antigen on the tumor, have been shown to contribute to the lack of efficacy of CAR-T-cells in many patients. Optimizing the manufacturing process by laboratory-based engineering approaches, such as memory T-cells enrichment, dural CAR development and specific gene editing, is essential to improve the quality of CAR-T-cell product, thereby enhancing its capacity of tumor clearance and in vivo persistence.
Enhancing CAR T-cell Potency by Genome Engineering
Although the natural basis of CAR T-cell efficacy as laid out in the previous sections presents the foundation of immunogene therapies with CAR T-cells (and likely other systems that depend on a sustained tumor control), CAR T-cell engineering may also impact cell function as recently reported 66,78. CAR T-cells produced with lentivirus display quasi-random integration of the vector throughout the genome, introducing the potential for genomic activation or disruption events79,80. Although the majority of CAR T-cells generated in this process are polyclonal 66,79, the CAR T-cell population undergoes rapid changes after infusion due to, among other factors, selective expansion of CAR T-cell clones for reasons that are currently poorly understood 66,79,81. In most patients a multitude of clones contribute to the anti-tumor response 79,81. Two recently published reports concern a clonal CD8+ CAR T-cell expansion in two patients, in whom the CAR was shown by sequencing vector integration sites to have integrated into the CBL and TET2 gene loci. In the case of the TET2 integration, the patient’s CAR T-cell population underwent delayed expansion accompanied by tumor clearance, complete remission status, and contraction of the clonal population.66 The CBL-integrated clone underwent a similar, albeit less dramatic expansion process78. CBL knockdown had been previously associated with decreased T-cell activation thresholds, reduced reliance on co-stimulation, and decreased sensitivity to PD-1 inhibition, which could represent mechanisms for the therapeutic effect 82–85. These cases highlight how lentiviral integrations can significantly impact CAR T-cell growth, persistence, and effector function. Further insight into the fate of CAR T-cells was provided by analyzing the CAR vector integration site landscape in the infusion product and post-infusion aliquots of 58 CLL and ALL patients, demonstrating that CAR-mediated gene disruptions frequently occur in proliferation-augmenting pathways 79. These findings suggest that such gene disruptions may be as important as T-cell quality and CAR design in the outcomes observed with CAR T-cell therapy 79.
Additionally, targeted CAR integrations have revealed locus-specific regulation and protective effects. For instance, CAR expression from the T-cell receptor-α (TRAC) locus optimized CAR expression and protected cells from exhaustion compared to integration in other sites 86. The genomic landscape of the CAR transgene cassette can therefore play a significant role in how individual CAR T-cells function. Unique cases such as the TET2 and CBL loci integration events are informative not only on how genome regulation can influence CAR expression and function, but also in terms of novel regulators of these functions. The identification of TET2 disruption as an enhancer of T-cell persistence has sparked a wide array of research focused on knocking out TET2 to improve CAR function and to determine the mechanisms underlying this selective advantage.
Natural killer cells have also been engineered to express a B cell targeting CAR combined with constitutive secretion of IL-15 87. Pre-clinical studies have similarly demonstrated a beneficial effect of CAR T-cells co-expressing IL-15 88. Based on these findings, several clinical trials have been launched to evaluate T-cells engineered to express this cytokine in conjunction with a tumor-targeting CAR. However, IL-15 was separately demonstrated to drive antigen-independent growth of T-cells, resulting in a pre-leukemic disorder in mice 89,90. Therefore, the addition of a safety switch to the CAR and IL-15 construct should allow for the control of the infused cells, as indicated in the design of one of these trials (NCT03721068), which targets GD2 in brain cancers using anti-GD2 CAR with IL-15 and the iCaspase 9 safety switch. Moreover, preclinical studies have shown augmented anti-tumor efficacy of IL-18 co-expressing T-cells in a CD19-redirected T-cell model 91. Similar combination therapies have been shown to jointly blunt tumor function and boost T-cell potency 92–95. Next-generation CAR T-cell therapies incorporating such engineering approaches are expected to further raise the therapeutic index.
Tumor-redirected T-cells encounter numerous inhibitory signals in the tumor bed, most notoriously transforming growth factor-beta (TGFβ) 96. Dominant negative TGFβR-engineered receptor CAR T-cells showed augmented potency against a solid tumor model 97, leading to the development of an ongoing clinical trial to target prostate cancers with a prostate-specific maturation antigen-specific CAR T-cell (NCT03089203).
Tumor resistance to CAR T-cell therapy
Extensive clinical data have revealed mechanisms by which tumor cells escape CAR T-cell targeting and informed engineering advances to overcome this. The anti-CD19 CAR was shown to require minute quantities of target antigen to display full effector function 98 and, therefore, the tumor cells could only escape this pressure via antigen loss 25,59,60,99–101 or antigen masking 61. Routine analyses revealed that a pediatric ALL patient treated with the murine anti-CD19 CAR relapsed with the original disease two months post-treatment 25. In the ensuing months and years, similar patterns of relapse were observed from routine correlative studies, which were later confirmed by clinical pathology 25,59,60,102. The molecular basis of these and similar cases of antigen loss-related anti-CD19 CAR T-cell therapy were shown to be related to the acquisition of open reading frame-disrupting mutations in the target antigen, compounded by altered mRNA splicing in tumor cells 59,60. Others similarly observed antigen-negative relapse in CD19-directed CAR T-cell therapies 30,38,99. Additionally, MLL-rearranged leukemias displayed lineage switch-related relapse with loss of CD19 protein expression 99,100, further illustrating how the immense immune pressure exerted by CD19-specific CAR T-cells mediates a Darwinian selection of the malignant cell pool. These examples, demonstate the impact of T-cell and tumor cell physiology on clinical responses to single antigen-directed CAR T-cell therapies. This knowledge has been used to prevent relapse through a bispecific CAR T-cell that recognizes two antigens present on the tumor surface. This anti-CD19/anti-CD22 bispecific CAR T-cell has been successfully used to treat an adult ALL patient who remains disease-free for more than a year post-therapy 103. Antigen loss has now also been observed in a patient on CD22-targeting CAR treatment 104, whereas others have shown that downregulation was sufficient to evade CART22 treatment 105.
The CAR Design
As described above, many factors independent of the CAR itself impact therapeutic efficacy. Correlative studies by various groups targeting the same tumor-associated antigen, e.g. CD19, with a scFv derived from the same monoclonal antibody, e.g. FMC63, but different spacer domains, cosignaling domains etc., have allowed the identification of several “pain points” and success stories of chimeric receptors (Figure 4). First, it has become obvious that costimulation has to be engineered into the CAR, as even the transduction of memory T-cells could not rescue a first generation CAR 106. Hence, around the time that CD28 costimulation was discovered as an essential component to memory T-cell formation and effector differentiation 2, second generation CARs were developed that included this domain 107. However, comparative clinical studies to demonstrate the differential impact of CD28 and other cosignaling domains on effector and memory function in vivo are lacking and would be useful, because despite the CAR-contained CD28 driving a profound effector differentiation, it can also render the T-cells dysfunctional with loss of persistence 108.
Figure 4.
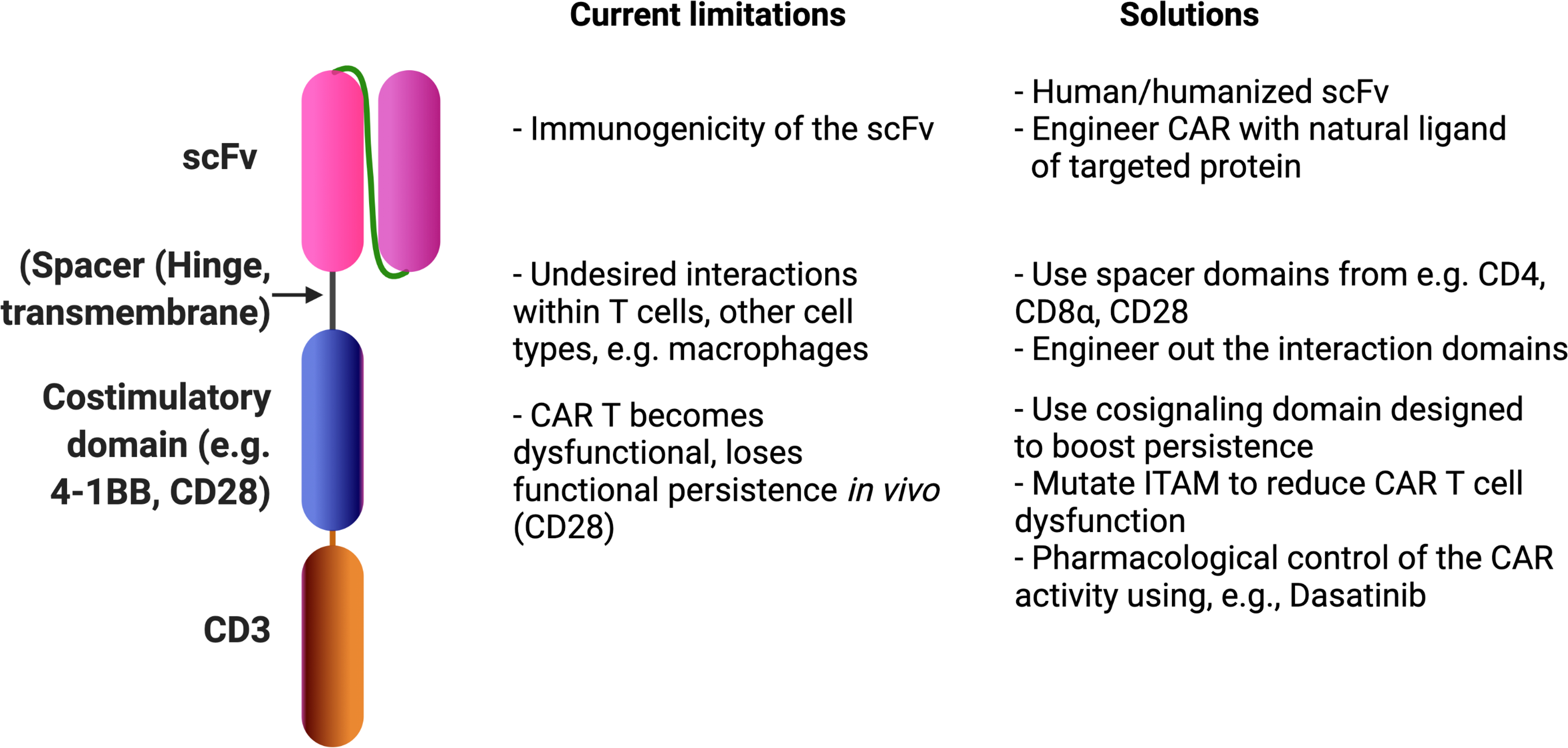
CAR design limitations that affect clinical responses following CAR T-cell treatment and potential solutions. Most CARs are made up of an tumor-associated antigen-binding scFv fragment (e.g. CD19, fused in-frame with a T-cell signaling domain), enhanced with a co-stimulatory domain (e.g. CD28 or 4–1BB) that is separated from the scFv by a spacer sequence. The design of this synthetic receptor affects various aspects of its in vivo performance and ultimately clinical responses. Additionally, small molecules such as Dasatinib may tone dysfunction-inducing CAR signaling 195.
Early data also revealed the profound impact of the spacer domain on CAR T-cell function (reviewed in 109). Most early generation CARs, including those in first generation CAR designs 5,110, used scFv derived from mouse antibodies. T-cells discern minute differences between cancerous and normal cells, and a single difference in amino acid residues can induce a robust immune response against this non-self entity 111–113. This same selective threat elimination machinery deletes recombinant proteins containing minimal sequence divergence from the native protein just as efficiently as foreign threats 114,115. It should therefore come as no surprise that suicide genes 116 and CARs incorporating non-human sequences are readily targeted by the immune system 30,31,117–119. Moreover, the poor expansion of re-infused CAR T-cells 31,55,110,120–122 correlated with the detection of patient-derived T-cell epitopes in the CAR 30,31. The field therefore is moving away from incorporating non-human tumor targeting moieties into the CAR 62. That being said, the remarkable response rates with a non-human CAR in multiple myeloma recently suggests that deep molecular remissions are possible (see below).
Extending CD19 CAR-T therapy beyond CD19+ malignancies
Although multiple myeloma derives from plasma cells, the terminal stage of B cell differentiation, myeloma precursor cells may express CD19. Pilot studies suggested a potential benefit to targeting CD19 in myeloma 123, yet little or no activity was apparent in the vast majority of treated patients, indicating that an alternative target antigen is needed to address this disease with CAR T-cell therapy98. Myeloma cells uniformly express B cell maturation antigen (BCMA)(Fig. 1), leading to the development of BCMA-specific CAR T-cells. Currently 90 relevant clinical trials are listed on ClinicalTrials.gov, with a few moving forward towards their commercial roll-out (see Tables 1 and 2 for constructs and trials furthest along in their development).
Table 1.
Summary of BCMA targeted CAR structures
| Manufacturer | CAR name | Gene delivery system | Species of antigen binding domain | Structure of antigen binding domain | hinge and transmembrane domain | Signaling domain | Satety switch |
|---|---|---|---|---|---|---|---|
| National Cancer Institute | CAR-BCMA | Retroviral vector | mouse | scFv | CD8α | CD28-CD3ξ | No |
| Bluebird Bio/Celgene | Idecabtagene Vicleucel / bb2121 | lentiviral vector | mouse | scFv | CD8α | 4–1BB-CD3ξ | No |
| bb21217 | lentiviral vector | mouse | scFv | CD8α | 4–1BB-CD3ξ | No | |
| HRAIN Biotechnology | BCMA CAR-T | Retroviral vector | mouse | scFv | NA | 4–1BB-CD3ξ | EGFRt |
| Nanjing Legend / Janssen | Ciltacabtagene autoleucel / LCAR-B38M | lentiviral vector | alpaca | VHH | CD8α | 4–1BB-CD3ξ | No |
| University of Pennslyvania | CART-BCMA | lentiviral vector | human | scFv | CD8α | 4–1BB-CD3ξ | No |
| Memorial Sloan Kettering Cancer Center | MCARH171 | Retroviral vector | human | scFv | CD8α | 4–1BB-CD3ξ | EGFRt |
| Memorial Sloan Kettering Cancer Center | JCARH25 | lentiviral vector | human | scFv | CD28 | 4–1BB-CD3ξ | No |
| Fred Hutchinson Cancer Research Center | FCARH143 | lentiviral vector | human | scFv | NA | 4–1BB-CD3ξ | EGFRt |
| CARsgen Therapeutics | CT053 | lentiviral vector | human | scFv | NA | 4–1BB-CD3ξ | No |
| IASO Biotherapeutics | CT103A | lentiviral vector | human | scFv | CD8α | 4–1BB-CD3ξ | No |
| Poseida Therapeutics | P-BCMA-101 | piggyBac™ DNA Modification System | human | Centyrin™ | NA | 4–1BB-CD3ξ | Yes (activated by Rimiducid) |
Table 2.
Summary of BCMA CAR engineered autologous T cells monodrug clinical trial in treating relapse and refractory multiple myeloma
| Manufacturer | Name of product | Clinical trial registered No. | Year of data updated | No. of pts evaluated | Enrollment based on BCMA expression | No. of lines of prior therapies | Disease burden at time of infusion | Conditioning therapy | Infusion dose | Efficacy | References | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Overall response rate | Stringent CR and CR rate | VGPR rate | median OS (month) | median PFS (month) | |||||||||||
| National Cancer Institute | CAR-BCMA | NCT02215967 | 2018 | 16 | Yes | 9.5 in average (range: 3 to 19) | r/r cases with BCMA uniformly expressed on tumor cells including extramedullary diseases.40% patients carried high risk cytogentics. | cyclophosphamide and fludarabine | 9×106 CAR+ T cells/kg | 81% | 13% | 50% | NA | 7.8 | 137 |
| Bluebird Bio / Celgene | Idecabtagene Vicleucel / bb2121 | NCT02658929 | 2020 | 62 | Yes | above 3 | 44% r/r cases had ≥50% bone marrow CD138+ plasma cells | cyclophosphamide and fludarabine | (50/150/450/800)×106 CAR+ T cells in total | 76% | 39% | 26% | 34.2 | 8.8 | 138 |
| bb21217 | NCT03274219 | 2020 | 46 | Yes | 6 in average (range: 3 to 17) | 57% r/r cases were triple refractory | cyclophosphamide and fludarabine | (150/300/450)×106 CAR+ T cells in total | 55% | 18% | 30% | NA | NA | 139 | |
| HRAIN Biotechnology | BCMA CAR-T | NCT03093168 | 2019 | 44 | No | above 2 | 19.6% cases had extramedullary plasmacytoma | cyclophosphamide and fludarabine | 9×106 CAR+ T cells/kg | 80% | 41% | 18% | Not reached | 15 | 140 |
| Nanjing Legend / Janssen | Ciltacabtagene autoleucel / LCAR-B38M | NCT03090659 | 2018 | 57 | Yes | 3 in average (range: 1 to 9) | 51% r/r cases had ≥40% tumor BCMA expression. Patients with extramedullary involvements were included. 37% of patients were in Stage III diease. | cyclophosphamide | (0.07–2.1)×106 CAR+ T cells/kg | 88% | 68% | 5% | Not reached | 15 | 143 |
| NCT03090659, ChiCTRONH17012285 | 2019 | 17 | Yes | 4.6 in average (range: 3 to 11) | 88% r/r cases had >70% tumor BCMA expression and 29% having extramedullary disease. 38% patients carried high risk cytogentics. | cyclophosphamide with or without fludarabine | (0.21–1.52)×106 CAR+ T cells/kg | 88% | 76% | 12% | Not reached | 12 | 141 | ||
| NCT03548207 | 2020 | 97 | No | 6 in average (range: 3 to 18) | 87.6% r/r cases were triple refractory. | cyclophosphamide and fludarabine | (0.5–1.0)×106 CAR+ T cells/kg | 95% | 56% | 32% | Not reached | Not reached | 144 | ||
| University of Pennslyvania | CART-BCMA | NCT02546167 | 2019 | 25 | No | 7 in average (range: 3 to 13) | median 65% myeloma cells on bone marrow biopsy. 28% patients had extramedullary disease, and 96% carred high risk cytogentics. | cyclophosphamide or no conditioning therapy | (10–500)×106 CAR+ T cells in total | 48% | 25% | 20% | 17 | 2,2,4mo in 3 cohorts, respectively | 127 |
| Memorial Sloan Kettering Cancer Center | MCARH171 | NCT03070327 | 2018 | 11 | Yes | 6 in average (range: 4 to 14) | 82% patients had high risk cytogenetics | cyclophosphamide with or without fludarabine | (72/137/475/818)×106 CAR+ T cells in total | 64% | NA | NA | NA | NA | 128 |
| Memorial Sloan Kettering Cancer Center | Orvacabtagene Autoleucel / JCARH25 | NCT03430011 | 2018 | 8 | No | 10 in average (range: 4 to 15) | 50% patients had high risk cytogenetics | cyclophosphamide and fludarabine | (50/150)×106 CAR+ T cells in total | 100% | 38% | 25% | NA | NA | 129 |
| 2020 | 44 | No | 6 in average (range: 3 to 8) | NA | cyclophosphamide and fludarabine | (300/450/600)×106 CAR+ T cells in total | 91% | 39% | 25% | Not reached | Not reached | 130 | |||
| Fred Hutchinson Cancer Research Center | FCARH143 | NCT03338972 | 2018 | 11 | Yes | 8 in average (range: 6 to 11) | The median percentage of bone marrow plasma cells was 58% (range 20% to >80%), and 100% patients had high risk cytogenetics. | cyclophosphamide and fludarabine | (50/150)×106 CAR+ T cells in total | 100% | 36% | 46% | NA | NA | 131 |
| CARsgen Therapeutics | CT053 | NCT03716856, NCT03302403, NCT03380039 | 2020 | 24 | Yes | 4.5 in average (range: 2 to 11) | 41.7% had extramedullary involvement | cyclophosphamide and fludarabine | (50/100/150/180)×106 CAR+ T cells in total | 88% | 79% | NA | NA | 18.8 | 132 |
| NCT03975907 | 2020 | 12 | No | 6 in average (range: 3 to 7) | 14.2% had extramedullary disease, and 35.7% had high-risk cytogenetics. | cyclophosphamide and fludarabine | (100/150)×106 CAR+ T cells in total | 100% | 42% | 25% | NA | NA | 134 | ||
| NCT03915184 | 2020 | 10 | No | 6 in average (range: 3 to 11) | 93% were triple refractory, 36% had extramedullary disease, and 64% had high-risk cytogenetics | cyclophosphamide and fludarabine | (150–300)×106 CAR+ T cells in total | 100% | 40% | 10% | NA | NA | 133 | ||
| IASO Biotherapeutics | CT103A | ChiCTR1800018137 | 2019 | 16 | NA | above 3 | 25% relapsed after a prior murine BCMA CAR-T therapy and 31.3% patients had extramedullary disease and/or plasma cell leukemia | cyclophosphamide and fludarabine | (1/3/6/8)×106 CAR+ T cells/kg | 100% | 75% (in 8 cases beyond 6 months) | 25% (in 8 cases beyond 6 months) | NA | NA | 135 |
| Poseida Therapeutics | P-BCMA-101 | NCT03288493 | 2020 | 34 | No | 7 in average (range: 3 to 18) | NA | cyclophosphamide and fludarabine | (0.75–15)×106 CAR+ T cells/kg | 57% | NA | NA | NA | NA | 136 |
BCMA has been targeted by various groups using a diverse array of chimeric receptors (reviewed in 124,125). Although human anti-BCMA CARs gained traction in myeloma126–135, non-human derived BCMA CARs, with an anti-BCMA murine136–139 - or alpaca- immunoglobulin140–143 chain are further along in clinical trials (Table 2). Most of these products include 4–1BB as a cosignaling domain which, at least in CD19 targeting CARs.
Lymphodepletion using cyclophosphamide and fludarabine prior to adoptive T-cell transfer further boosts CAR T-cell expansion 144 by depleting cytokine sinks 145 and immune suppressive cells146,147. Although response rates vary widely among different BCMA-specific CAR T-cell products, the biggest challenge remains durability of response, with patients appearing to ultimately progress regardless of product 141.
The mechanisms that underlie myeloma resistance to CART-BCMA therapy are coming to light through correlative analysis of biobanked specimens from the early phase clinical trials. Comparisons are difficult to make across trials, institutions and therapies, even though they all target the same myeloma-associated antigen, as differences in the cell manufacturing process, vector used, CAR design and the trial participant selection criteria, among other factors, are likely to affect outcome. Modulation of BCMA expression may also play a role. Early studies preselected patients based on expression of BCMA. Although to date no significant association between baseline BCMA expression and clinical response to CART-BCMA therapy has been reported in the published literature, several studies have observed a reduction in BCMA expression following therapy, which may be contributing to resistance 126. The mechanism of BCMA downregulation in myeloma is not entirely understood, but this protein is naturally shed from the cell surface by the gamma-secretase protease complex148–150. The resulting increased concentrations of soluble BCMA could also block CAR binding to the native, cell-bound protein, thereby limiting the clinical impact of CART-BCMA cells further. Preliminary results of a clinical trial that included patients who had failed prior BCMA targeted therapy and combined a γ-secretase inhibitor (JSMD194) with a low dose of BCMA CAR-T-cells, reported a 100% response rate151.
T-cell-intrinsic mechanisms similar to those seen with CD19-specific CAR T-cells in ALL and CLL may also be contributing to resistance. In this latter setting, patient T-cells expressing a fully human, BBζ-signaling CAR exhibited the most dramatic expansion kinetics in complete responders, whereas non-responders exhibited little expansion in the first month after infusion. This led to the discovery of an early memory T-cell subset in apheresed, i.e. pre-CAR engineering, T-cells that is associated with responses in CLL 41. Similarly, data from a phase I study of BCMA-specific CAR-T-cell therapy, expansion and persistence of the CAR T-cells in non-responders was significantly lower than that observed in responders 67,126,152. Again, the frequency of naïve-like, early memory T-cells within the apheresis product used to generated the CAR T-cells show a correlation with early engraftment. Although prospective studies using selected subsets of T-cells are necessary to confirm the role of these T-cells on outcome, these data suggest that some resistance to therapy may be intrinsic to the T-cell product.
CAR-T-cell therapy for solid tumors
Although CAR T-cells can mediate deep and durable cancer remission in B-cell malignancies, achieving comparable clinical responses in non-hematopoietic solid cancers remains a daunting task. Nevertheless, a complete response to CAR T-cell therapy of recurrent multifocal glioblastoma was achieved using multiple intracavitary and intraventricular infusions of autologous T-cells genetically-redirected to interleukin-13 receptor alpha 2 (IL13Rα2) 153, laying the foundation for additional investigations of how to apply effective CAR T-cell therapy in this and other non-hematopoietic solid cancers 5,110,154–156. CAR T-cell trials have established that deep, durable remissions with CAR-engineered cells, correlate with a minimal proportion of early memory T-cells in pre- and post-CAR engineering T-cells. Critical features include early memory T-cell differentiation in responding patients and absence or low level of T-cell dysfunction, glycolysis, effector cell differentiation, and exhaustion 41. These findings were validated in functional studies and in additional cohorts of leukemias, but also myeloma and non-Hodgkin’s lymphoma67. CAR T-cells targeting solid tumor antigens may have a different set of requirements to achieve efficacy than those targeting B-lineage malignancies. In addition to identifying appropriate target antigens, these requirements include the need for CAR T-cells to (i) traffic to sites of disease, (ii) migrate through tumor endothelial and stromal barriers before infiltrating into tumors, (iii) broadly attack cancer cells in the face of heterogeneous antigen expression, and (iv) thrive in a harsh tumor microenvironment (TME) characterized by hypoxia, oxidative stress, nutrient deprivation, acidic pH, as well as many immunosuppressive soluble cytokines and factors, overexpression of inhibitory molecules with coordinate expression of inhibitory receptors on T-cells, and the presence of an array of immune cells with immunosuppressive function, including Tregs, TAMs, MDSCs and TANs (Figure 3). Ultimately, CAR T-cell therapy may achieve greater efficacy in patients harboring solid tumors once approaches are developed that address each of these barriers together.
An expanding cadre of tumor-specific and tumor-associated antigens (TAAs) that could be targeted using CAR T-cell therapy in non-hematopoietic solid cancers have been identified, including mesothelin, folate receptor alpha, HER2, IL13Rα2, EGFRvIII, claudin 18.2, MUC1, Glypican-2, carboxy-anhydrase-IX (CAIX), and others. Nevertheless, identification of an antigen with restricted expression on solid cancer cells has been challenging. Ideally, CAR T-cells should be highly specific for a tumor-restricted antigen, expressed uniformly and at high levels on cancer cells, but not on vital healthy tissue. The importance of antigen exclusivity was demonstrated in CAR T-cell trials targeting tumor-associated antigens (TAAs) such as HER2 and CAIX that are expressed by both cancer cells and normal tissues, and which resulted in severe toxicity 110,157. The need for consistent antigen expression was illustrated in a clinical trial targeting mutant EGFRvIII, a CAR target antigen with highly restricted but heterogeneous expression in glioblastoma (GBM). Although intravenous T-cell infusion resulted in CAR T-cell trafficking to the brain with accompanied antigen-directed activity against EGFRvIII+ cancer cells, the heterogeneous EGFRvIII expression and potential antigen loss resulted in the outgrowth of antigen-negative disease 155. In some cases, targeting antigens with more restricted and uniform expression in tumors, or those preferentially expressed on organs that are not essential for patient survival, such as follicle stimulating hormone receptor 158, may pave the way toward broader and safer antitumor activity. Nevertheless, heterogeneous TAA expression is common in solid tumors, highlighting the need to develop multi-antigen targeting approaches or strategies that improve epitope spreading and engagement of endogenous antitumor immunity. Evidence already exists for epitope spreading and bolstering of endogenous immunity in clinical trials and in preclinical models of CAR T-cells in solid tumor 159,160, suggesting antigen spreading may be necessary to improve activity. As an alternative approach to address both antigen heterogeneity and the threat of antigen loss, so-called universal immune receptors (UIRs) were created (reviewed in 161). These CARs do not directly recognize the tumor antigen, but rather recognize a tag, such as biotin 162, on an antigen-targeted ligand (e.g. an antibody, scFv fragment) that serves as an immunologic bridge between the CAR and the TAA. UIRs allow the modified T-cells to recognize multiple distinct TAAs simultaneously or sequentially, thus addressing both heterogeneity and TAA loss observed with monospecific CARs, with the added benefit of dose-dependent control of T-cell activity. Clinical trials of UIR T-cells are ongoing (e.g. NCT03680560, NCT03266692, NCT03189836). Another approach, referred to as dual or tandem CARs, allows CAR T-cells to recognize two or more distinct antigens rather than one. Proof of principle has been established in solid tumor models using a HER2/MUC1 bispecific CAR for breast cancer cells in vitro 163, a HER2/IL-13Ra2 bispecific CAR for the treatment of a glioma xenograft in vivo 164, and a EGFR/EpCAM/HER2 tri-specific against Raji lymphoma cells engineered to express these TAAs165. Alternatively, diversification of TAAs recognized by single CAR T-cell products for solid tumor treatment may be achieved using SynNotch systems for conditional expression a second CAR following engagement of a primary CAR with a cognate TAA, thereby allowing for potential localized expression of a CAR specific for a distinct antigen at the site of primary target encounter 166. An alternative approach would be through bicistronic vectors for engineered co-expression of a CAR specific for one antigen and a soluble bispecific T-cell engager specific for a second antigen 167.
Although for hematopoietic cancers intravenous (IV) infusion of CAR T-cells may target cancer cells in natural immune cell environments such as the blood, lymph nodes and bone marrow, it remains challenging to deliver CAR T-cells targeting solid tumors to distant tumor deposits. In some cases, direct intratumoral or regional delivery of T-cells may facilitate and improve T-cell infiltration and antitumor activity, particularly for compartmentalized cancers 153,168,169. Lymphodepleting chemotherapy as a preconditioning regimen may also augment CAR T-cell accumulation in solid tumors after IV infusion. Following IV administration of indium-111 labeled tumor-infiltrating lymphocytes (TILs) to patients with metastatic melanoma, the cells rapidly accumulated in the lungs, liver and spleen before progressively localizing in tumor deposits 170. In these trials, TIL accumulation was enhanced with prior lymphodepletion and associated with improved clinical response to treatment 170,171. Still, the natural trafficking of T-cells to tumors requires that they respond to chemokines produced in the TME 172, and that tumor-derived chemokines be matched to the expression of the appropriate chemokine receptors on the infused T-cells to permit trafficking 173. Although most CAR T-cells do not naturally express cognate receptors for the chemokines produced by tumors, it is possible to engineer matched chemokine receptor expression to achieve enhanced infiltration and killing of solid tumors 174–176. CAR T-cells may also be outfitted to produce chemokine ligands, such as CCL19 and other factors, to foster chemokine-receptor-dependent recruitment of endogenous T-cells and dendritic cells to tumor sites when infused without prior lymphodepletion 177.
CAR T-cells trafficking to solid tumor sites also encounter formidable physical barriers that can both block T-cell infiltration and disable T-cell function. Major barriers include the fibrotic tumor stroma comprised of extracellular matrix (ECM) and cancer associated fibroblasts (CAFs), and the abnormal vasculature at the tumor site. Solid malignancies, such as pancreatic, ovarian and breast cancers, often contain fibrotic tumor stroma that may impede effective delivery of drug, including CAR T-cells. CAR T-cells naturally express low levels of enzymes that degrade ECM components, but engineering the expression of heparanase was shown to improve their capacity to degrade ECM proteoglycans, thereby promoting CAR T-cell entry into stroma-rich tumors and antitumor activity 178.
CAFs contribute to ECM remodeling, modulate tumor angiogenesis and promote metastasis, with CAF depletion fostering endogenous antitumor immunity in an autochthonous model of pancreatic ductal adenocarcinoma 179. Thus, engineering CAR T-cells against fibroblast activation protein (FAP), which is expressed by CAFs and myofibroblasts, was shown to target stromal CAFs and inhibit cancer progression without significant toxicity in multiple solid tumors 180. However, FAP-targeted CARs also recognized multipotent bone marrow stromal cells, resulting in lethal bone toxicity and cachexia in other tumor models 181.
CAR T-cells can also be designed to target and disrupt the tumor vasculature to allow T-cell infiltration and restrict the flow of blood and nutrients to solid tumors. For instance, targeting VEGFR-2 using CARs can augment T-cell infiltration and inhibit the progression of different types of vascularized syngeneic solid tumors 182,183. CARs specific for PSMA can ablate PSMA+ vessels and limit tumor progression in vivo through indirect loss of tumor cells, related to the disruption of the vasculature 184, and CARs targeting the angiogenic integrin, αvβ3, on the vascular endothelium can disrupt tumor vessels and suppress tumor outgrowth 185. CAR T-cells may also be combined with anti-vasculature agents, including anti-VEGF or PGE2 antibodies 186, anti-TEM-1/endosialin immunotoxin 187, or agents targeting molecules on the tumor endothelium, such as Fas-L, which establishes a tumor endothelial death barrier and kills incoming effector CD8 T-cells 186. Together, these findings provide the rationale for the further investigation and use of stroma-disrupting strategies as both preparative and combinatorial regimens to augment T-cell entry into solid tumors in TAA-targeted CAR T-cell trials.
In the stroma and the tumor bed, CAR T-cells contend with overexpression of inhibitory checkpoint ligands with coordinate expression of inhibitory receptors on T-cells, immunosuppressive soluble cytokines and factors, various immunosuppressive cell types, and a hypoxic and nutrient-deprived environment. Both tumor cells and immune cells in the TME can regulate CAR T-cell activation through the expression of inhibitory signals that block T lymphocyte activation and function, thereby circumventing otherwise effective immune control of tumor progression.
Future prospects
Over the past decade an astounding series of proof-of-concept trials have taken place, wth validation of early results in phase II trials 39,45,188–191 leading to the approval of CD19-specific CAR T-cell therapies for select B cell malignancies. Separately, insight into the biology of CRS has led to biomarker-driven trials (NCT02906371) and the discovery and validation of a novel biomarker profile of this potentially lethal toxicity 58. Additional observations from routine and translational studies revealed mechanisms of resistance and response, and the identification of the natural basis of successful and failed CAR T-cell therapy 41,65,67. Novel therapies started to incorporate small molecules which proved to augment T-cell function and simultaneously inhibit the malignant population 35,93,95,192. Combination trials also targeted more than one antigen, either on the same target as with CD19 and CD22, or precursor and progeny of the tumor as with CD19 and CD20, CD22, or BCMA190,193,194. The next few years are likely to witness the increased efficacy of CAR T-cells for solid tumors, a major current focus in this field. However, a better understanding and monitoring of the tumor would be essential for CAR-T-cell therapy to be offered to patients in early stages of their disease, before genomic instability and evolution of the tumor complicate treatment.
Acknowledgements
This work was supported by funding from the National Institutes of Health (R01CA241762 and R01GM138908, to JJM), Stand up to Cancer (SU2C) Convergence 2.0 Grant (to JJM), the Parker Institute for Cancer Immunotherapy (JJM and MAC), and the National Natural Science Foundation of China (81970189), the National Visiting Scholar Program from China Scholarship Council, and the Guang-Ci Visiting Scholar Program from Rui Jin Hospital Affiliated to Shanghai Jiao Tong University (to JX).
Footnotes
Conflict of interest: JJM, DJP, and MCM are inventors on several patents issued and pending in the field of CAR T-cell therapy for cancer, which are assigned to the University of Pennsylvania. Under the University of Pennsylvania’s policies, JJM, DJP and MCM may or currently receive royalties from licensing of these patent rights.
References
- 1.Bird RE et al. Single-chain antigen-binding proteins. Science 242, 423–426 (1988). [DOI] [PubMed] [Google Scholar]
- 2.June CH, Ledbetter JA, Gillespie MM, Lindsten T & Thompson CB T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Molecular and cellular biology 7, 4472–4481 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brentjens RJ et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med 9, 279–286, doi: 10.1038/nm827 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Mullis KB The unusual origin of the polymerase chain reaction. Scientific American 262, 56–65 (1990). [DOI] [PubMed] [Google Scholar]
- 5.Kershaw MH et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clinical Cancer Research 12, 6106–6115 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamers CH et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. Journal of Clinical Oncology (2016). [DOI] [PubMed] [Google Scholar]
- 7.Eshhar Z, Waks T, Gross G & Schindler DG Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A 90, 720–724, doi: 10.1073/pnas.90.2.720 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sadelain M Chimeric antigen receptors: a paradigm shift in immunotherapy. (2017).
- 9.Filley AC, Henriquez M & Dey M CART Immunotherapy: Development, Success, and Translation to Malignant Gliomas and Other Solid Tumors. Frontiers in Oncology 8, doi: 10.3389/fonc.2018.00453 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milone MC et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy In Vivo. Mol Ther. 17, 1453–1464 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carpenito C et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proceedings of the National Academy of Sciences 106, 3360–3365 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y et al. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. The Journal of Immunology 183, 5563–5574 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Savoldo B et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. The Journal of clinical investigation 121, 1822–1826 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedmann-Morvinski D, Bendavid A, Waks T, Schindler D & Eshhar Z Redirected primary T cells harboring a chimeric receptor require costimulation for their antigen-specific activation. Blood 105, 3087–3093 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Brocker T Chimeric Fv-zeta or Fv-epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood 96, 1999–2001 (2000). [PubMed] [Google Scholar]
- 16.Mitsuyasu RT et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood 96, 785–793 (2000). [PubMed] [Google Scholar]
- 17.Scholler J et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med 4, 132ra153, doi: 10.1126/scitranslmed.3003761 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uckun F et al. Detailed studies on expression and function of CD19 surface determinant by using B43 monoclonal antibody and the clinical potential of anti- CD19 immunotoxins. Blood 71, 13–29, doi: 10.1182/blood.V71.1.13.13 (1988). [DOI] [PubMed] [Google Scholar]
- 19.Ishiura N et al. Differential phosphorylation of functional tyrosines in CD19 modulates B‐lymphocyte activation. European journal of immunology 40, 1192–1204 (2010). [DOI] [PubMed] [Google Scholar]
- 20.LeBien TW & Tedder TF B lymphocytes: how they develop and function. Blood 112, 1570–1580 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kochenderfer JN et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 116, 4099–4102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalos M et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Science translational medicine 3, 95ra73–95ra73 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porter DL, Levine BL, Kalos M, Bagg A & June CH Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. New England Journal of Medicine 365, 725–733 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brentjens RJ et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 118, 4817–4828 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grupp SA et al. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. New England Journal of Medicine 368, 1509–1518 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maude S et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med 371, 1507–1517, doi: 10.1056/NEJMoa1407222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kochenderfer JN et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. Journal of Clinical Oncology 33, 540 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porter DL et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Science translational medicine 7, 303ra139–303ra139 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee DW et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. The Lancet 385, 517–528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turtle CJ et al. CD19 CAR–T cells of defined CD4+: CD8+ composition in adult B cell ALL patients. The Journal of clinical investigation 126, 2123–2138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turtle CJ et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor–modified T cells. Science translational medicine 8, 355ra116–355ra116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schuster SJ et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. New England Journal of Medicine 377, 2545–2554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neelapu SS et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. New England Journal of Medicine 377, 2531–2544 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kochenderfer JN et al. Lymphoma remissions caused by anti-CD19 chimeric antigen receptor T cells are associated with high serum interleukin-15 levels. Journal of Clinical Oncology 35, 1803 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turtle CJ et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor–modified T cells after failure of ibrutinib. Journal of Clinical Oncology 35, 3010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Locke FL et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Molecular Therapy 25, 285–295 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melenhorst JJ et al. (Am Soc Hematology, 2018). [Google Scholar]
- 38.Park JH et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. New England Journal of Medicine 378, 449–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maude SL et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. New England Journal of Medicine 378, 439–448 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Porter DL et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 7, 303ra139, doi: 10.1126/scitranslmed.aac5415 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fraietta JA et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 24, 563–571, doi: 10.1038/s41591-018-0010-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neelapu SS et al. (American Society of Hematology; Washington, DC, 2019). [Google Scholar]
- 43.Baird JH et al. CD22-Directed CAR T-Cell Therapy Induces Complete Remissions in CD19-Directed CAR-Refractory Large B-Cell Lymphoma. Blood, doi: 10.1182/blood.2020009432 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bukhari A et al. Rapid relapse of large B-cell lymphoma after CD19 directed CAR-T-cell therapy due to CD-19 antigen loss. Am J Hematol 94, E273–e275, doi: 10.1002/ajh.25591 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Schuster SJ et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. New England Journal of Medicine 380, 45–56 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Schuster SJ et al. Sustained disease control for adult patients with relapsed or refractory diffuse large B-cell lymphoma: an updated analysis of Juliet, a global pivotal phase 2 trial of tisagenlecleucel. Blood 132, 1684–1684 (2018). [Google Scholar]
- 47.Wang M et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. New England Journal of Medicine 382, 1331–1342, doi: 10.1056/NEJMoa1914347 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin Z et al. The severe cytokine release syndrome in phase I trials of CD19-CAR-T cell therapy: a systematic review. Annals of hematology 97, 1327–1335 (2018). [DOI] [PubMed] [Google Scholar]
- 49.Lee DW et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biology of Blood and Marrow Transplantation 25, 625–638 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ajina A & Maher J Strategies to Address Chimeric Antigen Receptor Tonic Signaling. Mol Cancer Ther 17, 1795–1815, doi: 10.1158/1535-7163.Mct-17-1097 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maus MV et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune effector cell-related adverse events. Journal for ImmunoTherapy of Cancer 8, e001511 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neelapu SS et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nature reviews Clinical oncology 15, 47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stadtmauer EA et al. CRISPR-engineered T cells in patients with refractory cancer. Science, eaba7365, doi: 10.1126/science.aba7365 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hay KA et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 130, 2295–2306, doi: 10.1182/blood-2017-06-793141 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Turtle CJ et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest 126, 2123–2138, doi: 10.1172/JCI85309 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davila ML et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Science translational medicine 6, 224ra225–224ra225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kadauke S et al. Early Administration of Tocilizumab (Toci) for the Prevention of Grade 4 Cytokine Release Syndrome (CRS) after CD19-directed CAR T-cell Therapy (CTL019). Cytotherapy 21, e2–e3 (2019). [Google Scholar]
- 58.Teachey DT et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer discovery 6, 664–679 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sotillo E et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer discovery 5, 1282–1295, doi: 10.1158/2159-8290.cd-15-1020 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Orlando EJ et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat Med 24, 1504–1506, doi: 10.1038/s41591-018-0146-z (2018). [DOI] [PubMed] [Google Scholar]
- 61.Ruella M et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med 24, 1499–1503, doi: 10.1038/s41591-018-0201-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maude SL et al. (American Society of Clinical Oncology, 2016). [Google Scholar]
- 63.Singh N et al. Impaired death receptor signaling in leukemia causes antigen-independent resistance by inducing CAR T cell dysfunction. Cancer discovery, CD-19–0813, doi: 10.1158/2159-8290.CD-19-0813 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mueller KT et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood 130, 2317–2325, doi: 10.1182/blood-2017-06-786129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Finney OC et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. The Journal of clinical investigation 129, 2123–2132 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fraietta JA et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 558, 307–312, doi: 10.1038/s41586-018-0178-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang M et al. (American Society of Hematology; Washington, DC, 2019). [Google Scholar]
- 68.Milone MC et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Molecular Therapy 17, 1453–1464 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aguirre-Ghiso JA Models, mechanisms and clinical evidence for cancer dormancy. Nature Reviews Cancer 7, 834–846 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.White E & DiPaola RS The double-edged sword of autophagy modulation in cancer. Clinical cancer research 15, 5308–5316 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sosa MS, Bragado P & Aguirre-Ghiso JA Mechanisms of disseminated cancer cell dormancy: an awakening field. Nature Reviews Cancer 14, 611–622 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Palma M et al. T cells in chronic lymphocytic leukemia display dysregulated expression of immune checkpoints and activation markers. Haematologica 102, 562–572, doi: 10.3324/haematol.2016.151100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chong EA et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood 129, 1039–1041, doi: 10.1182/blood-2016-09-738245 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ren J et al. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res 23, 2255–2266, doi: 10.1158/1078-0432.CCR-16-1300 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.John LB et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res 19, 5636–5646, doi: 10.1158/1078-0432.Ccr-13-0458 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Newick K et al. Augmentation of CAR T-cell Trafficking and Antitumor Efficacy by Blocking Protein Kinase A Localization. Cancer Immunol Res 4, 541–551, doi: 10.1158/2326-6066.CIR-15-0263 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Renner K et al. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front Immunol 8, 248, doi: 10.3389/fimmu.2017.00248 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shah NN et al. Clonal expansion of CAR T cells harboring lentivector integration in the CBL gene following anti-CD22 CAR T-cell therapy. Blood Adv 3, 2317–2322, doi: 10.1182/bloodadvances.2019000219 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nobles CL et al. CD19-targeting CAR T cell immunotherapy outcomes correlate with genomic modification by vector integration. J Clin Invest, doi: 10.1172/jci130144 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bushman FD Retroviral Insertional Mutagenesis in Humans: Evidence for Four Genetic Mechanisms Promoting Expansion of Cell Clones. Mol Ther 28, 352–356, doi: 10.1016/j.ymthe.2019.12.009 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sheih A et al. Clonal kinetics and single-cell transcriptional profiling of CAR-T cells in patients undergoing CD19 CAR-T immunotherapy. Nat Commun 11, 1–13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Naramura M, Kole HK, Hu RJ & Gu H Altered thymic positive selection and intracellular signals in Cbl-deficient mice. Proc Natl Acad Sci U S A 95, 15547–15552, doi: 10.1073/pnas.95.26.15547 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Peer S, Baier G & Gruber T Cblb-deficient T cells are less susceptible to PD-L1-mediated inhibition. Oncotarget 8, 41841–41853, doi: 10.18632/oncotarget.18360 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schmitz ML Activation of T cells: releasing the brakes by proteolytic elimination of Cbl-b. Sci Signal 2, p e38, doi: 10.1126/scisignal.276pe38 (2009). [DOI] [PubMed] [Google Scholar]
- 85.Stromnes IM et al. Abrogating Cbl-b in effector CD8(+) T cells improves the efficacy of adoptive therapy of leukemia in mice. J Clin Invest 120, 3722–3734, doi: 10.1172/jci41991 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Eyquem J et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117, doi: 10.1038/nature21405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu E et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. New England Journal of Medicine 382, 545–553, doi: 10.1056/NEJMoa1910607 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lanitis E et al. Optimized gene engineering of murine CAR-T cells reveals the beneficial effects of IL-15 coexpression. Journal of Experimental Medicine 218, doi: 10.1084/jem.20192203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mishra A et al. Aberrant overexpression of IL-15 initiates large granular lymphocyte leukemia through chromosomal instability and DNA hypermethylation. Cancer Cell 22, 645–655 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fehniger TA et al. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J Exp Med 193, 219–232 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hu B et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep 20, 3025–3033 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fraietta JA et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 127, 1117–1127, doi: 10.1182/blood-2015-11-679134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gill SI et al. Prospective Clinical Trial of Anti-CD19 CAR T Cells in Combination with Ibrutinib for the Treatment of Chronic Lymphocytic Leukemia Shows a High Response Rate. Blood 132, 298–298, doi: 10.1182/blood-2018-99-115418 (2018). [DOI] [Google Scholar]
- 94.Gauthier J et al. Feasibility and efficacy of CD19-targeted CAR T cells with concurrent ibrutinib for CLL after ibrutinib failure. Blood, The Journal of the American Society of Hematology 135, 1650–1660 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kater AP & Melenhorst JJ CAR-T and ibrutinib vs CLL: sequential or simultaneous? Blood, The Journal of the American Society of Hematology 135, 1611–1612 (2020). [DOI] [PubMed] [Google Scholar]
- 96.Gorelik L & Flavell RA Immune-mediated eradication of tumors through the blockade of transforming growth factor-β signaling in T cells. Nature Medicine 7, 1118–1122, doi: 10.1038/nm1001-1118 (2001). [DOI] [PubMed] [Google Scholar]
- 97.Kloss CC et al. Dominant-Negative TGF-beta Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol Ther 26, 1855–1866, doi: 10.1016/j.ymthe.2018.05.003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Garfall AL et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N Engl J Med 373, 1040–1047, doi: 10.1056/NEJMoa1504542 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gardner R et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 127, 2406–2410, doi: 10.1182/blood-2015-08-665547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jacoby E et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun 7, 12320, doi: 10.1038/ncomms12320 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang Z et al. Point mutation in CD19 facilitates immune escape of B cell lymphoma from CAR-T cell therapy. Journal for immunotherapy of cancer 8, e001150, doi: 10.1136/jitc-2020-001150 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gardner R et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 127, 2406–2410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jia H et al. Haploidentical CD19/CD22 bispecific CAR-T cells induced MRD-negative remission in a patient with relapsed and refractory adult B-ALL after haploidentical hematopoietic stem cell transplantation. Journal of Hematology & Oncology 12, 57, doi: 10.1186/s13045-019-0741-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shalabi H et al. Sequential loss of tumor surface antigens following chimeric antigen receptor T-cell therapies in diffuse large B-cell lymphoma. Haematologica, haematol. 2017.183459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fry TJ et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nature medicine 24, 20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Louis CU et al. Antitumor activity and long-term fate of chimeric antigen receptor–positive T cells in patients with neuroblastoma. Blood, The Journal of the American Society of Hematology 118, 6050–6056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Finney HM, Akbar AN & Lawson AD Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCRζ chain. The Journal of Immunology 172, 104–113 (2004). [DOI] [PubMed] [Google Scholar]
- 108.Long AH et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature medicine 21, 581–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hong M, Clubb JD & Chen YY Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell, doi: 10.1016/j.ccell.2020.07.005 (2020). [DOI] [PubMed] [Google Scholar]
- 110.Lamers CH et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol 24, e20–22, doi:24/13/e20 [pii] 10.1200/JCO.2006.05.9964 (2006). [DOI] [PubMed] [Google Scholar]
- 111.Den Haan J et al. Identification of a graft versus host disease-associated human minor histocompatibility antigen. Science 268, 1476–1480 (1995). [DOI] [PubMed] [Google Scholar]
- 112.Den Haan JM et al. The minor histocompatibility antigen HA-1: a diallelic gene with a single amino acid polymorphism. Science 279, 1054–1057 (1998). [DOI] [PubMed] [Google Scholar]
- 113.Bevan MJ The major histocompatibility complex determines susceptibility to cytotoxic T cells directed against minor histocompatibility antigens. J Exp Med 142, 1349–1364 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lamberth K et al. Post hoc assessment of the immunogenicity of bioengineered factor VIIa demonstrates the use of preclinical tools. Science Translational Medicine 9, eaag1286, doi: 10.1126/scitranslmed.aag1286 (2017). [DOI] [PubMed] [Google Scholar]
- 115.Mahlangu JN et al. Changes in the amino acid sequence of the recombinant human factor VIIa analog, vatreptacog alfa, are associated with clinical immunogenicity. Journal of Thrombosis and Haemostasis 13, 1989–1998, doi: 10.1111/jth.13141 (2015). [DOI] [PubMed] [Google Scholar]
- 116.Berger C, Flowers ME, Warren EH & Riddell SR Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood 107, 2294–2302, doi: 10.1182/blood-2005-08-3503 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Xu J et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc Natl Acad Sci U S A 116, 9543–9551, doi: 10.1073/pnas.1819745116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jensen MC et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant 16, 1245–1256, doi: 10.1016/j.bbmt.2010.03.014 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wagner DL et al. Immunogenicity of CAR T cells in cancer therapy. Nature Reviews Clinical Oncology, 1–15 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kershaw MH et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 12, 6106–6115, doi: 10.1158/1078-0432.Ccr-06-1183 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lamers CH et al. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 117, 72–82, doi: 10.1182/blood-2010-07-294520 (2011). [DOI] [PubMed] [Google Scholar]
- 122.Maude SL et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371, 1507–1517, doi: 10.1056/NEJMoa1407222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hajek R, Okubote SA & Svachova H Myeloma stem cell concepts, heterogeneity and plasticity of multiple myeloma. Br J Haematol 163, 551–564, doi: 10.1111/bjh.12563 (2013). [DOI] [PubMed] [Google Scholar]
- 124.Mikkilineni L & Kochenderfer JN CAR T cell therapies for patients with multiple myeloma. Nature Reviews Clinical Oncology, doi: 10.1038/s41571-020-0427-6 (2020). [DOI] [PubMed] [Google Scholar]
- 125.D’Agostino M & Raje N Anti-BCMA CAR T-cell therapy in multiple myeloma: can we do better? Leukemia 34, 21–34, doi: 10.1038/s41375-019-0669-4 (2020). [DOI] [PubMed] [Google Scholar]
- 126.Cohen AD et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest 129, 2210–2221, doi: 10.1172/JCI126397 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mailankody S et al. Clinical Responses and Pharmacokinetics of MCARH171, a Human-Derived Bcma Targeted CAR T Cell Therapy in Relapsed/Refractory Multiple Myeloma: Final Results of a Phase I Clinical Trial. Blood 132, 959–959, doi: 10.1182/blood-2018-99-119717 (2018). [DOI] [Google Scholar]
- 128.Mailankody S et al. JCARH125, Anti-BCMA CAR T-cell Therapy for Relapsed/Refractory Multiple Myeloma: Initial Proof of Concept Results from a Phase 1/2 Multicenter Study (EVOLVE). Blood 132, 957–957, doi: 10.1182/blood-2018-99-113548 (2018). [DOI] [Google Scholar]
- 129.Mailankody S et al. (American Society of Clinical Oncology, 2020). [Google Scholar]
- 130.Green DJ et al. Fully Human Bcma Targeted Chimeric Antigen Receptor T Cells Administered in a Defined Composition Demonstrate Potency at Low Doses in Advanced Stage High Risk Multiple Myeloma. Blood 132, 1011–1011, doi: 10.1182/blood-2018-99-117729 (2018). [DOI] [Google Scholar]
- 131.Hao S et al. Two-Year Follow-up of Investigator-Initiated Phase 1 Trials of the Safety and Efficacy of Fully Human Anti-Bcma CAR T Cells (CT053) in Relapsed/Refractory Multiple Myeloma. Blood 136, 27–28, doi: 10.1182/blood-2020-140156 (2020). [DOI] [Google Scholar]
- 132.Kumar SK et al. Results from Lummicar-2: A Phase 1b/2 Study of Fully Human B-Cell Maturation Antigen-Specific CAR T Cells (CT053) in Patients with Relapsed and/or Refractory Multiple Myeloma. Blood 136, 28–29, doi: 10.1182/blood-2020-139802 (2020). [DOI] [Google Scholar]
- 133.Chen W et al. Results from Lummicar-1: A Phase 1 Study of Fully Human B-Cell Maturation Antigen-Specific CAR T Cells (CT053) in Chinese Subjects with Relapsed and/or Refractory Multiple Myeloma. Blood 136, 49–50, doi: 10.1182/blood-2020-140727 (2020). [DOI] [Google Scholar]
- 134.Li C et al. Efficacy and Safety of Fully Human Bcma Targeting CAR T Cell Therapy in Relapsed/Refractory Multiple Myeloma. Blood 134, 929–929, doi: 10.1182/blood-2019-128468 (2019). [DOI] [Google Scholar]
- 135.Costello CL et al. Phase 1/2 Study of the Safety and Response of P-BCMA-101 CAR-T Cells in Patients with Relapsed/Refractory (r/r) Multiple Myeloma (MM) (PRIME) with Novel Therapeutic Strategies. Blood 136, 29–30, doi: 10.1182/blood-2020-142695 (2020). [DOI] [Google Scholar]
- 136.Brudno JN et al. T cells genetically modified to express an anti–B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. Journal of Clinical Oncology 36, 2267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lin Y et al. Idecabtagene Vicleucel (ide-cel, bb2121), a BCMA-Directed CAR T Cell Therapy, in Patients with Relapsed and Refractory Multiple Myeloma: Updated Results from Phase 1 CRB-401 Study. Blood 136, 26–27, doi: 10.1182/blood-2020-134324 (2020). [DOI] [Google Scholar]
- 138.Alsina M et al. Updated Results from the Phase I CRB-402 Study of Anti-Bcma CAR-T Cell Therapy bb21217 in Patients with Relapsed and Refractory Multiple Myeloma: Correlation of Expansion and Duration of Response with T Cell Phenotypes. Blood 136, 25–26, doi: 10.1182/blood-2020-140410 (2020). [DOI] [Google Scholar]
- 139.Fu W Sr. et al. Efficacy and Safety of CAR-T Therapy with Safety Switch Targeting Bcma for Patients with Relapsed/Refractory Multiple Myeloma in a Phase 1 Clinical Study. Blood 134, 3154–3154, doi: 10.1182/blood-2019-127608 (2019). [DOI] [Google Scholar]
- 140.Xu J et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proceedings of the National Academy of Sciences 116, 9543–9551 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang B-Y et al. (American Society of Hematology; Washington, DC, 2019). [Google Scholar]
- 142.Zhao W-H et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. Journal of hematology & oncology 11, 141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Madduri D et al. Results from CARTITUDE-1: A Phase 1b/2 Study of JNJ-4528, a CAR-T Cell Therapy Directed Against B-Cell Maturation Antigen (BCMA), in Patients with Relapsed and/or Refractory Multiple Myeloma (R/R MM). Blood 134, 577–577, doi: 10.1182/blood-2019-121731 (2019).31416814 [DOI] [Google Scholar]
- 144.Haas AR et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol Ther 27, 1919–1929, doi: 10.1016/j.ymthe.2019.07.015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gattinoni L et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med 202, 907–912, doi: 10.1084/jem.20050732 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Baba J et al. Depletion of radio-resistant regulatory T cells enhances antitumor immunity during recovery from lymphopenia. Blood 120, 2417–2427, doi: 10.1182/blood-2012-02-411124 (2012). [DOI] [PubMed] [Google Scholar]
- 147.Kodumudi KN, Weber A, Sarnaik AA & Pilon-Thomas S Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. The Journal of Immunology 189, 5147–5154 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pont MJ et al. gamma-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 134, 1585–1597, doi: 10.1182/blood.2019000050 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Green DJ et al. (American Society of Hematology; Washington, DC, 2019). [Google Scholar]
- 150.Laurent SA et al. γ-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun 6, 1–12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cowan AJ et al. (American Society of Hematology; Washington, DC, 2019). [Google Scholar]
- 152.Raje N et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. New England Journal of Medicine 380, 1726–1737 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Brown CE et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med 375, 2561–2569, doi: 10.1056/NEJMoa1610497 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kandalaft LE, Powell DJ & Coukos G A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. Journal of translational medicine 10, 157 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.O’Rourke DM et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 9, doi: 10.1126/scitranslmed.aaa0984 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Castellarin M, Watanabe K, June CH, Kloss CC & Posey AD Driving cars to the clinic for solid tumors. Gene therapy 25, 165–175 (2018). [DOI] [PubMed] [Google Scholar]
- 157.Morgan RA et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 18, 843–851, doi:mt201024 [pii] 10.1038/mt.2010.24 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Urbanska K, Stashwick C, Poussin M & Powell DJ Jr. Follicle-Stimulating Hormone Receptor as a Target in the Redirected T-cell Therapy for Cancer. Cancer Immunol Res 3, 1130–1137, doi: 10.1158/2326-6066.CIR-15-0047 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Beatty GL et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2, 112–120, doi: 10.1158/2326-6066.CIR-13-0170 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Evans RA et al. Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight 1, doi: 10.1172/jci.insight.88328 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Minutolo NG, Hollander EE & Powell DJ Jr. The Emergence of Universal Immune Receptor T Cell Therapy for Cancer. Front Oncol 9, 176, doi: 10.3389/fonc.2019.00176 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Urbanska K et al. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res 72, 1844–1852, doi: 10.1158/0008-5472.CAN-11-3890 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Wilkie S et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol 32, 1059–1070, doi: 10.1007/s10875-012-9689-9 (2012). [DOI] [PubMed] [Google Scholar]
- 164.Hegde M et al. Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Invest 126, 3036–3052, doi: 10.1172/JCI83416 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Balakrishnan A et al. Multispecific Targeting with Synthetic Ankyrin Repeat Motif Chimeric Antigen Receptors. Clin Cancer Res, doi: 10.1158/1078-0432.CCR-19-1479 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Roybal KT et al. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 167, 419–432 e416, doi: 10.1016/j.cell.2016.09.011 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Choi BD et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol 37, 1049–1058, doi: 10.1038/s41587-019-0192-1 (2019). [DOI] [PubMed] [Google Scholar]
- 168.Song DG et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4–1BB). Cancer Res 71, 4617–4627, doi: 10.1158/0008-5472.CAN-11-0422 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tchou J et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol Res 5, 1152–1161, doi: 10.1158/2326-6066.CIR-17-0189 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Fisher B et al. Tumor localization of adoptively transferred indium-111 labeled tumor infiltrating lymphocytes in patients with metastatic melanoma. J Clin Oncol 7, 250–261, doi: 10.1200/JCO.1989.7.2.250 (1989). [DOI] [PubMed] [Google Scholar]
- 171.Pockaj BA et al. Localization of 111indium-labeled tumor infiltrating lymphocytes to tumor in patients receiving adoptive immunotherapy. Augmentation with cyclophosphamide and correlation with response. Cancer 73, 1731–1737, doi: 10.1002/1097-0142(19940315)73:6<1731::aid-cncr2820730630>3.0.co;2-h (1994). [DOI] [PubMed] [Google Scholar]
- 172.Harlin H et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 69, 3077–3085, doi: 10.1158/0008-5472.CAN-08-2281 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Brown CE et al. Tumor-derived chemokine MCP-1/CCL2 is sufficient for mediating tumor tropism of adoptively transferred T cells. J Immunol 179, 3332–3341, doi: 10.4049/jimmunol.179.5.3332 (2007). [DOI] [PubMed] [Google Scholar]
- 174.Moon EK et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res 17, 4719–4730, doi: 10.1158/1078-0432.CCR-11-0351 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Craddock JA et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. 33, 780–788 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Di Stasi A et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 113, 6392–6402, doi: 10.1182/blood-2009-03-209650 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Adachi K et al. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol 36, 346–351, doi: 10.1038/nbt.4086 (2018). [DOI] [PubMed] [Google Scholar]
- 178.Caruana I et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med 21, 524–529, doi: 10.1038/nm.3833 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Feig C et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A 110, 20212–20217, doi: 10.1073/pnas.1320318110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wang LC et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res 2, 154–166, doi: 10.1158/2326-6066.CIR-13-0027 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Tran E et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med 210, 1125–1135, doi: 10.1084/jem.20130110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chinnasamy D et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J Clin Invest 120, 3953–3968, doi: 10.1172/JCI43490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chinnasamy D et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res 18, 1672–1683, doi: 10.1158/1078-0432.CCR-11-3050 1078–0432.CCR-11–3050 [pii] (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Santoro SP et al. T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol Res 3, 68–84, doi: 10.1158/2326-6066.CIR-14-0192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Fu X, Rivera A, Tao L & Zhang X Genetically modified T cells targeting neovasculature efficiently destroy tumor blood vessels, shrink established solid tumors and increase nanoparticle delivery. Int J Cancer 133, 2483–2492, doi: 10.1002/ijc.28269 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Motz GT et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med 20, 607–615, doi: 10.1038/nm.3541 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Guo Y et al. Tumour endothelial marker 1/endosialin-mediated targeting of human sarcoma. Eur J Cancer 90, 111–121, doi: 10.1016/j.ejca.2017.10.035 (2018). [DOI] [PubMed] [Google Scholar]
- 188.Neelapu SS et al. (American Society of Clinical Oncology, 2019). [Google Scholar]
- 189.Munshi NC et al. Idecabtagene vicleucel (ide-cel; bb2121), a BCMA-targeted CAR T-cell therapy, in patients with relapsed and refractory multiple myeloma (RRMM): Initial KarMMa results. Journal of Clinical Oncology 38, 8503–8503, doi: 10.1200/JCO.2020.38.15_suppl.8503 (2020). [DOI] [Google Scholar]
- 190.Yan Z et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: a single-arm, phase 2 trial. The Lancet Haematology 6, e521–e529 (2019). [DOI] [PubMed] [Google Scholar]
- 191.Locke FL et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. The lancet oncology 20, 31–42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Gill S et al. (American Society of Clinical Oncology, 2017). [Google Scholar]
- 193.Shah NN et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nature Medicine 26, 1569–1575, doi: 10.1038/s41591-020-1081-3 (2020). [DOI] [PubMed] [Google Scholar]
- 194.Pan J et al. Sequential CD19–22 CAR T therapy induces sustained remission in children with r/r B-ALL. Blood, The Journal of the American Society of Hematology 135, 387–391 (2020). [DOI] [PubMed] [Google Scholar]
- 195.Weber EW et al. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv 3, 711 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]