

Abstract

Since the approval of three hydroxamic acid-based HDAC inhibitors as anticancer drugs, such functional groups acquired even more notoriety in synthetic medicinal chemistry. The ability of hydroxamic acids (HAs) to chelate metal ions makes this moiety an attractive metal binding group—in particular, Fe(III) and Zn(II)—so that HA derivatives find wide applications as metalloenzymes inhibitors. In this minireview, we will discuss the most relevant features concerning hydroxamic acid derivatives. In a first instance, the physicochemical characteristics of HAs will be summarized; then, an exhaustive description of the most relevant methods for the introduction of such moiety into organic substrates and an overview of their uses in medicinal chemistry will be presented.
1. Introduction
Hydroxamic acid (HA) derivatives represent a group of relevant scaffolds that find widespread application among the chemical sciences.1 The hydroxamic acid moiety, thanks to its ability to chelate metal ions, such as Fe(III) and Zn(II), has been extensively investigated as a pharmacophoric group of many metalloprotease inhibitors, such as Marimastat, a potent broad-spectrum peptidomimetic matrix metalloprotease inhibitor developed by British Biotech.2 On the other hand, the strong chelating capability of Deferoxamine has resulted in the approval of such a drug for the treatment of iron overdose.3 Although their applicability has been significantly investigated in several fields of medicinal chemistry, the largest application of HA derivatives has been found as histone deacetylase (HDAC) inhibitors. HDACs are key enzymes involved in cell cycle regulation that catalyze the hydrolysis of acetyl l-lysine residues.4 HDAC isoforms are upregulated in various types of cancer, and currently, three hydroxamic acid-based HDAC inhibitors are approved for the treatment of T-cell lymphoma or multiple myeloma (Belinostat, Panobinostat, and Vorinostat) (Figure 1).4 Thanks to their wide use, a large number of synthetic approaches for obtaining HAs have been developed over time.
Figure 1.

Chemical structures of Marimastat, a potent peptidomimetic matrix metalloprotease inhibitor, and Belinostat, Panobinostat, and Vorinostat (SAHA), three hydroxamic acid-based HDAC inhibitors approved for the treatment of T-cell lymphoma or multiple myeloma.
2. Physical and Chemical Properties
Hydroxamic acids are N-hydroxy amides, but unlike common amides, which exhibit high stability, they are distinctly reactive.5 They can be found in two different tautomeric forms, as depicted in Figure 2a: the keto form 1a is the most stable tautomer observed in acidic medium, while the iminol form 1b prevails in basic conditions.6 Hydroxamic acids are weak acids (Figure 2b).7 They can be considered diprotic acids: in water solvent, the hydroxyl group of HAs loses the first proton with a pKa ranging between 8.5 and 9.5, while deprotonation in DMSO mainly involves the loss of the hydrogen from the −NH group. HAs are smoothly soluble in alkaline solutions due to the stability of the hydroxamate anion. Electron withdrawing groups (EWG) increase the acidity of HAs, and the substitution of the −NH proton with an electron donating group (EDG), such as alkyl or aryl, decreases the pKa (Figure 2b). The second deprotonation occurs under drastic conditions and furnishes the hydroximate dianion, with a dissociation constant outside the conventional pH range (Figure 2b).7
Figure 2.

(a) Representation of the two tautomeric forms of hydroxamic acids. Form 1a prevails in acid conditions, while 1b prevails in basic conditions. (b) Deprotonation of HAs under basic conditions yields the hydroxamate anion. A second deprotonation forms the hydroximate dianion. The acidity of HAs increases with the substitution with EWG and the replacement of the −NH proton with alkyl or aryl groups. In aqueous solution, the HAs predominantly assume the more stable trans form; interconversion into the cis form occurs during chelation.
HAs are able to chelate metal ions, such as Fe(III) and Zn(II). During complexation, they assume a cis geometry that allows them to establish a stronger interaction with the cation. They form bidentate O,O′ complexes with transition metals, intensely coloring the metal and HA solutions. HAs possess marked affinity for Fe(III), as evidenced from the high stability of the formed complexes8 and the presence in nature of siderophores, capable of storing Fe(III) inside cells.9 Vorinostat (suberoylanilide hydroxamic acid, SAHA) establishes a very stable tris-hydroxamate complex with Fe(III) in water (Figure 3).10 On the other hand, SAHA yields bis-hydroxamate complexes with Zn(II) both in the solid state and in solution, although Zn(II) possesses a lower affinity for HAs compared to Fe(III) (Figure 3).
Figure 3.

Complex of suberoylanilide hydroxamic acid (SAHA) with Zn(II) and Fe(III), respectively.
3. Synthetic Approaches to HAs
Given the remarkable importance of HAs, several synthetic approaches were proposed to obtain such derivatives.11 To date, many versatile synthetic strategies are available to convert different functional groups into hydroxamic acids. The primary source for obtaining HAs is hydroxylamine (Figure 4), almost always provided and used in aqueous solution. There are various commercial solutions of hydroxylamine in water, as well as of hydroxylamine in the form of hydrochloric salt. Furthermore, the use of protected hydroxylamines, such as ethers or silyl ethers, is becoming increasingly popular, showing the great advantage of synthesizing hydroxamic acids in high yields and avoiding unpleasant side products. The most intuitive method for obtaining HAs relies on the use of carboxylic acids as starting materials, suitably activated as an ester or acyl chloride, to carry out a nucleophilic acyl substitution reaction with hydroxylamine (Figure 4). The higher nucleophilic character of N, combined with the greater stability of the resulting amide compared to the ester, produces almost exclusively hydroxamic acid, with respect to the opposite reaction. However, many other functional groups are easily converted into hydroxamic acids in one or more steps. This paragraph will provide an overview of the most important methods for synthesizing HAs based on the nature of the starting material.
Figure 4.

Some examples of hydroxylamines among those mostly used in the literature for the synthesis of HAs. The simplest method to obtain HAs concerns the reaction with hydroxylamine after proper activation of the carboxylic group.
Starting from carboxylic acids, it is possible to obtain HAs using protected hydroxylamines following conventional coupling reactions in the presence of EDCI/NEt3 (Scheme 1). The subsequent deprotection of hydroxylamine depends on the nature of the protecting group. The silyl ethers are efficiently removed by treatment with 2–3 equiv of tetra-n-butylammonium fluoride (TBAF) in THF or TFA (25% in DCM)12 at room temperature, while O-benzyl hydroxamates can be deprotected by catalytic hydrogenation with quantitative yields. The trityl group is effectively removed in TFA, while hydroxamic acid, such as THP ether can be deprotected by treatment with catalytic TsOH in MeOH or with fluoride sources (selectfluor).13
Scheme 1. Carboxylic Acid Approach to HAs Using Protected Hydroxylamines.

Given the low reactivity of the free carboxylic moiety, an efficient method for obtaining HAs lies in the reaction via acyl chlorides, an extremely more reactive functional group (Scheme 2a). Subsequent substitution of the halogen with hydroxylamine (10 equiv) forms the amide bond rapidly. The attack of hydroxylamine occurs in anhydrous conditions, employing dry solvents, as residual water could hydrolyze the highly reactive acyl halide, and in the presence of a base (TEA, Pyridine) that traps the HCl formed during the process. The carboxylic acids can conveniently be converted into the corresponding acyl chlorides in high yields in the presence of thionyl chloride, tosyl chloride, oxalyl chloride, or cyanuric chloride.14 Among all functional groups that can be easily converted into hydroxamic acids, the most versatile reagents are esters (Scheme 2b). Given their stability, methyl and ethyl esters can be chosen as starting materials to obtain HAs. Methyl and ethyl esters react rather smoothly with aqueous hydroxylamine solutions in the presence of a base (KOH or NaOH) to provide the corresponding hydroxamates. Unfortunately, this methodology provides the corresponding carboxylic acids as main byproducts, even if in small amounts. One way to avoid this unpleasant event is to make the ester react with hydroxylamine hydrochloride in alcoholic solutions of KOH or an alcoholic solution of MeONa.15 Another convenient strategy is to use catalytic amounts of KCN to accelerate the reaction between ethyl ester and hydroxylamine in the absence of a base.16,17
Scheme 2. Reactions of Hydroxylamine with Acyl Chlorides and Esters.

HAs can also be obtained from amides. An excess of alkaline aqueous solution of hydroxylamine converts amides into HAs. With respect to the procedures based on the use of ester starting materials, such hydrolytic conditions have the advantage that the stability to hydrolysis of the amide function reduces the formation of side products. Another interesting option for obtaining HAs from amides lies in the use of bacterial amidases (Scheme 3). P. aeruginosa amidase efficiently hydrolyzes the amide bond at neutral pH, and in the presence of hydroxylamine, the corresponding carboxylic acid is converted into HA in high yields. Biocatalytic transformation of nicotinic amide, carried out exploiting the acyltransferase activity of Bacillus smithii strain IITR6b2, leads to the corresponding hydroxamic acid at neutral pH in 40 min, with an efficiency of 94%.11
Scheme 3. Biocatalytic Synthesis of Hydroxamic Acids from Amides.

Aldehyde represents another important functional group that can be easily converted into hydroxamic acid through different reactions (Scheme 4).11 The conversion of aromatic aldehydes via oxidation in the Na2N2O3 water solution rapidly yields the corresponding product. Another approach consists of a metal-free oxidative amidation of aldehydes, using N-hydroxysuccinimide and cobalt diacetate as the catalyst for electron transfer. Byproducts, such as carboxylic acids, have been observed, and this method was not appropriate for α,β-unsaturated aldehydes. Furthermore, aldehyde amidation via NHS ester formation, using a catalytic amount of Cu(OAc)2·H2O, tert-butyl hydrogen peroxide (70% aqueous solution) as an oxidant agent and acetonitrile as solvent was efficiently performed.18 A photochemical approach has been described by Papadopoulos et al., which considers a reaction of the aldehyde derivative with diisopropyl azodicarboxylate (DIAD) and phenylglyoxylic acid (10 mol %) as photocatalyst. After 2 h, hydroxylamine hydrochloride and triethylamine were added to obtain the corresponding hydroxamic acid in high yield and without using metal catalyst.19 Moreover, Porcheddu and collaborators20 described an alternative way to obtain hydroxamic acids using a novel solid-phase technique. The solid supported hydroxylamine reagent was prepared starting from a pyridine solution of hydroxylamine hydrochloride, which was stirred with polystyrene sulfonyl chloride at room temperature. The resin formed was suspended in THF and treated with a solution of the aldehyde starting material in the presence of MeONa in MeOH to afford the corresponding HA.20
Scheme 4. HAs Synthesis from Aldehydes.

4. Medicinal Chemistry Applications of HAs
4.1. HAs as Enzymatic Inhibitors
The ability of the hydroxamic acid moiety to efficiently coordinate metals has been widely exploited for the development of inhibitors targeting metal-bearing enzymes.21 In particular, considerable medicinal chemistry research efforts have been addressed for the identification of HA derivatives inhibiting human HDACs, a family of enzymes involved in the regulation of several biological pathways in cells, including tumor-related processes.22 In the past years, a plethora of structurally different HA compounds targeting HDAC isoforms have been reported, some of them undergoing clinical trials and entering also in therapeutic use (e.g., Panobinostat, Vorinostat, and Belinostat). Their activity mainly resides in the ability of HAs to coordinate the catalytic zinc ion in the enzyme binding site, as well as on the presence of specific structural decorations at the “linker” and “cap” regions driving isoform selectivity.23 The formation of HA-Zn(II) complexes generally occurs via a bidentate mechanism (Figure 5, panel a), although monodentate coordination was also observed in some crystal structures of inhibitors bearing a phenyl HA group.22,24
Figure 5.
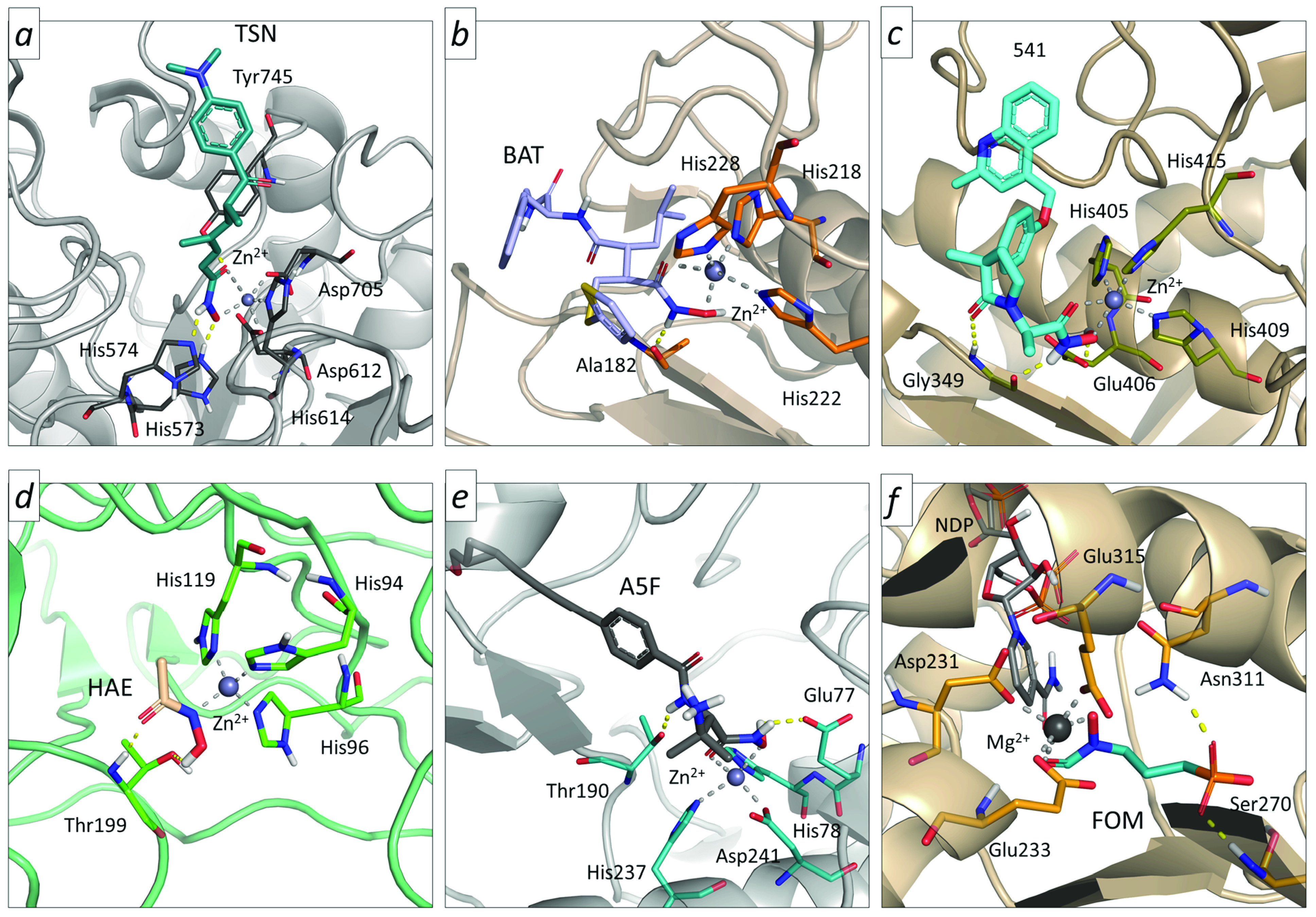
Binding mode of selected HA derivatives into HDAC6 (panel a, trichostatin – TSN, PDB code: 5WGI), MMP-12 (panel b, batimastat – BAT, PDB code: 1JK3), ADAM-17 (panel c, IK682–541, PDB code: 2FV5), hCAII (panel d, acetohydroxamic acid – HAE, PDB code: 1AM6), LpxC (panel e, A5F, PDB code: 6MOO), and DXR (panel f, fosmidomycin– FOM, PDB code: 3AU9).
HA derivatives have also been largely investigated for targeting the members of the MMPs family, which are zinc- and calcium-dependent endopeptidases participating in extracellular matrix remodeling.25 Indeed, considering that MMP aberrant activity is involved in the physiopathology of several disorders, including inflammation, arthritis, cancer, and brain diseases,25 drug development on these targets has been continuously pursued over the past decades, resulting in hundreds of inhibitors bearing the HA pharmacophore. Currently reported HA inhibitors bind to the Zn(II) in the catalytic cleft of MMPs via a bidentate coordination (Figure 5, panel b), resulting in binding site occlusion and thus inhibition of catalytic activity. Although most of the developed MMPs inhibitors present a HA metal binding group (MTG), none of them are currently used in therapeutic regiments, Marimastat and Batimastat reaching up to phase III clinical trials. Medicinal chemistry efforts have also been addressed for the development of HA derivates targeting the zinc-containing ADAMs (a disintegrin and metalloproteinase),21 which belong to the metzincin subfamily of adamalysins. Several small molecule inhibitors targeting ADAMs have been reported in recent years, which can potentially be used to treat cancer, Alzheimer’s disease, and rheumatoid arthritis. Indeed, ADAMs inhibitors bind to the Zn(II) in the enzyme active site via a bidentate coordination (Figure 5, panel c). Despite the initial promising expectations, none of the reported ADAMs HA inhibitors reached the market, mainly because of nonoptimal selectivity profiles. Research efforts have also been devoted to the design of HA inhibitors of 5-lipoxygenase (5-LO), which is a member of the lipoxygenase family acting as mediator of inflammatory immune responses. Such an enzyme requires Fe(III) for catalytic activity.21 Interestingly, HA inhibitors coordinating the Fe(III) ion of 5-LO present a “reverse” type of connectivity with respect to inhibitors of HDACs, ADAMs, and MMPs, the main scaffold of the compounds being attached to the nitrogen atom of the hydroxamic moiety. Several HA and hydroxyurea derivatives have been reported for this target, with Zileuton being approved for the treatment of chronic asthma.
HAs have been considerably less explored in other targets. An example of this comes from human carbonic anhydrases (hCA), a family of zinc metalloenzymes that includes more than 12 isoforms participating to the regulation of various physiological processes in cells.26 The development of isoform-selective hCA inhibitors has gained considerable interest to treat glaucoma and more recently cancer. Interestingly, results of a crystallographic study revealed that the acetohydroxamic acid binds to hCAII through a monodentate coordination, with the nitrogen atom of the hydroxamic acid interacting with Zn(II) in deprotonated form (Figure 5, panel d).27 Moreover, the utility of HA derivatives can be extended to bacterial targets. In particular, HAs have been extensively studied to target the UDP-3-O-Acyl-N-acetylglucosamine deacetylase (LpxC), which is a Zn-dependent deacetylase essential for the lipid A biosynthesis in Gram-negative bacteria. LpxC has received increased attention for the development of broad-spectrum antibacterial drugs, with this target being highly conserved among a number of bacteria species. Interestingly, the majority of reported LpxC inhibitors bear a hydroxamic acid moiety (e.g., the clinical candidate ACHN-975) binding to the catalytic zinc through a bidentate coordination (Figure 5, panel e), while other structural decorations were introduced in their scaffolds to obtain a broader spectrum of activity.21 HA derivatives have also been reported to target DXR (1-deoxy-d-xylulose 5-phosphate reductoisomerase), which is an enzyme playing a central role in the synthesis of isoprenoids for several pathogenic bacteria and Plasmodium falciparum. Such a target requires NADPH (nicotinamide adenine dinucleotide phosphate) and two Mg2+ or Mn2+ ions to exert catalytic activity, even though the mechanism by which it converts DXP (1-deoxy-d-xylulose 5-phosphate) to MEP (2-C-methyl-d-erythritol-4-phosphate) has yet to be completely understood. Most reported DXR inhibitors present the hydroxamic acid group with a “reverse” type of connectivity, or it is framed into ring systems (e.g., FR900098 and DXRi-4, respectively).17 Crystallographic experiments on fosmidomycin revealed that the hydroxamate group coordinates the Mg2+ (or Mn2+) cation of DXR in a bidentate fashion (Figure 5, panel f), as done by HA derivates with canonical connectivity. A considerable number of “reverse” HA derivatives have been reported over the past decades, but none of them have been marketed. In 2018, Nikolaou et al. also reported the development of hydroxamic acids as Autotaxin (ATX) inhibitors.28 ATX is a family member of ectonucleotide pyrophosphatases/phosphodiesterases with the ability to catalyze the hydrolysis of pyrophosphate or phosphodiester bonds in nucleotides, with implications in several chronic inflammatory conditions, such as arthritis and pulmonary fibrosis, and also in cancer.
4.2. Mutagenicity of HAs
The safety profile is one of the main issues to consider during the development of HAs. Indeed, compounds bearing the hydroxamic acid moiety may show mutagenic properties in vitro, due to their interactions with DNA.29 The most widely accepted hypothesis for the mutagenicity of hydroxamic acids involves the transformation of hydroxamate into the respective isocyanate by Lossen rearrangement,30 and intramolecular nucleophilic 1,2-shift (Figure 6). Studies conducted on bacterial acetyl-CoA activity have highlighted how hydroxamic acids can be activated into their respective O-acetyl derivatives, under physiological conditions. Furthermore, the presence of Zn(II) should mediate the dehydration of hydroxamic acids with the formation of an unsaturated complex with an anionic acetohydroxamate, water, and acetylnitrene. Then, nitrene undergoes rearrangement to the corresponding isocyanate.
Figure 6.

Lossen rearrangement.
To prevent the mutagenicity issues associated with hydroxamate function, alternative ZBGs, such as carboxylic acid, thiol, o-aminoanilide, 2-mercaptoacetamides, oxadiazoles, and trifluoromethyloxadiazoles, are currently under investigation (Figure 7). However, the mutagenicity of hydroxamic acid derivatives depends on the nature of associated acyl residue. For example, Vorinostat was endowed with a weak mutagenic property, with the absence of chromosome aberrations in human lymphocytes.
Figure 7.

Most common HAs bioisosters.
5. Conclusions
Hydroxamic acid is one of the most interesting moieties in medicinal chemistry, due to its ability to chelate metal ions, such as Fe(III) and Zn(II). This ability is exploited for the development of various compounds able to inhibit different metalloenzymes, such as HDACs, CAs, MMPs, ADAMs, etc. In order to develop HA-based compounds, several synthetic approaches have been developed, starting from different functional groups, such as carboxylic acids, amides, acyl chlorides, esters, and aldehydes. Their mutagenicity appears to be modulated by decoration of the HA and/or substitution with HA bioisosters. Importantly, some HA derivatives have reached the market and others are currently under clinical evaluation, demonstrating their key role in drug discovery.
Acknowledgments
This work was supported by a grant from the Associazione Italiana per la Ricerca sul Cancro [AIRC IG 23635].
Author Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
References
- Muri E. M.; Nieto M. J.; Sindelar R. D.; Williamson J. S. Hydroxamic acids as pharmacological agents. Curr. Med. Chem. 2002, 9 (17), 1631–1653. 10.2174/0929867023369402. [DOI] [PubMed] [Google Scholar]
- Millar A. W.; Brown P. D.; Moore J.; Galloway W. A.; Cornish A. G.; Lenehan T. J.; Lynch K. P. Results of single and repeat dose studies of the oral matrix metalloproteinase inhibitor marimastat in healthy male volunteers. Br. J. Clin. Pharmacol. 1998, 45 (1), 21–26. 10.1046/j.1365-2125.1998.00639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann K.; Picciotti M.; Spevack T.; Durbin D. Management of acute iron overdose. Clin. Pharm. 1989, 8 (6), 428–440. [PubMed] [Google Scholar]
- Zhao C.; Dong H.; Xu Q.; Zhang Y. Histone deacetylase (HDAC) inhibitors in cancer: a patent review (2017-present). Expert Opin. Ther. Pat. 2020, 30 (4), 263–274. 10.1080/13543776.2020.1725470. [DOI] [PubMed] [Google Scholar]
- Keth J.; Johann T.; Frey H. Hydroxamic acid: an underrated moiety? Marrying bioinorganic chemistry and polymer science. Biomacromolecules 2020, 21 (7), 2546–2556. 10.1021/acs.biomac.0c00449. [DOI] [PubMed] [Google Scholar]
- Kakkar R.Theoretical studies on hydroxamic acids. In Hydroxamic Acids, Gupta S. P., Ed; Springer Verlag: Heidelberg, 2013; pp 19–53. [Google Scholar]
- Fazary A. E. Thermodynamic studies on the protonation equilibria of some hydroxamic acids in NaNO3 solutions in water and in mixtures of water and dioxane. J. Chem. Eng. Data 2005, 50 (3), 888–895. 10.1021/je0496185. [DOI] [Google Scholar]
- Kurzak B.; Kozłowski H.; Farkas E. Hydroxamic and aminohydroxamic acids and their complexes with metal ions. Coord. Chem. Rev. 1992, 114 (2), 169–200. 10.1016/0010-8545(92)85002-8. [DOI] [Google Scholar]
- Neilands J. Siderophores: structure and function of microbial iron transport compounds. J. Biol. Chem. 1995, 270 (45), 26723–26726. 10.1074/jbc.270.45.26723. [DOI] [PubMed] [Google Scholar]
- Griffith D. M.; Szőcs B.; Keogh T.; Suponitsky K. Y.; Farkas E.; Buglyó P.; Marmion C. J. Suberoylanilide hydroxamic acid, a potent histone deacetylase inhibitor; its X-ray crystal structure and solid state and solution studies of its Zn (II), Ni (II), Cu (II) and Fe (III) complexes. J. Inorg. Biochem. 2011, 105 (6), 763–769. 10.1016/j.jinorgbio.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Ganeshpurkar A.; Kumar D.; Singh S. K. Strategies for the synthesis of hydroxamic acids. Curr. Org. Synth. 2018, 15 (2), 154–165. 10.2174/1570179414666170614123508. [DOI] [Google Scholar]
- Zagni C.; Citarella A.; Oussama M.; Rescifina A.; Maugeri A.; Navarra M.; Scala A.; Piperno A.; Micale N. Hydroxamic acid-based Histone Deacetylase (HDAC) inhibitors bearing a pyrazole scaffold and a cinnamoyl linker. Int. J. Mol. Sci. 2019, 20 (4), 945. 10.3390/ijms20040945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Wong C.-H. An efficient method for the cleavage of p-methoxybenzylidene (PMP), tetrahydropyranyl (THP) and 1, 3-dithiane protecting groups by Selectfluor. Tetrahedron Lett. 2002, 43 (22), 4037–4039. 10.1016/S0040-4039(02)00740-2. [DOI] [Google Scholar]
- Giacomelli G.; Porcheddu A.; Salaris M. Simple one-flask method for the preparation of hydroxamic acids. Org. Lett. 2003, 5 (15), 2715–2717. 10.1021/ol034903j. [DOI] [PubMed] [Google Scholar]
- Giacomini E.; Nebbioso A.; Ciotta A.; Ianni C.; Falchi F.; Roberti M.; Tolomeo M.; Grimaudo S.; Cristina A. D.; Pipitone R. M. Novel antiproliferative chimeric compounds with marked histone deacetylase inhibitory activity. ACS Med. Chem. Lett. 2014, 5 (9), 973–978. 10.1021/ml5000959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Géraldy M.; Morgen M.; Sehr P.; Steimbach R. R.; Moi D.; Ridinger J.; Oehme I.; Witt O.; Malz M.; Nogueira M. S.; Koch O.; Gunkel N.; Miller A. K. Selective inhibition of Histone Deacetylase 10: Hydrogen bonding to the gatekeeper residue is implicated. J. Med. Chem. 2019, 62 (9), 4426–4443. 10.1021/acs.jmedchem.8b01936. [DOI] [PubMed] [Google Scholar]
- Ho C. Y.; Strobel E.; Ralbovsky J.; Galemmo R. A. Improved solution- and solid-phase preparation of hydroxamic acids from esters. J. Org. Chem. 2005, 70 (12), 4873–4875. 10.1021/jo050036f. [DOI] [PubMed] [Google Scholar]
- Pilo M.; Porcheddu A.; De Luca L. A copper-catalysed amidation of aldehydes via N-hydroxysuccinimide ester formation. Org. Biomol. Chem. 2013, 11 (47), 8241–8246. 10.1039/c3ob41440j. [DOI] [PubMed] [Google Scholar]
- Papadopoulos G. N.; Kokotos C. G. Photoorganocatalytic one-pot synthesis of hydroxamic acids from aldehydes. Chem. - Eur. J. 2016, 22 (20), 6964–6967. 10.1002/chem.201600333. [DOI] [PubMed] [Google Scholar]
- Porcheddu A.; Giacomelli G. Angeli– Rimini’s reaction on solid support: A new approach to hydroxamic acids. J. Org. Chem. 2006, 71 (18), 7057–7059. 10.1021/jo061018g. [DOI] [PubMed] [Google Scholar]
- Chen A. Y.; Adamek R. N.; Dick B. L.; Credille C. V.; Morrison C. N.; Cohen S. M. Targeting metalloenzymes for therapeutic intervention. Chem. Rev. 2019, 119 (2), 1323–1455. 10.1021/acs.chemrev.8b00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho T. C. S.; Chan A. H. Y.; Ganesan A. Thirty years of HDAC inhibitors: 2020 insight and hindsight. J. Med. Chem. 2020, 63 (21), 12460–12484. 10.1021/acs.jmedchem.0c00830. [DOI] [PubMed] [Google Scholar]
- Linciano P.; Benedetti R.; Pinzi L.; Russo F.; Chianese U.; Sorbi C.; Altucci L.; Rastelli G.; Brasili L.; Franchini S. Investigation of the effect of different linker chemotypes on the inhibition of histone deacetylases (HDACs). Bioorg. Chem. 2021, 106, 104462. 10.1016/j.bioorg.2020.104462. [DOI] [PubMed] [Google Scholar]
- Porter N. J.; Mahendran A.; Breslow R.; Christianson D. W. Unusual zinc-binding mode of HDAC6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (51), 13459–13464. 10.1073/pnas.1718823114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker M.; Floyd C. D.; Brown P.; Gearing A. J. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 1999, 99 (9), 2735–76. 10.1021/cr9804543. [DOI] [PubMed] [Google Scholar]
- Alterio V.; Di Fiore A.; D’Ambrosio K.; Supuran C. T.; De Simone G. Multiple binding modes of inhibitors to Carbonic Anhydrases: How to design specific drugs targeting 15 different isoforms?. Chem. Rev. 2012, 112 (8), 4421–4468. 10.1021/cr200176r. [DOI] [PubMed] [Google Scholar]
- Scolnick L. R.; Clements A. M.; Liao J.; Crenshaw L.; Hellberg M.; May J.; Dean T. R.; Christianson D. W. Novel binding mode of hydroxamate inhibitors to human Carbonic Anhydrase II. J. Am. Chem. Soc. 1997, 119 (4), 850–851. 10.1021/ja963832z. [DOI] [Google Scholar]
- Nikolaou A.; Ninou I.; Kokotou M. G.; Kaffe E.; Afantitis A.; Aidinis V.; Kokotos G. Hydroxamic acids constitute a novel class of autotaxin inhibitors that exhibit in vivo efficacy in a pulmonary fibrosis model. J. Med. Chem. 2018, 61 (8), 3697–3711. 10.1021/acs.jmedchem.8b00232. [DOI] [PubMed] [Google Scholar]
- Shen S.; Kozikowski A. P. Why hydroxamates may not be the best histone deacetylase inhibitors—what some may have forgotten or would rather forget?. ChemMedChem 2016, 11 (1), 15. 10.1002/cmdc.201500486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M.; Alsarraf J.; Araji N.; Tranoy-Opalinski I.; Renoux B.; Papot S. The Lossen rearrangement from free hydroxamic acids. Org. Biomol. Chem. 2019, 17 (22), 5420–5427. 10.1039/C9OB00789J. [DOI] [PubMed] [Google Scholar]