

Abstract
The aim of this study was to investigate the function and regulatory mechanism of long non-coding RNA (lncRNA) X-inactive-specific transcript (XIST) in cerebral ischemic stroke (CIS). The impact of lncRNA XIST on CIS was evaluated in acute CIS patients, middle cerebral artery occlusion (MCAO) mice, and oxygen-glucose deprivation and restoration brain endothelial cells. Our results demonstrated that the expression of lncRNA XIST decreased during the early stages of CIS but then increased in the later stages in CIS patients and ischemic models in vivo and in vitro. In addition, the serum levels of lncRNA XIST negatively correlated with severity of neurological impairment of CIS patients. Further studies exhibited that lncRNA XIST regulated the expression of proangiogenic factor-integrin α5 (Itgα5) and anti-inflammation factor-Kruppel-like transcription factor 4 (KLF4) by targeting microRNA-92a (miR-92a). Silencing of lncRNA XIST impaired angiogenesis and exacerbated cerebral vascular injury following CIS, leading to larger infarcts and worse neurological deficits in transient MCAO mice. Mechanistic analysis revealed that lncRNA XIST modulated angiogenesis and alleviated cerebral vascular injury following CIS through mediating the miR-92a/Itgα5 or KLF4 axis, respectively. These data indicate that lncRNA XIST confers protection against CIS, providing a valuable target for future prevention and treatment of CIS.
Keywords: cerebral ischemic stroke, lncRNA, miR-92a, angiogenesis, blood-brain barrier
Graphical abstract
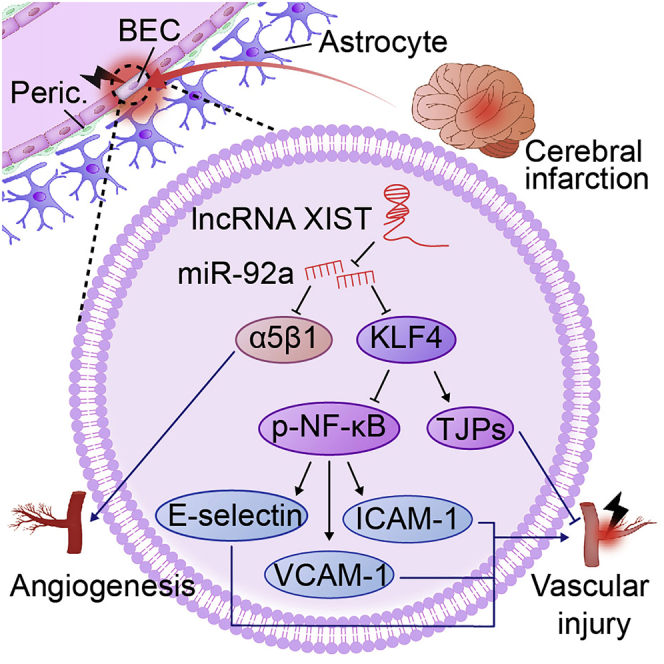
This study revealed that lncRNA XIST confers protection against cerebral ischemic injury, and the serum levels of lncRNA XIST mirror the severity of cerebral ischemic stroke (CIS), providing a potential biomarker for predicting the prognosis of CIS and also providing a valuable target for future prevention and treatment of CIS.
Introduction
Angiogenesis has been shown to be critical in improving neurological functional recovery after cerebral ischemic stroke (CIS);1,2 therefore, induction of angiogenesis is considered as a promising therapeutic strategy for CIS.
We and others have shown that extracellular matrix (ECM) protein fibronectin and its integrin receptors α5β1 and αvβ3 are strongly upregulated on angiogenic vessels in the ischemic penumbra and that the α5β1 integrin plays an essential role in promoting angiogenesis under hypoxic or ischemic conditions.3, 4, 5, 6, 7 However, like other proangiogenic factors,8 integrin α5 (Itgα5) has the potential to exaggerate blood-brain barrier (BBB) breakdown9,10 and worsen secondary neuronal injury, offsetting its beneficial angiogenic effects during the early stages of CIS.10
Interesting, immediately after the ischemic insult, many spontaneous protective mechanisms are activated to maintain cell homeostasis.11,12 The Kruppel-like transcription factor 4 (KLF4) is an evolutionarily conserved zinc finger-containing transcription factor involved in a variety of cellular functions by activating or repressing the transcriptional activity of multiple genes.13 There is evidence that KLF4 is required for the maintenance of endothelial and vascular integrity in the adult animal.14 More recently, we found that the serum level of KLF4 is negatively correlated with infarct volume in the CIS patients. High expression of KLF4 is always associated with relatively less vascular endothelial inflammation response in the ischemic hemisphere. Moreover, KLF4 can alleviate CIS-induced cerebral vascular injury by regulating endothelial expressions of inflammatory cell adhesion molecules (CAMs), nuclear factor κB (NF-κB), and tight junction proteins (TJPs),11,12 indicating that KLF4 confers vascular protection against cerebral ischemic injury.
MicroRNAs (miRNAs) are small non-coding RNAs that regulate gene expression by binding to target messenger RNAs (mRNAs), leading to translational repression or degradation. Our previous study demonstrated that the serum level of miRNA-92a (miR-92a) is upregulated in CIS patients with poor collaterals, and miR-92a level is negatively correlated with collateral circulation of the patients with CIS.15 Inhibition of miR-92a was found to lead to enhanced blood vessel growth and functional recovery in mouse models of limb ischemia and myocardial infarction. miR-92a appears to target mRNAs corresponding to several proangiogenic proteins, including Itgα5.16 In addition, knockdown of miR-92a in human arterial endothelial cells resulted in partial rescue from cytokine-induced proinflammatory marker expression that was attributable to enhanced KLF4 expression.17 Thus, miR-92a may serve as a valuable therapeutic target for angiogenesis and vascular protection.
Recently, increasing evidence has confirmed that long non-coding RNAs (lncRNAs) act as competing endogenous RNAs by interacting with miRNAs and regulating the expression of the miRNA target protein.18, 19, 20 In light of this, we conducted a bioinformatics analysis, which predicted a binding interaction between miR-92a and lncRNA X-inactive specific transcript (XIST). As a new-found lncRNA, lncRNA XIST has been shown to participate in the angiogenesis in several human diseases.21, 22, 23 However, its function and regulatory mechanism in CIS are poorly understood. The aim of the current study was to investigate the impact of lncRNA XIST on vascular remodeling and repair after CIS and uncover the underlying mechanisms of lncRNA XIST in modulating brain endothelial cell (BEC) function and angiogenesis after CIS.
Results
Demographic and clinical characteristics
Seventy-seven patients with CIS and sixty healthy controls were included in this study. The patients and the controls’ clinical characteristics are presented in Table S1.
The frequency rate of hypercholesterolemia, hypertension, diabetes, and atrial fibrillation in the stroke patients was significantly higher than that of the controls (p < 0.05 or p < 0.01). However, no significant difference was observed between the two groups in terms of age, gender, and nicotine/alcohol use and with regard to antiplatelet or statin treatments before stroke (Table S1).
The expression of lncRNA XIST is upregulated after CIS, and serum level of lncRNA XIST negatively correlates with severity of neurological impairment of CIS patients
To investigate the involvement of lncRNA XIST in CIS, blood samples from acute CIS patients were collected 48 h and 7 days after onset of the acute CIS event. As shown in Figure 1A, qRT-PCR revealed that the serum levels of lncRNA XIST in acute CIS patients decreased markedly at day 2 (48 h) (p < 0.001) but increased significantly at day 7 after ischemic stroke (p < 0.01) as compared to healthy controls.
Figure 1.

The expression of lncRNA XIST is upregulated and correlates with NIHSS scores after CIS
(A) Serum levels of lncRNA XIST in acute CIS patients and healthy controls were measured using qRT-PCR. Results are expressed as the mean ± standard deviation. Note that the serum levels of lncRNA XIST in CIS patients (n = 77) decreased at day 2 but increased at day 7 after ischemic onset as compared to healthy controls (n = 60). (B) The Pearson analyses revealed that serum levels of lncRNA XIST were significantly negatively correlated with NIHSS scores at day 7 after ischemic onset in acute CIS patients (n = 77). (C) The expression of lncRNA XIST in the ipsilateral ischemic cerebral cortex from sham-operated mice (sham; S, control) or mice at days 0, 1, 2, 4, and 7 post-ischemia was examined by qRT-PCR. Results are expressed as the mean ± standard deviation (n = 6 per experimental group). Note that the expression of lncRNA XIST decreased during the first 2 days post-ischemia but then increased between 4 and 7 days post-ischemia. (D) qRT-PCR analysis for lncRNA XIST levels after BECs was subject to OGD/R. Results are expressed as the mean ± standard deviation (n = 4 per experimental group). Note that within the first 6 h of OGD, lncRNA XIST levels decreased but then by 24–72 h, lncRNA XIST levels markedly increased compared to pre-OGD control levels. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 compared with control.
To further elucidate its clinical significance as a biomarker in CIS, lncRNA XIST levels were assessed in relation to National Institutes of Health Stroke Scale (NIHSS) scores obtained at day 7 after onset of the acute CIS event. The Pearson analyses displayed that serum levels of lncRNA XIST were significantly negatively correlated with NIHSS scores at day 7 after ischemic onset in the acute CIS patients (r = −0.636, p < 0.001) (Figure 1B).
Animal and cell-based models demonstrated similar trends in lncRNA XIST expression levels as those seen in human subjects (Figures 1C and 1D). As shown in Figure 1C, during the first 2 days following the ischemic insult, the expression of lncRNA XIST in the ipsilateral ischemic cerebral cortex decreased (p < 0.05 at day 1; p < 0.01 at day 2) but then increased significantly between 4 and 7 days post-ischemia in comparison with controls (p < 0.05 at day 4; p < 0.001 at day 7). In a parallel in vitro approach, we also investigated the influence of oxygen-glucose deprivation and restoration (OGD/R) on lncRNA XIST expression in bEnd3 cells using qRT-PCR. This revealed that within the first 6 h of OGD, lncRNA XIST levels decreased immediately (p < 0.05 or p < 0.01), but then by 24–72 h, lncRNA XIST levels were remarkably elevated compared to pre-OGD control levels (all p < 0.001) (Figure 1D).
Silencing of lncRNA XIST aggravates cerebral ischemia-induced brain damage
Based on the above findings that lncRNA XIST negatively correlated with the severity of neurological impairment of CIS patients, we speculated that lncRNA XIST might be playing a key role in alleviating ischemic brain damage following CIS. To confirm this, intraventricular injection of small hairpin RNA (shRNA)-lncRNA XIST (si-XIST) was used to knock down the expression of lncRNA XIST in the brain of middle cerebral artery occlusion (MCAO) mice. As shown in Figures 2A−2D, si-XIST-treated mice showed a larger cerebral infarct volume (Figures 2B−2C) at day 7 post-ischemic reperfusion (si-XIST versus negative controlshRNA [si-Ctl]: p < 0.01) and a more severe neurological deficit than did si-Ctl-treated mice in response to cerebral ischemic insults (si-XIST versus si-Ctl: p < 0.001 for day 2, p < 0.01 for day 4, and p < 0.05 for day 7) (Figure 2D). These data suggested that lncRNA XIST could alleviate cerebral ischemia-induced brain damage.
Figure 2.

Silencing of lncRNA XIST aggravates cerebral ischemia-induced brain damage in vivo
(A) Efficiency of shRNA-lncRNA XIST (si-XIST) in brain tissues of mice (n = 6). ∗∗∗p < 0.001. (B) Representative images of TTC staining from brain sections of sham or the negative control (NC) shRNA (si-Ctl) and si-XIST-treated mice at day 7 post-ischemic reperfusion. (C) Quantification of infarct volume after 7 days post-ischemic reperfusion. Results are expressed as the mean ± standard deviation and analyzed by one-way ANOVA (n = 6 mice per experimental group). Note that no infarct was seen in the control (sham) brain. Compared to the si-Ctl-treated mice, si-XIST-treated mice exhibited significantly larger infarct volumes at day 7 post-ischemic reperfusion. ∗∗p < 0.01. (D) The neurological functional performance was assessed using Modified Neurological Severity Score (mNSS) evaluation before and 2, 4, and 7 days after MCAO (n = 12 mice per experimental group). Note that si-XIST-treated mice had worse functional recovery than did si-Ctl-treated mice at days 2, 4, and 7 post-ischemic reperfusion. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001 versus si-Ctl-treated mice.
Silencing of lncRNA XIST impairs angiogenesis and exacerbates cerebral vascular injury following CIS by inhibiting the expression of Itgα5 or KLF4
In our previous studies, we demonstrated that fibronectin and its receptor α5β1 integrin are strongly upregulated on angiogenic vessels in the ischemic penumbra and that the α5β1 integrin plays an essential role in promoting angiogenesis under hypoxic or ischemic conditions.4,5,7,10,24 We also found that KLF4 confers vascular protection against cerebral ischemic injury by ameliorating vascular endothelial inflammation and regulating TJP expression following CIS.11 In the current study, we wanted to examine whether lncRNA XIST had impacts on the expressions of Itgα5 and KLF4 and consequently influenced the angiogenesis and cerebral vascular injury following CIS. To study this process, we use immunofluorescent (IF) or dual-IF staining to evaluate the expressions of Itgα5, KLF4, CD31/Ki67, E-selectin, vascular CAM 1 (VCAM-1), inter-CAM 1 (ICAM-1), Claudin-5, and zonula occludens-1 (ZO-1) in the ischemic hemisphere from sham-operated mice (sham) or si-Ctl and si-XIST-treated mice at day 7 post-ischemic reperfusion. This showed that compared to si-Ctl-treated mice, the number of Itgα5-, KLF4-, Claudin-5-, and ZO-1-positive events and CD31/Ki67 dual-positive cells/field in the ischemic penumbra was all significantly reduced (si-XIST versus si-Ctl: p < 0.05 for Itgα5, p < 0.01 for KLF4, p < 0.05 for Claudin-5, p < 0.05 for ZO-1, and p < 0.05 for CD31/Ki67 dual-positive cells), but the number of three CAMs, including E-selectin-, VCAM-1-, and ICAM-1-positive events/field, was remarkably induced in the si-XIST-treated mice after 7 days reperfusion (si-XIST versus si-Ctl: p < 0.05 for E-selectin, p < 0.05 for VCAM-1, and p < 0.01 for ICAM-1) (Figures 3A and 3B).
Figure 3.

Silencing of lncRNA XIST inhibits angiogenesis and increases cerebral vascular injury following CIS in vivo
(A) Images show the IF or dual-IF staining for Itgα5, KLF4, CD31 with Ki67, three CAMs (E-selectin, VCAM-1, and ICAM-1), Claudin-5, and ZO-1 in the ischemic hemisphere taken from sham or the negative control shRNA (si-Ctl) and shRNA-lncRNA XIST (si-XIST)-treated mice at day 7 post-ischemic reperfusion. Scale bar, 100 μm (insets, 30 μm). (B) Quantification of Itgα5, KLF4, CD31/Ki67, E-selectin, VCAM-1, ICAM-1, Claudin-5, and ZO-1 expressions. Results are expressed as the mean ± standard deviation and analyzed by one-way ANOVA (n = 6 mice per experimental group). Note that compared to si-Ctl-treated mice, the number of Itgα5-, KLF4-, Claudin-5-, and ZO-1-positive events and CD31/Ki67 dual-positive cells was all significantly reduced, but the number of three CAMs, including E-selectin-, VCAM-1-, and ICAM-1-positive events, was all remarkably induced in the si-XIST-treated mice at day 7 post-ischemic reperfusion. ##p < 0.01 and ###p < 0.001 versus control (sham); ∗p < 0.05 and ∗∗p < 0.01 versus si-Ctl-treated mice.
Consistent with in vivo studies, in vitro studies revealed that bEnd3 cells transfected with si-XIST showed decreased expressions of Itgα5, KLF4, and TJPs (Claudin-5 and ZO-1) (si-XIST versus si-Ctl: p < 0.01 for Itgα5; p < 0.01 for KLF4; p < 0.01 for Claudin-5; and p < 0.001 for ZO-1) but increased expressions of three CAMs including E-selectin, VCAM-1, and ICAM-1 relative to the si-Ctl-treated cells at 24 h restoration from OGD (si-XIST versus si-Ctl: all p < 0.001 for E-selectin, VCAM-1, and ICAM-1) (Figures 4A−4F, 4H, and 4I).
Figure 4.

The influence of silencing of lncRNA XIST on the migration and tube formation and the expression of Itgα5, KLF4, three CAMs, phosphorylation of (p)-NF-κB, and TJPs of bEnd3 cells under OGD/R conditions
(A) Representative images of western blot for the expression of Itgα5, KLF4, three CAMs (E-selectin, VCAM-1, and ICAM-1), p-NF-κB, and TJPs (Claudin-5 and ZO-1) in bEnd3 cells transfected with the negative control siRNA (si-Ctl) or siRNA-lncRNA XIST (si-XIST) at 24 h restoration from OGD. The NO-OGD/R cells served as a control. (B−I) Bar graphs show the quantitative analyses of western blots as ratios of Itgα5/β-actin (B), KLF4/β-actin (C), E-selectin/β-actin (D), VCAM-1/β-actin (E), ICAM-1/β-actin (F), p-NF-κB/total NF-κB (G), Claudin-5/β-actin (H), and ZO-1/β-actin (I) (n = 4 per experimental group). Note that the expressions of Itgα5, KLF4, and TJPs (Claudin-5 and ZO-1) in the si-XIST-treated bEnd3 cells were markedly reduced, but the expressions of three CAMs including E-selectin, VCAM-1, and ICAM-1 and the p-NF-κB were significantly elevated relative to the si-Ctl-treated group at 24 h restoration from OGD. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. (J) Representative images of cell migration and capillary tube formation of bEnd3 cells transfected with si-Ctl or si-XIST after OGD/R treatment. Scale bar, 400 μm for migration; 500 μm for tube formation. (K) Quantification of cell migration of bEnd3 cells. The relative migration distance was quantified using ImageJ software and expressed as the ratio to the values of the NO-OGD/R cells (control). (L) Quantification of tube formation of bEnd3 cells. Data represent the mean ± standard deviation and are analyzed by one-way ANOVA (n = 4 per experimental group). Note that the migration and capillary-like tube formation of the si-XIST-treated bEnd3 cells were both significantly induced after OGD/R relative to the NO-OGD/R control, but this effect was significantly inhibited by silencing of lncRNA XIST in bEnd3 cells. ∗p < 0.05 and ∗∗p < 0.01.
Induction of CAMs including E-selectin and VCAM-1 has previously been shown to be mediated by the tumor necrosis factor α (TNF-α)-NF-κB pathway in endothelial cells.25 In a recent study, we observed that KLF4 inhibited TNF-α-induced activation of NF-κB to alleviate the cerebral ischemia-induced cerebral vascular inflammation.11 As the above findings revealed that lncRNA XIST regulated expression of KLF4, we wondered whether lncRNA XIST had any influence on the activation of NF-κB following CIS. As expected, small interfering RNA (siRNA)-mediated knockdown of lncRNA XIST in bEnd3 cells showed increased phosphorylation (p) of -NF-κB relative to the si-Ctl-treated cells at 24 h restoration from OGD (si-XIST versus si-Ctl: p < 0.001) (Figure 4G), suggesting lncRNA XIST modulates the activation of NF-κB following CIS.
Because endothelial cell migration and tube formation constitute an important process in blood vessel formation, we also tested whether lncRNA XIST has any effect on migration and tube formation of bEnd3 cells under OGD/R conditions. We found that silencing of lncRNA XIST significantly decreased the migration and tube formation of bEnd3 cells relative to the si-Ctl-treated group after OGD/R treatment (si-XIST versus si-Ctl: p < 0.05 for migration; p < 0.01 for tube formation) (Figures 4J−4L). To further examine whether lncRNA XIST impaired the angiogenesis by inhibiting the expression of Itgα5, bEnd3 cells were co-transfected with si-XIST and Itgα5 and then were subjected to 4 h of OGD. From the western blot results, we found that Itgα5 expression in the si-XIST + α5-transfected group was significantly higher than that in the si-XIST + mock-transfected group at 24 h restoration from OGD (Figures 5A and 5B). Of interest, in this rescue experiment, bEnd3 cells co-transfected with si-XIST and Itgα5 displayed increased migration and tube formation than the si-XIST + mock-transfected group under OGD/R conditions (si-XIST + α5 group versus si-XIST + mock group: p < 0.05 for migration; p < 0.01 for tube formation) (Figures 5C−5E). Similarly, bEnd3 cells co-transfected with si-XIST and KLF4 showed significantly higher levels of Claudin-5 and ZO-1 (si-XIST + KLF4 group versus si-XIST + mock group: p < 0.05 for Claudin-5; p < 0.01 for ZO-1) but markedly reduced levels of E-selectin, VCAM-1, ICAM-1, and p-NF-κB (si-XIST + KLF4 group versus si-XIST + mock group: p < 0.05 for E-selectin; p < 0.01 for VCAM-1, ICAM-1, and p-NF-κB) relative to the si-XIST + mock-transfected group at 24 h restoration from OGD (Figures 5F−5M).
Figure 5.

Overexpression of Itgα5 or KLF4 reverses the effect of lncRNA XIST on the migration and tube formation and the expression of three CAMs, p-NF-κB, and TJPs of bEnd3 cells under OGD/R conditions
(A) Representative images of western blot for the expression of Itgα5 in bEnd3 cells co-transfected with si-XIST and Itgα5 overexpression plasmid at 24 h restoration from OGD. (B) Bar graphs show the quantitative analyses of western blots as ratios of Itgα5/β-actin (n = 4 per experimental group). (C) Representative images of cell migration and capillary tube formation of bEnd3 cells co-transfected with si-XIST and Itgα5 overexpression plasmid after OGD/R treatment. Scale bar, 400 μm for migration; 500 μm for tube formation. (D and E) Quantification of cell migration (D) and tube formation (E) of bEnd3 cells (n = 4 per experimental group). (F) Representative images of western blot for the expression of KLF4, three CAMs (E-selectin, VCAM-1, and ICAM-1), p-NF-κB, and TJPs (Claudin-5 and ZO-1) in bEnd3 cells co-transfected with si-XIST and KLF4 overexpression plasmid at 24 h restoration from OGD. (G−M) Bar graphs show the quantitative analyses of western blots as ratios of KLF4/β-actin (G), E-selectin/β-actin (H), VCAM-1/β-actin (I), ICAM-1/β-actin (J), p-NF-κB/total NF-κB (K), Claudin-5/β-actin (L), and ZO-1/β-actin (M) (n = 4 per experimental group). Note that bEnd3 cells co-transfected with si-XIST and Itgα5 displayed increased migration and tube formation than the si-XIST + mock-transfected group under OGD/R conditions, whereas bEnd3 cells co-transfected with si-XIST and KLF4 showed significantly higher levels of Claudin-5 and ZO-1 but markedly reduced levels of E-selectin, VCAM-1, ICAM-1, and p-NF-κB relative to the si-XIST + mock-transfected group at 24 h restoration from OGD. ∗p < 0.05 and ∗∗p < 0.01.
Collectively, these data suggest that silencing of lncRNA XIST impairs angiogenesis and increases the cerebral vascular injury following CIS by inhibiting the expression of Itgα5 or KLF4.
miR-92a is involved in the process of lncRNA XIST regulating angiogenesis and alleviating cerebral vascular injury
With the consideration of the miR-92a potential regulatory effect on Itgα5 or KLF4,16,17 we investigated whether miR-92a participates in the process of lncRNA XIST regulating angiogenesis and alleviating cerebral vascular injury. DIANA-LncBase version (v.) 2 was used for identify miR-92a, which could potentially bind to lncRNA XIST. As illustrated in Figure S1A, lncRNA XIST contains a potential binding site for miR-92a, and miR-92a could potentially bind to 3′ UTR of Itgα5 or KLF4. An RNA fluorescence in situ hybridization (FISH) assay showed that lncRNA XIST colocalized with miR-92a, and miR-92a had a colocalization relationship with Itgα5 or KLF4 in bEnd3 cells (Figure S1B). We then determined the expression of miR-92a in the ipsilateral ischemic cerebral cortex of MCAO mice and found that cerebral ischemia induced a strong increase in the expression of miR-92a in the ischemic hemisphere, reaching a peak at day 2 and then declining at day 4 (Figure S1C). Consistent with these findings, OGD/R induced expression of miR-92a on bEnd3 cells in a similar manner in vitro (Figure S1D). qRT-PCR analyses showed that upregulation or downregulation of miR-92a can be achieved by transfection with miR-92a mimics (miR-92a-M) or inhibitors (miR-92a-I) in bEnd3 cells in normoxia (Figure S1E) or at 24 h restoration of OGD (Figure S1F). The dual-luciferase report assay displayed that lncRNA XIST directly interacted with miR-92a (Figure S1G). Next, we evaluated how the miR-92a level varied with knockdown of lncRNA XIST by siRNA. As shown in Figure S1H, qRT-PCR analysis revealed that in the setting of lncRNA XIST knockdown, miR-92a levels were significantly elevated in bEnd3 cells at 24 h restoration from OGD (si-XIST versus si-Ctl: p < 0.01), suggesting lncRNA XIST decreased the expression of miR-92a after OGD/R. In addition, we employed a dual-luciferase report assay to validate interaction between miR-92a and Itgα5 or KLF4 and found that miR-92a-M reduced the luciferase activity of Itgα5-wild type (WT) or KLF4-WT but not of Itgα5 mutant (Mut) or KLF4 Mut (Figures S1I and S1J). The western blot analyses demonstrated that overexpression of miR-92a decreased, but inhibition of miR-92a increased the expression of Itgα5 and KLF4 in the bEnd3 cells at 24 h restoration from OGD (Figures S1K−S1M). These results indicate miR-92a directly targets Itgα5 or KLF4 after OGD/R.
To examine the role of miR-92a in lncRNA XIST mediating cerebral vascular injury following CIS, bEnd3 cells were co-transfected with the si-Ctl or si-XIST and/or miR-92a-I for 48 h and then subjected to 4 h of OGD followed by 24 h of restoration. The western blot analysis revealed that compare to the si-Ctl + negative control inhibitor (NC-I)-treated group, silencing of lncRNA XIST notably reduced the expressions of KLF4 and TJPs (Claudin-5 and ZO-1) (the si-XIST + NC-I group versus si-Ctl + NC-I group: p < 0.001 for KLF4; p < 0.01 for Claudin-5; p < 0.001 for ZO-1) but markedly increased expression of three CAMs (E-selectin, VCAM-1, and ICAM-1) and p-NF-κB (the si-XIST + NC-I group versus si-Ctl + NC-I group: all p < 0.001 for E-selectin, VCAM-1, ICAM-1, and p-NF-κB) in bEnd3 cells at 24 h restoration from OGD. However, these effects were significantly rescued by inhibiting the levels of miR-92a in the bEnd3 cells (p < 0.05 for KLF4; p < 0.05 for Claudin-5; p < 0.01 for ZO-1; p < 0.01 for E-selectin; p < 0.001 for VCAM-1, ICAM-1, and p-NF-κB) (Figures 6A and 6C−6I). These results suggest that lncRNA XIST regulates the expression of KLF4, CAMs, NF-κB as well as TJPs in bEnd3 cells after OGD/R via negatively modulating miR-92a expression.
Figure 6.

lncRNA XIST alleviates a vascular endothelial inflammation response and regulates migration and tube formation of bEnd3 cells under OGD/R conditions by targeting miR-92a
(A) Representative images of western blot for the expression of Itgα5, KLF4, E-selectin, VCAM-1, ICAM-1, p-NF-κB, Claudin-5, and ZO-1 in bEnd3 cells co-transfected with the si-Ctl or si-XIST and/or miR-92a inhibitor (miR-92a-I) at 24 h restoration from OGD. (B−I) Bar graphs show the quantitative analyses of western blots as ratios of Itgα5/β-actin (B), KLF4/β-actin (C), E-selectin/β-actin (D), VCAM-1/β-actin (E), ICAM-1/β-actin (F), p-NF-κB/total NF-κB (G), Claudin-5/β-actin (H), and ZO-1/β-actin (I) (n = 4 per experimental group). Note that the expressions of Itgα5, KLF4, and TJPs (Claudin-5 and ZO-1) in the si-XIST + NC inhibitor (NC-I)-treated bEnd3 cells markedly decreased, but the expressions of three CAMs including E-selectin, VCAM-1, and ICAM-1 and the p-NF-κB were significantly elevated relative to the si-Ctl + NC-I-treated group at 24 h restoration from OGD. However, these effects were significantly rescued by inhibiting the levels of miR-92a in the bEnd3 cells. ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. (J) Representative images of cell migration and capillary tube formation of bEnd3 cells transfected with si-Ctl or si-XIST and/or miR-92a-I after OGD/R treatment. Scale bar, 400 μm for migration; 500 μm for tube formation. (K and L) Quantification of cell migration (K) and tube formation (L) of bEnd3 cells (n = 4 per experimental group). Note that inhibition of miR-92a led to an increased migration and capillary-like tube formation of bEnd3 cells under OGD/R conditions, and miR-92a inhibition could reverse the inhibitory effects of XIST silencing on migration and tube formation of bEnd3 cells after OGD/R. ∗p < 0.05 and ∗∗p < 0.01.
We also found that inhibition of miR-92a led to an increased expression of Itgα5 and migration of bEnd3 cells after 24 h of wound scratch healing and enhanced capillary-like tube formation of bEnd3 cells after OGD/R treatment (the si-Ctl + miR-92a-I group versus si-Ctl + NC-I group: p < 0.01 for Itgα5; p < 0.05 for migration and tube formation). When lncRNA XIST was downregulated, the expression of Itgα5, the migration, and tube formation of bEnd3 cells in the si-XIST-treated group all markedly decreased relative to the si-Ctl-treated controls (the si-XIST + NC-I group versus si-Ctl + NC-I group: p < 0.001 for Itgα5; p < 0.01 for migration and tube formation), whereas inhibition of miR-92a remarkably reversed the inhibitory effects of lncRNA XIST silencing on expression of Itgα5, migration, and tube formation of bEnd3 cells after OGD/R (the si-XIST + miR-92a-I group versus si-XIST + NC-I group: all p < 0.05 for expression of Itgα5, migration, and tube formation) (Figures 6A, 6B, and 6J−6L). These results indicate that miR-92a is involved in the role lncRNA XIST of regulating angiogenesis via mediation of Itgα5 after OGD/R.
Discussion
In the current study, for the first time, we examined whether and how lncRNA XIST exerts its protective role against CIS. Our main findings were as follows: (1) the expression of lncRNA XIST is upregulated after CIS, and serum level of lncRNA XIST negatively correlates with severity of neurological impairment of CIS patients; (2) silencing of lncRNA XIST aggravates cerebral ischemia-induced brain damage; (3) silencing of lncRNA XIST impairs angiogenesis and exacerbates cerebral vascular injury following CIS by inhibiting the expression of Itgα5 and KLF4; and (4) miR-92a is involved in the process of lncRNA XIST regulating angiogenesis and alleviating cerebral vascular injury. Taken together, these data indicate that lncRNA XIST induces angiogenesis as well as alleviates cerebral vascular injury following CIS by mediating the miR-92a/Itgα5 or KLF4 axis, suggesting a novel approach for the treatment of CIS.
Silencing of lncRNA XIST impairs angiogenesis following CIS via mediation of miR-92a/Itgα5 axis
lncRNA XIST has been shown to participate in hypoxia-induced cell proliferation, migration, and tube formation.21 There is also evidence that knockdown of lncRNA XIST could inhibit the angiogenesis of glioma22 and chronic compressive spinal cord injury.23 Consistent with these reports, in the current study, we demonstrated that silencing of lncRNA XIST impaired angiogenesis following CIS in vitro and in vivo.
We noticed that the proangiogenic factor Itgα5 in BECs was correspondingly reduced after the expression of lncRNA XIST was diminished following CIS. However, of interest, in the rescue experiment, overexpression of Itgα5 in BECS could reverse the inhibiting effect of lncRNA XIST silencing on the migration and tube formation of bEnd3 cells under the ischemic condition. These results suggest that lncRNA XIST regulates angiogenesis following CIS by mediating BEC expression of Itgα5.
As inhibition of miR-92a was found to lead to enhanced blood vessel growth and functional recovery in mouse models of limb ischemia and myocardial infarction by targeting Itgα5,16 we wonder whether miR-92a was involved in the process of lncRNA XIST regulating angiogenesis via mediation of Itgα5 after CIS. To confirm this, first, we used RNA FISH, luciferase reporter assay, and co-transfection of si-XIST with miR-92a-I experiments to demonstrate that lncRNA XIST regulated the expression of Itgα5 by targeting miR-92a.
Subsequently, in functional research, we found that inhibition of miR-92a led to an increased migration as well as enhanced tube formation of bEnd3 cells in response to OGD/R, and inhibition of miR-92a remarkably reversed the inhibitory effects of lncRNA XIST silencing on migration and tube formation of bEnd3 cells after OGD/R. These results indicate that lncRNA XIST regulates angiogenesis through mediation of miR-92a following CIS.
We also noticed that the expressions of lncRNA XIST and miR-92a were both induced in the ischemic hemisphere or BECs following CIS, but their peak timing was different: when the expression of lncRNA XIST reached a peak, the expression of miR-92a drastically declined. It seems likely that the enhanced lncRNA XIST suppresses the cerebral vascular endothelial expression of miR-92a after CIS. This was further supported by the knockdown experiment that inhibition of lncRNA XIST significantly increased the levels of miR-92a in bEnd3 cells at 24 h restoration from OGD.
Silencing of lncRNA XIST exacerbates cerebral vascular injury following CIS by regulating the miR-92a/KLF4 axis
Early after the onset of CIS, endothelial cells of the arterial wall lose their tight junction and express inflammatory CAMs such as E-selectin, ICAM-1, and VCAM-1.26, 27, 28 These CAMs are supposed to participate in angiogenesis,29, 30, 31 and they are also widely believed to affect not only vascular permeability but also the vascular responses to the changes in the perivascular environment.32 In the current study, we found that three of the inflammatory CAMs including E-selectin, ICAM-1, and VCAM-1 were all elevated upon inhibition of lncRNA XIST following CIS in vitro and in vivo. However, in the same condition, silencing of lncRNA XIST impaired angiogenesis and deteriorated the BBB injury, indicating that the increased CAMs do not promote angiogenesis but act to exacerbate cerebral vascular injury following CIS.
The CAMs such as E-selectin are known to be expressed in proliferating endothelial cells under noninflammatory conditions, but it can also be induced by inflammatory stimuli.33,34 Induction of CAMs including E-selectin and VCAM-1 has previously been shown to be mediated by the TNF-α-NF-κB pathway in endothelial cells.25 In the current study, we found that siRNA-mediated knockdown of lncRNA XIST in bEnd3 cells led to increased p-NF-κB in response to OGD/R, suggesting that lncRNA XIST modulates the activation of NF-κB following CIS. These results, to some extent, explain why the proliferated BECs (CD31/Ki67 dual-positive cells) in the ischemic penumbra were significantly reduced, but CAMs were increased after the expression of lncRNA XIST was diminished following CIS in vivo.
In addition, during the early stages of angiogenesis, degradation of the ECM and separation of neighboring endothelial cells are crucial initiating steps, which suggests that newly formed vessels may transiently increase vascular permeability,35 which could result in aggravation of BBB damage caused by CIS.
A recent study showed that endothelial cell-specific Itgα5 deficiency increases the BBB integrity in an experimental mouse model of CIS,9 implying that inhibition of the fibronectin-α5β1 integrin signaling axis could attenuate the BBB permeability.
With the consideration of the inhibitory effects of silencing lncRNA XIST on Itgα5 expression and angiogenesis of BECs, we investigated whether inhibiting lncRNA XIST had any beneficial impact on the cerebral vascular injury after CIS. Unexpectedly, diminishing the expression of lncRNA XIST did not ameliorate but exacerbated the cerebral vascular injury after CIS, as evidenced by increased expression of three CAMs and p-NF-κB and decreased expression of TJPs including Claudin-5 and ZO-1 in vitro and in vivo. We further found that the underlying mechanism was related to the reduced expression of KLF4 induced by silencing of lncRNA XIST in BECs after CIS. As reported, KLF4 alleviates cerebral vascular injury by ameliorating vascular endothelial inflammation and regulating TJP expression following CIS;11 therefore, the decreased expression of KLF4 in BECs would result in the augmentation of cerebral vascular injury following CIS. This was further supported by the findings that overexpression of KLF4 significantly reversed the damaging effect of silencing of lncRNA XIST on cerebral blood vessels after OGD/R. These results suggest lncRNA XIST alleviates cerebral vascular injury following CIS by regulating BEC expression of KLF4.
It was reported that knockdown of miR-92a in human arterial endothelial cells resulted in partial rescue from cytokine-induced proinflammatory marker expression that was attributable to enhanced KLF4 expression.17 As lncRNA XIST has a potential regulatory effect on miR-92a, it is possible that lncRNA XIST suppresses the cerebral vascular injury following CIS through inhibition of miR-92a. In the current study, our RNA FISH assay, luciferase reporter assay, and co-transfection of si-XIST with miR-92a-I experiments revealed that lncRNA XIST regulated the expression of KLF4 by targeting miR-92a. We also found that inhibition of miR-92a rescues the augmented cerebral vascular injury induced by silencing of lncRNA XIST after OGD/R, indicating miR-92a is involved in the role of lncRNA XIST alleviating cerebral vascular injury following CIS through mediation of KLF4 expression.
Silencing of lncRNA XIST aggravates ischemia-induced brain damage, and the levels of lncRNA XIST mirror the severity of ischemic stroke
Evidence displayed that expression of CAMs is associated with cerebral infarct size.26 The activated NF-κB and increased CAMs are essential factors involved in ischemia-induced BBB damage.36 They promote migration of immune cells across the BBB within the cerebral parenchyma, which further exacerbates the brain tissue damage and leads to brain swelling and expansion of the infarction from the penumbra.37
Based on the above findings that silencing lncRNA XIST increased expression of three CAMs and p-NF-κB and decreased expression of TJPs under ischemic condition in vitro and in vivo, we speculated that lncRNA XIST might be playing a key role in alleviating ischemic brain damage following CIS. As expected, knockdown of lncRNA XIST increased the cerebral infarct volume and aggravated the neurological deficit following CIS.
We also noticed that the serum levels of lncRNA XIST in CIS patients decreased markedly at day 2 but increased significantly at day 7 after the onset of stroke. Furthermore, lncRNA XIST expression in the ischemic brain or BECs from an animal or cell-based model demonstrated similar trends as those seen in human subjects. Importantly, the Pearson analyses displayed that serum levels of lncRNA XIST were significantly negatively correlated with NIHSS scores at day 7 after ischemic onset in acute CIS patients, indicating lncRNA XIST exerts a protective role against cerebral ischemic injury.
In conclusion, our results demonstrate that lncRNA XIST confers protection against cerebral ischemic injury, and it can regulate angiogenesis and alleviate cerebral vascular injury following CIS by mediating the miR-92a/Itgα5 or KLF4 axis. We provide evidence that serum levels of lncRNA XIST might be used as a potential biomarker for predicting the prognosis of acute CIS and also provide a proof of concept for the further translational validation of overexpression of lncRNA XIST for the treatment of CIS patients in clinical practice in the future.
Limitations and future directions
Our study had several limitations. First, in the current study, only silencing experiments were performed for in vitro and in vivo study. In the next set of experiments, overexpressing experiments should be employed to directly investigate the impact of lncRNA XIST on the angiogenesis and cerebral injury after CIS. Second, as stroke is a sexually dimorphic disease, with differences between males and females observed both clinically and in the laboratory,38 the female mice should be investigated in the future. Third, future validation of the expression of lncRNA XIST, miR-92a, Itgα5, and KLF4 in human tissue will be helpful to better understand their intricate relationships following CIS.
Materials and methods
Patients
Seventy-seven patients with first-ever anterior circulation acute CIS were recruited from June 1, 2019, to December 31, 2020, at the Gongli Hospital, The Second Military Medical University, Shanghai, China. The included patients had to be admitted within 48 h from the onset of stroke or transient ischemic attack (TIA). Diagnosis had to be corroborated by a neurologist’s investigation and cerebral magnetic resonance imaging (MRI). Exclusion criteria were intracranial hemorrhage, pregnancy, presenting with a stroke with an undetermined time of onset, cancer, hematological diseases, severe renal or liver failure, recent myocardial infarction (less than 3 months previously), and ongoing treatment with anti-inflammatory drugs. Stroke severity was assessed using the NIHSS. Sixty age- and sex-matched healthy individuals were selected as the controls.
All enrolled patients provided written, informed consent. The study was performed in accordance with the principles of the Helsinki Declaration and was approved by the Ethics Committee of Gongli Hospital, The Second Military Medical University, Shanghai, China.
Animal treatment
Male C57BL/6 mice weighing 20−25 g (8−10 weeks of age) at the time of surgery were used for all experiments. Mice were housed individually in a regulated environment of humidity and temperature (12 h light/dark cycle, lights on at 8:00 a.m.) with standard mouse diet and water. To evaluate the role of lncRNA XIST in cerebral ischemia, adeno-associated virus 9 carrying the si-XIST (Genechem, Shanghai, China) or si-Ctl (5 × 1012 vg/mL in 5 μL PBS) was intraventricularly administered into the right lateral ventricle of the mice under anesthesia as previously described.39,40
3 weeks after injection, the si-Ctl or si-XIST-treated mice were subjected to intraluminal right MCAO under pentobarbital anesthesia to induce focal cerebral ischemia as described previously.11 After 90 min of MCAO, the blood flow was restored by removing the filament. Sham animals (controls) were subjected to the same procedure but did not receive either intraventricular administration or MCAO. Mice were allowed to recover and were euthanized at day 7 post-ischemia.
The animal study protocol was approved by the Animal Care and Use Committee of Gongli Hospital, The Second Military Medical University, Shanghai, China.
Assessment of neurological deficit and infarct volume
Neurobehavioral tests were assessed blindly before and 2, 4, and 7 days after MCAO using the Modified Neurological Severity Score (mNSS) (n = 12/group), as previously described.10 The severity score was graded at a scale from 0 to 14, in which 0 represents normal, and a higher score indicates a more severe injury.
After functional evaluation of neurological deficits, mice were euthanized at day 7 post-ischemia. The brains were removed and sliced into 2 mm coronal sections, which were stained with 2% triphenyl tetrazolium chloride (TTC) at room temperature for 10 min. The stained brain sections were captured with a digital camera. The infarct area of each brain was measured in a blinded manner, using SigmaScan Pro5 image analysis software. The total volumes of both the contralateral and ipsilateral hemisphere were measured, and the infarct percentage was calculated as percent contralateral structure to avoid mis-measurement secondary to edema.10
Cell culture and treatment
Immortalized mouse BECs of the bEnd3 cell line were obtained from Shanghai Bioleaf Biotech. The si-XIST (5′-UAUUAUAACAGUAAGUCUGAU-3′), si-Ctl, miR-92a-M, NC mimics (NC-M), miR-92a-I, and NC-I were designed and synthesized by RiboBio (Guangzhou, China). As previously reported,10,11 the murine Itga5 (GenBank: NM_010577.4) or KLF4 (GenBank: NM_010637.3) coding sequence was cloned into a pCDNA3.1 plasmid vector (Invitrogen, Carlsbad, CA, USA) through EcoRI and XhoI sites, and the pcDNA3.1 empty vector acted as NC.
For cell transfection, bEnd3 cells were cultured in 6-well plates at a density of 1 × 105 cells per well and transfected with siRNAs (si-XIST or si-Ctl), miR-92a-M, NC-M, miR-92a-I, NC-I, or sequencing-verified constructs alone or in combination using Lipofectamine 3000 (Invitrogen) in accordance with the manufacturer’s protocol. After 48 h of transfection, the cells were collected and used for further analysis.
To mimic ischemia-like conditions in vitro, bEnd3 cell cultures were exposed to OGD/R.11 In brief, 48 h after transfection, bEnd3 cell cultures were subjected to ischemia-like injury through OGD for 4 h by placing cultures in an anaerobic chamber (Forma; Thermo Scientific, Asheville, NC, USA) with an atmosphere of 5% CO2 and 95% N2 in a deoxygenated, glucose-free balanced salt solution (BSS0). After 4 h of OGD, cultures were returned to control conditions (restoration) by adding 5.5 mM glucose to the media under normoxic conditions. Control cultures (no injury) were incubated with a BSS containing 5.5 mM glucose (BSS5.5). All cultures were maintained in a humidified 37°C incubator.
Immunohistochemistry studies and antibodies
Mice were euthanized by perfusion with ice-cold saline, and the brains were rapidly dissected and stored at −80°C. IF studies were performed as previously described on 10 μm-thick frozen coronal sections.10,11 The following monoclonal antibodies from BD Pharmingen (La Jolla, CA, USA) were used in this study: phycoerythrin (PE)-conjugated rat anti-mouse Itgα5 (CD49e) (clone 5H10-27, 1:100) and fluorescein isothiocyanate (FITC)-conjugated rat anti-mouse CD31 (platelet endothelial cell adhesion molecule-1 [PECAM-1]) (clone MEC13.3, 553372, 1:200). The mouse anti-E-selectin monoclonal antibody (SC-137054, 1:100), mouse anti-ICAM-1 monoclonal antibody (SC-8439, 1:100), and mouse anti-VCAM-1 monoclonal antibody (SC-13160, 1:100) were obtained from Santa Cruz Biotechnology. The rabbit anti-KLF4 polyclonal antibody (ab129473, 1:500) and rabbit polyclonal antibodies against Ki67 (ab15580, 1:800) were obtained from Abcam. The rat anti-ZO-1 monoclonal antibody (R40.76, MABT11, 1:100; Merck Millipore, Darmstadt, Germany), mouse anti-Claudin 5 monoclonal antibody (4C3C2, 35-2500, 1:200; Invitrogen, Camarillo, CA, USA), and Alexa Fluor 488-conjugated goat anti-rat secondary antibody were obtained from Invitrogen (Carlsbad, CA, USA). Cy3-conjugated goat anti-rat secondary antibody was gotten from EarthOx (Millbrae, CA, USA). Cy3-conjugated goat anti-rabbit and anti-mouse secondary antibody were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). The NCs for staining and confocal imaging were used to confirm a coexistence of the vessel proteins.
Quantification of the number of positive cells for the different antigens was performed as previous reported.10,11 In brief, images of the region of interest were acquired using a ×20 objective on a Leica TCS SP5 II microscope to determine the number of positive events per field of view (FOV). A minimum of three serial brain sections per mouse was selected for analysis of each antigen and matched between mice so that the approximate position of sections used for IF staining was equivalent between different experimental conditions. Three images were taken from the ischemic penumbra, including cortex and striatum, as well as ischemic core of each brain section, and quantified by eye for the number of positive events per FOV. The number of antigen-positive events per FOV for each section was calculated as the mean of total numbers obtained from the three regions. These averages of three brain sections were used for statistical analysis for each mouse.
FISH
lncRNA XIST, miR-92a, Itgα5, and KLF4 probes were designed and synthesized by RiboBio (Guangzhou, China), and the probe sequences are available upon request. The probe signals were detected with a FISH kit (RiboBio, Guangzhou, China) according to the manufacturer’s instructions. Briefly, bEnd3 cells were fixed in 4% formaldehyde for 15 min at room temperature. After prehybridization in PBS, the cells were hybridized in hybridization solution at 37°C overnight. Then, cell nuclei underwent counterstaining by utilizing DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride) staining. Images were captured using a confocal microscope (Leica, Wetzlar, Germany).
Dual-luciferase reporter assay
The binding sites between lncRNA XIST and miR-92a, as well as miR-9a and Itgα5 or KLF4, were predicted using DIANA-LncBase v.2. A WT and Mut lncRNA XIST sequence and a WT and Mut 3′ UTR fragment of Itgα5 or KLF4 containing the putative miR-92a binding site were synthesized by Genechem (Shanghai, China). The constructs were cloned into the pGL3 luciferase reporter vector (Genechem, Shanghai, China) to generate XIST-WT, XIST-Mut, Itgα5-WT, Itgα5-Mut, KLF4-WT, and KLF4-Mut luciferase reporter systems. The mutations were confirmed by sequencing. Then, the luciferase reporter plasmids were co-transfected with miR-92a-M or NC-M (GenePharma, Shanghai, China) into the HEK293T cells. After 48 h of transfection, the luciferase activity was determined using the dual-luciferase reporter assay system (Promega, Madison, WI, USA) according to the manufacturer’s instructions. The relative luciferase activity was normalized to Renilla luciferase internal control.
RNA extraction and quantitative real-time PCR
Quantitative real-time PCR analysis was used to determine the expression of lncRNA XIST and miR-92a in CIS patients’ blood, brain tissue, or cultured cells. The blood samples were drawn from the forearms of acute CIS patients 48 h and 7 days after onset of the acute CIS event (n = 77), as well as the healthy controls (n = 60). The brain samples of the mice were taken from the ipsilateral ischemic cerebral cortex at different time points of reperfusion after MCAO (n = 6), and cell lysates were obtained from cultured bEnd3 cells at different time points of restoration following OGD (n = 4/group). Total RNA and miRNA were extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA), and the miRNeasy Mini Kit (QIAGEN, Hilden, Germany), respectively. RNA of a patient’s sample was reverse transcribed into cDNA using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific). For mouse sample and bEnd3 cells, the cDNA was synthesized using the PrimeScript RT Reagent Kit (Takara, Japan) for lncRNA, and the miScript II RT kit (QIAGEN, Germany) was used to convert RNA to cDNA for miRNA, respectively. Quantitative real-time PCR was conducted using the miScript SYBR Green PCR kit (QIAGEN, Germany) and an Applied Biosystems StepOnePlus Real-Time PCR system. The primer sequences used were as follows: lncRNA XIST forward, 5′-CTGCTGATCATTTGGTGGTGT-3′ and reverse, 5′-CTCTGCCTGACCTGCTATCAT-3′ (for patients’ sample); lncRNA XIST forward, 5′-TGCTCCACTTGAGACGGAAC-3′ and reverse, 5′-GGCTTCTGAGGTAGGGAGGA-3′ (for mouse sample and bEnd3 cell); miR-92a forward, 5′-ACACTCCAGCTGGGTATTGCACTTGTCCCG-3′ and reverse, 5′-TGGTGTCGTGGAGTCG-3′ (for patients’ sample); miR-92a forward, 5′-TATTGCACTTGTCCCGGCCTG-3′ (for mouse sample and bEnd3 cell); cel-miR-39-3p forward, 5′-ACACTCCAGCTGGGTCACCGGGTGTAAATC-3′ and reverse, 5′-TGGTGTCGTGGAGTCG-3′ (for patients’ sample). In addition, the sequences of miR-92a reverse universal and U6 primers (for mouse sample and bEnd3 cell) were provided from the QIAGEN kit. The average cycle threshold (Ct) value was normalized using the cel-miR-39-3p or U6 signal. Relative transcript levels were calculated using the 2−ΔΔCt method. Each lncRNA/miRNA level was expressed as the fold increase over the level of healthy control, sham control, or NO-OGD/R control group.
Western blot analysis
24 h after restoration from OGD, bEnd3 cells were harvested and lysed with lysis buffer (1% Nonidet P-40 [NP-40], 50 mM Tris HCl, pH 8.0, 150 mM sodium chloride) supplemented with protease and phosphatase inhibitor cocktails. Protein concentration was determined using the bicinchoninic acid (BCA) protein assay kit (Eppendorf-Bio Photometer, Germany). Western blotting and semiquantitative analyses were performed as described previously.10,11 The following primary antibodies were purchased from Invitrogen (Carlsbad, CA, USA): Armenian hamster anti-ICAM-1 monoclonal antibody (3E2B, MA5405, 1:20), rabbit anti-NF-κB polyclonal antibody (51-3500, 1:1,000), rabbit anti-p-NF-κB polyclonal antibody (PA5-37658, 1:1,000), rabbit anti-E-selectin monoclonal antibody (15, MA5-29785, 1:1,000), rabbit anti-VCAM-1 monoclonal antibody (SA05-04, MA5-31965, 1:1,000), rabbit anti-KLF4 (PA5-27440, 1:5,000), rabbit anti-Claudin-5 polyclonal antibody (34-1600, 1:170), and rabbit anti-ZO-1 polyclonal antibody (61-7300, 1:1,000). The rabbit anti-Itgα5 polyclonal antibody (AB1928, 1:1,000) was gotten from Merck Millipore (Darmstadt, Germany), and β-actin was obtained from Neomarker (Fremont, CA, USA; 1:1,000,). Within each sample, levels of proteins were first normalized to the level of β-actin and then expressed as the fold increase over the level of the NO-OGD/R control group.
Migration assay and tube formation assay
Migration assay and tube formation assay were analyzed as previously reported.6 For the scratch wound healing assay, after 48 h of transfection, bEnd3 cells in 6-well tissue-culture plates (5 × 105 cells per well) were subjected to OGD for 4 h, followed by 24 h of restoration. Then, the cells were scratched and left to heal for 24 h at normoxia. Photographs were taken at the same site 0 and 24 h after the injury. The healing of the wounds was assessed by measuring the wound gap.
To detect tube formation capacity, bEnd3 cells were added to Matrigel-coated 24-well plates at a density of 1 × 105 cells/well after transfection and OGD/R treatment and incubated in normal condition for 8 h. Then, four representative fields were selected for imaging, and the average total areas of complete tubes formed by cells per unit area were compared. The experiments were undertaken in triplicate and were blindly analyzed.
Statistical analysis
Categorical variables were expressed as counts (percentage) and continuous variables as mean ± standard deviation unless otherwise indicated. Statistical significance was assessed by the t test, chi-square test, and one- or two-way analysis of variance (ANOVA), and a Bonferroni post hoc test was used to test multiple comparisons. Correlations were assessed using the Pearson’s method. All statistical analyses were performed with SPSS (v.16.0; SPSS, Chicago, IL, USA) and significance was defined as p < 0.05.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (no. 81771328, 82171462), Key Specialty Construction Project of the Shanghai Municipal Commission of Health and Family Planning (no. ZK2019A08), Program for the Development of Science and Technology of Pudong Science and Technology Committee of Shanghai (no. PKJ2019-Y20), “Chunhui Plan” of Ministry of Education of China (no. HLJ2019012), and Heilongjiang Province Postdoctoral Startup Foundation (no. LBH-Q19033).
Author contributions
C.W., J.D., and J.S. conducted the experiments. S.H. analyzed the data. F.W., X.Z., and Y.F. performed the neurological evaluation and contributed to the data interpretation. D.P. assisted with the cell culture and western blot. L.L. conceived and designed the experiments, analyzed and interpreted the data, and wrote the manuscript. All authors read and approved the final manuscript.
Declaration of interests
All sources of funding for the research declare that they have no competing financial or personal interests and that none of the authors’ institutions have contracts relating to this research through which they may stand to gain financially now or in the future. The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.06.025.
Supplemental information
References
- 1.Ruan L., Wang B., ZhuGe Q., Jin K. Coupling of neurogenesis and angiogenesis after ischemic stroke. Brain Res. 2015;1623:166–173. doi: 10.1016/j.brainres.2015.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen J., Venkat P., Zacharek A., Chopp M. Neurorestorative therapy for stroke. Front. Hum. Neurosci. 2014;8:382. doi: 10.3389/fnhum.2014.00382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.del Zoppo G.J., Milner R. Integrin-matrix interactions in the cerebral microvasculature. Arterioscler. Thromb. Vasc. Biol. 2006;26:1966–1975. doi: 10.1161/01.ATV.0000232525.65682.a2. [DOI] [PubMed] [Google Scholar]
- 4.Li L., Liu F., Welser-Alves J.V., McCullough L.D., Milner R. Upregulation of fibronectin and the α5β1 and αvβ3 integrins on blood vessels within the cerebral ischemic penumbra. Exp. Neurol. 2012;233:283–291. doi: 10.1016/j.expneurol.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li L., Welser-Alves J., van der Flier A., Boroujerdi A., Hynes R.O., Milner R. An angiogenic role for the α5β1 integrin in promoting endothelial cell proliferation during cerebral hypoxia. Exp. Neurol. 2012;237:46–54. doi: 10.1016/j.expneurol.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pang D., Wang L., Dong J., Lai X., Huang Q., Milner R., Li L. Integrin α5β1-Ang1/Tie2 receptor cross-talk regulates brain endothelial cell responses following cerebral ischemia. Exp. Mol. Med. 2018;50:1–12. doi: 10.1038/s12276-018-0145-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun J., Yu L., Huang S., Lai X., Milner R., Li L. Vascular expression of angiopoietin1, α5β1 integrin and tight junction proteins is tightly regulated during vascular remodeling in the post-ischemic brain. Neuroscience. 2017;362:248–256. doi: 10.1016/j.neuroscience.2017.08.040. [DOI] [PubMed] [Google Scholar]
- 8.Croll S.D., Ransohoff R.M., Cai N., Zhang Q., Martin F.J., Wei T., Kasselman L.J., Kintner J., Murphy A.J., Yancopoulos G.D., Wiegand S.J. VEGF-mediated inflammation precedes angiogenesis in adult brain. Exp. Neurol. 2004;187:388–402. doi: 10.1016/j.expneurol.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 9.Roberts J., de Hoog L., Bix G.J. Mice deficient in endothelial α5 integrin are profoundly resistant to experimental ischemic stroke. J. Cereb. Blood Flow Metab. 2017;37:85–96. doi: 10.1177/0271678X15616979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang L., Zhang X., Liu X., Feng G., Fu Y., Milner R., Li L. Overexpression of α5β1 integrin and angiopoietin-1 co-operatively promote blood-brain barrier integrity and angiogenesis following ischemic stroke. Exp. Neurol. 2019;321:113042. doi: 10.1016/j.expneurol.2019.113042. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X., Wang L., Han Z., Dong J., Pang D., Fu Y., Li L. KLF4 alleviates cerebral vascular injury by ameliorating vascular endothelial inflammation and regulating tight junction protein expression following ischemic stroke. J. Neuroinflammation. 2020;17:107. doi: 10.1186/s12974-020-01780-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang H., Xi X., Zhao B., Su Z., Wang Z. KLF4 protects brain microvascular endothelial cells from ischemic stroke induced apoptosis by transcriptionally activating MALAT1. Biochem. Biophys. Res. Commun. 2018;495:2376–2382. doi: 10.1016/j.bbrc.2017.11.205. [DOI] [PubMed] [Google Scholar]
- 13.Ghaleb A.M., Yang V.W. Krüppel-like factor 4 (KLF4): What we currently know. Gene. 2017;611:27–37. doi: 10.1016/j.gene.2017.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sangwung P., Zhou G., Nayak L., Chan E.R., Kumar S., Kang D.W., Zhang R., Liao X., Lu Y., Sugi K., et al. KLF2 and KLF4 control endothelial identity and vascular integrity. JCI Insight. 2017;2:e91700. doi: 10.1172/jci.insight.91700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng B., Wu D., Sun J., Huang S., Wu F., Gu X., Zhu W., Zhou F., Li L. The correlation between miRNAs levels and collateral pathway in the patients with acute cerebral infraction. J. Apoplexy Nervous Dis. 2016;33:100–103. [Google Scholar]
- 16.Bonauer A., Carmona G., Iwasaki M., Mione M., Koyanagi M., Fischer A., Burchfield J., Fox H., Doebele C., Ohtani K., et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–1713. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- 17.Fang Y., Davies P.F. Site-specific microRNA-92a regulation of Kruppel-like factors 4 and 2 in atherosusceptible endothelium. Arterioscler. Thromb. Vasc. Biol. 2012;32:979–987. doi: 10.1161/ATVBAHA.111.244053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cesana M., Cacchiarelli D., Legnini I., Santini T., Sthandier O., Chinappi M., Tramontano A., Bozzoni I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147:358–369. doi: 10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X., Hou L., Huang W., Gao Y., Lv X., Tang J. The Mechanism of Long Non-coding RNA MEG3 for Neurons Apoptosis Caused by Hypoxia: Mediated by miR-181b-12/15-LOX Signaling Pathway. Front. Cell. Neurosci. 2016;10:201. doi: 10.3389/fncel.2016.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tay Y., Rinn J., Pandolfi P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–352. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu C., Bai X., Liu C., Hu Z. Long noncoding RNA XIST participates hypoxia-induced angiogenesis in human brain microvascular endothelial cells through regulating miR-485/SOX7 axis. Am. J. Transl. Res. 2019;11:6487–6497. [PMC free article] [PubMed] [Google Scholar]
- 22.Yu H., Xue Y., Wang P., Liu X., Ma J., Zheng J., Li Z., Li Z., Cai H., Liu Y. Knockdown of long non-coding RNA XIST increases blood-tumor barrier permeability and inhibits glioma angiogenesis by targeting miR-137. Oncogenesis. 2017;6:e303. doi: 10.1038/oncsis.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng X., Xu J., Yu Z., Xu J., Long H. LncRNA Xist Contributes to Endogenous Neurological Repair After Chronic Compressive Spinal Cord Injury by Promoting Angiogenesis Through the miR-32-5p/Notch-1 Axis. Front. Cell Dev. Biol. 2020;8:744. doi: 10.3389/fcell.2020.00744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang H., Huang Q., Wang F., Milner R., Li L. Cerebral ischemia-induced angiogenesis is dependent on tumor necrosis factor receptor 1-mediated upregulation of α5β1 and αVβ3 integrins. J. Neuroinflammation. 2016;13:227. doi: 10.1186/s12974-016-0697-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collins T., Read M.A., Neish A.S., Whitley M.Z., Thanos D., Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- 26.Frijns C.J., Kappelle L.J. Inflammatory cell adhesion molecules in ischemic cerebrovascular disease. Stroke. 2002;33:2115–2122. doi: 10.1161/01.str.0000021902.33129.69. [DOI] [PubMed] [Google Scholar]
- 27.O’Carroll S.J., Kho D.T., Wiltshire R., Nelson V., Rotimi O., Johnson R., Angel C.E., Graham E.S. Pro-inflammatory TNFα and IL-1β differentially regulate the inflammatory phenotype of brain microvascular endothelial cells. J. Neuroinflammation. 2015;12:131. doi: 10.1186/s12974-015-0346-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizuma A., Yenari M.A. Anti-Inflammatory Targets for the Treatment of Reperfusion Injury in Stroke. Front. Neurol. 2017;8:467. doi: 10.3389/fneur.2017.00467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bischoff J. Cell adhesion and angiogenesis. J. Clin. Invest. 1997;99:373–376. doi: 10.1172/JCI119168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim T.K., Park C.S., Na H.J., Lee K., Yoon A., Chung J., Lee S. Ig-like domain 6 of VCAM-1 is a potential therapeutic target in TNFα-induced angiogenesis. Exp. Mol. Med. 2017;49:e294. doi: 10.1038/emm.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langston W., Chidlow J.H., Jr., Booth B.A., Barlow S.C., Lefer D.J., Patel R.P., Kevil C.G. Regulation of endothelial glutathione by ICAM-1 governs VEGF-A-mediated eNOS activity and angiogenesis. Free Radic. Biol. Med. 2007;42:720–729. doi: 10.1016/j.freeradbiomed.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodrigues S.F., Granger D.N. Blood cells and endothelial barrier function. Tissue Barriers. 2015;3:e978720. doi: 10.4161/21688370.2014.978720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo J., Paranya G., Bischoff J. Noninflammatory expression of E-selectin is regulated by cell growth. Blood. 1999;93:3785–3791. [PubMed] [Google Scholar]
- 34.Bischoff J., Brasel C., Kräling B., Vranovska K. E-selectin is upregulated in proliferating endothelial cells in vitro. Microcirculation. 1997;4:279–287. doi: 10.3109/10739689709146791. [DOI] [PubMed] [Google Scholar]
- 35.Vallon M., Chang J., Zhang H., Kuo C.J. Developmental and pathological angiogenesis in the central nervous system. Cell. Mol. Life Sci. 2014;71:3489–3506. doi: 10.1007/s00018-014-1625-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu P.Y., Zhang Z., Liu Y., Tang X.L., Shu S., Bao X.Y., Zhang Y., Gu Y., Xu Y., Cao X. TMEM16A Inhibition Preserves Blood-Brain Barrier Integrity After Ischemic Stroke. Front. Cell. Neurosci. 2019;13:360. doi: 10.3389/fncel.2019.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yilmaz G., Granger D.N. Leukocyte recruitment and ischemic brain injury. Neuromolecular Med. 2010;12:193–204. doi: 10.1007/s12017-009-8074-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turtzo L.C., McCullough L.D. Sex-specific responses to stroke. Future Neurol. 2010;5:47–59. doi: 10.2217/fnl.09.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qu J., Zhao H., Li Q., Pan P., Ma K., Liu X., Feng H., Chen Y. MST1 Suppression Reduces Early Brain Injury by Inhibiting the NF-κB/MMP-9 Pathway after Subarachnoid Hemorrhage in Mice. Behav. Neurol. 2018;2018:6470957. doi: 10.1155/2018/6470957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gong Y., Mu D., Prabhakar S., Moser A., Musolino P., Ren J., Breakefield X.O., Maguire C.A., Eichler F.S. Adenoassociated virus serotype 9-mediated gene therapy for x-linked adrenoleukodystrophy. Mol. Ther. 2015;23:824–834. doi: 10.1038/mt.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.