

Abstract
Cell-to-cell transmission of α-synuclein (α-syn) pathology is considered to underlie the spread of neurodegeneration in Parkinson's disease (PD). Previous studies have demonstrated that α-syn is secreted under physiological conditions in neuronal cell lines and primary neurons. However, the molecular mechanisms that regulate extracellular α-syn secretion remain unclear. In this study, we found that inhibition of monoamine oxidase-B (MAO-B) enzymatic activity facilitated α-syn secretion in human neuroblastoma SH-SY5Y cells. Both inhibition of MAO-B by selegiline or rasagiline and siRNA-mediated knock-down of MAO-B facilitated α-syn secretion. However, TVP-1022, the S-isomer of rasagiline that is 1000 times less active, failed to facilitate α-syn secretion. Additionally, the MAO-B inhibition-induced increase in α-syn secretion was unaffected by brefeldin A, which inhibits endoplasmic reticulum (ER)/Golgi transport, but was blocked by probenecid and glyburide, which inhibit ATP-binding cassette (ABC) transporter function. MAO-B inhibition preferentially facilitated the secretion of detergent-insoluble α-syn protein and decreased its intracellular accumulation under chloroquine-induced lysosomal dysfunction. Moreover, in a rat model (male Sprague Dawley rats) generated by injecting recombinant adeno-associated virus (rAAV)-A53T α-syn, subcutaneous administration of selegiline delayed the striatal formation of Ser129-phosphorylated α-syn aggregates, and mitigated loss of nigrostriatal dopaminergic neurons. Selegiline also delayed α-syn aggregation and dopaminergic neuronal loss in a cell-to-cell transmission rat model (male Sprague Dawley rats) generated by injecting rAAV-wild-type α-syn and externally inoculating α-syn fibrils into the striatum. These findings suggest that MAO-B inhibition modulates the intracellular clearance of detergent-insoluble α-syn via the ABC transporter-mediated non-classical secretion pathway, and temporarily suppresses the formation and transmission of α-syn aggregates.
SIGNIFICANCE STATEMENT The identification of a neuroprotective agent that slows or stops the progression of motor impairments is required to treat Parkinson's disease (PD). The process of α-synuclein (α-syn) aggregation is thought to underlie neurodegeneration in PD. Here, we demonstrated that pharmacological inhibition or knock-down of monoamine oxidase-B (MAO-B) in SH-SY5Y cells facilitated α-syn secretion via a non-classical pathway involving an ATP-binding cassette (ABC) transporter. MAO-B inhibition preferentially facilitated secretion of detergent-insoluble α-syn protein and reduced its intracellular accumulation under chloroquine-induced lysosomal dysfunction. Additionally, MAO-B inhibition by selegiline protected A53T α-syn-induced nigrostriatal dopaminergic neuronal loss and suppressed the formation and cell-to-cell transmission of α-syn aggregates in rat models. We therefore propose a new function of MAO-B inhibition that modulates α-syn secretion and aggregation.
Keywords: ABC transporter, aggregation, α-synuclein, monoamine oxidase, Parkinson's disease, secretion
Introduction
In individuals with Parkinson's disease (PD), the administration of levodopa improves motor impairments and allows patients to engage in independent activities for longer periods of time than those who do not receive this treatment (Fahn et al., 2004; Poewe et al., 2010). However, the motor abilities of patients with PD eventually undergo severe deterioration (Cilia et al., 2014). Thus, the identification of a neuroprotective agent that slows or stops the progression of motor impairments in PD is urgently needed. Motor impairments in PD are caused by a loss of dopaminergic neurons in the substantia nigra, which is the pathologic hallmark of this disease. PD is also pathologically characterized by the presence of intracytoplasmic proteinaceous inclusions, known as Lewy bodies (LBs) and Lewy neurites (Eriksen et al., 2003; Lee and Trojanowski, 2006). These inclusions are primarily composed of fibrillar α-synuclein (α-syn) aggregates (Spillantini et al., 1997). In PD, cell-to-cell transmission of α-syn aggregates is considered to underlie the spread of neuronal damage throughout the brain (Goedert et al., 2017; Steiner et al., 2018). Accumulating data indicate that α-syn can transfer between cells both in vitro and in vivo. Two hypotheses have been raised to explain the cell-to-cell transmission of α-syn. The first proposes that α-syn fibrils are transported by the intercellular trafficking of lysosomes via tunneling nanotubes (Abounit et al., 2016). The second hypothesizes that α-syn is secreted into the extracellular space as exosomes or free proteins via a non-classical pathway, independently of endoplasmic reticulum (ER)/Golgi transport (Jang et al., 2010). The presence of endogenous α-syn protein in human cerebrospinal fluid and murine interstitial fluid supports the physiological secretion of α-syn into the extracellular space (Borghi et al., 2000; Yamada and Iwatsubo, 2018). Extracellular α-syn plays a key role in the cell-to-cell transmission of its aggregates. However, it is unclear how α-syn secretion is regulated. It is also unknown whether the manipulation of this process can reduce the cell-to-cell transmission of α-syn and neurodegeneration.
Monoamine oxidase (MAO)-A and MAO-B are enzymes that catalyze the oxidative deamination of monoamines in the outer mitochondrial membrane (Shih, 2004). Although MAO-B is mainly distributed in astrocytes, it is also expressed in dopaminergic neurons (Kang et al., 2018). In positron emission tomography, MAO-B expression increases slightly with aging in healthy individuals (Fowler et al., 1997). Additionally, MAO-B levels are higher in the brains of PD patients than in those of healthy controls (Kang et al., 2018). Previous studies have also demonstrated a pathologic link between α-syn and MAO-B via the generation of the MAO-B enzymatic DA metabolite, 3,4-dihydroxyphenylacetaldehyde (DOPAL), in PD (Burke et al., 2008). Increased MAO levels promote the conversion of DA to DOPAL, leading to the production of ROS through the oxidation of DOPAL to its quinone derivative. DOPAL and its quinone derivative then stimulate α-syn oligomerization under oxidative conditions.
In this study, we demonstrated that the inhibition of MAO-B enzymatic activity facilitated α-syn secretion into the extracellular space via the non-classical pathway in dopaminergic SH-SY5Y cells. The increased secretion of α-syn caused by MAO-B inhibitors, selegiline and rasagiline, was able to be blocked by probenecid, glyburide, and reserpine, which inhibit ATP-binding cassette (ABC) transporters (Andrei et al., 1999; Zhou et al., 2001; Flieger et al., 2003; Loebinger et al., 2008; Frye et al., 2009; Liu et al., 2014). Furthermore, SH-SY5Y cells treated with MAO-B inhibitor preferentially secreted detergent-insoluble α-syn protein with reduction in its intracellular accumulation under condition of lysosomal dysfunction induced by chloroquine. We also revealed that the inhibition of MAO-B by selegiline delayed the formation and cell-to-cell transmission of α-syn aggregates in rat models by recombinant adeno-associated virus (rAAV)-mediated expression of α-syn.
Materials and Methods
Cell line, chemical treatments, siRNA transfection, and LDH assay
SH-SY5Y cell line stably expressing wild-type α-syn (wt-aS/SH #4) was maintained as described previously (Machiya et al., 2010). Reagents were purchased from Sigma unless otherwise stated. Selegiline was provided from FP Pharmaceutical Corporation. Rasagiline mesylate and TVP-1022 were purchased from Abcam and MedChemExpress, respectively. To assess effects of MAO-B inhibition on the α-syn expression, parental SH-SY5Y cells were treated with selegiline for 24 h. Also, parental SH-SY5Y cells were treated with 100 μm cycloheximide (CHX) in the absence or presence of selegiline for 24 h (Machiya et al., 2010). For ER/Golgi transport inhibition, wt-aS/SH cells were treated with 1 mm MAO-B inhibitor (selegiline or rasagiline) and 7 μm brefeldin A for 6 h. For ABC transporter inhibition, wt-aS/SH cells were treated with 1 mm MAO-B inhibitor and 10 μm probenecid for 24 h. Treatment with reserpine was performed for 24 h or 48 h. Also, the cells were preincubated in medium containing 5 μm glyburide for 24 h, and then incubated in medium containing 5 μm glyburide plus 1 mm MAO-B inhibitor for 24 h. For lysosome inhibition, wt-aS/SH cells were treated with 1 mm MAO-B inhibitor and 100 μm chloroquine for 48 h. In all experiments, cells were treated with the same volume of vehicle as a control.
For siRNA transfection, ∼20% confluent cells in 6-well plates were transfected with siRNA oligonucleotides (final concentration at 10 nm, Silencer Select RNAi, Thermo Fisher Scientific) using RNAiMAX reagent (Thermo Fisher Scientific) according to the manufacturer's protocol. We used 21 nucleotide-long siRNAs for knock-down of MAO-A (5′-GGACCAACCCAAAACAGAAtt-3′) or MAO-B (5′-GGACCAACCCAGAAUCGUAtt-3′). As a non-silencing control, the Silencer Select Negative Control #1 siRNA was used. At 72 h after transfection, the medium was discarded, and the cells were re-transfected with siRNA. At 48 h after the second transfection, the cells were used for chemical treatments. In siRNA transfection, cell viability was assessed using Cell Counting Kit-8 (Dojindo Molecular Technologies) according to the manufacturer's protocol.
To assess cell membrane damage, LDH assay was performed using the Cytotoxicity LDH Assay Kit-WST (Dojindo Molecular Technologies). Cells were treated with the same conditions as the corresponding experiments. As a positive control, lysis buffer containing Triton X-100 was added to cells.
Preparation of cell lysates and conditioned media (CMs)
For preparation of cell lysates, cells were suspended in buffer A [20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, 10% glycerol, 1 × protease inhibitor cocktail (Roche Diagnostic), 1 mm EDTA, 5 mm NaF, 1 mm Na3VO4, 1 × phosSTOP (Roche Diagnostic)], sonicated at 30W for 1 s 5 times, and kept on ice for 30 min (Machiya et al., 2010). After centrifugation at 12,000 × g for 30 min at 4°C, the resulting supernatant was collected and stored at −80°C until use. For preparation of CM, wt-aS/SH cells were replaced from growth media to Opti-MEM (Thermo Fisher Scientific). To perform TCA-precipitation, collected CM was centrifuged at 6000 × g for 5 min to remove cell debris. Immediately, CM was added with 1/4 volume of 100% TCA, incubated for 30 min on ice, and centrifuged at 14,000 × g for 5 min. The pellet was washed three times with 300 µl of cold acetone, air dried, and dissolved in 100 µl of Laemmli's sample buffer containing 2.5% β-mercaptoethanol.
Fractionation of cells and CMs
Cells were sonicated on ice in buffer A and centrifuged at 100,000 × g for 30 min at 4°C (Arawaka et al., 2017). The supernatant was collected as 1% Triton X-100 soluble fraction. After washing pellets with buffer A, resultant pellets were dissolved in the equal aliquot of the solution containing 8 m urea and 2% SDS and centrifuged at 100,000 × g for 30 min. The supernatant was collected as 1% Triton X-100 insoluble fraction. To separate secreted proteins into 1% Triton X-100 soluble and insoluble fractions, TCA-precipitated pellet was sequentially extracted with buffer A and the solution containing 8 m urea and 2% SDS according to the same protocol as described above.
Western blotting
Equal amounts of denatured samples were subjected to SDS-PAGE on 13.5% polyacrylamide gels and then transferred to PVDF membranes (Millipore) as described previously (Sasaki et al., 2015). Briefly, the transferred membrane was incubated in PBS (10 mm phosphate, 137 mm NaCl, 2.7 mm KCl) containing 4% paraformaldehyde (PFA). After incubation, the membrane was washed in Tris-buffered saline (TBS; 25 mm Tris-HCl, pH 7.4, 137 mm NaCl, and 2.7 mm KCl) containing 0.05% (v/v) Tween 20 (TBS-T) for 10 min 3 times. The membrane was blocked by TBS-T containing 5% skim milk for 30 min, incubated in TBS-T containing 2.5% skim milk and primary antibody overnight at 4°C, and further incubated in the same buffer containing the corresponding secondary antibody overnight at 4°C. To visualize the signal, membranes were treated with ECL plus (Thermo Fisher Scientific) for detection of insoluble α-syn protein of cell lysates and α-syn of CM. In other signals, membranes were treated with ECL (Thermo Fisher Scientific). Signals were recorded using a CCD camera, VersaDog 5000 (Bio-Rad) or Fusion FX7 (Vilbert Lourmat). Levels of α-syn were estimated by semiquantifying band intensities with Quantity One software (Bio-Rad). The following antibodies were used: anti-α-syn (1:2500, Syn-1, BD Transduction Laboratories), anti-β-actin (1:10,000, AC-15, Sigma), anti-MAO-A (1:5000, EPR7101, Abcam) and anti-MAO-B (1:5000, EPR7102, Abcam) antibodies.
Rat rAAV-mediated α-syn expression and transmission models
Experiments using rats had been approved by the Animal Subjects Committees of Yamagata University (#27075) and Osaka Medical and Pharmaceutical University (#21002-A). We unilaterally injected 1.36 × 109 genome copies of the rAAV2-familial PD-linked A53T human α-syn vector into the substantia nigra of nine-week-old male Sprague Dawley rats as described previously (Sato et al., 2011).
Recombinant α-syn proteins were purified as described previously (Nonaka et al., 2005). To obtain α-syn preformed fibrils (PFF), purified α-syn proteins were incubated with agitation at 37°C in 30 mm Tris-HCl buffer (pH 7.4) for 18 d. Fibril formation was monitored by thioflavin-T assay. PFF were collected by centrifugation at 300,000 × g for 1 h followed by dilution in the sterilized Tris-HCl buffer with brief sonication. We unilaterally injected 1.09 × 109 genome copies of the rAAV2-wild-type α-syn vector into the substantia nigra of nine-week-old male Sprague Dawley rats using Hamilton Neuro syringe. After two weeks of rAAV injection, the solution containing 10 µg of PFF was injected into the ipsilateral striatum at the following coordinates: 0.2 mm posterior to and 3.3 mm left of bregma and 4.7 mm ventral to dura.
Immunohistochemistry and immunofluorescence
Immunohistochemical staining was performed on free-floating sections as described previously (Sato et al., 2011). Briefly, rats were transcardially perfused with physiological saline followed by 4% PFA/PBS. Their brains were removed and postfixed in 8% PFA/PBS containing 4% sucrose for 48 h. The brains were equilibrated stepwise with 7.5%, 15%, and 30% sucrose. These brains were coronally sectioned on a freezing microtome at a thickness of 30 µm. Sections were collected in ten series to be regularly spaced at intervals of 300 µm from each other. For immunohistochemical staining, sections were permeabilized with 0.1% Triton X-100/PBS for 10 min and treated with 0.3% hydrogen peroxide for 5 min. After blocking in 3% normal goat serum for 30 min, the sections were incubated with the primary antibody with gentle shaking at 4°C overnight. After washing, the sections were incubated with the appropriate biotinylated secondary antibody for 1 h, followed by avidin-biotin-peroxidase complex (Vector Laboratories) for 1 h. The sections were visualized using 3,3′-diaminobenzidine. The following primary antibodies were used: anti-human α-syn (1:200, LB509, Thermo Fisher Scientific), anti-TH (1:2000, AB152, Millipore), anti-dopamine transporter (DAT; 1:2000, sc-1433, Santa Cruz Biotechnology), anti-GFP (1:500, A11122, Thermo Fisher Scientific), and anti-Ser129-phosphorylated (pS129) α-syn (1:200, EYPSYN-01, courtesy of Eisai) antibodies.
For immunofluorescent staining, PFA-fixed sections were blocked with TBS-T containing 5% skim milk for 1 h, and then incubated with a mixture of anti-TH (1:400, LNC1, mouse monoclonal, Cell Signaling Technology) and anti-pS129 α-syn (1:500, rabbit monoclonal, EP1536Y, Abcam) antibodies overnight at 4°C. After washing, sections were incubated with a mixture of Alexa Fluor 488 donkey anti-mouse IgG (H + L) (1:500, Jackson ImmunoResearch) and Alexa Fluor 594 donkey anti-rabbit IgG (H + L) (1:500, Jackson ImmunoResearch) for 2 h at RT. Images were acquired on laser-scanning confocal microscope (TCS SP8, Leica).
Assessment of nigral cell number and striatal optical density
To count nigral TH-positive neurons, we used sections covering the entire substantia nigra from caudal (bregma ∼−4.60 mm) to rostral (bregma ∼−6.60 mm) boundaries according to the rat brain atlas (Sato et al., 2011; Arawaka et al., 2017). This contained six to seven serial sections for each animal. After immunostaining, an unbiased stereological estimation was performed by an optical fractionator method using Stereo Investigator software (MicroBrightField). The region of interest was traced and sampled using an Olympus BX50 microscope at magnifications of 4× and 20×, respectively. The counting parameters were the x-y sampling grid size (180 × 180 µm), the counting frame size (160 × 160 µm), the dissector height (6 µm), and the guard-zone thickness (3 µm). The obtained counts were verified to archive a Gunderson's coefficient of error <0.10.
To assess striatal dopaminergic nerve fibers, we measured their optical densities (Sato et al., 2011; Arawaka et al., 2017). After immunostaining, the nearest section to bregma was determined according to the rat brain atlas. We analyzed nine sections from ∼+1.2 to ∼–1.2 mm relative to bregma. The optical densities of immunoreactive fibers were measured using ImageJ software (version 1.45s, National Institutes of Health). Nonspecific background intensities were simultaneously assessed by measuring the optical densities of cerebral cortical areas and were subtracted from the total values.
Assessment of striatal Ser129-phosphorylated α-syn-positive aggregates
To assess α-syn aggregates, we counted the number of pS129 α-syn-positive aggregates in the striatum (Sato et al., 2011; Arawaka et al., 2017). In sections stained with anti-pS129 α-syn antibodies, we analyzed the injected sides of three sections, including +0.3, 0, and –0.3 mm relative to bregma, by the optical fractionator method using Stereo Investigator software. The region of interest was traced and sampled using an Olympus BX50 microscope at magnifications of 4× and 10×, respectively. By setting the x-y sampling grid size equal to the counting frame size (330 × 330 µm), we scanned the whole area of the striatum on the section. We counted dot-like, rod-like or round structures positive for anti-pS129 α-syn antibodies. These structures were morphologically defined as axonal swellings and dystrophic terminals.
Cylinder test and measurement of striatal DA contents
At eight weeks after viral injection, cylinder test was performed as described previously (Oueslati et al., 2013; Boix et al., 2015). The number of wall contacts was counted by one observer blinded to the treatment given and one non-blinded observer. Rats failing to reach 10 contacts were excluded from the analyses (Ztaou et al., 2016). The striatal DA contents were measured as described previously (Kasai et al., 2017). The left (lesioned side) or right (intact side) striatum was individually homogenized in 0.2 m perchloric acid containing 100 pg/µl isoproterenol as an internal standard. The homogenates were kept on ice for 30 min followed by centrifugation at 15,000 × g for 20 min at 4°C, and each supernatant was filtrated through a 0.45-µm filter membrane. The supernatants were stored at −80°C until measurement. The DA content was measured in a high-performance liquid chromatography-electrochemical detection system (Eicom Corp.). Each sample was injected into a C18 reverse-phase column (Eicompak SC-5ODS) conditioned at 25°C. The mobile phase comprising 0.1 m acetic acid-citric acid buffer (pH 3.5), 15% (v/v) methanol, 190 mg/l sodium 1-octanesulfonate, and 5 mg/l ethylenediaminetetraacetic acid was delivered at a flow rate of 0.5 ml/min. The applied potential was set at +750 mV versus Ag/AgCl. The content of monoamines and their metabolites was calculated using standard curves.
Statistics
Statistical analysis was performed using the SPSS software (version 17, IBM). Comparison of two groups was performed by unpaired t test. Multiple comparisons above three groups were performed by one-way or two-way ANOVA with Bonferroni's post hoc test, when the variances were homogenous. In case of non-homogeneity of variances, comparisons were performed by Welch-ANOVA with Games–Howell post hoc test. To analyze the data of pS129 α-syn aggregates by two-way ANOVA, they were log-transformed to achieve homogeneity of variance. Ratios of the striatal DA contents in the intact and lesioned sides were analyzed by one-way ANOVA with Tukey's post hoc test. Data are expressed as mean ± SD; p < 0.05 was considered statistically significant.
Results
Inhibition of MAO-B enzymatic activity modulates α-syn secretion in SH-SY5Y cells
To explore the effects of MAO-B on α-syn metabolism in cells, we investigated whether MAO-B inhibition affected the expression of α-syn protein. In parental SH-SY5Y cells, treatment with MAO-B inhibitor selegiline did not alter the intracellular levels of endogenous α-syn (Fig. 1A). However, when the effects of selegiline on the metabolic fate of α-syn were assessed using a CHX-chase experiment, treatment with CHX for 24 h reduced the levels of α-syn protein to 0.67 ± 0.15-fold compared with those in untreated cells (p < 0.001), and the α-syn levels were further reduced by the addition of 1 μm (0.44 ± 0.14-fold reduction, p = 0.022) or 1 mm (0.39 ± 0.16-fold reduction, p = 0.003) of selegiline (Fig. 1B). These findings suggest that selegiline facilitates the intracellular clearance of α-syn, and that this effect is weaker than that of α-syn protein synthesis. To test the possibility that selegiline might affect α-syn secretion in cells, we next examined α-syn protein levels in CM using wt-aS/SH cells. Treatment with selegiline for 24 h increased the extracellular levels of α-syn to 2.24 ± 0.51-fold at 1 nm (p = 0.016), 3.25 ± 0.51-fold at 1 μm (p < 0.001), and 7.32 ± 0.34-fold at 1 mm (p < 0.001; Fig. 1C). β-Actin signals were not detected in CM, and treatment with selegiline did not alter extracellular LDH release (Fig. 1D). These findings indicate that α-syn signals in CM are not caused by contamination of intracellular proteins or cell membrane damage. Similar to selegiline, treatment with a different MAO-B inhibitor, rasagiline, significantly increased the extracellular levels of α-syn to 2.08 ± 0.60-fold at 1 μm (p = 0.008) and 2.69 ± 0.69-fold at 1 mm (p < 0.001; Fig. 1E). In contrast, treatment with TVP-1022, the relatively inactive (∼1000 times less active than rasagiline) S-isomer of rasagiline, failed to increase α-syn secretion (Youdim et al., 2001; Fig. 1F). Rasagiline and TVP-1022 did not affect extracellular LDH release (Fig. 1G). siRNA-mediated knock-down of MAO-B significantly increased α-syn secretion to 1.62 ± 0.34-fold (p = 0.014), while knock-down of MAO-A had no such effect (Fig. 1H). MAO-B knock-down did not affect cell viability. These findings indicate that the inhibition of MAO-B enzymatic activity modulates α-syn secretion.
Figure 1.
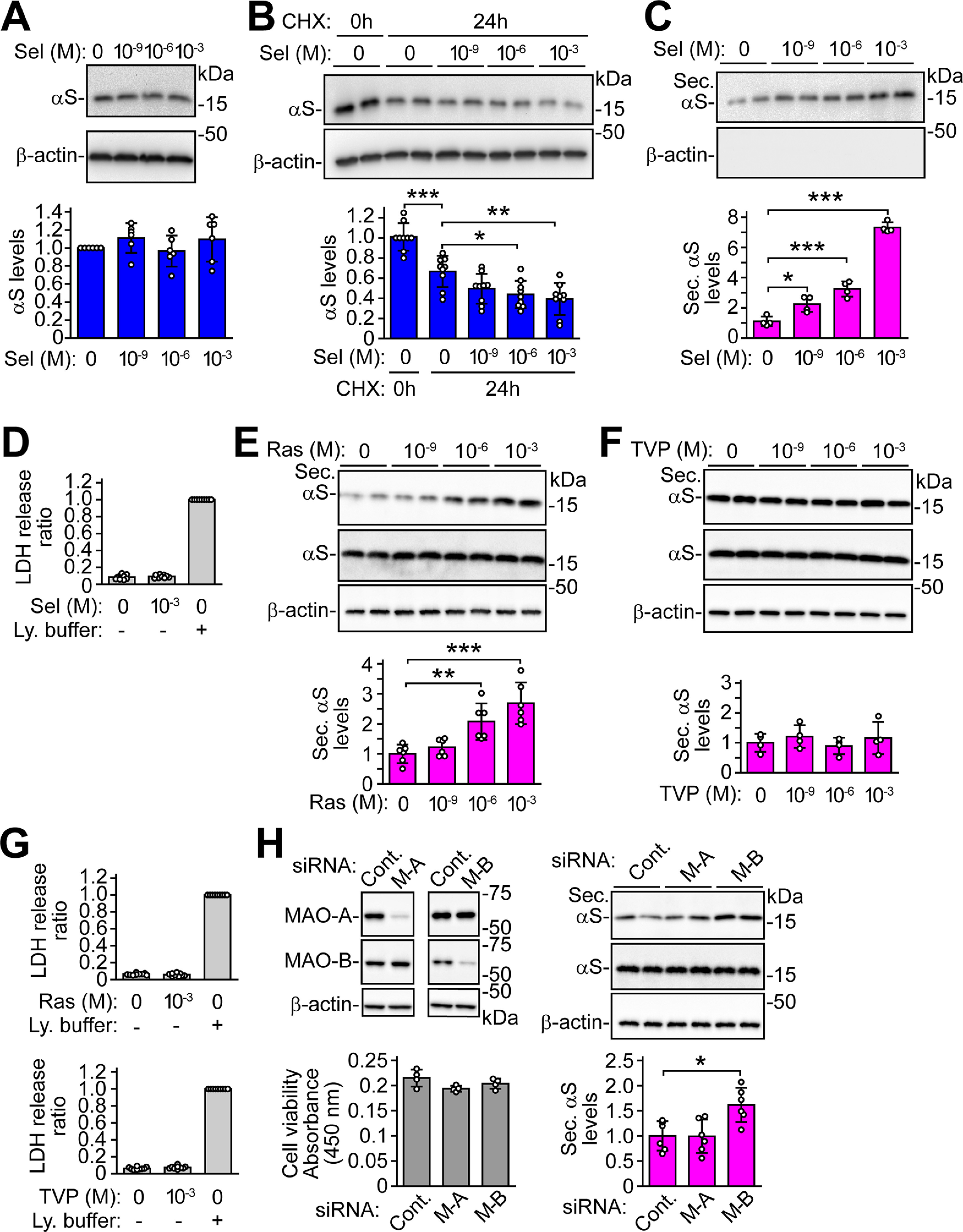
MAO-B inhibition facilitates α-syn secretion in SH-SY5Y cells. Western blottings of TCA-precipitated CM and cell lysates are labeled by Syn-1 antibody. β-Actin is used as a loading control. A, Parental SH-SY5Y cells were treated with selegiline for 24 h (n = 6). Cell lysates are blotted. Data were analyzed by Welch-ANOVA (F(3,20) = 1.272, p = 0.311) with Games–Howell post hoc test. B, CHX chase experiments of parental SH-SY5Y cells with and without selegiline. The intracellular levels of α-syn were analyzed by Western blotting of cell lysates with Syn-1 antibody (n = 9; F(4,40) = 26.140, p < 0.001, ANOVA). C, wt-aS/SH cells were treated with selegiline for 24 h. The extracellular levels of α-syn were analyzed by Western blotting of CM with Syn-1 antibody (n = 4; F(3,12) = 159.338, p < 0.001, ANOVA). D, Assessment of LDH release in selegiline-treated cells (n = 9). E, After treating wt-aS/SH cells with rasagiline for 24 h, CM and cell lysates were blotted (n = 6; F(3,20) = 14.391, p < 0.001, ANOVA). F, After treating wt-aS/SH cells with TVP-1022 for 24 h, CM and cell lysates were blotted (n = 4; F(3,12) = 0.546, p = 0.660, ANOVA). G, Assessment of LDH release in rasagiline-treated and TVP-1022-treated cells (n = 9). H, MAO-A or MAO-B knock-down wt-aS/SH cells were incubated in Opti-MEM for 24 h. The extracellular levels of α-syn were analyzed by Western blotting of CM with Syn-1 antibody (n = 6; F(2,15) = 7.394, p = 0.006, ANOVA). Left upper and middle panels show blots of cell lysates with MAO-A and MAO-B antibodies, respectively. Right upper and middle panels show blots of CM and cell lysates with Syn-1, respectively. Left lower graph shows cell viability in knock-down cells (n = 4). A–C, E, F, H, Graphs show the ratios of relative intensities of α-syn signals to controls. D, G, Graphs show LDH release ratios to lysis buffer-treated cells as a positive control. Data represent mean ± SD. B–H, Data were analyzed by one-way ANOVA with Bonferroni's post hoc test; *p < 0.05, **p < 0.01, ***p < 0.001. Control, Cont; cycloheximide, CHX; MAO-A, M-A; MAO-B, M-B; lysis buffer, Ly. buffer; rasagiline, Ras; secreted α-syn, Sec. αS; selegiline, Sel; TVP-1022, TVP.
MAO-B inhibition modulates α-syn secretion via a non-classical pathway involving an ABC transporter
α-Syn has been demonstrated to be secreted via a non-classical pathway (Jang et al., 2010). To explore whether MAO-B inhibition facilitates α-syn secretion via this pathway, wt-aS/SH cells were co-treated with MAO-B inhibitors and brefeldin A, which interferes with classical ER/Golgi transport (Jang et al., 2010). Selegiline-induced and rasagiline-induced increases in α-syn secretion were unaffected by 7 μm brefeldin A treatment (Fig. 2A,B). From experiments using pharmacological inhibition of ABC transporter by probenecid, glyburide, and reserpine, the secretion of non-classically secreted proteins including interleukin 1β (IL-1β), macrophage migration inhibitory factor (MIF), and Y-box protein-1 (YB-1) has been shown to involve ABC transporters (Andrei et al., 1999; Flieger et al., 2003; Loebinger et al., 2008; Frye et al., 2009). We therefore investigated whether MAO-B inhibition-induced α-syn secretion involved an ABC transporter. In wt-aS/SH cells, treatment with 10 μm probenecid for 24 h suppressed the increase in α-syn secretion caused by 1 mm selegiline (p < 0.001) or rasagiline (p = 0.049; Fig. 2C,D). Similarly, treatment with 5 μm glyburide also reduced the increase in α-syn secretion caused by 1 mm selegiline (p = 0.002) or rasagiline (p < 0.001; Fig. 2E,F). In these experiments, the intracellular levels of α-syn were not altered, and these chemical treatments did not affect LDH release (Fig. 2A–F). Additionally, treatment with 10 μm reserpine for 24 h significantly blocked α-syn secretion to 0.47 ± 0.13-fold compared with untreated cells (p = 0.030; Fig. 3A). In MAO-B knock-down wt-aS/SH cells, an increase in α-syn secretion was inhibited by reserpine (p < 0.001; Fig. 3A). Treatment with reserpine also suppressed the increase in α-syn secretion induced by selegiline (p = 0.001) or rasagiline (p < 0.001; Fig. 3B,C). However, intracellular α-syn expression and LDH release were not altered (Fig. 3A–C). These findings suggest that MAO-B inhibition facilitates α-syn secretion via the non-classical pathway, and that an ABC transporter is involved in this secretion.
Figure 2.

MAO-B inhibition facilitates α-syn secretion via the non-classical ABC transporter-mediated pathway. Western blot analysis of TCA-precipitated CM and cell lysates with Syn-1 antibody. β-Actin was used as a loading control. A, B, Effect of brefeldin A on MAO-B inhibitor-induced α-syn secretion. wt-aS/SH cells were treated with 1 mm selegiline (A) or 1 mm rasagiline (B) in the absence and presence of 7 μm brefeldin A for 6 h (n = 3 per group). C, D, Effect of probenecid on MAO-B inhibitor-induced α-syn secretion. wt-aS/SH cells were treated with 1 mm selegiline (C; n = 4; F(2,9) = 65.197, p < 0.001, ANOVA) or 1 mm rasagiline (D; n = 3; F(2,6) = 10.362, p = 0.011, ANOVA) in the absence and presence of 10 μm probenecid for 24 h. E, F, Effect of glyburide on MAO-B inhibitor-induced α-syn secretion. After pretreating wt-aS/SH cells with vehicle or 5 μm glyburide for 24 h, cells were treated with 1 mm selegiline (E; n = 3; F(2,6) = 77.896, p < 0.001, ANOVA) or 1 mm rasagiline (F; n = 3; F(2,6) = 64.275, p < 0.001, ANOVA) in the absence and presence of 5 μm glyburide for 24 h. C–F, Middle graphs show the ratios of relative intensities of α-syn signals to controls. A–F, Right graphs show LDH release ratios to lysis buffer-treated cells (n = 6). Data represent mean ± SD. Data were analyzed by one-way ANOVA with Bonferroni's post hoc test; *p < 0.05, **p <0.01, ***p <0.001. Brefeldin A, Bre; Glyburide, Gly; lysis buffer, Ly. buffer; probenecid, Pro; rasagiline, Ras; secreted α-syn, Sec. αS; selegiline, Sel.
Figure 3.
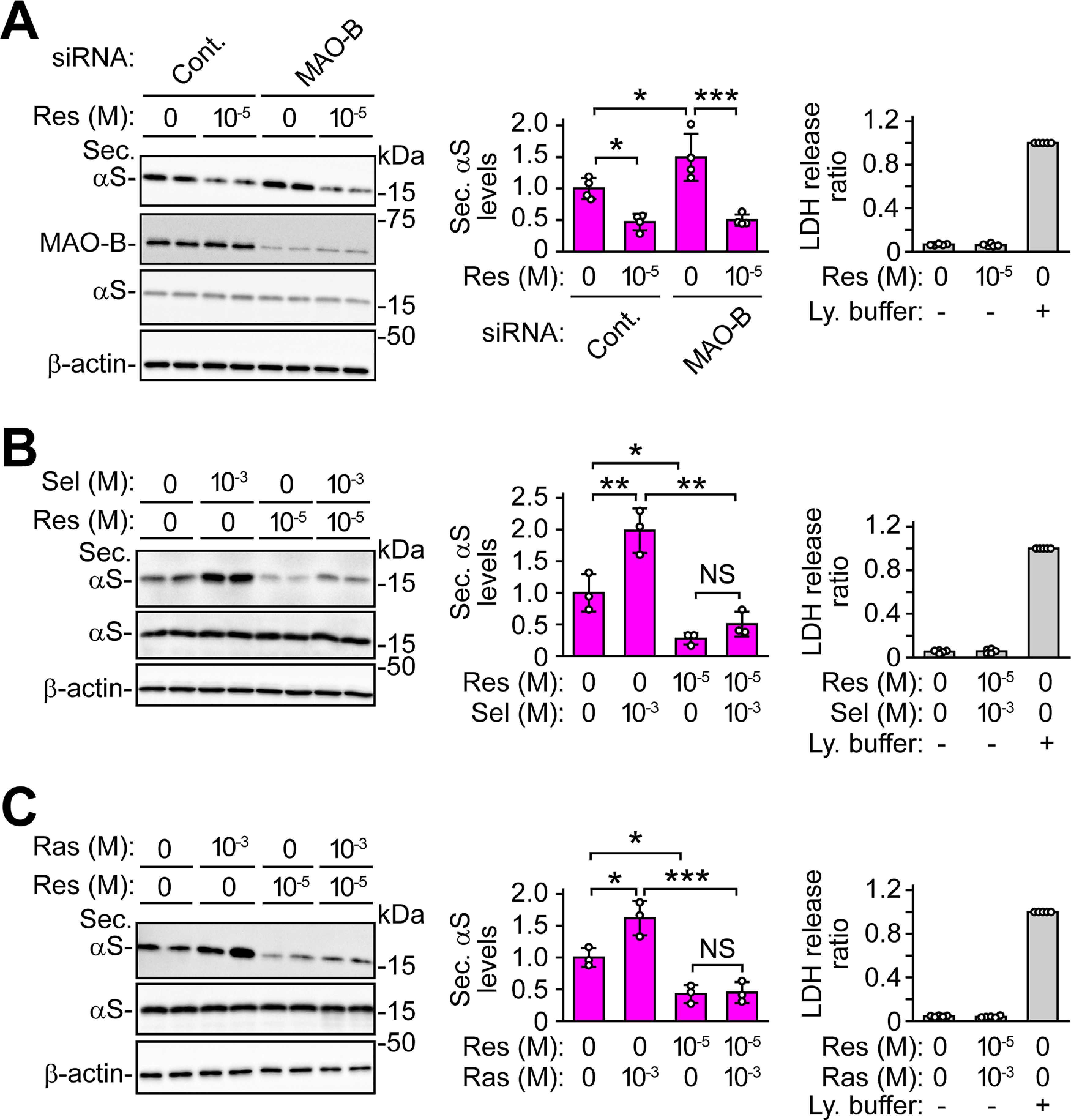
Effects of reserpine on MAO-B inhibition-induced α-syn secretion. Western blot analysis of TCA-precipitated CM and cell lysates with Syn-1 antibody. β-Actin was used as a loading control. A, MAO-B knock-down wt-aS/SH cells were treated with 10 μm reserpine for 24 h (n = 4; F(3,12) = 19.555, p < 0.001, ANOVA). Blots of cell lysates are labeled by Syn-1, MAO-B, and β-actin antibodies. B, wt-aS/SH cells were treated with 1 mm selegiline and 10 μm reserpine for 48 h (n = 3; F(3,8) = 26.584, p < 0.001, ANOVA). C, wt-aS/SH cells were treated with 1 mm rasagiline and 10 μm reserpine for 24 h (n = 3; F(3,8) = 26.760, p < 0.001, ANOVA). A–C, Middle graphs show the ratios of relative intensities of α-syn signals to controls. Right graphs show LDH release ratios to lysis buffer-treated cells (n = 5). Data represent mean ± SD. Data were analyzed by one-way ANOVA with Bonferroni's post hoc test; *p < 0.05, **p < 0.01, ***p < 0.001. Lysis buffer, Ly. buffer; Not significant, NS; rasagiline, Ras; reserpine, Res; secreted α-syn, Sec. αS; selegiline, Sel.
MAO-B inhibition preferentially facilitates the secretion of detergent-insoluble α-syn protein
To investigate how selegiline affects the secretion of α-syn protein under stress conditions, we treated wt-aS/SH cells with 1 mm selegiline in the absence and presence of a lysosome inhibitor, chloroquine, at 100 μm for 48 h. We then fractionated the cells into 1% Triton X-100 soluble and insoluble proteins. The intracellular levels of 1% Triton X-100-soluble α-syn protein were similar between selegiline-treated or chloroquine-treated cells and untreated cells (Fig. 4A). However, the intracellular levels of 1% Triton X-100-insoluble α-syn protein were significantly decreased to 0.52 ± 0.14-fold in selegiline-treated cells (p = 0.013), while they were increased to 1.71 ± 0.13-fold in chloroquine-treated cells (p = 0.001; Fig. 4B). In cells co-treated with selegiline and chloroquine, selegiline treatment blocked the chloroquine-induced increase (p = 0.002). We next investigated the extracellular secretion of 1% Triton X-100-soluble and Triton X-100-insoluble α-syn proteins. The extracellular levels of 1% Triton X-100-soluble α-syn protein were similar between the selegiline-treated or chloroquine-treated cells and untreated cells (Fig. 4C). In contrast, the extracellular levels of 1% Triton X-100-insoluble α-syn protein were significantly elevated to 10.95 ± 2.35-fold in selegiline-treated cells (p = 0.048) and 18.58 ± 5.57-fold in chloroquine-treated cells (p = 0.001). In cells co-treated with selegiline and chloroquine, the extracellular levels of 1% Triton X-100-insoluble α-syn protein were further elevated to 28.67 ± 6.47-fold (p = 0.044 compared with chloroquine-treated cells; Fig. 4D). In the LDH assay, treatment with chloroquine increased LDH release to ∼30% of that of lysis buffer-treated cells, suggesting that chloroquine-induced membrane damage may affect the elevation of 1% Triton X-100-insoluble α-syn in CM (Fig. 4E). However, LDH release after co-treatment with chloroquine and selegiline was similar to that after treatment with chloroquine only, indicating that selegiline facilitated α-syn secretion without additional damage of cell membranes. These findings suggest that selegiline preferentially facilitates the secretion of detergent-insoluble α-syn protein under lysosomal inhibition, resulting in the prevention of intracellular α-syn accumulation.
Figure 4.

Effects of MAO-B inhibition on secretion and intracellular accumulation of detergent-insoluble α-syn protein under lysosomal dysfunction. wt-aS/SH cells were treated with 100 μm chloroquine and 1 mm selegiline for 48 h. The levels of α-syn in 1% Triton X-100 soluble fractions of cells (A; n = 4; F(3,12) = 0.081, p = 0.969, ANOVA), 1% Triton X-100 insoluble fractions of cells (B; n = 4; F(3,12) = 31.578, p < 0.001, ANOVA), 1% Triton X-100 soluble fractions of CM (C; n = 4; F(3,12) = 3.058, p = 0.070, ANOVA), and 1% Triton X-100 insoluble fractions of CM (D; n = 4; F(3,12) = 27.908, p < 0.001, ANOVA) were analyzed by Western blotting. β-Actin was used as a loading control of 1% Triton X-100 soluble fractions of cells. Graphs show the ratios of relative intensities of α-syn signals to controls. E, Assessment of LDH release in selegiline and chloroquine-treated cells (n = 9; F(3,32) = 45.271, p < 0.001, ANOVA). Graphs show LDH release ratios to lysis buffer-treated cells. Data represent mean ± SD and were analyzed by one-way ANOVA with Bonferroni's post hoc test; *p < 0.05, **p < 0.01, ***p < 0.001. Chloroquine, Chl; conditioned medium, CM; lysis buffer, Ly. buffer; secreted α-syn, Sec. αS; selegiline, Sel.
The effects of selegiline on the extracellular and intracellular levels of 1% Triton X-100-insoluble α-syn protein were significantly blocked by the addition of reserpine (extracellular levels, p = 0.026; intracellular levels, p = 0.021; Fig. 5A). Conversely, the effects of 100 μm chloroquine on the extracellular and intracellular levels of 1% Triton X-100-insoluble α-syn protein were not significantly altered by the addition of reserpine (Fig. 5B). These findings suggest that MAO-B inhibition preferentially facilitates the secretion of detergent-insoluble α-syn protein via the ABC transporter-mediated pathway, and this secretory pathway is distinct from one induced by lysosomal dysfunction.
Figure 5.

Distinct involvement of ABC transporter between MAO-B inhibition-induced and lysosomal dysfunction-induced α-syn secretion. Western blot analysis of 1% Triton X-100 soluble fractions of CM (left upper blots of each panel), 1% Triton X-100 insoluble fractions of CM (right upper blots of each panel), 1% Triton X-100 soluble fractions of cells (left middle blots of each panel), and 1% Triton X-100 insoluble fractions of cells (right lower blots of each panel) with Syn-1 antibody. β-Actin was used as a loading control of 1% Triton X-100 soluble fractions of cells. A, wt-aS/SH cells were treated with 1 mm selegiline in the absence and presence of 10 μm reserpine for 48 h (comparison of secreted insoluble α-syn levels, n = 3, F(2,6) = 16.883, p = 0.003, ANOVA; comparison of intracellular insoluble α-syn levels, n = 5, F(2,12) = 38.356, p < 0.001, ANOVA). B, wt-aS/SH cells were treated with 100 μm chloroquine in the absence and presence of 10 μm reserpine for 48 h (comparison of secreted insoluble α-syn levels, n = 4, F(2,9) = 52.850, p < 0.001, ANOVA; comparison of intracellular insoluble α-syn levels, n = 3, F(2,6) = 46.223, p < 0.001, ANOVA). Graphs show the ratios of relative intensities of α-syn signals to controls. Data represent mean ± SD and were analyzed by one-way ANOVA with Bonferroni's post hoc test; *p < 0.05, **p < 0.01, ***p < 0.001. Chloroquine, Chl; not significant, NS; reserpine, Res; secreted α-syn, Sec. αS; selegiline, Sel.
Effects of MAO-B inhibition on α-syn-induced dopaminergic neuronal loss in rats
To elucidate the effects of MAO inhibition on α-syn-induced neurotoxicity in vivo, we injected rAAV-A53T human α-syn into the rat substantia nigra and repeatedly administered selegiline (10 mg/kg, subcutaneous injection, once a day) to rats from 3 d before the viral injection to the day before killing (Fig. 6A). This dose of selegiline has been demonstrated to inhibit both MAO-B and MAO-A activities in the rodent brain. In the vehicle-treated group, immunohistochemistry using the human α-syn antibody (LB509) showed that A53T α-syn was expressed in neurons of the substantia nigra pars compact and its surrounding area to a lesser extent at two weeks after viral injection (Fig. 6B). The immunoreactivities with LB509 antibody decreased at eight weeks after viral injection, but they were seen in nigral neurons during the observation period. Also, nigral TH-positive neurons were gradually reduced after the rAAV-A53T α-syn injection (Fig. 6B). At 8 and 12 weeks after the viral injection, selegiline-treated rats had significantly higher percentages of remaining nigral TH-positive neurons compared with the vehicle-treated rats (p < 0.001 each; Fig. 6C). Similarly, selegiline treatment significantly reduced the loss of striatal TH-positive fibers compared with vehicle treatment (p < 0.001 each). To examine whether the rAAV-mediated gene transfer itself causes artificial damage in this model, we performed an injection of rAAV-GFP into rats. The rAAV-mediated expression of GFP led to ∼15% loss of nigral TH-positive neurons (Fig. 6D). However, it did not result in the progressive loss of nigral TH-positive neurons between two and eight weeks after the viral injection. Similarly, the numbers of nigral DAT-positive neurons in rAAV-GFP rats were comparable between two and eight weeks after the viral injection.
Figure 6.

MAO-B inhibition attenuates α-syn-induced dopaminergic neuronal loss in rats. A, Experiment schedule. Selegiline was resolved in saline before use. The selegiline solution was subcutaneously injected at a dose of 10 mg/kg once daily from 3 d before the viral injection to the day before killing. As a control, the same volume of vehicle was simultaneously injected in a separate set of rats. Rats were killed at two weeks (vehicle, n = 5; selegiline, n = 6), four weeks (vehicle, n = 4; selegiline, n = 5), eight weeks (vehicle, n = 4; selegiline, n = 4), and 12 weeks (vehicle, n = 8; selegiline, n = 4) after rAAV-A53T α-syn injection. B, Immunohistochemical analysis of rat midbrains injected rAAV-A53T α-syn. Left panels show anti-human α-syn antibody (LB509)-stained images at low (scale bar: 2 mm) and high magnifications (scale bar: 100 μm). Right panels show anti-TH antibody-stained sections (scale bar: 2 mm). C, Assessment of nigrostriatal dopaminergic neuronal damage by rAAV-A53T α-syn injection. Left graph shows percentages of the remaining nigral TH-positive neurons in the lesioned sides to those in the intact sides. A two-way ANOVA demonstrated significant main effects for the drug (F(1,32) = 26.901, p < 0.001) and for time (F(3,32) = 75.607, p < 0.001), with a significant drug × time interaction (F(3,32) = 8.814, p < 0.001). In the vehicle-treated rats, the numbers of TH-positive neurons in intact sides were 25,265.0 ± 1306.5 and 21,855.8 ± 4146.9 at 8 and 12 weeks after viral injection, respectively. In the selegiline-treated rats, the numbers of TH-positive neurons in intact sides were 19,553.9 ± 3387.2 and 21,484.0 ± 2388.3 at 8 and 12 weeks after viral injection, respectively. Right graph shows percentages of the remaining TH-positive fibers in the striatum. A two-way ANOVA demonstrated significant main effects for the drug (F(1,32) = 75.728, p < 0.001) and for time (F(3,32) = 141.373, p < 0.001), with a significant drug × time interaction (F(3,32) = 16.381, p < 0.001). D, Assessment of nigral dopaminergic neuronal damage by rAAV-GFP injection. Rats were killed two and eight weeks later (n = 5 per group). Graph shows percentages of remaining nigral TH or DAT-positive neurons in the lesioned sides to those in the intact sides. E, Assessment of motor impairment by rAAV-A53T α-syn injection (n = 7 per group). Cylinder test was performed at eight weeks after rAAV-A53T α-syn injection. Left and right graphs show total forepaw touches and percentages of right paw touches to total paw ones, respectively. F, Striatal DA contents at eight weeks after rAAV-A53T α-syn injection (n = 7 per group). Left and right graphs show the striatal DA contents and the ratios of DA contents in the lesioned sides to those in the intact sides, respectively. Data represent mean ± SD and were analyzed by two-way ANOVA with Bonferroni's post hoc test (C), unpaired t test (E and right graph of F), and one-way ANOVA with Tukey's post hoc test (F, left graph). Data represent mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.001. Dopamine, DA; dopamine transporter, DAT; α-syn, αS; tyrosine hydroxylase, TH.
Eight weeks after the viral injection, the cylinder test was used to assess the effects of selegiline on motor impairment. In the vehicle-treated group, significant asymmetry of forepaw touches, with reduced use of the right side (contralateral to the viral-injected side), was observed in rAAV-A53T α-syn rats. Although the total forepaw touches were comparable between the vehicle-treated and selegiline-treated groups, the percentage of right paw touches to total paw touches in the selegiline-treated group was 1.94 ± 0.54-fold higher than that in the vehicle-treated group (t(12) = 2.945, p = 0.012, unpaired t test), indicating that selegiline prevents A53T mutant α-syn-induced motor impairment (Fig. 6E). Treatment with selegiline also increased the DA content in the lesioned and intact sides through the inhibition of MAO activity (Fig. 6F). The ratio of DA in the lesioned side to that in the intact side was 3.18 ± 2.26-fold higher in the selegiline-treated group than in the vehicle-treated group (t(7.182) = 2.429, p = 0.045, unpaired t test), indicating that selegiline treatment leads to retained DA content in the lesioned side by preventing dopaminergic neuronal loss.
Effects of MAO-B inhibition on the formation of α-syn aggregates in rats
In this rat model, we next examined the number of pS129 α-syn-positive aggregates in the striatum. We defined α-syn aggregates as pS129 α-syn-positive dot-like, round, or rod-like abnormal structures in neurites. In this study, we did not assess α-syn aggregate formation in the substantia nigra because the overexpression of α-syn induced diffuse immunoreactivity to pS129 α-syn in the somata. At two, four, and eight weeks after rAAV-53T α-syn injection, selegiline treatment reduced the formation of pS129 α-syn-positive aggregates to 0.53 ± 0.19-fold (p = 0.023), 0.55 ± 0.19-fold (p = 0.018), and 0.54 ± 0.14-fold (p = 0.030), respectively, compared with vehicle treatment (Fig. 7A,B). However, this effect of selegiline was lost at 12 weeks after viral injection. These findings suggest that treatment with selegiline can delay the formation of striatal pS129 α-syn-positive aggregates. Immunofluorescent analysis of the striatum at 12 weeks after viral injection demonstrated that pS129 α-syn-positive aggregates were formed in TH-positive neurons, supporting the relationship between the expression of α-syn and loss of nigrostriatal dopaminergic neurons (Fig. 7C).
Figure 7.

MAO-B inhibition delays the formation of pS129 α-syn aggregates. Rats were killed at two weeks (vehicle, n = 4; selegiline, n = 6), four weeks (vehicle, n = 4; selegiline, n = 5), eight weeks (vehicle, n = 4; selegiline, n = 4), and 12 weeks (vehicle, n = 8; selegiline, n = 4) after rAAV-A53T α-syn injection. A, Immunohistochemical analysis of striatal pS129 α-syn aggregates. Micrographs show pS129 α-syn aggregates of upper and middle portions of the dorsal lateral striatum at 4, 8, and 12 weeks after viral injection. Scale bar: 100 μm. B, Graph shows the number of pS129 α-syn aggregates. A two-way ANOVA demonstrated significant main effects for the drug (F(1,31) = 8.619, p = 0.006) and for time (F(3,31) = 162.594, p < 0.001), with a significant drug × time interaction (F(3,31) = 4.204, p = 0.013). Data were log-transformed and analyzed by two-way ANOVA with Bonferroni's post hoc test. Data represent mean ± SD; *p < 0.05. C, Immunofluorescent analysis of striatal pS129 α-syn aggregates at 12 weeks after rAAV-A53T α-syn injection. Left and middle micrographs show the sections stained with anti-TH (green) and anti-pS129 α-syn (red) antibodies, respectively. Right micrograph shows the merged image. Arrowheads indicate TH-positive and pS129 α-syn-positive aggregates. Scale bar: 25 μm. Tyrosine hydroxylase, TH.
Effects of MAO-B inhibition on cell-to-cell transmission of α-syn protein in rats
To assess whether MAO-B inhibition modulates the cell-to-cell transmission of α-syn, we injected rAAV-wild-type human α-syn into the rat substantia nigra, and then inoculated α-syn PFF into the ipsilateral striatum two weeks later (Fig. 8A). After 4 d, we began to administer selegiline (10 mg/kg, subcutaneous injection) and repeatedly administered it once a day until the day before killing. At six weeks post-PFF injection, the number of striatal pS129 α-syn aggregates in the selegiline-treated group was 0.55 ± 0.14-fold lower than that in the vehicle-treated group (p = 0.007; Fig. 8B). However, at eight weeks post-PFF injection, the numbers of pS129 α-syn aggregates were similar between the two groups (p = 0.704). The percentage of remaining nigral TH-positive neurons in the selegiline-treated group was 1.62 ± 0.25-fold higher than that in the vehicle-treated group at six weeks post-PFF injection (p = 0.001), but this effect was lost at eight weeks post-PFF injection (p = 0.221; Fig. 8C). The inoculation of TBS-soluble α-syn protein yielded a faint pS129 α-syn immunoreactivity around the needle insertion area, while the inoculation of saline did not (Fig. 8B). We also inoculated PFF into rats that were not injected with rAAV-wild-type α-syn. At six weeks after PFF injection, these rats showed very few pS129 α-syn-positive structures in the striatum (Fig. 8B). This finding indicates that PFF facilitates the formation of pS129 α-syn aggregates by reacting with rAAV-mediated expression of wild-type α-syn in this rat model. Additionally, the reduction in nigral TH-positive neurons after inoculation with saline or TBS-soluble α-syn was significantly weaker than that after inoculation with PFF (Fig. 8C). These findings suggest that treatment with selegiline delays striatal PFF-stimulated aggregation and nigral dopaminergic neuronal loss during a short period.
Figure 8.

MAO-B inhibition delays α-syn aggregation in the rat transmission model. A, Experiment schedule. At 4 d post-PFF injection, we began to subcutaneously administer selegiline (10 mg/kg) and repeatedly administered it once a day until the day before killing. As a control, the same volume of vehicle was simultaneously injected in a separate set of rats. Rats were killed at six weeks (vehicle, n = 5; selegiline, n = 5) and eight weeks (vehicle, n = 5; selegiline, n = 6 for analysis of pS129 α-syn aggregation or n = 4 for analysis of nigral TH-positive neuron viability) after PFF inoculation. B, Effect of selegiline on the formation of pS129 α-syn aggregates in the rat transmission model. Micrographs show anti-pS129 α-syn antibody-stained striatal sections at six weeks after PFF inoculation and eight weeks after PFF, saline or purified soluble α-syn protein inoculation in rAAV-wild-type α-syn-injected rats, and at six weeks after PFF inoculation in non-rAAV-injected rats. Graph shows the number of pS129 α-syn aggregates. A two-way ANOVA demonstrated significant main effects for drug (F(1,17) = 6.279, p = 0.023) and for time (F(1,17) = 89.970, p < 0.001), with no significant drug × time interaction (F(1,17) = 3.890, p = 0.065). Scale bars: 100 μm. C, Graph shows remaining nigral TH-positive neurons. A two-way ANOVA demonstrated significant main effects for drug (F(1,15) = 14.379, p = 0.002) and for time (F(1,15) = 14.197, p = 0.002), with no significant drug × time interaction (F(1,15) = 3.746, p = 0.072). The numbers of TH-positive neurons in intact sides were 18,018.1 ± 1749.9 and 15,060.1 ± 1690.8 at six weeks post-PFF injection in the vehicle-treated and selegiline-treated rats, respectively. Data represent mean ± SD and were analyzed by two-way ANOVA with Bonferroni's post hoc test; **p < 0.01. α-Syn, αS; tyrosine hydroxylase, TH.
Discussion
Although most secretory proteins are released via the classical ER/Golgi exocytotic pathway, some inflammatory mediators are known to be released via the non-classical ER/Golgi-independent pathway (Kim et al., 2018). These molecules commonly lack an amino-terminal or transmembrane signal peptide sequence for ER targeting. This non-classical secretion is insensitive to brefeldin A, which interferes with ER/Golgi transport (Flieger et al., 2003; Frye et al., 2009). α-Syn lacks the ER target signal and is secreted in a brefeldin A-insensitive manner in SH-SY5Y cells (Jang et al., 2010). In this study, we found that the inhibition of MAO-B enzymatic activity facilitated extracellular secretion of α-syn via the brefeldin A-insensitive non-classical pathway in SH-SY5Y cells, and that an ABC transporter was involved in this pathway. Among several export machineries of the non-classical pathway, ABC transporter-mediated secretion has been identified in the release of IL-1β, MIF, and YB-1 (Andrei et al., 1999; Flieger et al., 2003; Frye et al., 2009). ABC transporter-mediated secretion includes non-vesicular and vesicular transports (Tarling et al., 2013). In the non-vesicular transport, substrates are released from the cytosol to the extracellular space via cell membrane-embedded ABC transporters. In the vesicular transport, substrates are incorporated into vesicles, such as lysosomes, endosomes, and peroxisomes, via ABC transporters, and these vesicles export substrates into the extracellular space. It is unclear as to the transport system in α-syn secretion facilitated by MAO-B inhibition. However, ABC transporters are a potential target to control the clearance of misfolded proteins from neurons and brains (Abuznait and Kaddoumi, 2012). In the central nervous system, ABC transporters are localized at the different cell types including neurons, astrocytes, endothelial cells of the blood-brain barrier (BBB) and parenchymal cells. Previous studies have shown that ABC transporters contribute to the clearance of amyloid β peptides (Aβ) across the BBB, and alteration in expression and function of these transporters causes Aβ accumulation in the brain of Alzheimer's disease (AD; Abuznait and Kaddoumi, 2012). Also, approximately half of human ABC transporters are associated with ATP-dependent translocation of lipids or lipid-related compounds (Tarling et al., 2013). α-Syn binds phospholipids, and this interaction facilitates formation of α-syn fibrils (Comellas et al., 2012). Familial PD-linked mutant E46K α-syn has been shown to increase phospholipid binding and fibril formation (Choi et al., 2004). The interaction with lipids may increase the affinity of α-syn to ABC transporters. We revealed that selegiline preferentially facilitated the secretion of detergent-insoluble α-syn with a decrease in its intracellular accumulation under chloroquine-induced lysosomal dysfunction, and these effects were blocked by reserpine treatment. These findings suggest that MAO-B inhibition stimulate the clearance of insoluble α-syn protein via the ABC transporter-mediated non-classical secretion pathway under pathologic conditions (Fig. 9). Although the exact mechanisms underlying the effect of MAO-B inhibition on ABC transporter-mediated α-syn secretion are unclear, increased levels of MAO-B substrates may drive the secretion of α-syn. A previous study has reported that DA treatment promoted accumulation of α-syn oligomers in intracellular vesicles and stimulated the secretion of α-syn oligomers (Lee et al., 2011). The increase in DA contents by MAO-B inhibition may stimulate generation of misfolded α-syn protein, facilitating α-syn secretion via the ABC transporter-mediated pathway. Alternatively, MAO-B metabolizes β-phenylethylamine (β-PEA) more preferentially than MAO-A (Finberg and Rabey, 2016). β-PEA activates trace amine-associated receptor 1 (TAAR1), and this activation is coupled with the mobilization of intracellular calcium (Navarro et al., 2006; Rutigliano et al., 2017). Release of AD-related tau protein occurs in response to calcium influx-dependent changes in neuronal excitability in primary cortical neuron cultures (Pooler et al., 2013). α-Syn is also released in response to calcium influx (Yamada and Iwatsubo, 2018). These findings hypothesize that MAO-B inhibition may facilitate exocytosis of vesicles containing ABC transporters through the increase in calcium influx by β-PEA-mediated activation of TAAR1. It should be noted that α-syn secretion did not show a plateau by treatment with selegiline at high concentration. This may be explained by introducing a factor besides MAO-B inhibition. Pharmacological active metabolites of selegiline may additively enhance the effects of selegiline on α-syn secretion. Further studies are required to elucidate the relationship between MAO-B inhibition and facilitation of α-syn secretion.
Figure 9.

A model of MAO-B inhibition effects on α-syn clearance. Many proteins are secreted via the classical ER/Golgi-dependent pathway, while α-syn is secreted via the non-classical ER/Golgi-independent pathway. Impairment of lysosomal degradation causes solubility change of α-syn protein from the soluble form to the insoluble one, and promotes intracellular accumulation of insoluble α-syn. MAO-B inhibition preferentially facilitates secretion of insoluble α-syn into the extracellular space via the ABC transporter-mediated non-classical pathway. Consequently, MAO-B inhibition mitigates intracellular accumulation of insoluble α-syn. There are two candidate transport systems, non-vesicular (a) and vesicular transports (b), in the ABC transporter-mediated non-classical pathway.
MAO-B has been demonstrated to activate asparagine endopeptidase by direct binding to α-syn, and it facilitated generation of α-syn toxic fragments (Kang et al., 2018). Additionally, rasagiline attenuated nigrostriatal dopaminergic neuronal loss in rats injected rAAV-α-syn. To elucidate whether MAO-B inhibition affects α-syn aggregation in vivo, we investigated the effects of selegiline on α-syn aggregation and neurotoxicity in rats. We revealed that selegiline had protective effects against α-syn-induced neurotoxicity and delayed the formation of α-syn aggregates in the rats injected rAAV-A53T α-syn. A previous study has reported that selegiline delayed the fibril formation of α-syn by extending the lag phase of aggregation in vitro and proposed that selegiline interfered with early nuclei formation as well as fibril elongation, albeit to a lesser extent (Braga et al., 2011). It is therefore possible that selegiline reduces the formation of α-syn aggregates through its direct interaction with α-syn protein. However, in the present study, a suppressive effect of selegiline on the formation of striatal pS129 α-syn-positive aggregates was not observed at 12 weeks after the viral injection. This finding raises the following question: why was the suppressive effect of selegiline on α-syn aggregation attenuated in the advanced stages of α-syn pathology? It seems difficult to answer this question by considering the direct anti-aggregation properties of selegiline. The aforementioned study has also demonstrated that the inhibitory effect of selegiline was abolished by adding small fibril pieces as seeds into the aggregation reaction (Braga et al., 2011). However, this result was inconsistent with our finding that selegiline delayed the cell-to-cell transmission of α-syn aggregates. This question may be explained by two possibilities. First, in vehicle-treated rats, the number of α-syn aggregates may be reduced as neuronal loss progresses, because LBs have been demonstrated to disappear with neuronal loss (Greffard et al., 2010). Alternatively, selegiline may be unable to resist the formation of α-syn aggregates in the advanced stages of α-syn pathology. Although there are no data demonstrating that MAO-B inhibition facilitates α-syn secretion in vivo, MAO-B inhibition may lead to the spread of misfolded α-syn protein into the extracellular space. In support of this idea, secreted tau has been shown to enhance the propagation of tau pathology in transgenic mice (Wu et al., 2016). It is thus conceivable that MAO-B inhibition promotes cell-to-cell transmission of α-syn aggregates by facilitating the secretion of misfolded α-syn protein in the advanced stages of α-syn pathology. Conversely, this action of MAO-B inhibition may open a combination therapy with active and passive immunization against α-syn. Active immunization using α-syn mimicking short peptides persistently produced antibodies and reduced accumulation of α-syn oligomers in axons and synapses by activating microglial clearance in transgenic mouse models (Mandler et al., 2014). MAO-B inhibition may facilitate effective capture of extracellular α-syn by microglia and antibodies to block cell-to-cell transmission of α-syn pathology in PD.
In summary, we have demonstrated that MAO-B inhibition affects the non-classical secretion of α-syn in vitro and potentially acts as a temporary suppressor of the accumulation and transmission of α-syn aggregates in vivo. Our results provide support for the concept that MAO-B inhibition may exert undescribed actions.
Footnotes
We thank Kaori Koga and Tomomi Seino for preparing sections.
This is a cooperative study with FP Pharmaceutical Corporation. This work was designed independently of FP Pharmaceutical Corporation. The authors declare no other competing financial interests.
References
- Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L, Olivo-Marin JC, Melki R, Zurzolo C (2016) Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J 35:2120–2138. 10.15252/embj.201593411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abuznait AH, Kaddoumi A (2012) Role of ABC transporters in the pathogenesis of Alzheimer's disease. ACS Chem Neurosci 3:820–831. 10.1021/cn300077c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei C, Dazzi C, Lotti L, Torrisi MR, Chimini G, Rubartelli A (1999) The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol Biol Cell 10:1463–1475. 10.1091/mbc.10.5.1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arawaka S, Sato H, Sasaki A, Koyama S, Kato T (2017) Mechanisms underlying extensive Ser129-phosphorylation in α-synuclein aggregates. Acta Neuropathol Commun 5:48. 10.1186/s40478-017-0452-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boix J, Padel T, Paul G (2015) A partial lesion model of Parkinson's disease in mice–characterization of a 6-OHDA-induced medial forebrain bundle lesion. Behav Brain Res 284:196–206. 10.1016/j.bbr.2015.01.053 [DOI] [PubMed] [Google Scholar]
- Borghi R, Marchese R, Negro A, Marinelli L, Forloni G, Zaccheo D, Abbruzzese G, Tabaton M (2000) Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson's disease and normal subjects. Neurosci Lett 287:65–67. 10.1016/s0304-3940(00)01153-8 [DOI] [PubMed] [Google Scholar]
- Braga CA, Follmer C, Palhano FL, Khattar E, Freitas MS, Romão L, Di Giovanni S, Lashuel HA, Silva JL, Foguel D (2011) The anti-Parkinsonian drug selegiline delays the nucleation phase of α-synuclein aggregation leading to the formation of nontoxic species. J Mol Biol 405:254–273. 10.1016/j.jmb.2010.10.027 [DOI] [PubMed] [Google Scholar]
- Burke WJ, Kumar VB, Pandey N, Panneton WM, Gan Q, Franko MW, O'Dell M, Li SW, Pan Y, Chung HD, Galvin JE (2008) Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol 115:193–203. 10.1007/s00401-007-0303-9 [DOI] [PubMed] [Google Scholar]
- Choi W, Zibaee S, Jakes R, Serpell LC, Davletov B, Crowther RA, Goedert M (2004) Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett 576:363–368. 10.1016/j.febslet.2004.09.038 [DOI] [PubMed] [Google Scholar]
- Cilia R, Akpalu A, Sarfo FS, Cham M, Amboni M, Cereda E, Fabbri M, Adjei P, Akassi J, Bonetti A, Pezzoli G (2014) The modern pre-levodopa era of Parkinson's disease: insights into motor complications from sub-Saharan Africa. Brain 137:2731–2742. 10.1093/brain/awu195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comellas G, Lemkau LR, Zhou DH, George JM, Rienstra CM (2012) Structural intermediates during α-synuclein fibrillogenesis on phospholipid vesicles. J Am Chem Soc 134:5090–5099. 10.1021/ja209019s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen JL, Dawson TM, Dickson DW, Petrucelli L (2003) Caught in the act: alpha-synuclein is the culprit in Parkinson's disease. Neuron 40:453–456. 10.1016/s0896-6273(03)00684-6 [DOI] [PubMed] [Google Scholar]
- Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A, Olanow CW, Tanner C, Marek K (2004) Levodopa and the progression of Parkinson's disease. N Engl J Med 351:2498–2508. [DOI] [PubMed] [Google Scholar]
- Finberg JP, Rabey JM (2016) Inhibitors of MAO-A and MAO-B in psychiatry and neurology. Front Pharmacol 7:340. 10.3389/fphar.2016.00340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flieger O, Engling A, Bucala R, Lue H, Nickel W, Bernhagen J (2003) Regulated secretion of macrophage migration inhibitory factor is mediated by a non-classical pathway involving an ABC transporter. FEBS Lett 551:78–86. 10.1016/s0014-5793(03)00900-1 [DOI] [PubMed] [Google Scholar]
- Fowler JS, Volkow ND, Wang GJ, Logan J, Pappas N, Shea C, MacGregor R (1997) Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol Aging 18:431–435. 10.1016/S0197-4580(97)00037-7 [DOI] [PubMed] [Google Scholar]
- Frye BC, Halfter S, Djudjaj S, Muehlenberg P, Weber S, Raffetseder U, En-Nia A, Knott H, Baron JM, Dooley S, Bernhagen J, Mertens PR (2009) Y-box protein-1 is actively secreted through a non-classical pathway and acts as an extracellular mitogen. EMBO Rep 10:783–789. 10.1038/embor.2009.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Masuda-Suzukake M, Falcon B (2017) Like prions: the propagation of aggregated tau and α-synuclein in neurodegeneration. Brain 140:266–278. 10.1093/brain/aww230 [DOI] [PubMed] [Google Scholar]
- Greffard S, Verny M, Bonnet AM, Seilhean D, Hauw JJ, Duyckaerts C (2010) A stable proportion of Lewy body bearing neurons in the substantia nigra suggests a model in which the Lewy body causes neuronal death. Neurobiol Aging 31:99–103. 10.1016/j.neurobiolaging.2008.03.015 [DOI] [PubMed] [Google Scholar]
- Jang A, Lee HJ, Suk JE, Jung JW, Kim KP, Lee SJ (2010) Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem 113:1263–1274. 10.1111/j.1471-4159.2010.06695.x [DOI] [PubMed] [Google Scholar]
- Kang SS, Ahn EH, Zhang Z, Liu X, Manfredsson FP, Sandoval IM, Dhakal S, Iuvone PM, Cao X, Ye K (2018) α-Synuclein stimulation of monoamine oxidase-B and legumain protease mediates the pathology of Parkinson's disease. EMBO J 24:e98878. 10.15252/embj.201798878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai S, Yoshihara T, Lopatina O, Ishihara K, Higashida H (2017) Selegiline ameliorates depression-like behavior in mice lacking the CD157/BST1 gene, a risk factor for Parkinson's disease. Front Behav Neurosci 11:75. 10.3389/fnbeh.2017.00075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Gee HY, Lee MG (2018) Unconventional protein secretion - new insights into the pathogenesis and therapeutic targets of human diseases. J Cell Sci 131:jcs213686. 10.1242/jcs.213686 [DOI] [PubMed] [Google Scholar]
- Lee HJ, Baek SM, Ho DH, Suk JE, Cho ED, Lee SJ (2011) Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp Mol Med 43:216–222. 10.3858/emm.2011.43.4.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Trojanowski JQ (2006) Mechanisms of Parkinson's disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron 52:33–38. 10.1016/j.neuron.2006.09.026 [DOI] [PubMed] [Google Scholar]
- Liu C, Qu L, Lian S, Tian Z, Zhao C, Meng L, Shou C (2014) Unconventional secretion of synuclein-gamma promotes tumor cell invasion. FEBS J 281:5159–5171. 10.1111/febs.13055 [DOI] [PubMed] [Google Scholar]
- Loebinger MR, Giangreco A, Groot KR, Prichard L, Allen K, Simpson C, Bazley L, Navani N, Tibrewal S, Davies D, Janes SM (2008) Squamous cell cancers contain a side population of stem-like cells that are made chemosensitive by ABC transporter blockade. Br J Cancer 98:380–387. 10.1038/sj.bjc.6604185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiya Y, Hara S, Arawaka S, Fukushima S, Sato H, Sakamoto M, Koyama S, Kato T (2010) Phosphorylated alpha-synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner. J Biol Chem 285:40732–40744. 10.1074/jbc.M110.141952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandler M, Valera E, Rockenstein E, Weninger H, Patrick C, Adame A, Santic R, Meindl S, Vigl B, Smrzka O, Schneeberger A, Mattner F, Masliah E (2014) Next-generation active immunization approach for synucleinopathies: implications for Parkinson's disease clinical trials. Acta Neuropathol 127:861–879. 10.1007/s00401-014-1256-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro HA, Gilmour BP, Lewin AH (2006) A rapid functional assay for the human trace amine-associated receptor 1 based on the mobilization of internal calcium. J Biomol Screen 11:688–693. 10.1177/1087057106289891 [DOI] [PubMed] [Google Scholar]
- Nonaka T, Iwatsubo T, Hasegawa M (2005) Ubiquitination of alpha-synuclein. Biochemistry 44:361–368. 10.1021/bi0485528 [DOI] [PubMed] [Google Scholar]
- Oueslati A, Schneider BL, Aebischer P, Lashuel HA (2013) Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo. Proc Natl Acad Sci USA 110:E3945–E3954. 10.1073/pnas.1309991110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poewe W, Antonini A, Zijlmans JC, Burkhard PR, Vingerhoets F (2010) Levodopa in the treatment of Parkinson's disease: an old drug still going strong. Clin Interv Aging 5:229–238. 10.2147/cia.s6456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP (2013) Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep 14:389–394. 10.1038/embor.2013.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutigliano G, Accorroni A, Zucchi R (2017) The case for TAAR1 as a modulator of central nervous system function. Front Pharmacol 8:987. 10.3389/fphar.2017.00987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A, Arawaka S, Sato H, Kato T (2015) Sensitive western blotting for detection of endogenous Ser129-phosphorylated α-synuclein in intracellular and extracellular spaces. Sci Rep 5:14211. 10.1038/srep14211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Arawaka S, Hara S, Fukushima S, Koga K, Koyama S, Kato T (2011) Authentically phosphorylated α-synuclein at Ser129 accelerates neurodegeneration in a rat model of familial Parkinson's disease. J Neurosci 31:16884–16894. 10.1523/JNEUROSCI.3967-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih JC (2004) Cloning, after cloning, knock-out mice, and physiological functions of MAO A and B. Neurotoxicology 25:21–30. 10.1016/S0161-813X(03)00112-8 [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840. 10.1038/42166 [DOI] [PubMed] [Google Scholar]
- Steiner JA, Quansah E, Brundin P (2018) The concept of alpha-synuclein as a prion-like protein: ten years after. Cell Tissue Res 373:161–173. 10.1007/s00441-018-2814-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarling EJ, de Aguiar Vallim TQ, Edwards PA (2013) Role of ABC transporters in lipid transport and human disease. Trends Endocrinol Metab 24:342–350. 10.1016/j.tem.2013.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JW, Hussaini SA, Bastille IM, Rodriguez GA, Mrejeru A, Rilett K, Sanders DW, Cook C, Fu H, Boonen RA, Herman M, Nahmani E, Emrani S, Figueroa YH, Diamond MI, Clelland CL, Wray S, Duff KE (2016) Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 19:1085–1092. 10.1038/nn.4328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Iwatsubo T (2018) Extracellular α-synuclein levels are regulated by neuronal activity. Mol Neurodegener 13:9. 10.1186/s13024-018-0241-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youdim MB, Gross A, Finberg JP (2001) Rasagiline [N-propargyl-1R(+)-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B. Br J Pharmacol 132:500–506. 10.1038/sj.bjp.0703826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Schuetz JD, Bunting KD, Colapietro AM, Sampath J, Morris JJ, Lagutina I, Grosveld GC, Osawa M, Nakauchi H, Sorrentino BP (2001) The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat Med 7:1028–1034. 10.1038/nm0901-1028 [DOI] [PubMed] [Google Scholar]
- Ztaou S, Maurice N, Camon J, Guiraudie-Capraz G, Kerkerian-Le Goff L, Beurrier C, Liberge M, Amalric M (2016) Involvement of striatal cholinergic interneurons and M1 and M4 muscarinic receptors in motor symptoms of Parkinson's disease. J Neurosci 36:9161–9172. 10.1523/JNEUROSCI.0873-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]