

Abstract
Multi-nucleotide variants (MNVs) represent an important type of genetic variation and have biological and clinical significance. To simulate MNVs, we designed four dual-mutation base editors combining hA3A(Y130F), TadA8e(V106W), and protospacer adjacent motif (PAM)-flexible SpRY and selected cytosine and adenine base editor-SpRY (CABE-RY), which had the best editing performance, for further study. Characterization and comparison showed that CABE-RY had a smaller DNA editing window and lower RNA off-target edits than the corresponding single base editors. Thus, we have established a versatile tool to efficiently simulate MNVs over the genome, which could be very useful for functional studies on MNVs in humans.
Keywords: CRISPR, dual-mutation base editor, MNVs, PAM-flexible, simultaneous A/C conversions
Graphical abstract
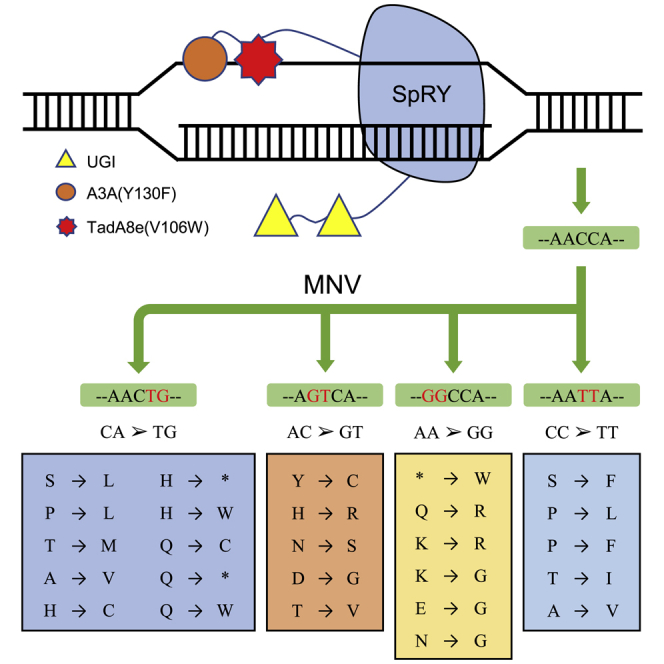
Tao et al. established a new dual-mutation base editor (CABE-RY) to efficiently simulate multi-nucleotide variants (MNVs). Further characterization and comparison showed that CABE-RY had a smaller DNA editing window and lower RNA off-target edits than the corresponding single base editors, providing a new tool for functional studies on MNVs in humans.
Introduction
Sequencing technologies have rapidly advanced our understanding of human genetic variants. Many disease-associated variants have been identified in patients. Single-nucleotide variants (SNVs), which represent one type of variant, are usually included in genetic evaluations with phenotypic information.1 Other kinds of genetic variants may also have influences on diseases. Therefore, precisely elucidating how SNVs contribute to causal relationships between genotypes and phenotypes remains a major challenge.
Multi-nucleotide variants (MNVs) represent another variant type that is considered to be related to disease. For example, Kaplanis et al.2 found significant enrichment of MNVs in genes associated with diagnosing developmental disorders (DDD), showing that MNVs are on average more harmful than SNVs. Usually, there are two or more nearby variants on the same haplotype in an individual.2,3 When nearby variants are within the same codon, the amino acid changes are different than if the separate SNVs are annotated independently. Due to neglect of MNVs, most existing variant callers mistake MNVs for SNVs with incorrect amino acid change predictions,4,5 which probably hampers scientific research and clinical practice.
Recently, the widespread application of CRISPR-based gene-editing technologies has revolutionized genetics and disease research. CRISPR-based base editors, which are derived by fusing deoxynucleoside deaminase to the nickase Cas9 and are recruited by a guide RNA to their target DNA region, efficiently make C-to-T or A-to-G nucleotide changes, improving simulation of SNVs to enable exploration of the relationship between genotype and phenotype. To further expand the editing capabilities of base editors, dual-mutation base editors, which combine cytosine base editors (CBEs) and adenine base editors (ABEs), have been successfully developed.6, 7, 8, 9, 10 Theoretically, dual-mutation base editors make simultaneous A/C conversions, the most frequent adjacent dinucleotide MNVs,2 more conveniently than single-mutation base editors. However, existing dual-mutation base editors have narrow application ranges due to protospacer adjacent motif (PAM) restriction. Although prime editors (PEs) can theoretically simulate most variants, including MNVs,11 the efficiency of PEs is low, especially in vivo.12 Therefore, better dual-mutation base editors that can simulate MNVs need to be developed.
SpRY is a mutant of SpCas9 that is highly PAM-compatible.13 The near-PAMless SpCas9 shows powerful advantages in PAM recognition. However, although it exhibits robust activity on sites with NRN PAMs (where R is A or G), it exhibits low activity on those with NYN PAMs (where Y is C or T). Considering that one of the limitations of using existing dual-mutation base editors is that they require target recognition of the NGG motif in the genome, in this study, we used SpRY to construct a dual-mutation base editor with minimal PAM restriction. To this end, we fused hA3A(Y130F), which has efficient C-to-T editing, including in G/C rich regions,14 and TadA8e(V106W), which has the highest efficiency of A-to-G editing among editors developed to date,15 to the N terminus of SpRY to produce a dual-mutation base editor named cytosine and adenine base editor-SpRY (CABE-RY). With this tool, we effectively performed simultaneous A/C conversion and successfully simulated MNVs.
Results
Necessity of a dual-mutation base editor with minimal PAM restriction for simulating MNVs
An increasing number of MNVs have been discovered and deposited in the Genome Aggregation Database (gnomAD).16 To better understand the biology of MNVs, we analyzed the MNV mutation type in gnomAD. The results showed that there was more simultaneous A/C conversion than AA or CC conversion, particularly in two adjacent mutations (Figure 1A). When two nearby mutations are located in the same codon, they may have a different functional impact on the protein than the individual mutations (Figure S1A). Indeed, among a total of 31,575 MNVs, 52.5% of them resulted in different missense mutations from those caused by the individual mutations, while 5.8% of MNVs rescued nonsense mutations caused by the individual mutations, and 1.3% of MNVs produced nonsense mutations that were not caused by the individual mutations (Figure 1B). This difference may affect understanding of functional research or clinical diagnosis of genetic diseases. The dual-mutation base editor allows us to better understand the functional impacts of MNVs by simulating these MNVs.
Figure 1.

Necessity of a dual-mutation base editor with minimal PAM restriction for simulating MNVs
(A) Substitution pattern of MNVs from gnomAD. The x coordinate is the distance between two SNVs. (B) Differences in functional impacts on the protein between MNVs and individual SNPs. (C) Heatmap showing the amino acid changes that can be simulated by the dual-mutation base editor. (D) The PAM-flexible dual-mutation base editor has a broadened scope to target MNVs within the same codon. A broadened editing window (positions 3–10) was used to calculate the targeting scope.
Ideally, a dual-mutation base editor should be able to effectively simulate 70 amino acid substitutions (Figure 1C), including six that cannot be achieved by existing CBEs and ABEs (Figure S1B). However, the previously reported dual-mutation base editors can only target sites with NGG PAMs, which greatly limits their application. Therefore, we speculated that the near-PAMless engineered CRISPR-Cas9 variant SpRY could be used to construct a PAM-flexible dual-mutation base editor without strict PAM restriction. We analyzed the editing scopes between a conventional NGG PAM editor and the PAM-flexible editor. Considering that dual-mutation editors usually have a broader editing window than conventional editors,9 we calculated the editable MNVs with an editing window spanning positions 3–10 (versus the common window spanning positions 4–8), which presumably increased the number of targetable MNVs. As expected, the PAM-flexible dual-mutation base editor was found to target ∼2.7 times the number of MNVs and specific codons than the conventional NGG PAM editor (Figure 1D; Figure S1C), suggesting that the PAM-flexible dual-mutation base editor is a potential tool with an expansive editing scope for modeling MNVs.
Establishment of a PAM-flexible dual-mutation base editor, CABE-RY
To create a PAM-flexible dual-mutation base editor, we combined SpRY with cytosine deaminase and adenine deaminase from the previously described hA3A(Y130F), which has efficient C-to-T editing, including in G/C rich regions,14 and TadA8e(V106W), which has the highest efficiency of A-to-G editing among editors developed thus far.15 Four constructs were generated based on the locations of SpRY and the two deaminases (Figure 2A; Sequence S1).
Figure 2.

Establishment of a PAM-flexible dual-mutation base editor, CABE-RY, in HEK293T cells
(A) Schematic diagram for construction of four dual-mutation base editors, CABE-1, CABE-2, CABE-3, and CABE-4. (B and C) Comparison of the C-to-T (B) and A-to-G (C) editing efficiencies of the four CABE editors. The horizontal black lines represent the mean editing efficiencies at 48 endogenous sites for each CABE editor. Each dot represents the editing efficiency of each edited base (n = 3 independent replicates). The error bars represent the standard error of the mean (SEM) values. (D and E) Mean C-to-T (D) and A-to-G (E) editing efficiency plots for the four CABE editors with different PAMs. The data are presented for each edited base at 48 endogenous sites (n = 3 independent replicates). The error bars represent the SEM values. (F) Schematic overview of CABE-RY. hA3A(Y130F)-TadA8e(V106W) is linked to the N terminus of SpRY.
Then, we compared the on-target editing efficiency of the four constructs across 48 endogenous sites. Each single-guide RNA (sgRNA), together with four individual constructs, was cotransfected into HEK293T cells. The editing efficiency was analyzed by Sanger sequencing, and the results showed that all 4 editors induced simultaneous A/C conversions. The mean efficiency of CABE-1 was higher than that of CABE-2 (21.5% versus 17.71% for A-to-G; 21.8% versus 14.88% for C-to-T). The mean A-to-G editing efficiency of CABE-3 was far lower than that of CABE-1 (5.4% versus 21.5%), while the mean C-to-T editing efficiency of CABE-4 was far lower than that of CABE-1 (11.1% versus 21.8%) (Figures 2B and 2C). We further analyzed the C-to-T and A-to-G editing activities of CABE-1 at sites of different PAMs (Figures 2D and 2E). Similarly, the CABE-1 had the best performance. Therefore, we chose CABE-1, in which hA3A(Y130F)-TadA8e(V106W) was fused to the N terminus of SpRY, for further study and named it CABE-RY (Figure 2F).
Determination of the on-target DNA editing efficiency of CABE-RY
Next, amplicon deep sequencing was used for the target sites to thoroughly characterize CABE-RY performance. Related simultaneous A/C conversions of some sites are shown in Figure 3A. The C-to-T editing window (positions 4–14) of CABE-RY was narrower than that of hA3A(Y130F)-RY (positions 1–14) (Figure S2A). Similarly, the A-to-G editing window (positions 3–9) was slightly smaller than that of ABE8e(V106W)-RY (positions 3–11) (Figure 3B; Figure S2B). Analysis of the 48 targets containing both As and Cs in the editing window revealed that the C-to-T editing efficiency was 46.9%, while the A-to-G editing efficiency was 48.3%. Then, we examined the C-to-T and A-to-G editing activities of CABE-RY in different PAMs. For NYN PAMs, we observed that the mean activities of both C-to-T and A-to-G editing were lower than those for NRN PAMs (36.7% versus 58.0% for C-to-T; 38.8% versus 59.8% for A-to-G) (Figure 3C; Figures S3A–S3C). This is consistent with the characteristics of SpRY.13 In addition, CABE-RY had a low insertion or deletion (indel) frequency of 1.60% (Figure S3D).
Figure 3.

Determination of the on-target DNA editing efficiency of CABE-RY in HEK293T cells
(A) Relative C-to-G and A-to-T base editing efficiencies of CABE-RY with different PAMs at 16 human genomic target DNA sites (n = 3 independent replicates). The error bars represent the SEM values. (B) Sequencing analysis of the base editing window of CABE-RY across NNN PAMs in HEK293T cells. The error bars represent the SEM values. The data are presented for each edited base separately at 48 endogenous sites (n = 3 independent replicates). (C) Aggregate distribution of C-to-T (red) and A-to-G (blue) edits made across the editing window with CABE-RY. The error bars represent the SEM values. The data are presented for each edited base in the editing window at 48 endogenous sites.
We further compared the editing efficiency between CABE-RY and coexpressed ABE8e(V106W)-RY and hA3A(Y130F)-RY in HEK293T cells (Figures S4A–S4C) and found that CABE-RY showed a slightly higher editing efficiency than the two coexpressed base editors (54.7% versus 47.8%). Additionally, robust base editing of CABE-RY was also observed in murine N2a cells (29.4%) (Figure S4D), suggesting that CABE-RY works universally for different species.
Determination of the off-target RNA editing efficiency of CABE-RY
One of the concerns regarding base editors is RNA off-target mutagenesis.17 To detect transcriptome-wide RNA off-target effects, we performed RNA sequencing (RNA-seq) on HEK293T cells coexpressing CABE-RY, ABE8e(V106W)-RY, hA3A(Y130F)-RY, and GFP (negative control) with a sgRNA targeting NAT SITE3 (Table S1), which had high DNA on-target editing efficiency (Figure 4A). As expected, ABE8e(V106W)-RY introduced thousands of RNA edits in the transcriptome, while hA3A(Y130F)-RY had a slight effect (Figure 4B; Figure S5A). Interestingly, the number of RNA off-target edits of CABE-RY was one-third that of ABE8e(V106W)-RY (Figure 4B; Figure S5A). In addition, these editors did not cause significant differences in gene expression at the whole-transcriptome level (Figures S5B–S5D).
Figure 4.

Determination of the off-target RNA editing efficiency of CABE-RY
(A) Heatmaps showing the on-target editing frequencies of GFP (control), ABE8e(V106W), hA3A(Y130F), and CABE-RY in NAT SITE3. A-to-G edits are indicated in pink, and C-to-T edits are indicated in blue. (B) Total number of RNA off-target edits detected in RNA-seq experiments for GFP (control), ABE8e(V106W), hA3A(Y130F), or CABE-RY. The error bars represent the SEM values. (C and D) Jitter plots showing the efficiencies of A-to-I (C) or C-to-U (D) RNA off-target editing. The data shown are from two independent replicates. The number of edits is represented on the top.
Then, we further analyzed the specific types of RNA off-target edits and found that the main off-target edit type for CABE-RY and ABE8e(V106W)-RY was A-to-I RNA editing, while that of hA3A(Y130F)-RY was C-to-U RNA editing (Figures 4C and 4D). Interestingly, CABE-RY showed lower A-to-I RNA editing than the other editors (nearly 3,000) and undetectable C-to-U RNA editing (comparable to the editing of the GFP control) (Figures 4C and 4D). A possible explanation of the performance may be the embedding of the deaminase domains in nCas9 for minimization of off-target effects of base editors;18 in addition, the combination of hA3A(Y130F) and TadA8e(V106W) may create possible steric hindrance to reduce off-target edits.
Successful simulation of MNVs by CABE-RY in HEK293T cells
Given these findings, we tested ten sites in gnomAD and directly compared the editing efficiency of CABE-RY with that of ABE8e(V106W)-RY or hA3A(Y130F)-RY in HEK293T cells. Compared with those of the single base editors, the editing efficiencies for C-to-T and A-to-G editing of CABE-RY were slightly lower (25.8% versus 31.4% for C-to-T, 38.1% versus 43.9% for A-to-G) (Figure 5A). However, CABE-RY created more mutation types and amino acid types than the single base editors (Figures S6 and S7) and showed high simultaneous A/C conversion efficiency (31.8%) in terms of simulating MNVs, which is unfeasible for a single base editor. It is worth noting that CABE-RY induced bystander edits like those induced by ABE and CBE. To better understand how bystander edits confound MNV simulation using CABE-RY, we first analyzed the potential bystanders flanking two adjacent MNVs with gnomAD (Figure S8) and found that 18.9% of MNVs may not be affected by bystanders, while 14.4% of MNVs may be affected by bystanders. Thus, CABE-RY is still a potentially useful tool for modeling MNVs.
Figure 5.

Evaluation of MNV simulation by CABE-RY in HEK293T cells
(A) Heatmaps showing the A-to-G (green) and C-to-T (orange) editing frequencies of ABE8e(V106W), hA3A(Y130F), and CABE-RY at ten sites (n = 3 independent replicates). The number and the letter at the bottom represent the related position and the respective base in the protospacer sequence. (B) Proportion of mutation types edited by CABE-RY (left) and PE3 (right) in different sites. Data are analyzed by deep sequencing and summarized from three independent experiments. Perfect MNV (orange) represents the mutation type that just alters the target MNV site without bystanders. Others (green) represent the mutation types that just alters the target MNV site within bystanders. WT (gray) represents unedited type. (C) Comparison of simulated perfect MNV efficiency between CABE-RY and PE3 on 10 endogenous sites. The error bars represent the SEM values.
PEs can theoretically simulate most variants, including MNVs, with no bystander edits. Therefore, we compared the simulation efficiency between PE3 and CABE-RY. A pegRNA with a 13 nt prime binding site (PBS) length and a 12 nt reverse transcriptase (RT) template length was designed as suggested.19 As expected, both CABE-RY and PE3 successfully induced MNVs, but CABE-RY exhibited significantly higher simulation efficiency than PE3 (31.8% versus 2.8%; Figure 5B). Notably, perfect simulation of MNVs without bystander edits was successfully achieved in 8.7% of cases on average (Figure 5C). These results demonstrate that CABE-RY is a good tool for simulating MNVs.
To further explore the accuracy of MNV simulation by CABE-RY, we analyzed the proportion of perfectly simulated MNVs. The results showed that the influence of bystander edits was site dependent. Some sites retained a relatively high percentage of perfect MNV simulations (35.47% for the WDR90 site, 13.47% for the SYNM), while some sites had significantly reduced proportions of perfect MNV simulations (Figure S6). Taken together, the results indicate that CABE-RY can efficiently simulate MNVs and is a versatile tool for dissection of the functions of MNVs.
Discussion
The data in gnomAD reveal that simultaneous A/C conversion has a higher mutation frequency than AA or CC conversion, particularly in two adjacent mutations for MNVs, and this mutation type can be simulated by reported dual-mutation base editors.6, 7, 8, 9, 10 However, the published dual-mutation base editors are restricted to sites with NGG PAMs, which greatly limits their application. Here, we constructed a dual-mutation base editor, CABE-RY, by combining hA3A(Y130F), TadA8e(V106W), and SpRY. This editor was found to effectively induce simultaneous A/C conversion over the whole genome with a sgRNA after successful verification. Characterized by the presence of SpRY, CABE-RY edits many more sites than existing dual-mutation editors with minimal PAM restriction.
We further characterized the editing of CABE-RY and revealed that CABE-RY efficiently edited DNA (46.9% efficiency for C-to-T edits and 48.3% efficiency for A-to-G edits). Interestingly, CABE-RY induced less RNA off-target mutagenesis, including less A-to-I RNA editing and undetectable C-to-U RNA editing, than the other editors tested. To illustrate that CABE-RY is an appropriate tool to simulate MNVs, we compared the simulation efficiency between PE3 and CABE-RY. CABE-RY exhibited higher simulation efficiency than PE3 (31.8% versus 2.8%), even when we focused only on perfect MNV (8.7% versus 2.0%). Here, we have confirmed that CABE-RY can efficiently generate simultaneous A/C conversions. Nevertheless, several concerns remain. For example, self-targeting of sgRNA is unavoidable, and it may lead to wrong sgRNA generation, resulting in reduction of editing efficiency and even unpredictable targeting. Meanwhile, CABE-RY exhibits robust activities with NRN PAMs but lower activities with NYN PAMs, which limits the use of CABE-RY over the genome. Considering other SpCas9 variants may have stronger affinity with NYN PAMs, such as FnCas9RHA on YG PAMs,20 M44 on TTTN PAMs,21 AsCas12a-K949A on TTTV PAMs, and 22 Nme2Cas9 on NNNNCC PAMs,23 we can choose these SpCas9 variants instead of SpRY, which may improve editing efficiency at specific sites. Nevertheless, to create a fully PAMless Cas9 in the future is desired. In fact, ortholog mining and protein engineering have produced many PAM variants. The combination of these strategies may further relieve PAM restrictions and create a truly PAMless Cas9.24
In addition, we also compared CABE-RY editing frequencies with those of single base editors. Our results demonstrated that compared with a single base editor, CABE-RY has several advantages. First, CABE-RY can efficiently generate simultaneous A/C conversions to simulate MNVs. Second, CABE-RY can generate more mutation types than single base editors; thus, it can effectively simulate 70 amino acid substitutions, including six that cannot be achieved by existing single base editors, which is helpful for understanding disease-related mutations. For example, the unique p.Cys91His mutation has been discovered in Niemann-Pick disease and p.Gln326Trp in intervertebral disc disease.25,26 In addition, it’s reported that simultaneous A/C conversion induced higher hemoglobin subunit gamma (HBG) reactivation.9 In conclusion, CABE-RY is a versatile tool for the field of gene editing and is especially useful for simulating MNVs.
Materials and methods
Cell culture
HEK293T cells were purchased from the American Type Culture Collection (ATCC). The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. We maintained the cell lines at 37°C in a 5% CO2 cell culture incubator.
Plasmid construction
To construct base editor expression plasmids, human codon-optimized DNA sequences of hA3A(Y130F), TadA8e(V106W), and UGI were synthesized by GenScript and cloned into the pCMV-SpRY(D10A) backbone containing C-terminal-fused EGFP and blasticidin (BSD). To construct sgRNA expression vectors, synthesized oligos were annealed and ligated into a BsaI-digested sgRNA expression vector (pGL3-U6-sgRNA-mCherry plasmid). To construct pegRNA expression vectors, synthesized oligos were annealed and ligated into pegRNA expression vector (pGL3-U6-pegRNA-mCherry plasmid). Sequences of sgRNA and pegRNA constructs used in this work are listed in Tables S1 and S2.
Analysis of on-target editing
To evaluate editing efficiency, HEK293T cells were seeded into 24-well plates 1 day before the analysis. The base editor expression plasmids (1,000 ng) and corresponding sgRNA plasmids (500 ng) were cotransfected using Lipofectamine 2000 (Life Technologies) as the manufacturer’s protocol recommended. Seventy-two hours after transfection, ∼10,000 cells with dual fluorescence signals (GFP and mCherry) were collected by fluorescence-activated cell sorting (FACS) to improve efficiency (Figure S9), and these cells were harvested for genomic DNA extraction using QuickExtract DNA Extraction Solution (Lucigen) according to the manufacturer’s protocols. The genomic regions encompassing the target sites were amplified from the genomic DNA with Phanta Max Super-Fidelity DNA polymerase (Vazyme, P505-03). The primers used are listed in Table S3. The PCR products were analyzed by Sanger sequencing or high-throughput sequencing as indicated. For Sanger sequencing, the chromatograms were quantified using EditR. For high-throughput sequencing, the PCR products were sequenced on an Illumina HiSeq X Ten (2 × 150 paired-end) at the Novogene Bioinformatics Institute (Beijing, China). The sequencing data were analyzed using CRISPResso2.
Analysis of RNA off-target editing
HEK293T cells were seeded into 6-cm dishes and transfected with 4 μg of CABE-RY, hA3A(Y130F)-RY, ABE8e(V106W)-RY, and GFP (control) plasmids and 2 μg of sgRNA expression vector using Lipofectamine 2000 at ∼70% confluency. Two days after transfection, the top 15% of the GFP signal-positive cells were harvested by FACS. RNA was immediately extracted using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. The RNA samples were subjected to deep sequencing (∼20 million reads per sample) on an Illumina HiSeq X Ten platform (2 × 150 paired-end) at the Novogene Bioinformatics Institute (Beijing, China). The clean data were first mapped to the human reference genome (version: hg38) with annotations from GENCODE version 30 by STAR software (version 2.5.1). After removing duplicates, GATK HaplotypeCaller (version 4.1.2) was used to identify and filter the edits. All edits were verified, and the efficiency was calculated using the bam-readcount program with the parameters -q 20 -b 30. Importantly, for the reference allele in the wild-type sample, the depth of a given edit had to be least 10×, and all the edits had to be present in at least 99% of reads.
Statistics and reproducibility
All statistical analyses mentioned above were performed on data from at least 3 biologically independent experiments (n = 3). The data shown in this research were statistically analyzed by unpaired two-tailed Student’s t test using GraphPad Software (GraphPad Prism 8). A p value smaller than 0.05 was considered to indicate statistical significance.
Data availability
The high-throughput sequencing data have deposited. The accession code is NCBI Sequence Read Archive database: PRJNA688630. All other data are available upon reasonable request.
Acknowledgments
We thank members of Huang lab and Xu lab for helpful discussions. We also thank the Molecular and Cell Biology Core Facility (MCBCF) at the School of Life Science and Technology, ShanghaiTech University, for providing technical support. This work is supported by the National Science Foundation of China (81830004) and the Leading Talents of Guangdong Province Program (608285568031).
Author contributions
X.H. and X.X. conceived, designed, and supervised the project. W.T. and Q.L. performed most experiments with the help of other authors. S.H. analyzed and interpreted the data. W.T., Q.L., and X.H. wrote the paper with inputs from all authors.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.07.016.
Contributor Information
Xiangmin Xu, Email: xixm@smu.edu.cn.
Xingxu Huang, Email: huangxx@shanghaitech.edu.cn.
Supplemental information
References
- 1.Hay E.H.A., Utsunomiya Y.T., Xu L., Zhou Y., Neves H.H.R., Carvalheiro R., Bickhart D.M., Ma L., Garcia J.F., Liu G.E. Genomic predictions combining SNP markers and copy number variations in Nellore cattle. BMC Genomics. 2018;19:441. doi: 10.1186/s12864-018-4787-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaplanis J., Akawi N., Gallone G., McRae J.F., Prigmore E., Wright C.F., Fitzpatrick D.R., Firth H.V., Barrett J.C., Hurles M.E., Deciphering Developmental Disorders study Exome-wide assessment of the functional impact and pathogenicity of multinucleotide mutations. Genome Res. 2019;29:1047–1056. doi: 10.1101/gr.239756.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., et al. Exome Aggregation Consortium Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li C., Zhang R., Meng X., Chen S., Zong Y., Lu C., Qiu J.L., Chen Y.H., Li J., Gao C. Targeted, random mutagenesis of plant genes with dual cytosine and adenine base editors. Nat. Biotechnol. 2020;38:875–882. doi: 10.1038/s41587-019-0393-7. [DOI] [PubMed] [Google Scholar]
- 7.Grünewald J., Zhou R., Lareau C.A., Garcia S.P., Iyer S., Miller B.R., Langner L.M., Hsu J.Y., Aryee M.J., Joung J.K. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat. Biotechnol. 2020;38:861–864. doi: 10.1038/s41587-020-0535-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie J., Huang X., Wang X., Gou S., Liang Y., Chen F., Li N., Ouyang Z., Zhang Q., Ge W., et al. ACBE, a new base editor for simultaneous C-to-T and A-to-G substitutions in mammalian systems. BMC Biol. 2020;18:131. doi: 10.1186/s12915-020-00866-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang X., Zhu B., Chen L., Xie L., Yu W., Wang Y., Li L., Yin S., Yang L., Hu H., et al. Dual base editor catalyzes both cytosine and adenine base conversions in human cells. Nat. Biotechnol. 2020;38:856–860. doi: 10.1038/s41587-020-0527-y. [DOI] [PubMed] [Google Scholar]
- 10.Sakata R.C., Ishiguro S., Mori H., Tanaka M., Tatsuno K., Ueda H., Yamamoto S., Seki M., Masuyama N., Nishida K., et al. Base editors for simultaneous introduction of C-to-T and A-to-G mutations. Nat. Biotechnol. 2020;38:865–869. doi: 10.1038/s41587-020-0509-0. [DOI] [PubMed] [Google Scholar]
- 11.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y., Li X., He S., Huang S., Li C., Chen Y., Liu Z., Huang X., Wang X. Efficient generation of mouse models with the prime editing system. Cell Discov. 2020;6:27. doi: 10.1038/s41421-020-0165-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walton R.T., Christie K.A., Whittaker M.N., Kleinstiver B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. 2020;368:290–296. doi: 10.1126/science.aba8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X., Li J., Wang Y., Yang B., Wei J., Wu J., Wang R., Huang X., Chen J., Yang L. Efficient base editing in methylated regions with a human APOBEC3A-Cas9 fusion. Nat. Biotechnol. 2018;36:946–949. doi: 10.1038/nbt.4198. [DOI] [PubMed] [Google Scholar]
- 15.Richter M.F., Zhao K.T., Eton E., Lapinaite A., Newby G.A., Thuronyi B.W., Wilson C., Koblan L.W., Zeng J., Bauer D.E., et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020;38:883–891. doi: 10.1038/s41587-020-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. Genome Aggregation Database Consortium The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou C., Sun Y., Yan R., Liu Y., Zuo E., Gu C., Han L., Wei Y., Hu X., Zeng R., et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. 2019;571:275–278. doi: 10.1038/s41586-019-1314-0. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y., Zhou C., Huang S., Dang L., Wei Y., He J., Zhou Y., Mao S., Tao W., Zhang Y., et al. A Cas-embedding strategy for minimizing off-target effects of DNA base editors. Nat. Commun. 2020;11:6073. doi: 10.1038/s41467-020-19690-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim H.K., Yu G., Park J., Min S., Lee S., Yoon S., Kim H.H. Predicting the efficiency of prime editing guide RNAs in human cells. Nat. Biotechnol. 2021;39:198–206. doi: 10.1038/s41587-020-0677-y. [DOI] [PubMed] [Google Scholar]
- 20.Hirano H., Gootenberg J.S., Horii T., Abudayyeh O.O., Kimura M., Hsu P.D., Nakane T., Ishitani R., Hatada I., Zhang F., et al. Structure and Engineering of Francisella novicida Cas9. Cell. 2016;164:950–961. doi: 10.1016/j.cell.2016.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu R.M., Liang L.L., Freed E., Chang H., Oh E., Liu Z.Y., Garst A., Eckert C.A., Gill R.T. Synthetic chimeric nucleases function for efficient genome editing. Nat. Commun. 2019;10:5524. doi: 10.1038/s41467-019-13500-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao L., Cox D.B.T., Yan W.X., Manteiga J.C., Schneider M.W., Yamano T., Nishimasu H., Nureki O., Crosetto N., Zhang F. Engineered Cpf1 variants with altered PAM specificities. Nat. Biotechnol. 2017;35:789–792. doi: 10.1038/nbt.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edraki A., Mir A., Ibraheim R., Gainetdinov I., Yoon Y., Song C.Q., Cao Y., Gallant J., Xue W., Rivera-Pérez J.A., Sontheimer E.J. A Compact, High-Accuracy Cas9 with a Dinucleotide PAM for In Vivo Genome Editing. Mol. Cell. 2019;73:714–726.e4. doi: 10.1016/j.molcel.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Collias D., Beisel C.L. CRISPR technologies and the search for the PAM-free nuclease. Nat. Commun. 2021;12:555. doi: 10.1038/s41467-020-20633-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hollak C.E., de Sonnaville E.S., Cassiman D., Linthorst G.E., Groener J.E., Morava E., Wevers R.A., Mannens M., Aerts J.M., Meersseman W., et al. Acid sphingomyelinase (Asm) deficiency patients in The Netherlands and Belgium: disease spectrum and natural course in attenuated patients. Mol. Genet. Metab. 2012;107:526–533. doi: 10.1016/j.ymgme.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 26.Annunen S., Paassilta P., Lohiniva J., Perälä M., Pihlajamaa T., Karppinen J., Tervonen O., Kröger H., Lähde S., Vanharanta H., et al. An allele of COL9A2 associated with intervertebral disc disease. Science. 1999;285:409–412. doi: 10.1126/science.285.5426.409. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The high-throughput sequencing data have deposited. The accession code is NCBI Sequence Read Archive database: PRJNA688630. All other data are available upon reasonable request.