

Abstract
Oncolytic virus therapy can increase the immunogenicity of tumors and remodel the immunosuppressive tumor microenvironment, leading to an increased antitumor response to immune-checkpoint inhibitors. Here, we investigated the therapeutic potential of G47Δ, a third-generation oncolytic herpes simplex virus type 1, in combination with immune-checkpoint inhibitors using various syngeneic murine subcutaneous tumor models. Intratumoral inoculations with G47Δ and systemic anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) antibody administration caused an enhanced antitumor activity when combined and worked synergistically. Conversely, the efficacy of G47Δ in combination with anti-programmed cell death protein-1 (PD-1) antibody was equivalent to that of the anti-PD-1 antibody alone in all murine models examined. The combination of intratumoral G47Δ and systemic anti-CTLA-4 antibody was shown to recruit effector T cells into the tumor efficiently while decreasing regulatory T cells. Furthermore, a wide range of gene signatures related to inflammation, lymphoid lineage, and T cell activation was highly upregulated with the combination therapy, suggesting the conversion of immune-insusceptible tumors to immune susceptible. The therapeutic effect proved tumor specific and long lasting. Immune cell subset depletion studies demonstrated that CD4+ T cells were required for synergistic curative activity. The results depict the dynamics of immune modulation of the tumor microenvironment and provide a clinical rationale for using G47Δ with immune checkpoint inhibitors.
Keywords: oncolytic virus therapy, G47Δ, CTLA-4, PD-1, immune-checkpoint inhibitors, herpes simplex virus type 1, tumor microenvironment, antitumor immunity, esophageal carcinoma, regulatory T cells
Graphical abstract
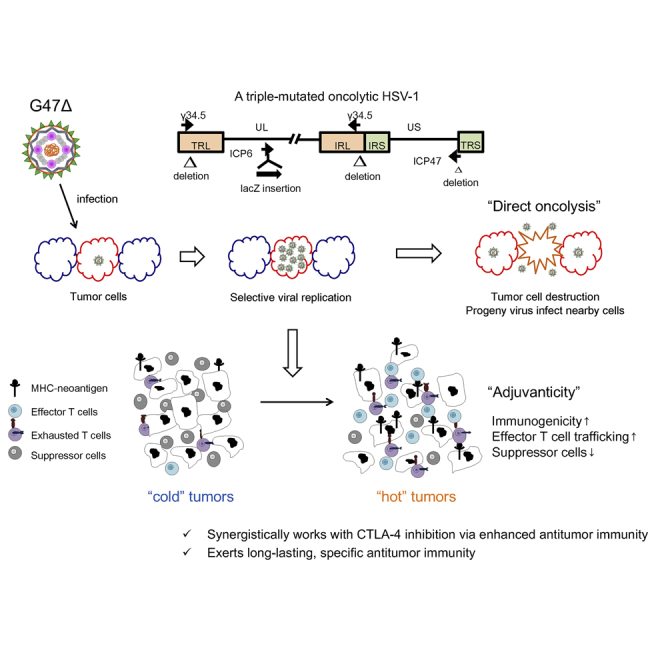
G47Δ, a third-generation oncolytic HSV-1, works synergistically with CTLA-4 inhibition, but not PD-1, in murine tumor models. The combination therapy causes increased effector T cells, decreased Tregs, and upregulated gene signatures related to immunogenicity within tumors. The therapeutic effect proved tumor specific and long lasting and requires CD4+ cells for synergy.
Introduction
The advent of cancer immunotherapy has caused a paradigm shift in the treatment of malignancies. Therapies using immune-checkpoint inhibitors (ICIs) targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed cell death protein-1 (PD-1), or programmed cell death ligand 1 (PD-L1) have proved to be efficacious for various cancers.1,2 However, abundant evidence has also revealed that durable objective responses to ICIs are limited to a fraction of patients.3,4 Further novel therapies are needed that can facilitate the priming of the immune system with a tumor neoantigen, lead effector T cell trafficking, and reverse the immunosuppressive tumor microenvironment (TME).5,6
Oncolytic viruses can selectively kill cancer cells, leading to bolstering of neoantigen-specific cytotoxic T cell responses.7,8 Further, oncolytic viruses potentially change immunogenically “cold” tumors to “hot” tumors, resulting in immune-mediated tumor eradication.9 Because of such capability to induce specific antitumor immunity, it appears logical to combine oncolytic viruses with ICIs for treating cancers. In fact, previous preclinical studies have revealed the therapeutic potential of various oncolytic viruses in combination with ICIs including CTLA-4 inhibition9, 10, 11, 12 and PD-1 inhibition.13,14 In addition, recent clinical trials have demonstrated promising results of the combination strategy.15,16
G47Δ is a triple-mutated, third-generation oncolytic herpes simplex virus type 1 (HSV-1) that was developed by adding another mutation, i.e., the deletion of the α47 gene, to the genome of a second-generation oncolytic HSV-1, G207.17 Because the α47 gene product inhibits the transporter associated with antigen presentation, the gene deletion prevents the downregulation of major histocompatibility complex (MHC) class I, which normally occurs in human cells after infection with HSV-1.18 Human melanoma cells infected with G47Δ were better at stimulating their matched tumor-infiltrating lymphocytes than those infected with G207.17 The enhanced immune-stimulation capability of G47Δ is therefore especially suited for combining with ICIs. The α47 deletion also places the late US11 gene under control of the immediate-early α47 promoter, resulting in enhanced viral replication capability in tumor cells.17 In preclinical studies, G47Δ exhibited robust antitumor efficacy while retaining excellent safety features.19, 20, 21 Notably, the investigator-initiated phase II clinical trial (UMIN-Clinical Trials Registry [CTR]: UMIN000015995) in patients with glioblastoma has recently been completed with good results (unpublished data), and G47Δ received a governmental approval as a new drug in Japan. G47Δ has also been used in clinical trials for prostate cancer (UMIN-CTR: UMIN000010463), olfactory neuroblastoma (Japan Registry of Clinical Trials: jRCTs033180325), and malignant pleural mesothelioma (Japan Registry of Clinical Trials: jRCTs033180326).
Here, we investigate the efficacy of G47Δ in combination with ICIs using various syngeneic murine tumor models and further elucidate the immunological mechanisms of the synergistic activities.
Results
Cytopathic effect and replication capability of G47Δ in murine cancer cells
Prior to investigating immunocompetent tumor models, we evaluated the cytopathic effects and replication capabilities of G47Δ in three murine cancer cell lines in vitro. In C57BL/6-derived murine esophageal carcinoma cell lines, AKR and HNM007, G47Δ at a multiplicity of infection (MOI) of 1.0 showed >70% cell destruction within 4 days after infection (Figure 1A). Both cell lines supported G47Δ replication at 24 h after infection at an MOI of 0.1, but the yields of progeny virus did not increase by 48 h (Figure 1B), possibly due to a low HSV-1 susceptibility of the mouse strain C57BL/6.22 The C3H-derived murine squamous cell carcinoma (SCC) cell line SCCVII was more susceptible to G47Δ than AKR and HNM007 and rapidly killed by G47Δ even at an MOI of 0.01 (Figure 1A). Higher virus yields were recovered from SCCVII cells compared with AKR or HNM007 cells (Figure 1B).
Figure 1.

In vitro and in vivo effects of G47Δ in murine carcinomas
(A) Cytopathic effects of G47Δ in vitro. Murine cancer cells were infected with G47Δ (AKR, HNM007, MOI of 0.1 [●] or 1 [○]; SCCVII, MOI of 0.01 [▲] or 0.1 [●]) or mock. Cell viability was expressed as a percentage of the mock-infected controls. G47Δ exhibited a good cytopathic effect in AKR and HNM007 cell lines at an MOI of 1.0 and in SCCVII at an MOI of 0.1. Data are presented as the mean of triplicates ± SD. (B) Virus yields of G47Δ in vitro. Murine cancer cells were infected with G47Δ at an MOI of 0.1, and recovered virus yields were determined at 24 and 48 h after infection. All murine cancer cell lines tested supported the replication of G47Δ to a certain extent. The results are presented as the mean of triplicates ± SD. (C) Four syngeneic murine subcutaneous tumor models (AKR [upper left], HNM007 [upper right], SCCVII [lower left], and B16-F10 [lower right]) were used. Established tumors, 5–6 mm in diameter, were inoculated with G47Δ (2 × 105 for SCCVII and 5 × 106 PFUs for others) or mock on days 0 and 3. The G47Δ treatment significantly inhibited the growth of subcutaneous tumors compared with the mock treatment in all models. The results are presented as the mean ± SEM (n = 7). One-way ANOVA followed by Dunnett’s test was used to determine statistical significance (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
Efficacy of G47Δ alone in syngeneic murine subcutaneous tumor models
The in vivo efficacy of G47Δ was studied in four syngeneic murine subcutaneous tumor models: AKR, HNM007, SCCVII, and C57BL/6-derived melanoma B16-F10. In all models, intratumoral injections with G47Δ significantly inhibited the growth of subcutaneous tumors compared with the mock treatment (AKR, p < 0.05 on day 21; HNM007, p < 0.01 on day 19; SCCVII, p < 0.01 on day 23; B16-F10, p < 0.01 on day 14; Figure 1C).
Efficacy of G47Δ in combination with ICIs
We examined whether the efficacy of G47Δ can be augmented when combined with systemic CTLA-4 or PD-1 inhibition. C57BL/6 mice harboring subcutaneous AKR tumors were treated with intratumoral injections with G47Δ (5 × 106 plaque-forming units [PFUs]) or mock in combination with intraperitoneal injections with the anti-CTLA-4 antibody (25 μg; Figure 2A), anti-PD-1 antibody (100 μg; Figure 2E), or isotype controls. G47Δ alone and CTLA-4 inhibition alone both caused significant delay in tumor growth compared with control (G47Δ versus control, p < 0.05; αCTLA-4 versus control, p < 0.01; Figure 2B). The combination therapy markedly inhibited the tumor growth compared with each monotherapy (versus G47Δ, p < 0.001; versus αCTLA-4, p < 0.01; Figure 2B), causing a cure in 5/8 animals (Figure 2C) and a significant prolongation of survival (p < 0.001 versus control; G47Δ, p < 0.01 versus αCTLA-4; Figure 2D). In this subcutaneous AKR tumor model, G47Δ and CTLA-4 inhibition worked synergistically, with a combination index (CI) of 0.67 on day 8 and 0.31 on day 12 (Table S2). However, in the same model, the efficacy of the combination of G47Δ and the anti-PD-1 antibody did not significantly differ from that of the anti-PD-1 antibody alone (Figures 2E−2H).
Figure 2.

Efficacy of G47Δ in combination with CTLA-4 or PD-1 inhibition in a murine subcutaneous AKR tumor model
(A−D) Effects of G47Δ or CTLA-4 inhibition, either alone or in combination, on tumor growth in the murine subcutaneous AKR tumor model. (A) Experimental design. C57BL/6 mice harboring unilateral subcutaneous AKR tumors were given intratumoral injections with G47Δ (5 × 106 PFUs on days 0 and 3) or mock in combination with intraperitoneal injections with the anti-CTLA-4 antibody (25 μg on days 0, 3, and 6) as indicated. (B) Delayed tumor growth was observed with either G47Δ (p < 0.05) or CTLA-4 inhibition (p < 0.01) alone, but the combination treatment was associated with a significant decrease in tumor growth compared with each monotherapy (versus G47Δ, p < 0.001; versus αCTLA-4, p < 0.01). The results are presented as the mean ± SEM (n = 8 per group). (C) Individual tumor growth curves of AKR tumors. The combination therapy achieved a cure in 5/8 animals. (D) Kaplan-Meier survival curves. The combination therapy resulted in significantly prolonged survival (p < 0.001 versus control; G47Δ, p < 0.01 versus αCTLA-4). (E−H) Efficacies of G47Δ and PD-1 inhibition, either alone or in combination, in the murine subcutaneous AKR tumor model. (E) Experimental design. C57BL/6 mice harboring subcutaneous AKR tumors were treated with G47Δ (5 × 106 PFUs) or mock together with intraperitoneal injections with the anti-PD-1 antibody (100 μg) as indicated. (F) Tumor growth was significantly inhibited by the anti-PD-1 antibody alone (versus control, p < 0.001). The efficacy of G47Δ combined with systemic PD-1 inhibition was equivalent to that of PD-1 inhibition alone. The results are presented as the mean ± SEM (n = 7 per group). (G) Individual tumor growth curves. The combination therapy did not yield a cure. (H) Kaplan-Meier survival curves. One-way ANOVA followed by Dunnett’s test was used for the comparisons of tumor growth. For survival analysis, the log-rank test followed by Holm’s sequential Bonferroni corrections was used to determine statistical significance (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ns, not significant).
Similar synergistic antitumor effects of G47Δ and CTLA-4 inhibition were observed in C57BL/6 mice harboring subcutaneous B16-F10 tumors and C3H mice bearing subcutaneous SCCVII tumors (Figure S2). Again, G47Δ in combination with PD-1 inhibition did not show a significant efficacy in the subcutaneous B16-F10 tumor model.
The combination therapy recruited effector T cells and reversed immunosuppressive TME
To study the types of immune cells within the tumor responding to the G47Δ and anti-CTLA-4 combination, AKR-harboring mice were treated as described in Figure 2A, tumors were harvested 7 days after the initial treatment, and the tumor-infiltrating lymphocytes analyzed. The combination therapy led to recruitment of more tumor-infiltrating CD3 cells than each monotherapy (p < 0.01 versus control; αCTLA-4, p < 0.05 versus G47Δ; Figure 3A), especially CD4+ T cells (p < 0.01 versus the other three groups; Figure 3B). G47Δ alone caused a significant increase in CD8+ T cells (versus control, p < 0.05), which, however, was not augmented by the combination with anti-CTLA-4 (Figure 3C). The total number of CD4+FoxP3+ regulatory T cells (Tregs) was significantly reduced by both G47Δ and CTLA-4 inhibition monotherapies (both p < 0.01 versus control; Figure 3D). Notably, the combination therapy markedly decreased the ratio of Tregs to the total CD4+ T cell population (p < 0.001 versus each group; Figure 3E) and significantly increased the CD8+ to Tregs ratio (p < 0.01 versus each group; Figure 3F). G47Δ and anti-CTLA-4 combination therapy significantly increased the proportion of CD3 cells and CD4+ T cells in the treated tumors compared with each monotherapy (p < 0.01 versus each group; Figure 3G). The combination therapy also significantly increased the proportion of CD8+ T cells compared with CTLA-4 inhibition alone (p < 0.01) but not with G47Δ monotherapy (p = 0.06). The combination therapy decreased the proportion of Tregs compared with control and G47Δ monotherapy but not with CTLA-4 inhibition alone (p = 0.2 versus anti-CTLA-4; Figure 3G). Overall, the combination therapy attracted more effecter T cells into the tumor and modified the immunosuppressive TME.
Figure 3.

Tumor-infiltrating lymphocytes in subcutaneous AKR tumors treated with the combination of G47Δ and CTLA-4 inhibition
C57BL/6 mice harboring subcutaneous AKR tumors were treated according to the schedule shown in Figure 2A. Tumor-infiltrating immune cells were analyzed by flow cytometry 7 days after the initial treatments. Comparisons of absolute numbers of (A) CD3 cells, (B) CD4+ T cells, (C) CD8+ T cells, and (D) Tregs (CD4+Foxp3+) per gram of tumor tissue. (E) The percentage of Tregs, gated on CD4+ T cells. (F) The ratio of CD8+ T cells to Tregs. (G) The percentage of CD3, CD8+, CD4+, and Tregs gated on CD45 cells. Results are representative of two independent experiments with 7 animals per group, and bars represent the SEM. One-way ANOVA followed by Dunnett’s test was used to determine statistical significance (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ns, not significant).
Intratumoral immune-related gene-expression changes induced by the combination therapy
Next, we analyzed the changes in expression of 44 immune-related genes related to inflammation, lymphocytes, activation markers, exhaustion signatures, and apoptosis. AKR-bearing mice were treated with G47Δ and CTLA-4 inhibition as described in Figure 2A, tumor tissues were harvested at 3 (early phase) and 7 (late phase) days after the initial treatments, and intratumoral mRNA expressions measured using quantitative PCR (qPCR). A number of genes related to inflammation and activation markers were upregulated by the combination therapy, but overall expression changes were modest on day 3 (Figure S3). On day 7, however, mice receiving the combination treatment showed marked changes in many of the immune-related gene expressions (Figure 4). Compared with G47Δ and CTLA-4 inhibition monotherapies (Figures 4A and 4B), most of the significant mRNA expression increases were detectable in the tumors subjected to the combination treatment (Figure 4C). In particular, the combination therapy greatly increased the gene expressions of PD-1, PD-L1, CTLA-4, Prf1, Gzmb, and Ifng/cxcr3 in subcutaneous AKR tumors (Figure 4C).
Figure 4.

Intratumoral immune-related gene-expression changes in subcutaneous AKR tumors 7 days after the initial treatments
Gene expression analyses from AKR tumors, focusing on selected mRNAs related to inflammation, lymphocytes, activation markers, exhaustion signatures, and apoptosis. C57BL/6 mice harboring subcutaneous AKR tumors were treated with intratumoral inoculations with G47Δ (5 × 106 PFUs) and intraperitoneal injections with an anti-CTLA-4 antibody (25 μg). The tumor tissues were harvested 7 days after the initial treatments, total RNA was extracted, cDNA was reverse transcribed, and gene expression analysis was performed using qPCR analysis. The details of the gene symbols are presented in Table S1. The fold change in expression of the indicated genes (A) with G47Δ treatment over control, (B) with CTLA-4 inhibition over control, (C) with the combination therapy over control, and (D) the combination therapy over CTLA-4 inhibition. The bar represents mean fold change + SEM (n = 6). The yellow bars represent mRNAs that were significantly upregulated (p < 0.05, fold change ≥ 2) as compared with the reference group. The blue bars show mRNAs that were significantly downregulated (p < 0.05, fold change < 0.5) as compared with the reference group. The expression data were normalized to the geometric mean of three housekeeping genes (Actb, Gapdh, and Hprt1). One-way ANOVA followed by Dunnett’s test was used to determine statistical significance. All experiments were performed twice, with six samples for each group.
Figure 5 shows the expressions of 12 representative genes in an early phase (day 3) and a late phase (day 7) of the combination treatment of subcutaneous AKR tumors. Genes related to inflammation (Ccl5, Il1a, and Il1b) were upregulated by the combination therapy on day 3 (p < 0.05 versus control; Figures 5A−5C, top), which was pronouncedly augmented on day 7 (p < 0.001 versus control and monotherapies; Figures 5A−5C, bottom). The lymphoid lineage genes (Cd4 and Cd8a) were commonly upregulated by the combination therapy on day 7 compared with other treatment groups (Cd4, p < 0.01; Cd8a, p < 0.05; Figures 5D and 5E). The increase in Foxp3 expression by the combination therapy was minimum on day 7 (Figure 5F), suggesting that the marked increase of Cd4 expression reflects the increase of effector CD4 (CD4+Foxp3−) rather than regulatory CD4 (CD4+Foxp3+) cells.
Figure 5.

Immune-related gene expressions in AKR tumors in early and late phases of the combination therapy
Expressions of the 12 genes in subcutaneous AKR tumors in an early phase (day 3) and a late phase (day 7) of the combination therapy. Comparisons of gene expressions related to (A−C) inflammation (Ccl5, Il1a, and Il1b), (D−F) lymphocytes (Cd4, Cd8a, and Foxp3), (G and H) Th1 response (Cxcl10 and Cxcr3), (I) Th2 response (Il2), (J) exhaustion marker (Cd274), and (K and L) T cell activation (Gzmb and Prf1). Top and bottom lines represent data on day 3 and day 7, respectively. The bar represents mean fold change + SEM (n = 6). One-way ANOVA followed by Dunnett’s test was used to determine statistical significance (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ns, not significant).
In contrast to a slight decrease of Cxcl10 (T helper cell type 1 [Th1]-related chemokine) expression (Figure 5G), a significant increase in Cxcr3 (the receptor of Th1-related chemokine) and Il2 (Th2-related cytokine) expressions was detected by the combination therapy on day 7 (Figures 5H and 5I). The expressions of the genes associated with T cell exhaustion (Cd274) and activation (Gzmb and Prf1) both increased by the combination therapy on day 7 (Figures 5J-5L). These results indicate that combination therapy significantly affected the expressions of genes related to T cell responses in the late phase.
The combination therapy elicits a specific and durable antitumor immune response
To investigate whether the combination therapy augments specific antitumor responses, splenocytes harvested from each treatment group were cultured together with AKR or Hepa1-6 (murine hepatoma derived from C57BL/6 mice) cells for 48 h, and the number of interferon (IFN)-γ spots was counted. The number of IFN-γ spots in response to AKR cells was significantly higher in the combination group than in other treatment groups (p < 0.001; Figures 6A and 6B). Notably, the significant increase in IFN-γ-secreting cells induced by the combination therapy was shown specific to AKR cells when compared with control Hepa1-6 cells (Figure 6C).
Figure 6.

The combination therapy enhanced specific antitumor immune responses
(A−C) An IFN-γ ELISpot assay of splenocytes from each treatment group. (A) The number of IFN-γ spots that responded to AKR cells was significantly higher in the combination group than in the other groups (p < 0.001). (B) A representative well from each group. (C) The number of IFN-γ-secreting spots stimulated by AKR cells, but not by Hepa1-6 cells, was significantly increased in the combination therapy group. The results are presented as the mean ± SEM (n = 7). (D and E) Rechallenge study. Mice (n = 5) in which subcutaneous AKR tumors were cured by the combination therapy were re-challenged on day 90 with one-fifth the number of AKR cells implanted in the contralateral hemisphere. As control, age-matched (3 months) naive mice were given the same challenge, i.e., the same dose of tumor cells (n = 5). (D) Growth of rechallenged AKR tumors in cured mice and naive mice. The results are presented as the mean ± SEM (n = 5). (E) Kaplan-Meier survival analysis. One-way ANOVA followed by Dunnett’s test (A), Student’s t test (C and D), and log-rank analysis (E) were used to determine the statistical significance of differences (∗∗p < 0.01; ns, not significant).
To examine the long-term persistence of antitumor memory, mice cured of subcutaneous AKR tumors by the combination therapy (n = 5) were reimplanted with AKR cells. Age-matched, naive C57BL/6 mice received the same tumor challenge, i.e., AKR cell dose, as controls. Whereas age-matched naive mice showed 100% engraftment of subcutaneous AKR tumors, all cured mice completely rejected the rechallenge with AKR cells, demonstrating the induction of an adaptive antitumor immune memory by the combination therapy (Figures 6D and 6E).
CD4+T cells are required for the enhanced efficacy of the combination therapy
To determine which immune cell components are required for the synergistic therapeutic effect of the combination therapy, the treatments were repeated in mice harboring subcutaneous AKR tumors under a depletion of CD4+, CD8+, or natural killer (NK) cells. The experimental schedules are shown in Figure S4. Adequate cell depletion of each cell subset was confirmed using flow cytometry of splenocytes (Figure S4).
A CD8+ T cell depletion decreased but did not abolish the enhanced efficacy of the combination therapy (0/7 cures; Figures 7A and 7B). In contrast, the efficacy of the combination therapy was completely abrogated by a depletion of CD4+ T cells (Figures 7C and 7D). The synergistic therapeutic effect and the survival benefit by the combination therapy were retained under a depletion of NK cells (1/7 cure; Figures 7E and 7F).
Figure 7.

Depletion of CD4+ T cells abrogated the enhanced efficacy of the combination therapy
The experimental designs are presented in Figure S4. (A and B) Tumor growth (A) and survival analysis (B) of CD8+ T cell depletion assay (n = 7). A CD8+ T cell depletion decreased but did not abolish the enhanced efficacy of the combination therapy (0/7 cures). (C and D) Tumor growth (C) and survival analysis (D) of CD4+ T cell depletion assay (n = 7). The efficacy of the combination therapy was completely abrogated by a depletion of CD4+ T cells. (E and F) Tumor growth (E) and survival analysis (F) of the NK cell depletion assay (n = 7). The synergistic therapeutic effect and the survival benefit by the combination therapy were retained under a depletion of NK cells (1/7 cure). The results are presented as the mean ± SEM. One-way ANOVA followed by Dunnett’s test was used for comparisons of tumor growth. For the survival analysis, the log-rank test followed by Holm’s sequential Bonferroni corrections was used to determine statistical significance (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ns, not significant).
Discussion
G47Δ is a novel therapeutic reagent that exhibits an enhanced replication capability in cancer cells with excellent safety features,17,19 a strong induction of specific antitumor immunity, and modifications of the immunosuppressive TME.10,21,23 The present study revealed the synergistic antitumor efficacy of G47Δ and CTLA-4 inhibition and the background immune modulation in the TME in murine tumor models. When combined, G47Δ and CTLA-4 inhibition caused increased effector CD4+ and CD8+ T cell infiltration into tumors and decreased the intratumoral Treg proportion.24,25 An increase in the CD8-to-Tregs ratio is known to associate with survival outcomes in patients with various malignancies.26,27
In an early phase (day 3) of the treatment, in the TME, genes related to inflammation (STAT3, CCl3, and CCl5) and macrophages (Cd68), were significantly upregulated in the G47Δ-treated group and further enhanced by the combination with CTLA-4 inhibition, presumably reflecting the host immune response to viral infection.21,28 Initial antivirus immune responses have been reported to potentiate the efficacy of oncolytic virus therapy.10,29 In a late phase (day 7), the combination therapy significantly increased the expression of CXCR3, a Th1-related chemokine receptor, which is a key mediator for orchestrating mature T cell infiltration into tumors in cooperation with CCL5.30 Also, a marked increase in expressions of activation marker genes (Gzmb, Prf1, Icos) and at the same time, an upregulation of inhibitory-immune checkpoints (Cd274, Pdcd1, Ctla-4) were observed. A similar finding was reported with a vaccinia virus.24
The augmentation of the efficacy by combining G47Δ with CTLA-4 inhibition was completely abrogated by a depletion of CD4+ T cells, but only partly diminished by a CD8+ T cell depletion. The importance of CD4+ T cells for maximizing the efficacy of cancer immunotherapy has been repeatedly reported.31,32 Supporting our results, it has been shown that the presence of intratumoral Th1-like CD4+ T cells correlates with therapeutic outcomes of ipilimumab33 and that the efficacy of ICIs and/or oncolytic viruses depends on CD4+ T cells.10,34 Our results, together with previous studies by others, highlight the importance of CD4+ T cells.10,34 However, the effects of the combination therapy were reduced also in CD8+- or NK cell-depleted animals compared to non-depleted animals, indicating a role of various immune cell subsets in conveying the observed efficacy. Some studies have suggested that CD8+ T cells, not CD4+ T cells, are essential for the therapeutic effect of oncolytic viruses in combination with ICIs.14,35 Innate immune responses are reported to be essential for the antitumor activity of oncolytic viruses. Investigators have demonstrated NK cells as important mediators of the antitumor effect associated with oncolytic virus therapy,11 especially in the early phase.36 M1-polarized macrophages are reportedly required for the synergistic activities of oncolytic HSV-1 and ICIs.10 Furthermore, Batf3-dependent dendritic cells are crucial for inducing systemic antitumor immunity in therapies using oncolytic HSV-1 combined with ICIs and oncolytic vaccinia virus.37,38 Precise mechanisms and crucial immune cell components underlying the efficacy of G47Δ and anti-CTLA-4 combination therapy remain to be elucidated.
CTLA-4 and PD-1 have different action mechanisms: CTLA-4 attenuates T cell activation in the priming phases, whereas PD-1 primarily attenuates T cell activity in peripheral tissues through cell-intrinsic mechanisms.33 However, one must be careful in interpreting the immunotherapeutic mechanisms for humans using findings in murine models, because it is well known that immune systems function differently in mouse, rat, and human.39, 40, 41 The present study shows that PD-1 inhibition does not enhance the efficacy of G47Δ in any of the murine tumor models examined, whereas various studies have reported that a combination of oncolytic virus therapy with PD-1/PD-L1 inhibition results in increased efficacy in different animal and treatment models.14,35,42,43 Immunological mechanisms are likely to differ between species and tumor models. Results obtained from preclinical studies do not necessarily predict the clinical antitumor activity in humans. However, a recent preclinical study showed that intratumoral administration of Talimogene laherparepvec (T-VEC), a double-mutated, second-generation oncolytic HSV-1, synergistically worked with systemic CTLA-4 inhibition via enhanced antitumor immunity in mouse models.12 The augmented antitumor effect observed in the preclinical study was verified in human trials: a phase I and a subsequent phase II clinical trial demonstrated that the combination of T-VEC and ipilimumab led to a greater efficacy without additional safety concerns than either T-VEC or ipilimumab alone in patients with melanoma.15,44 Such a fact that the findings of oncolytic HSV-1 in mice can be extended to humans implies that the synergistic efficacy of G47Δ and CTLA-4 inhibition is likely to be reproduced in human malignancies, which merits scrutiny in clinical studies.
In the present study, the treatment schedule may have played a key role. We started both CTLA-4 and PD-1 inhibition immediately after the initial G47Δ inoculation, although others have suggested that PD-1 inhibition might be advantageous when it is administered some days after initial oncolytic virus treatment.35,43 In contrast, a recent study showed that PD-1 inhibition synergistically worked with oncolytic virus therapy when it was initiated concomitantly with the oncolytic virus.14 It has been reported using mouse models that the use of oncolytic virus therapy ahead of ICI treatments sensitizes refractory cancer to an immune-checkpoint blockade.45,46 We recently showed that a neoadjuvant use of G47Δ enhanced the efficacy of radiofrequency ablation and ICI treatments in mice.47 The effects of different ICIs clearly depend on the immunogenicity of the tumor models used. In fact, a recent study using less immunogenic brain tumor models provided different results: not only CTLA-4 but also PD-1 inhibition enhanced the antitumor efficacy of G47Δ armed with murine interleukin (IL)-12.10 Systemic immune organization and the immune composition of the TME greatly differ between various murine tumor models, varying in the degree of immune infiltration and diversity.48 In our study, detailed analysis and comparison of immune compositions of AKR and other subcutaneous tumor models may lead to a potential mechanism associated with the synergistic activity of CTLA-4 but not PD-1 inhibition in the AKR tumors.
Previous studies demonstrated the discrepancy between the preclinical and clinical efficacy of anti-CTLA-4 immunotherapy.25,49,50 In particular, CTLA-4 inhibition has the capacity to selectively deplete intratumoral Tregs in preclinical murine models25,49 but does not have this effect in human tumors.50 Related to its effect on Tregs, the therapeutic efficacy of oncolytic vaccinia virus was augmented when combined with an anti-CTLA-4 antibody but not enhanced when used together with an anti-CD25 antibody, another Treg-depletion agent, indicating that the synergistic effect of CTLA-4 inhibition depends on its activation of CD8+ T cells rather than on depletion of Tregs.51 Further studies are required to elucidate the potentially important mechanism of CTLA-4 inhibition on Tregs. Furthermore, a recent clinical study showed anti-PD-1 immunotherapy to be more efficacious and less toxic than anti-CTLA-4 immunotherapy.52 In human, it has been shown that oncolytic herpes virus (T-VEC) in combination with PD-1 inhibition and/or CTLA-4 inhibition leads to increased efficacies.15,16 Whether the combination of G47Δ and CTLA-4 inhibition is useful for treating human malignancies awaits to be verified.
In conclusion, the results demonstrate the dynamics of immune modulation of TME when intratumoral G47Δ therapy is combined with systemic CTLA-4 inhibition in a murine tumor model and provide a rationale for the clinical use of G47Δ with ICIs. G47Δ converts immunogenically cold tumors to hot tumors and works synergistically with ICIs in eliciting a potent, adaptive, specific antitumor immune response, which may lead to a curative therapy for poorly immunogenic malignancies.
Materials and methods
Cell lines
Murine esophageal SCC cell lines, AKR and HNM007, both derived from C57BL/6 mice, were kindly provided by Dr. Nakagawa.53,54 SCCVII, a murine SCC cell line derived from a C3H mouse, was provided by Professor Yoshiaki Yura. Hepa1-6, a murine hepatoma cell line derived from a C57BL/6 mouse, was obtained from Japanese Collection of Research Bioresources (Osaka, Japan). Murine melanoma cell line B16-F10 and Vero (African green monkey kidney) cells were purchased from the American Type Culture Collection (Rockville, MD, USA). All cell lines were cultured according to the instructions provided by the suppliers.
Virus and antibodies
G47Δ was grown, purified, and titered on Vero cells using a plaque assay as described previously.17,55 Therapeutic mouse anti-CTLA-4 (clone 9H10) and anti-PD-1 (clone RMP1-14) antibodies were produced by Bio X Cell. Polyclonal Syrian hamster immunoglobulin G (IgG) and rat IgG2a were used as isotype controls for anti-CTLA-4 and anti-PD-1, respectively. Depletion of monoclonal antibodies for CD8 (clone 53-6.7), CD4 (clone GK1.5), and NK1.1 (clone PK136) was purchased from Bio X Cell.
Cytopathic effect and virus yield studies
In vitro cytopathic effect studies were performed as previously described.56 Briefly, cells were seeded onto six-well plates at 2 × 105 cells/well and incubated overnight at 37°C. The following day, the cells were infected with G47Δ at various MOIs (0.01, 0.1, or 1) or mock and further incubated at 34.5°C. The number of surviving cells was counted daily with a Coulter Counter (Beckman Coulter, Fullerton, CA, USA) and expressed as a percentage of mock-infected controls. For virus yield studies, the cells were seeded onto 6-well plates at 3 × 105 cells/well and incubated overnight at 37°C. The following day, triplicate wells were infected with G47Δ at an MOI of 0.1. At 24 and 48 h after infection, the cells were scraped into the medium and lysed by three cycles of freezing and thawing. The progeny virus was titered as described previously.56
Animal experiments
All animal experiment protocols were approved by the Committee for Ethics of Animal Experimentation and were in accordance with the Guideline for Animal Experiments in the University of Tokyo. 6-week-old female C57BL/6 and C3H mice were purchased from CLEA Japan (Tokyo, Japan). The mice were maintained under specific pathogen-free conditions and provided with sterile food, water, and cages.
Subcutaneous tumor models
Subcutaneous AKR, HNM007, and B16-F10 tumors were generated by inoculating 5 × 105 cells into the left flanks of 6-week-old female C57BL/6 mice. SCCVII subcutaneous tumors were established by inoculating 1 × 106 cells into the left flanks of 6-week-old female C3H mice. When tumors reached approximately 4−5 mm in diameter, the animals were randomized, and mock or G47Δ (5 × 106 PFUs for AKR and B16-F10 and 2 × 105 PFUs for SCCVII) in 20 μL of phosphate-buffered saline (PBS) containing 10% glycerol was inoculated into the left-flank tumors. Intratumoral G47Δ injection was repeated 3 days later. The tumor volume (length × width × height) at the starting point was approximately 40−50 mm3 and was measured twice a week. Mice were sacrificed when the maximum diameter of the tumor reached 24 mm.
The combination therapy
To evaluate the efficacy of G47Δ, anti-CTLA-4 antibody, anti-PD-1 antibody and their combinations in immunocompetent mice, unilateral AKR, SCCVII, and B16-F10 subcutaneous tumors were established. When the tumors reached approximately 4–5 mm in diameter (40−50 mm3 in tumor volume), the animals were randomized, and mock or G47Δ (5 × 106 PFUs for AKR and B16-F10 and 2 × 105 PFUs for SCCVII) in 20 μL of PBS containing 10% glycerol was inoculated into the left-flank tumors. The mock-infected extract was prepared from virus buffer-infected cells employing the same procedures as those used for the virus inoculum.23 Intratumoral G47Δ injection was repeated 3 days later. Concurrently, the mice received three intraperitoneal injections with an anti-CTLA-4 antibody (25, 50, and 100 μg for AKR, SCCVII, and B16-F10 tumors, respectively) or an anti-PD-1 antibody (200 μg) every 3 days. The amount of anti-CTLA-4 antibody and anti-PD-1 antibody was determined according to the results of preliminary experiments so that the amount would cause a moderate effect on the tumor growth when the antibody was used alone. The control groups received a corresponding dose of isotype antibody. Treatment schedules are presented in Figures 2 and S2. The tumor volume (length × width × height) was measured twice a week. The mice were euthanized when the maximum diameter of the tumor reached 24 mm or when signs of deterioration or acute weight loss were observed.
For in vivo drug-combination analysis, we employed the fractional product method.57,58 The efficacy of each monotherapy and the combination therapy were assessed as the fractional tumor volume (FTV). FTV was calculated based on the following equation: (the mean volume of the treated tumors)/(that of the control tumors). Then, the expected FTV of the combination therapy was estimated by multiplying the FTV by each monotherapy. The CI was derived from the ratio of observed FTV of the combination therapy to expected FTV of the combination therapy. CI < 1, CI = 1, and CI > 1 indicate a synergistic, an additive, and an antagonistic effect, respectively.
Rechallenge study
C57BL/6 mice, of which subcutaneous AKR tumors had been confirmed cured after treatment with G47Δ and anti-CTLA-4 antibody, were re-challenged subcutaneously with 1 × 105 AKR cells per mouse on day 90 after the initial treatment. As controls, naive, age-matched C57BL/6 mice received the same tumor challenge, i.e., at the same cell dose.
Flow cytometry
Mice with established AKR (5 × 105 cells) subcutaneous tumors were treated with the combination therapy (G47Δ + anti-CTLA-4 antibody) according to the schedule shown in Figure 2. 7 days later, the subcutaneous tumors were harvested, stained for immune markers, and analyzed using flow cytometry. Tumor-infiltrating cells were prepared with a Tumor Dissociation Kit (Miltenyi Biotec, Auburn, CA, USA) according to the manufacturer’s instructions. The total number of analyzed live cells was matched between the groups with flow count beads (Beckman Coulter, Fullerton, CA, USA). Zombie Yellow Fixable Viability Kit (BioLegend, San Diego, CA, USA) was used to stain dead cells. The cells were then pretreated with purified anti-mouse CD16/32 (2.4G2; BD Biosciences, Franklin Lakes, NJ, USA), stained with antibodies, and analyzed with a CytoFLEX (V5-B5-R3 configuration; Beckman Coulter, Fullerton, CA, USA). The following fluorescent-labeled antibodies were purchased from BioLegend (San Diego, CA, USA) and used for analysis: peridinin-chlorophyll-protein/cyanine 5.5 (PerCP/Cy5.5)-conjugated anti-CD45.2 (104), Brilliant Violet 785-conjugated anti-CD3 (17A2), PE-Cy7-conjugated anti-CD8 (53-6.7), fluorescein isothiocyanate (FITC)-conjugated anti-CD4 (RM4.5), and PE-conjugated anti-NK1.1 (PK136). For all channels, positive and negative cells were gated based on fluorescence minus one control. For intracellular staining, the cells were permeabilized and fixed using the FlowX FoxP3/Transcription Factor Fixation & Perm Buffer Kit (R&D Systems) before the addition of antibodies for 20 min at room temperature and then stained with allophycocyanin (APC)-conjugated anti-Foxp3 (MF23; BioLegend). The cells were kept in stabilizing fixative until acquisition. The gating strategy is shown in Figure S1. Foxp3 positivity was gated with the appropriate isotype controls. The data were analyzed with FlowJo software (version [v.]10.4; FlowJo).
Extraction of RNA and quantitative real-time PCR
For gene product analysis, mice bearing subcutaneous AKR tumors were inoculated twice intratumorally with G47Δ (on days 0 and 2) and/or injected twice intraperitoneally with an anti-CTLA-4 antibody (on days 0 and 2). On days 3 and 7, the tumors were excised and snap frozen in liquid nitrogen. The samples were homogenized, and RNA was extracted using the RNeasy Mini Kit (QIAGEN) and QIAcube according to the manufacturer’s instructions. RNA purity and yield were assessed using NanoDrop. RNA was reverse transcribed to cDNA using the ReverTra Ace qPCR RT Master Mix with gDNA Remover (Toyobo). The samples were stored at −20°C until RT-PCR was performed. Real-time qPCR was performed on 2 ng of cDNA from each sample (n = 6) using Custom TaqMan Array Card Assays (Thermo Fisher Scientific; the detail of the card is shown in Table S1) and the QuantStudio 7 Flex Real-Time PCR System (Thermo Fisher Scientific). The expression data for individual samples were normalized to the geometric mean of three housekeeping genes (HKGs): Actb, Gapdh, and Hprt1. The comparative threshold (Ct) values and fold changes in gene expression (relative quantification) were calculated using the Expression Suite software v.1.0.3 (Life Technologies, Carlsbad, CA, USA), a data analysis tool that utilizes the comparative Ct (ΔΔCt) method. Only genes with a |log2 fold change| of >1 and a p value ≤0.05 were considered to have changed significantly. All experiments were performed twice, with six samples for each group.
Enzyme-linked immunospot (ELISpot) assay
Mice with established subcutaneous AKR tumors were given the combination therapy (G47Δ + anti-CTLA-4 antibody) according to the schedule shown in Figure 2. Splenocytes of mice from each treatment group were collected 14 days after the initial virus treatment, and a mouse IFN-γ ELISpot assay was performed according to the manufacturer’s protocol (Mabtech, Nacka Strand, Sweden). Clear, 96-well medial suprainfundibular plates (MSIPs) pre-coated with monoclonal anti-mouse IFN-γ antibody AN18 (Mabtech) were used. Splenocytes (2 × 105) were co-cultured with either AKR (1 × 104) or Hepa1-6 (1 × 104) tumor cells at a 20:1 ratio for 48 h in αIFN-γ-coated wells. Naive splenocytes were used as controls. After incubation, the spots were detected using 1 μg/mL of biotinylated anti-mouse IFN-γ antibody R4-6A2-biotin. Specific spots were counted and analyzed using an ImmunoSpot Analyzer and ImmunoSpot software (Cellular Technology Limited [CTL], Cleveland, OH, USA).
Immune cell subset depletion studies
The experimental schedules of depletion studies are presented in Figure S4. For depletion of CD8+ and CD4+ immune cells, mice were injected intraperitoneally with 250 μg of monoclonal antibodies against CD8+ and CD4+ 1 day before the treatment, on the day of the treatment, and then every 5 days throughout the experiment. For depletion of NK cells, mice were injected intraperitoneally with 500 μg of an antibody against NK1.1 1 day before and 2 days after the tumor challenge, followed by injection of 250 μg every 5 days throughout the experiment.
Statistical analysis
All data were expressed as the mean ± standard deviation (SD) or the mean ± standard error of the mean (SEM). A two-tailed Student’s t test (for comparison of two groups; Figures 6C and 6D) or one-way ANOVA followed by Dunnett’s test (for comparison of three or more groups; Figures 1, 2, 3, 4, 5, 6A, and 7) was used to determine statistical significance, as appropriate. Survival curves were constructed using the Kaplan-Meier method, and the log-rank test (for comparisons of 2 groups) or log-rank test followed by Holm’s sequential Bonferroni corrections (for comparisons of 3 or more groups) was used to determine statistical significance, as appropriate. In the figures, standard symbols are used as follows: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001, as well as ns (not significant). Statistical analyses were carried out with JMP 13.0.0 (SAS Institute, Cary, NC, USA).
Acknowledgments
This research is supported in part by grants to T.T. from Practical Research for Innovative Cancer Control, Japan Agency for Medical Research and Development (AMED; grant number JP18ck0106416), and Translational Research Program, AMED (grant number JP20lm0203140).
Author contributions
K.S. and T.T. were involved with the conception and performance of experiments, statistical analysis, interpretation of results, and writing the manuscript. M.I. assisted with some of the experiments. M.T., H.I., and Y.S. were involved with the conception and design of experiments. All authors reviewed and edited the manuscript.
Declaration of interests
T.T. owns the patent right for G47Δ in multiple countries including Japan.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.05.004.
Supplemental information
References
- 1.Larkin J., Chiarion-Sileni V., Gonzalez R., Grob J.J., Cowey C.L., Lao C.D., Schadendorf D., Dummer R., Smylie M., Rutkowski P. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ribas A., Wolchok J.D. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pitt J.M., Vétizou M., Daillère R., Roberti M.P., Yamazaki T., Routy B., Lepage P., Boneca I.G., Chamaillard M., Kroemer G., Zitvogel L. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity. 2016;44:1255–1269. doi: 10.1016/j.immuni.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 4.Kalbasi A., Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020;20:25–39. doi: 10.1038/s41577-019-0218-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zappasodi R., Merghoub T., Wolchok J.D. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell. 2018;33:581–598. doi: 10.1016/j.ccell.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Allen E.M., Miao D., Schilling B., Shukla S.A., Blank C., Zimmer L., Sucker A., Hillen U., Foppen M.H.G., Goldinger S.M. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaufman H.L., Kohlhapp F.J., Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015;14:642–662. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Russell S.J., Barber G.N. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell. 2018;33:599–605. doi: 10.1016/j.ccell.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shekarian T., Sivado E., Jallas A.C., Depil S., Kielbassa J., Janoueix-Lerosey I., Hutter G., Goutagny N., Bergeron C., Viari A. Repurposing rotavirus vaccines for intratumoral immunotherapy can overcome resistance to immune checkpoint blockade. Sci. Transl. Med. 2019;11:eaat5025. doi: 10.1126/scitranslmed.aat5025. [DOI] [PubMed] [Google Scholar]
- 10.Saha D., Martuza R.L., Rabkin S.D. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell. 2017;32:253–267.e5. doi: 10.1016/j.ccell.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zamarin D., Holmgaard R.B., Subudhi S.K., Park J.S., Mansour M., Palese P., Merghoub T., Wolchok J.D., Allison J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014;6:226ra32. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moesta A.K., Cooke K., Piasecki J., Mitchell P., Rottman J.B., Fitzgerald K., Zhan J., Yang B., Le T., Belmontes B. Local Delivery of OncoVEXmGM-CSF Generates Systemic Antitumor Immune Responses Enhanced by Cytotoxic T-Lymphocyte-Associated Protein Blockade. Clin. Cancer Res. 2017;23:6190–6202. doi: 10.1158/1078-0432.CCR-17-0681. [DOI] [PubMed] [Google Scholar]
- 13.Kanaya N., Kuroda S., Kakiuchi Y., Kumon K., Tsumura T., Hashimoto M., Morihiro T., Kubota T., Aoyama K., Kikuchi S. Immune Modulation by Telomerase-Specific Oncolytic Adenovirus Synergistically Enhances Antitumor Efficacy with Anti-PD1 Antibody. Mol. Ther. 2020;28:794–804. doi: 10.1016/j.ymthe.2020.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zamarin D., Ricca J.M., Sadekova S., Oseledchyk A., Yu Y., Blumenschein W.M., Wong J., Gigoux M., Merghoub T., Wolchok J.D. PD-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. J. Clin. Invest. 2018;128:1413–1428. doi: 10.1172/JCI98047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chesney J., Puzanov I., Collichio F., Singh P., Milhem M.M., Glaspy J., Hamid O., Ross M., Friedlander P., Garbe C. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J. Clin. Oncol. 2018;36:1658–1667. doi: 10.1200/JCO.2017.73.7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ribas A., Dummer R., Puzanov I., VanderWalde A., Andtbacka R.H.I., Michielin O., Olszanski A.J., Malvehy J., Cebon J., Fernandez E. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell. 2017;170:1109–1119.e10. doi: 10.1016/j.cell.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Todo T., Martuza R.L., Rabkin S.D., Johnson P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA. 2001;98:6396–6401. doi: 10.1073/pnas.101136398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.York I.A., Roop C., Andrews D.W., Riddell S.R., Graham F.L., Johnson D.C. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell. 1994;77:525–535. doi: 10.1016/0092-8674(94)90215-1. [DOI] [PubMed] [Google Scholar]
- 19.Fukuhara H., Martuza R.L., Rabkin S.D., Ito Y., Todo T. Oncolytic herpes simplex virus vector g47delta in combination with androgen ablation for the treatment of human prostate adenocarcinoma. Clin. Cancer Res. 2005;11:7886–7890. doi: 10.1158/1078-0432.CCR-05-1090. [DOI] [PubMed] [Google Scholar]
- 20.Fukuhara H., Ino Y., Kuroda T., Martuza R.L., Todo T. Triple gene-deleted oncolytic herpes simplex virus vector double-armed with interleukin 18 and soluble B7-1 constructed by bacterial artificial chromosome-mediated system. Cancer Res. 2005;65:10663–10668. doi: 10.1158/0008-5472.CAN-05-2534. [DOI] [PubMed] [Google Scholar]
- 21.Sugawara K., Iwai M., Yajima S., Tanaka M., Yanagihara K., Seto Y., Todo T. Efficacy of a Third-Generation Oncolytic Herpes Virus G47Δ in Advanced Stage Models of Human Gastric Cancer. Mol. Ther. Oncolytics. 2020;17:205–215. doi: 10.1016/j.omto.2020.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lundberg P., Welander P., Openshaw H., Nalbandian C., Edwards C., Moldawer L., Cantin E. A locus on mouse chromosome 6 that determines resistance to herpes simplex virus also influences reactivation, while an unlinked locus augments resistance of female mice. J. Virol. 2003;77:11661–11673. doi: 10.1128/JVI.77.21.11661-11673.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Todo T., Martuza R.L., Dallman M.J., Rabkin S.D. In situ expression of soluble B7-1 in the context of oncolytic herpes simplex virus induces potent antitumor immunity. Cancer Res. 2001;61:153–161. [PubMed] [Google Scholar]
- 24.Chon H.J., Lee W.S., Yang H., Kong S.J., Lee N.K., Moon E.S., Choi J., Han E.C., Kim J.H., Ahn J.B. Tumor Microenvironment Remodeling by Intratumoral Oncolytic Vaccinia Virus Enhances the Efficacy of Immune-Checkpoint Blockade. Clin. Cancer Res. 2019;25:1612–1623. doi: 10.1158/1078-0432.CCR-18-1932. [DOI] [PubMed] [Google Scholar]
- 25.Selby M.J., Engelhardt J.J., Quigley M., Henning K.A., Chen T., Srinivasan M., Korman A.J. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 2013;1:32–42. doi: 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 26.Baras A.S., Drake C., Liu J.J., Gandhi N., Kates M., Hoque M.O., Meeker A., Hahn N., Taube J.M., Schoenberg M.P. The ratio of CD8 to Treg tumor-infiltrating lymphocytes is associated with response to cisplatin-based neoadjuvant chemotherapy in patients with muscle invasive urothelial carcinoma of the bladder. OncoImmunology. 2016;5:e1134412. doi: 10.1080/2162402X.2015.1134412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Twyman-Saint Victor C., Rech A.J., Maity A., Rengan R., Pauken K.E., Stelekati E., Benci J.L., Xu B., Dada H., Odorizzi P.M. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature. 2015;520:373–377. doi: 10.1038/nature14292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alvarez-Breckenridge C.A., Yu J., Price R., Wojton J., Pradarelli J., Mao H., Wei M., Wang Y., He S., Hardcastle J. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat. Med. 2012;18:1827–1834. doi: 10.1038/nm.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gujar S., Pol J.G., Kim Y., Lee P.W., Kroemer G. Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends Immunol. 2018;39:209–221. doi: 10.1016/j.it.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Dangaj D., Bruand M., Grimm A.J., Ronet C., Barras D., Duttagupta P.A., Lanitis E., Duraiswamy J., Tanyi J.L., Benencia F. Cooperation between Constitutive and Inducible Chemokines Enables T Cell Engraftment and Immune Attack in Solid Tumors. Cancer Cell. 2019;35:885–900.e10. doi: 10.1016/j.ccell.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alspach E., Lussier D.M., Miceli A.P., Kizhvatov I., DuPage M., Luoma A.M., Meng W., Lichti C.F., Esaulova E., Vomund A.N. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature. 2019;574:696–701. doi: 10.1038/s41586-019-1671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borst J., Ahrends T., Bąbała N., Melief C.J.M., Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018;18:635–647. doi: 10.1038/s41577-018-0044-0. [DOI] [PubMed] [Google Scholar]
- 33.Wei S.C., Levine J.H., Cogdill A.P., Zhao Y., Anang N.A.S., Andrews M.C., Sharma P., Wang J., Wargo J.A., Pe’er D., Allison J.P. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell. 2017;170:1120–1133.e17. doi: 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sato Y., Bolzenius J.K., Eteleeb A.M., Su X., Maher C.A., Sehn J.K., Arora V.K. CD4+ T cells induce rejection of urothelial tumors after immune checkpoint blockade. JCI Insight. 2018;3:e121062. doi: 10.1172/jci.insight.121062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajani K., Parrish C., Kottke T., Thompson J., Zaidi S., Ilett L., Shim K.G., Diaz R.M., Pandha H., Harrington K. Combination Therapy With Reovirus and Anti-PD-1 Blockade Controls Tumor Growth Through Innate and Adaptive Immune Responses. Mol. Ther. 2016;24:166–174. doi: 10.1038/mt.2015.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diaz R.M., Galivo F., Kottke T., Wongthida P., Qiao J., Thompson J., Valdes M., Barber G., Vile R.G. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- 37.Bommareddy P.K., Aspromonte S., Zloza A., Rabkin S.D., Kaufman H.L. MEK inhibition enhances oncolytic virus immunotherapy through increased tumor cell killing and T cell activation. Sci. Transl. Med. 2018;10:eaau0417. doi: 10.1126/scitranslmed.aau0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dai P., Wang W., Yang N., Serna-Tamayo C., Ricca J.M., Zamarin D., Shuman S., Merghoub T., Wolchok J.D., Deng L. Intratumoral delivery of inactivated modified vaccinia virus Ankara (iMVA) induces systemic antitumor immunity via STING and Batf3-dependent dendritic cells. Sci. Immunol. 2017;2:eaal1713. doi: 10.1126/sciimmunol.aal1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagar L.E., Salahudeen A., Constantz C.M., Wendel B.S., Lyons M.M., Mallajosyula V., Jatt L.P., Adamska J.Z., Blum L.K., Gupta N. Modeling human adaptive immune responses with tonsil organoids. Nat. Med. 2021;27:125–135. doi: 10.1038/s41591-020-01145-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jameson S.C., Masopust D. What Is the Predictive Value of Animal Models for Vaccine Efficacy in Humans? Reevaluating the Potential of Mouse Models for the Human Immune System. Cold Spring Harb. Perspect. Biol. 2018;10:a029132. doi: 10.1101/cshperspect.a029132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wagar L.E., DiFazio R.M., Davis M.M. Advanced model systems and tools for basic and translational human immunology. Genome Med. 2018;10:73. doi: 10.1186/s13073-018-0584-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Z., Ravindranathan R., Kalinski P., Guo Z.S., Bartlett D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017;8:14754. doi: 10.1038/ncomms14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fend L., Yamazaki T., Remy C., Fahrner C., Gantzer M., Nourtier V., Préville X., Quéméneur E., Kepp O., Adam J. Immune Checkpoint Blockade, Immunogenic Chemotherapy or IFN-α Blockade Boost the Local and Abscopal Effects of Oncolytic Virotherapy. Cancer Res. 2017;77:4146–4157. doi: 10.1158/0008-5472.CAN-16-2165. [DOI] [PubMed] [Google Scholar]
- 44.Puzanov I., Milhem M.M., Minor D., Hamid O., Li A., Chen L., Chastain M., Gorski K.S., Anderson A., Chou J. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016;34:2619–2626. doi: 10.1200/JCO.2016.67.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Samson A., Scott K.J., Taggart D., West E.J., Wilson E., Nuovo G.J., Thomson S., Corns R., Mathew R.K., Fuller M.J. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018;10:eaam7577. doi: 10.1126/scitranslmed.aam7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bourgeois-Daigneault M.C., Roy D.G., Aitken A.S., El Sayes N., Martin N.T., Varette O., Falls T., St-Germain L.E., Pelin A., Lichty B.D. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018;10:eaao1641. doi: 10.1126/scitranslmed.aao1641. [DOI] [PubMed] [Google Scholar]
- 47.Yamada T., Tateishi R., Iwai M., Koike K., Todo T. Neoadjuvant Use of Oncolytic Herpes Virus G47Δ Enhances the Antitumor Efficacy of Radiofrequency Ablation. Mol. Ther. Oncolytics. 2020;18:535–545. doi: 10.1016/j.omto.2020.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Allen B.M., Hiam K.J., Burnett C.E., Venida A., DeBarge R., Tenvooren I., Marquez D.M., Cho N.W., Carmi Y., Spitzer M.H. Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat. Med. 2020;26:1125–1134. doi: 10.1038/s41591-020-0892-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simpson T.R., Li F., Montalvo-Ortiz W., Sepulveda M.A., Bergerhoff K., Arce F., Roddie C., Henry J.Y., Yagita H., Wolchok J.D. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013;210:1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma A., Subudhi S.K., Blando J., Scutti J., Vence L., Wargo J., Allison J.P., Ribas A., Sharma P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3+ Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. 2019;25:1233–1238. doi: 10.1158/1078-0432.CCR-18-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rojas J.J., Sampath P., Hou W., Thorne S.H. Defining Effective Combinations of Immune Checkpoint Blockade and Oncolytic Virotherapy. Clin. Cancer Res. 2015;21:5543–5551. doi: 10.1158/1078-0432.CCR-14-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robert C., Schachter J., Long G.V., Arance A., Grob J.J., Mortier L., Daud A., Carlino M.S., McNeil C., Lotem M., KEYNOTE-006 investigators Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015;372:2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 53.Karakasheva T.A., Waldron T.J., Eruslanov E., Kim S.B., Lee J.S., O’Brien S., Hicks P.D., Basu D., Singhal S., Malavasi F., Rustgi A.K. CD38-Expressing Myeloid-Derived Suppressor Cells Promote Tumor Growth in a Murine Model of Esophageal Cancer. Cancer Res. 2015;75:4074–4085. doi: 10.1158/0008-5472.CAN-14-3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Predina J.D., Judy B., Aliperti L.A., Fridlender Z.G., Blouin A., Kapoor V., Laguna B., Nakagawa H., Rustgi A.K., Aguilar L. Neoadjuvant in situ gene-mediated cytotoxic immunotherapy improves postoperative outcomes in novel syngeneic esophageal carcinoma models. Cancer Gene Ther. 2011;18:871–883. doi: 10.1038/cgt.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mineta T., Rabkin S.D., Yazaki T., Hunter W.D., Martuza R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995;1:938–943. doi: 10.1038/nm0995-938. [DOI] [PubMed] [Google Scholar]
- 56.Todo T., Rabkin S.D., Sundaresan P., Wu A., Meehan K.R., Herscowitz H.B., Martuza R.L. Systemic antitumor immunity in experimental brain tumor therapy using a multimutated, replication-competent herpes simplex virus. Hum. Gene Ther. 1999;10:2741–2755. doi: 10.1089/10430349950016483. [DOI] [PubMed] [Google Scholar]
- 57.Matar P., Rojo F., Cassia R., Moreno-Bueno G., Di Cosimo S., Tabernero J., Guzmán M., Rodriguez S., Arribas J., Palacios J., Baselga J. Combined epidermal growth factor receptor targeting with the tyrosine kinase inhibitor gefitinib (ZD1839) and the monoclonal antibody cetuximab (IMC-C225): superiority over single-agent receptor targeting. Clin. Cancer Res. 2004;10:6487–6501. doi: 10.1158/1078-0432.CCR-04-0870. [DOI] [PubMed] [Google Scholar]
- 58.Li X.Q., Ouyang Z.G., Zhang S.H., Liu H., Shang Y., Li Y., Zhen Y.S. Synergy of enediyne antibiotic lidamycin and temozolomide in suppressing glioma growth with potentiated apoptosis induction. J. Neurooncol. 2014;119:91–100. doi: 10.1007/s11060-014-1477-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.